

Abstract
Although allogeneic bone marrow transplantation has been shown to prevent autoimmune diabetes in heavily irradiated NOD mice, similar procedure is not suitable for the treatment of patients with type 1 diabetes because of associated severe side effects. Therefore, we evaluated whether mouse newborn blood, equivalent to human umbilical cord blood, could be used for diabetes prevention without recipient preconditioning. To test this hypothesis, unconditioned, pre-diabetic female NOD mice were given a single injection of whole newborn blood derived from the allogeneic, diabetes-resistant mouse strain, C57BL/6. Transfusion of allogeneic newborn but not adult blood prevented diabetes incidence in a majority of treated mice for a prolonged period of time. This was accompanied by the release of insulin in response to a challenge with glucose. Invasive cellular infiltration of islets was also substantially reduced in these mice. Although newborn blood transfusion induced low level of hematopoietic microchimerism, it did not strictly correlate with amelioration of diabetes. Induction of genes implicated in diabetes such as, Il18, Tnfa, and Inos but not Il4, Il17 or Ifng was repressed in splenocytes derived from protected mice. Notably, expression of the transcription factor Tbet/Tbx21 but not Gata3 or Rorgt was upregulated in protected mice. These data indicate that allogeneic newborn blood transfusion can prevent diabetes in NOD mice associated with modulation of selected cytokine genes implicated in diabetes manifestation. The data presented herein provide the proof of principle for the utility of allogeneic umbilical cord blood transfusion to treat patients with autoimmune diabetes.
Introduction
Type 1 diabetes (T1D)3, an autoimmune disease, occurs in genetically susceptible individuals (1). NOD mice develop T1D spontaneously and serve as an invaluable model for studying the mechanisms involved in diabetes pathogenesis and possible treatment strategies (2). Since T lymphocytes are the primary effectors of T1D, they have been targeted in attempts to thwart autoimmunity. Transplantation of allogeneic bone marrow (3-6) or Sca-1+ hematopoietic stem cells (HSC) (7-8) derived from diabetes-resistant mice into heavily irradiated NOD mice has been shown to replace the host immune system completely and prevent autoimmunity. However, toxic pre-conditioning regimens including irradiation required for full allogeneic hematopoietic chimerization lead to graft-vs-host disease (GVHD) when reconstituted across MHC barriers and result in generalized immunosuppression (8).
Human umbilical cord blood at birth represents a particularly desirable source of transplantable cells over bone marrow since it contains large numbers of HSC, progenitor cells, and naïve T cells with reduced alloreactivity, and a limited capacity to cause GVHD (9). Although infusion of autologous T cells from cryopreserved cord blood units has been used in diabetic patients as a source rich in T regulatory cells, its efficacy to ameliorate autoimmune diabetes has not yet been elucidated (10). The diabetogenic potential of bone marrow cells was indicated by the ability of bone marrow to transfer diabetes from diabetic to non-diabetic HLA identical siblings (11). This is in line with studies in the NOD mouse model showing that the bone marrow retains the stemness to propagate diabetes (4, 7, 12). Although autologous umbilical cord blood had palliative effects (10), it is not an ideal choice for HSC to rest the immune system in T1D patients since like bone marrow, umbilical cord blood from T1D patients may have progenitors of autoreactive T lymphocytes that can perpetuate diabetes. On the other hand, HSC derived from umbilical cord blood of allogeneic, non-diabetic individuals may provide better protection against diabetes since they lack diabetogenic T lymphocytes. This concept needs to be evaluated in a preclinical model prior to the application of this promising strategy to treat T1D patients.
The NOD mouse model is not only useful for understanding the mechanisms of autoimmune diabetes but also for answering clinical questions that are not possible to address in humans (2). The mouse newborn blood (NBB) provides a useful model for human umbilical cord blood at birth for a number of reasons. Whereas the frequency of stem cell antigen-1+ (Sca-1+) HSC is similar in both NBB and bone marrow, only the NBB can repopulate the immune system of irradiated hosts without inducing GVHD across allogeneic barriers (13-15). In contrast to adult blood, NBB contains mostly immature T cells (CD4+ + CD8+) and a few single-positive, TCR+ CD4+ and TCR+ CD8+ cells (13-14). Therefore, mouse NBB appears to be an ideal source of somatic HSC that can be used for diabetes treatment. To test this possibility, we developed a clinically relevant model in which un-conditioned diabetes prone female NOD mice were transfused at prediabetic stage with NBB derived from diabetes resistant C57BL/6 mice. The data indicate that allogeneic NBB transfusion could prevent diabetes by halting the progression of inflammatory responses in islets and reducing the inflammatory cytokine expression. Within the limitations of the extrapolation of data obtained from animal models to clinical settings, these data suggest that allogeneic NBB transfusion may provide a potentially useful clinical strategy to treat T1D patients.
Materials and Methods
Mice
Female NOD/Ltj (H-2g7) and timed pregnant C57BL/6J (H-2b) mice were purchased from The Jackson Laboratories (Bar Harbor, ME) and housed in filter-top cages in temperature controlled and light-cycled rooms. The animal protocol was approved by the University of Illinois at Chicago Biological Resources Center and experiments were conducted according to the principles outlined by the Animal Welfare Act and the National Institutes of Health guidelines for the care and use of animals in biomedical research.
Blood collection
Neonatal blood was collected in the anti-coagulant, Alsever’s solution by a cardiac puncture from newborn mice up to 5 days of age. The volume of blood obtained from single newborn mouse varied between 10 to 50 μL. Blood from 8 to 13 newborn mice from the same litter was pooled per experiment. Adult blood was collected in Alsever’s solution from the tail vein.
Flow cytometry
After RBC lysis, nucleated cells were stained with monoclonal antibodies conjugated with FITC (CD3, CD8, CD25, DX5 and Kb), PE (CD4, CD11c, CD44, CD45R, CD45RA, CD62L, CD69 and NK1.1) or allophycocyanin (Sca-1, CD3, and mPDCA-1). All antibodies were obtained from eBioscience (San Diego, CA) except that CD4, CD34, CD44, CD45R and Kb antibodies were purchased from BD PharMingen (San Diego, CA), and anti-mPDCA-1 antibody was obtained from Miltenyi Biotec (Auburn, CA). Isotype matched normal mouse Ig conjugated with fluorochromes was used to determine the background staining of cells. Data were acquired on a BD LSR (Becton Dickinson) or a CyAn ADP flow cytometer (DakoCytomation). Off-line color compensation and data analysis were done using the FlowJo6.4.1 software (Tree Star, Mountain View, CA).
Treatment of mice and diabetes monitoring
A small aliquot of whole NBB or adult peripheral blood was treated with RBC lysis solution (eBioscience) and the total numbers of nucleated cells were counted in a hemocytomter. Known numbers of nucleated cells were stained to assess the frequency of Sca-1+ cells and this information was used to calculate the total numbers of Sca-1+ cells present in the NBB preparation. For injection into NOD recipients, whole blood was centrifuged and the numbers of nucleated cells were adjusted by resuspending in sterile HBSS (Invitrogen). Un-conditioned 12 wk old female NOD/Ltj mice were injected with newborn or adult C57BL/6 derived whole blood through a lateral tail vein.
Diabetes was monitored by determining glucose levels at weekly intervals in the tail vein blood, using a glucometer (Accu-Chek Advantage, Roche Diagnostics, Indianapolis, IN). Mice were considered diabetic when the blood glucose levels reached >250 mg/dL for two consecutive readings within the same wk of the initial diagnosis. For the glucose tolerance test, mice were fasted for 4 h and blood was drawn from the tail vein at 0, 15, 45 and 60 min after an i.p. injection of dextrose (2 g/kg body weight). Insulin levels in sera were measured using an Ultra Sensitive Mouse Insulin ELISA kit obtained from Crystal Chem (Downers Grove, IL, sensitivity range: 0.1-12.8 ng/ml).
Determination of chimerism
To determine chimerism by flow cytometry, tail vein blood was collected in Alsever’s solution at various time intervals after NBB transfusion. After RBC lysis, nucleated cells were stained with a FITC-conjugated antibody against Kb, the MHC class I antigen expressed by donor C57BL/6 cells. The cells were then analyzed by flow cytometry. At the end of the experiment, mice were killed and an aliquot of splenocytes pooled from 3-5 mice from the same group was stained with FITC conjugated anti-Kb antibody and assessed by flow cytometry.
To determine chimerism by RT-PCR, splenocytes were lysed in TRIzol (Invitrogen, CA) and total RNA was isolated. The first-strand cDNA was synthesized using the T-primed First-Strand Kit (Amersham Biosciences, Piscataway, NJ). PCR was performed with 1.0 μl of cDNA in a final volume of 25 μl containing 0.4 μM concentrations each of Kb (16) and β2-microglobulin (17) primer sets and AmpliTaq Gold PCR Master Mix (Applied Biosystems, Foster City, CA). Amplicons were electrophoresed on a 1.2% agarose gel in the presence of ethidium bromide, visualized under UV light, and imaged using a CCD camera (AlphaInnotech Gel Documentation System).
Assessment of gene expression by real-time RT-PCR
Spleen cells (5 × 106/ml) were stimulated with 100 ng of PMA and 1 μg of ionomycin in complete RPMI medium supplemented with antibiotics and 10% FBS (18). After overnight culture, cells were lysed in TRIzol and total RNA was extracted, treated with DNase using TURBO DNA-free kit (Applied Biosystems) and reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription kit (Applied Biosytems). Real-time quantitative RT-PCR was performed on an ABI Prism 7500 Real-Time PCR system (Applied Biosystems) using 1 μl of cDNA equivalent to 100 ng of RNA and 2X SYBR Premix Ex Taq (Perfect Real Time) reagent (Takara-Clontech). Primer set for mouse Gapdh was designed using the PRIMER EXPRESS version 2.0 software (Applied Biosciences): Forward -5’-TGG TGA AGG TCG GTG TGA AC-3’ and Reverse -5’-CCA TGT AGT TGA GGT CAA TGA AGG-3’. Primer sets for Il4, Il17, Il18, Ifng, Tnfa, Inos, Tbx21, Gata3, and Rorgt were designed according to previous publications (19-22). All primers were synthesized at Integrated DNA Technologies (Coralville, IA). Relative quantitation for all assays used Gapdh as the normalizer. Quantitation of gene expression in comparison to Gapdh was calculated using the comparative threshold cycle method (23). Sizes of amplicons and the absence of primer-dimer were verified by electrophoresis of PCR products on a 4% low melt agarose gel.
Determination of lymphokine secretion
Culture supernatant was obtained from splenocytes after overnight activation with PMA and ionomycin, as described above. The amounts of lymphokines were determined using ELISA kits obtained from eBioscience.
Histology and confocal microscopy
Pancreata were fixed in 10% buffered formalin and 6 μM sections were stained with H&E and observed under light microscope (Olympus BX51). At least 3 sections were analyzed per pancreas by 3 different investigators. The numbers of islets per section were counted and a score was assigned as follows: 1, No to mild peri-insulitis, 2, moderate infiltration (<50%) and 3, severe (>50%) and destructive infiltration.
Hydrated sections were blocked with 10% FBS, incubated with 1:100 concentration of guinea pig anti-insulin antibody (Zymed Laboratories, CA) followed by incubation with 1:1000 diluted TRITC labeled rabbit antisera against guinea pig Ig (Sigma-Aldrich, MO). Sections were counterstained with Hoechst and mounted in Vectashield mounting medium (Vector Laboratories, CA). Confocal images were obtained using a Zeiss LSM510 laser-scanning microscope and processed using the Zeiss LSM Image browser (4.0 version).
Statistics
Statistical analysis was performed by using an unpaired two-tailed Student’s t test (GraphPad Prism, San Diego, CA).
Results
Phenotypic characterization of NBB from C57B6 mice
Previous studies indicated that the frequency of Sca-1+ cells in ficoll-purified mononuclear cells obtained from adult bone marrow and NBB of B10.D2 mice ranged from 12 to 17%, while only <1% of the adult PBMC expressed this determinant (13-14). In contrast to bone marrow and adult blood, the NBB contained large numbers (40%) of immature, double-positive (CD4+CD8+) cells with only 3% of TCR-bearing mature cells (13-14). In order to confirm and extend these findings, blood was collected from newborn mice (n=50) of the diabetes-resistant C57BL/6 strain up to 5 days of age in the anti-coagulant, Alsever’s solution. After RBC lysis, nucleated cells were stained with various antibodies and analyzed by flow cytometry. A typical cytogram shown in Figure 1 indicated that three populations, designated as B, C and D, could be distinguished in NBB, based on forward angle and side scatter properties. This is in contrast to the commonly observed single population of cells displaying high forward light scatter in RBC lysed adult peripheral blood (not shown). Although the proportion of cells in Gate D varies between experiments (7 to 15% of total cells-Gate A), only the cells in Gate D expressed all of the determinants analyzed. The frequency of cells expressing the HSC marker, Sca-1 represented ~15% of cells in Gate D, which is comparable to those reported in ficoll purified mononuclear cells from neonatal B10.D2 mice (13-14). In contrast, adult PBMC from C57BL/6 mice contained <1% Sca-1+ cells (data not shown), consistent with previous observations in B10.D2 mice (13-14). As reported in B10.D2 mice, we also found that the NBB of C57BL/6 mice contained large numbers (~58%) of immature (CD4+CD8+) T cells with low frequency of CD3+ mature T cells (15%). Notably, we found that a majority (58%) of cells in Gate D were positive for CD44, a determinant ubiquitously expressed by a number of cell types (24) including the mesenchymal stem cells (25). In addition, a significant fraction (52%) of cells displaying low forward and high side scatter properties (Gate B) also expressed CD44 (Table I, line 4). Further analysis revealed that a significant fraction of (40%) cells in Gate D displayed CD62L, expressed by naïve T lymphocytes (Figure 1). Only a minor fraction (<10%) of cells expressed the MHC class I determinant, Kb as well as CD69, CD45R and CD45RA. Interestingly, the naturally occurring T regulatory cells of the phenotype CD4+CD25+ and NK1.1+ cells represented <10% of cells in Gate D. Dendritic cells expressing CD11c, Dex-5 and mPCDA determinants also constituted a small fraction of NBB. Finally, only a minor fraction of cells expressed CD34, a marker expressed by human but not mouse HSC (26).
Figure 1. Phenotype of NBB derived from C57BL/6 mice.
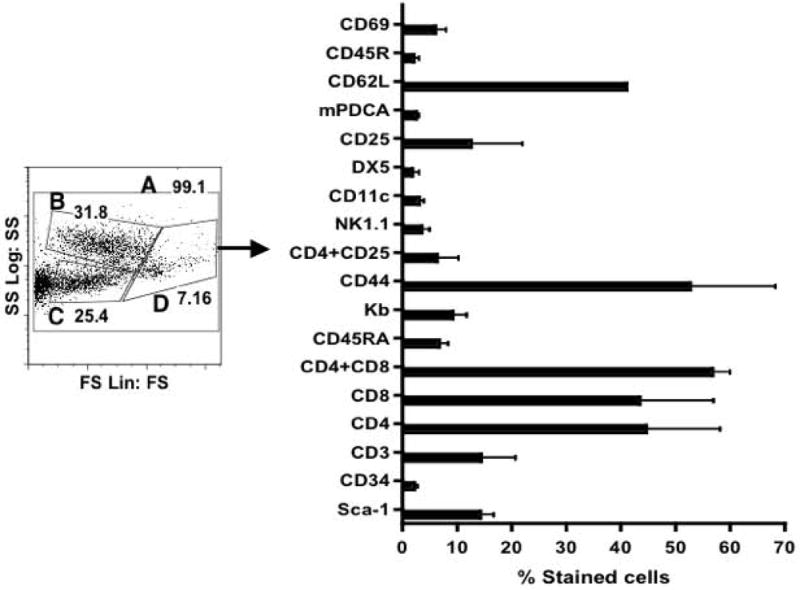
Peripheral blood was obtained from newborn mice up to 5 days of age in 5 different experiments (n=50). After RBC lysis, cells were stained with various antibodies and analyzed by flow cytometry. Cells were electronically gated based on forward angle light and side scatter properties. The expression of CD44 was detected in cells found in Gate B, C and D in various proportions (see Table I). Only cells in Gate D expressed all of the determinants examined.
Table I.
Phenotype of newborn blood from C57BL/6 mice*
| Phenotype | Gate A | Gate B | Gate C | Gate D | |
|---|---|---|---|---|---|
| 1 | Total Sca-1+ | 2.4 | 0.6 | 0.02 | 17.8 |
| 2 | Total Kb+ | 4.6 | 0.6 | 0.2 | 14.9 |
| 3 | Kb + Sca-1+ | 1.0 | 0.4 | 0.04 | 4.8 |
| 4 | Total CD44+ | 17.4 | 55.3 | 18.3 | 72.6 |
| 5 | CD44+ + Sca-1+ | 2.7 | 1.3 | 0.1 | 6.5 |
| 6 | Total CD62L+ | 7.3 | 1.43 | 0.3 | 60.3 |
| 7 | CD62L+ + Sca-1+ | 2.2 | 0.4 | 0.05 | 4.8 |
| 8 | Total CD3+ | 5.2 | 0.8 | 0.07 | 11.0 |
| 9 | CD3+ + Sca-1+ | 1.0 | 0.7 | 0.05 | 3.0 |
| 10 | Total CD4+ | 6.7 | 0.5 | 0.05 | 77.0 |
| 11 | CD4+ + Sca-1+ | 1.2 | 0.4 | 0.01 | 6.2 |
| 12 | Total CD8+ | 6.6 | 0.4 | 0.07 | 73.0 |
| 13 | CD8+ + Sca-1+ | 1.0 | 0.3 | 0.01 | 5.4 |
| 14 | Total CD25+ | 6.2 | 2.1 | 0.5 | 43.2 |
| 15 | CD25+ + Sca-1+ | 1.9 | 0.6 | 0.05 | 3.2 |
| 16 | Total DX5+ | 3.6 | 0.5 | 0.02 | 23.8 |
| 17 | DX5+ + Sca-1+ | 1.0 | 0.5 | 0.03 | 3.5 |
| 18 | Total CD69+ | 5.5 | 2.3 | 0.7 | 17.9 |
| 19 | CD69+ + Sca-1+ | 2.0 | 0.4 | 0.04 | 2.2 |
| 20 | Total CD45R+ | 6.0 | 1.2 | 0.3 | 10.4 |
| 21 | CD45R+ + Sca-1+ | 2.3 | 0.5 | 0.02 | 2.3 |
| 22 | Total CD45RA+ | 4.5 | 1.9 | 0.5 | 10.4 |
| 23 | CD45RA+ + Sca-1+ | 0.7 | 0.5 | 0.02 | 2.0 |
| 24 | Total NK1.1+ | 5.6 | 1.9 | 0.5 | 7.0 |
| 25 | NK1.1+ + Sca-1+ | 2.0 | 0.4 | 0.02 | 1.3 |
| 26 | Total CD11c+ | 5.3 | 2.3 | 0.6 | 7.6 |
| 27 | CD11c+ + Sca-1+ | 1.9 | 0.4 | 0.03 | 1.6 |
Phenotypic characterization of NBB derived from C57BL/6 mice by flow cytometry. Cells were stained either with single antibody or double stained with anti-Sca-1 and indicated antibodies. Data indicate total and double-positive cells among variously gated populations shown in Figure 1. Data are from a representative of 5 different experiments (n=50 newborn pups).
Further analysis showed that 3 to 5% of Sca-1+ cells co-expressed MHC class I, CD44, CD62L, CD3, CD4, CD8, CD25, and DX5 (Table I, lines: 3, 5, 7, 9, 11, 13, 15, and 17), consistent with the expression of Sca-1 in a variety of cell types (26). However, a modest proportion of cells expressing CD69, CD45R, CD45RA, NK1.1, and CD11c co-expressed the Sca-1 determinant (Table I, lines: 19, 21, 23, 25 and 27). Taken together, these data indicate that like B10.D2 mice (13-14), the NBB of diabetes resistant C57BL/6 mice contained immature T cells (CD4+ + CD8+, CD62L+) and more Sca-1+ cells than adult blood among mononuclear cells. Interestingly, CD44 appears to be ubiquitously expressed in the NBB without a large representation of suppressor cells such as, T regulatory cells (CD4+CD25+) and NK1.1 cells.
Transfusion of allogeneic NBB prevents diabetes in NOD mice
Since NBB contains Sca-1+ HSC and produces minimal GVHD in irradiated allogeneic adult mice (13-14), we reasoned that the NBB derived from diabetes-resistant mice could be used to substitute the mutated HSC, implicated in autoimmune diseases (27). To test this, 12 wk-old female prediabetic NOD mice (H-2g7, Mls-1c) were given a single i.v. injection of whole newborn blood derived from diabetes-resistant B6 mice (H-2b, Mls-1b). In 5 different experiments, each mouse was given whole NBB containing 25 to 50 × 106 nucleated cells. The numbers of Sca-1+ cells present in 25-50 × 106 nucleated cells ranged from 0.125 to 0.5 × 106 Sca-1+ cells, as assessed by flow cytometry (see Materials and Methods for details). In contrast to previous studies of allogeneic bone marrow and HSC transplantations (3-8), no recipient preconditioning was used. Cumulative data from 5 experiments shown in Figure 2 indicate that 80% of untreated female NOD mice became diabetic (non-fasting blood glucose: >250 mg/dL) by 28 wk of age. Transfusion of allogeneic NBB prevented the onset of diabetes significantly (untreated vs NBB transfused: P=0.006). Only 30% of treated mice showed aberrant glycemic control in contrast to 80 % of untreated NOD female mice. Protection against diabetes was not reversible as late as 38 wk of age (data not shown). No apparent GVHD, as indicated by weight loss, anemia, diarrhea, and general appearance, was observed in protected mice. In contrast to NBB, adult C57BL/6 PBMC containing similar numbers of Sca-1+ cells (25 × 106 mononuclear cells containing ~0.125 × 106 Sca-1+ cells) as NBB cells failed to prevent diabetes in NOD mice (untreated vs adult blood transfused: P= 0.54, NS).
Figure 2. Amelioration of diabetes in NOD mice by allogeneic NBB transfusion.
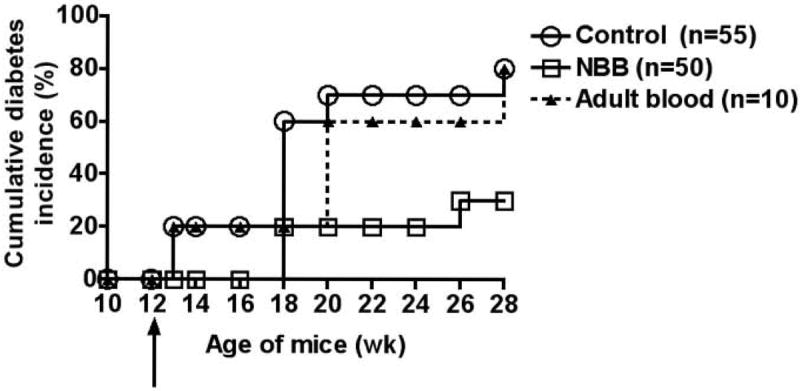
Incidence of diabetes (>250 mg/dL) in untreated NOD mice and those treated with allogeneic NBB or adult blood is shown. Arrow indicates the time of injection of cells. Statistical significance: untreated vs. newborn blood transfused, P=0.006, and untreated vs. adult blood transfused, P=0.54, NS.
In order to determine whether NBB transfusion can reverse overt diabetes, newly diagnosed diabetic mice (blood glucose levels ranging from 250 to 400 mg/dL) at 16 wk of age were given a single i.v. injection of 25 × 106 nucleated allogeneic NBB cells. Although diabetes was not fully reverted, 4 out of 5 treated mice maintained the same level of hyperglycemia and body weight, and survived longer than untreated, diabetic littermates till the end of the observation period, 32 wk of age (data not shown). Taken together, these data indicate that while transfusion of allogeneic NBB at the pre-diabetic stage prevented diabetes, it could only delay the progression of diabetes in newly diagnosed diabetic mice.
Restoration of normoglycemia and insulin release response in cured mice
Intraperitoneal glucose tolerance test performed at 28 to 32 wk of age (16 wk after NBB infusion) revealed that treated-non-diabetic mice displayed normal blood glucose clearance as untreated-non-diabetic mice (Figure 3A). In contrast, untreated-diabetic mice and those treated with allogeneic NBB but remained diabetic, displayed hyperglycemia, which did not change after glucose challenge. Serum insulin levels were increased by ~10-fold in protected mice after 45 min of a glucose challenge, as assessed by ELISA (Figure 3B). In untreated-non-diabetic mice, serum insulin levels were enhanced only by ~3-fold in response to a glucose challenge. In contrast, increases in insulin levels were barely observed in untreated-diabetic mice and in NBB treated-diabetic mice. These results indicate that allogeneic NBB transfusion can restore glycemic control by preserving the insulin-producing beta cell function.
Figure 3. Allogeneic NBB transfusion preserved the beta cell function.
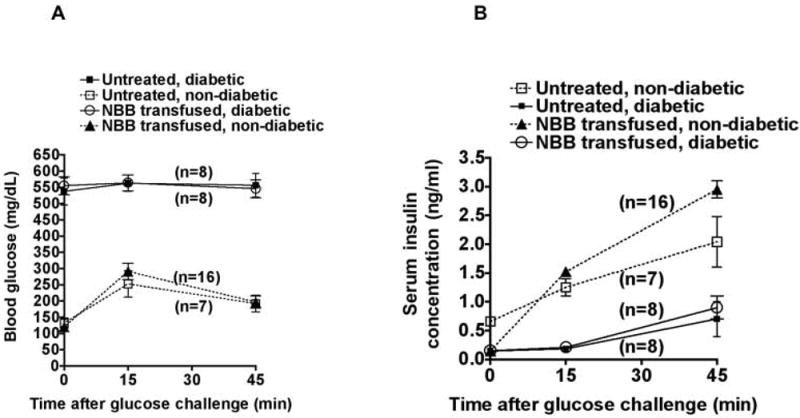
A. Intraperitoneal glucose tolerance test was performed at 28 to 32 wk of age. Blood glucose levels in various groups of mice challenged with glucose are shown. Values indicate mean +/- SD per group. Numbers of mice tested are indicated in parentheses.
B. Corresponding insulin levels of these mice are shown. Mean values +/- SD per group are indicated.
Reduction of invasive insulitis by allogeneic NBB transfusion
In the NOD mouse system, peri-insulitis, cellular infiltration at the periphery of islets is seen at 3 to 4 wk of age (28) and in our model, NBB transfusion was given at 12 wk of age, when the pathologic process was already under way. Pancreata of untreated-non-diabetic NOD mice displayed little or no cellular infiltration (Grade 1, Figure 4A) at 28 wk of age. As expected, islets of untreated-diabetic mice displayed moderate (Grade 2, Figure 4B) and invasive cellular infiltration (Grade 3, Figure 4C). Whereas little or no apparent loss of insulin-producing beta cells was found in pancreata of treated-non-diabetic mice with Grade 1 or Grade 2 pathology scores (Figure 4A & B, lower panel), beta cells without invasive cellular infiltration could not be identified in the pancreata of overtly diabetic mice (Figure 3C, lower panel). A majority (>70%) of islets in untreated-diabetic mice (n=3) had the highest level of cellular infiltration (Grade 3, Figure 4D). Interestingly, in mice that were treated with NBB and remained diabetic, 48% of islets displayed severe and 43% moderate cellular infiltration. Significantly, only 25% of islets examined from protected mice had severe cellular infiltration while 46% had no or minimal infiltration. Although the frequency of pancreata with Grade 1 pathology score in mice that were treated but remained diabetic was similar to those of treated-non-diabetic mice, pancreata of overtly diabetic mice had severe (Grade 3) cellular infiltration. These data indicate that amelioration of diabetes following transfusion with MHC- and minor histocompatibility antigen-mismatched NBB cells is due to the blockade of transition from mild to overt and invasive insulitis.
Figure 4. Reduction in invasive infiltration of islets in protected mice.
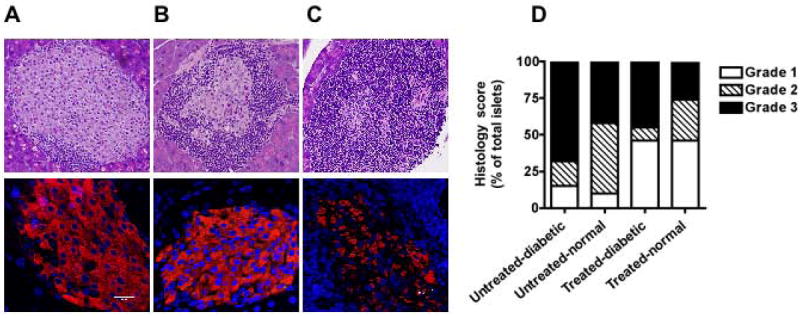
Top row shows H & E stained sections of pancreata displaying islets with no/minimal (Grade 1, A), moderate (Grade 2, B) and severe cellular infiltration (Grade 3, C) (original magnification × 200). Bottom row shows confocal images of islets displaying insulin-producing beta cells (red) in corresponding sections. Nuclei were stained with Hoechst (blue). Bar indicates 20 μM (original magnification × 630).
D. Histological scores of pancreata in indicated sets of mice are shown.
Lack of correlation between microchimerism and protection against diabetes
Previous studies showed an association between significant level of chimerism and protection against diabetes following transplantation of NOD mice with allogeneic bone marrow or purified Sca-1+ HSC performed under cytoreductive conditions (3-8, 12). One study suggested that <1% chimerism was sufficient for protection against diabetes following allogeneic bone marrow transplantation (29). Therefore, it was of interest to determine whether protection against diabetes following allogeneic NBB transfusion in un-irradiated NOD mice was associated with significant level of chimerism. Data shown in Figure 5A indicate the presence of donor-derived H-2Kb-expressing leukocytes in the peripheral blood of NOD recipients as detected by flow cytometry 5 wk after NBB transfusion. This microchimerism (<5% of allogeneic leukocytes) was transient and not detected at later time points in the peripheral blood. In order to ascertain whether a correlation exists between protection afforded by NBB transfusion and the level of chimerism, mice were killed between 30 and 38 wk of age and their spleens were analyzed for the expression of donor derived H-2Kb by flow cytometry. Data shown in Figure 5B indicate that the level of microchimerism found in splenocytes of mice that were protected from diabetes and those remained diabetic after NBB transfusion did not differ. In order to further analyze the correlation between microchimerism and protection against diabetes in NBB transfused mice, splenocytes were analyzed for the expression of donor-derived H-2Kb gene by RT-PCR. The data shown in Figure 5C indicate that out of 7 mice tested, three (N-2, N-6 and N-7) were strongly chimeric, while one (N-3) weakly expressed the B6-derived MHC class I gene (Figure 5C). Only two (N-2 and N-7) of these chimeric mice were protected from diabetes. Although chimerism could not be detected in another mouse (N-1) by RT-PCR, it did not develop diabetes. Induction of transient microchimerism in non-preconditioned mice is similar to blood transfusion-associated microchimerism in humans (30). While macrochimerism (>5%) (3-8, 12) and microchimerism (<1%) (29) induced by bone marrow transplantation or purified HSC in myeloablated recipients have been implicated in diabetes protection, NBB transfusion in un-conditioned NOD mice resulted in microchimerism that did not strictly correlate with protection against diabetes.
Figure 5. Relationship between microchimerism and protection against diabetes.
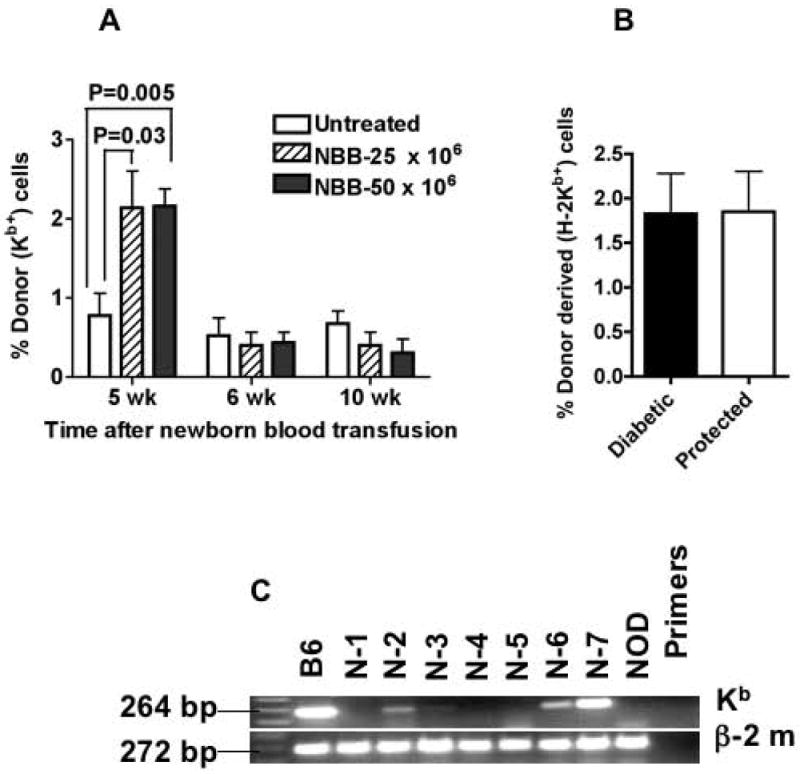
A. The presence of H-2Kb-expressing cells was determined by flow cytometry in the peripheral blood of untreated NOD mice and in those transfused with indicated numbers of mononuclear cells containing NBB at various time points. Statistical significance between groups is shown (n=8-10 mice per group).
B. Microchimerism was analyzed in splenocytes after 14 to 18 wk of NBB transfusion by flow cytometry. Data are shown for NBB transfused but remained diabetic and those protected from diabetes after NBB transfusion (n= 10 per group).
C. RT-PCR analysis of splenocytes obtained after 18 wk of NBB transfusion. B6: spleen cells from C57BL/6 positive control; N-1 through N-7: spleen cells from NOD mice transfused with C57BL/6 NBB; NOD: spleen cells from NOD mice. Amplicon sizes of Kb and β-2 microglobulin are shown.
Modulation of expression of selected genes by NBB transfusion
In order to further gain insights into the mechanisms of protection mediated by NBB transfusion, induction of cytokine genes implicated in diabetes was analyzed. In 5 separate experiments, spleens from 3-5 cured and diabetic mice were simultaneously harvested between 16 and 26 wk after NBB transfusion (at 28-38 wk of age). Splenocytes from each group were pooled and stimulated overnight with PMA and ionomycin or left untreated. Relative gene expression was analyzed concurrently in unstimulated and stimulated splenocytes from both non-diabetic and diabetic mice by real-time RT-PCR using Gapdh as the normalizer. The results indicated that the induction of Tnfa, implicated in diabetes (31-34) was most prominently reduced in splenocytes of treated, non-diabetic mice in all 5 experiments (Figure 6A). The mRNA expression of Il18, also implicated in autoimmune diabetes (35) was significantly reduced in treated-non-diabetic mice. Although in this experiment, Il4 was suppressed in protected mice, it is highly variable in different experiments. The expression of Inos, implicated in beta cell damage (36), was also reduced in protected mice. However, the expression levels of Il17 and Ifng were not compromised in NBB transfused-non-diabetic mice. Although the transcription factor Tbet, also known as Tbx21 has been implicated in diabetes manifestation (37), this was significantly upregulated in protected mice in all 5 experiments. The expression of Rorgt (38) and Gata3 (39), respectively critical for the transcription of Il17 and Il4, was not significantly enhanced in mice protected from diabetes by NBB transfusion.
Figure 6. Modulation of gene expression in NBB transfused and protected mice.
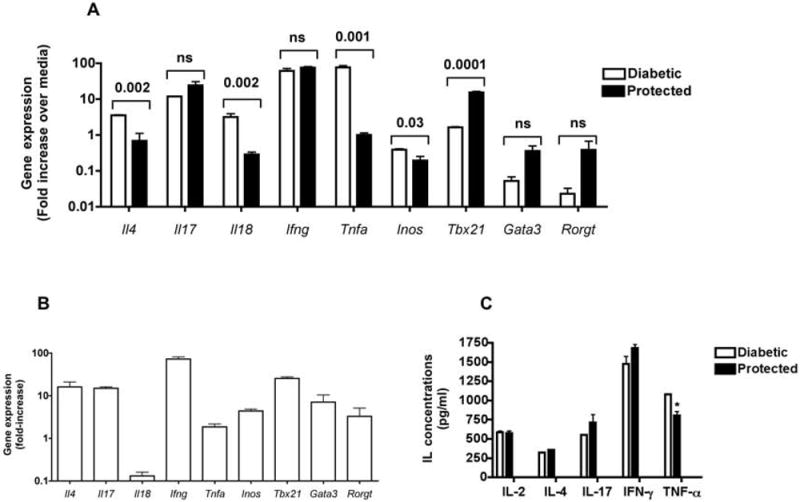
A. Spleens were harvested from NOD mice protected from diabetes after transfusion with NBB derived from C57BL/6 mice and from untreated-diabetic mice at 32 wk of age and stimulated with PMA and ionomycin. Unstimulated splenocytes cultured over night in complete media served as controls. Real-time RT-PCR analysis was performed and the relative levels of gene expression in stimulated vs. unstimulated cells were analyzed using Gapdh as the normalizer. Data shown are from a single experiment (mean+/-SD of triplicates) of a total of 5 experiments, each containing 3 to 5 mice per group. P values between protected and diabetic groups are indicated.
B. Prediabetic NOD mice were killed at 12 wk of age and their splenocytes were activated as described above. Relative gene expression in stimulated vs unstimulated cells was assessed using Gapdh as the normalizer. Data shown are from a single experiment (mean+/-SD of triplicates) in a total of 3 experiments, each containing 3 to 5 mice per group. The data shown are from an experiment analyzed simultaneously with that shown in A.
C. Splenocytes were obtained from NBB transfused-non-diabetic and those untreated-diabetic mice and stimulated with PMA and ionomycin or left untreated as described in A. After overnight culture, supernatants were obtained and tested for various cytokines by ELISA. Shown is mean +/- SE of duplicate samples from a representative of 3 independent experiments with similar results. * P= 0.05.
To further understand the implications of these findings, lymphokine gene expression was also assessed simultaneously in splenocytes of 12 wk old untreated-pre-diabetic mice and NBB transfused-diabetic, and non-diabetic mice at 32 to 38 wk of age. Representative results shown in Figure 6B indicate that splenocytes of untreated-pre-diabetic mice activated with PMA and ionomycin expressed lower levels of Il18 and Tnfa and higher levels of Ifng and Tbx21 in comparison to other genes simultaneously analyzed. Although the levels of gene expression in pre-diabetic mice cannot be directly compared to those of NBB treated, non-diabetic mice due to age differences, the pattern of gene expression was similar in both groups. Thus, the levels of Il18 and Tnfa mRNA were lower while the expression of Tbx21 was higher in both pre-diabetic and cured mice when compared to NBB-treated, diabetic mice (Figure 6A and B). We also observed lower expression of Il18 and Tnfa, and increased level of Tbx21 mRNA in a small group of untreated NOD mice (n=3) that did not develop diabetes at 32 wk of age (data not shown).
The amounts of lymphokines present in the supernatant of splenocytes stimulated with PMA and ionomycin were determined by ELISA. The data shown in Figure 6C indicate that the splenocytes from NBB transfused and protected mice produced less TNF-α protein, consistent with the downregulation of Tnfa mRNA. The secretion of IL-17 and IFN-γ proteins by splenocytes of protected mice was slightly enhanced. Collectively, these data indicate that NBB transfusion led to the prevention of diabetes accompanied by selective reduction in the expression of cytokines implicated in diabetes such as Il18 and Tnfa with a concomitant upregulation of Tbx21 mRNA.
Discussion
We have demonstrated here that transfusion of un-conditioned, prediabetic female NOD mice with allogeneic NBB but not adult blood prevented the onset of diabetes. However, NBB given after the disease manifestation delayed but did not revert diabetes. Abrogation of autoimmune diabetes was accompanied by the blockade of progression from prediabetes to overt diabetes and preservation of beta cell function. Although low level of microchimerism was induced by NBB transfusion, it poorly correlated with the level of protection against diabetes. Importantly, selective downregulation of inflammatory cytokine genes implicated in diabetes, such as Il18, Tnfa and Inos, and upregulation of the transcription factor Tbx21 were consistently seen in NOD mice protected from diabetes by NBB transfusion.
Although transfusion of newly diagnosed, preconditioned T1D patients with autologous CD34+ cells mobilized from the bone marrow had palliative effects (40), the long-term benefits of this treatment procedure remain obscure. Since transplantation of bone marrow from a diabetic sibling into another HLA identical non-diabetic sibling transferred diabetes (11), the progenitors present in the transferred autologous cord blood (10) or CD34+ cells mobilized from the bone marrow (40) are likely to reinstate the diabetogenic phenotype. Numerous studies demonstrated that transplantation of syngeneic T cell-depleted HSC (7) or bone marrow (4-5, 12) transferred diabetes into heavily irradiated NOD mice. These data are consistent with the notion that transplantation of non-diabetic, allogeneic but not autologous HSC containing progenitors of diabetogenic T lymphocytes is a better option for treatment of T1D patients. However, validation of allogeneic but not syngeneic NBB is potent in ameliorating spontaneous autoimmune diabetes in NOD mice will assure that allogeneic rather than autologous umbilical cord blood is a better option for treatment of T1D patients. Nevertheless, the preclinical model described herein provides the proof of principle for the notion that transfusion of neonatal whole blood derived from diabetes-resistant mice can provide robust protection against autoimmune diabetes without recipient preconditioning. In addition, our data shed light on the possible mechanisms involved in the abrogation of autoimmune diabetes by somatic stem cell therapy.
Previous studies showed that both bone marrow and NBB contain similar frequency (~15%) of Sca-1+ cells (13-14) and comparably reconstitute the entire hematopoietic system in irradiated allogeneic mice (13-15). Therefore, it is likely that the allogeneic NBB derived Sca-1+ HSC may partially reset the immune system in un-irradiated NOD mice and lead to the abrogation of diabetes. Differentiation of allogeneic, immature CD4+CD8+ neonatal T cells into mature T cells may also contribute to the altered immune system observed in NOD recipients. Since NBB contained large numbers of CD44+ cells, they may potentially differentiate into CD44+ mesenchymal stem cells and modestly suppress diabetes similar to those derived from the bone marrow (41). Other mechanisms include de novo generation of islets from pancreatic ductal cells (42), thwarting autoimmunity by inducing T cell tolerance (6), and boosting of the T regulatory cells implicated in the control of autoimmune diabetes in NOD mice (43-44).
Importantly, our data indicate that amelioration of diabetes by allogeneic NBB transfusion is associated with selective suppression of cytokines implicated in diabetes such as Tnfa (31-34), Il18 (35), and Inos (36) but not Il4 (34, 45) or Il17 (46). The expression of Gata3 and Rorgt, respectively crucial for the transcription of IL-4 (38) and IL-17 (39), was also not reduced in protected mice. These data are consistent with the lack of involvement of Il4 and Il17 in diabetes, suggested previously (47-49). The inducible expression levels of Tnfa appeared to be lower in protected mice as in pre-diabetic and untreated-non-diabetic NOD mice. Inasmuch as TNF-α is implicated in diabetes (31-34), its reduction may minimize the beta cell damage. In addition, the expression of Inos was diminished in protected mice. Nitric oxide synthesized from l-arginine by iNOS in activated macrophages as well as by beta cells exposed to inflammatory cytokines such as IL-1β and TNF-α in vitro is considered to be toxic to beta cells (50-51). Although a similar toxic role for nitric oxide remains to be established in vivo, our data suggest that iNOS suppression following NBB transfusion may contribute to protection against spontaneous diabetes.
Our data also demonstrate that protection afforded by NBB transfusion accompanies notable expression of the transcription factor Tbx21, similar to pre-diabetic and untreated-non-diabetic mice. The transcription factor Tbx21 was originally described to be critical for polarization of Th1 cells without influencing the production of IFN-γ by CD8+ T cells (52). The absence of Tbx21 in both innate and adaptive immune systems has been shown to prevent autoimmune diabetes in NOD mice, suggesting a pathogenic role for Tbx21 (37). In contrast, ablation of Tbx21 failed to influence insulitis in Balb/c mice (53). The basis for the discrepancy between these data remains obscure. Since Tbx21 regulates numerous immune system-associated genes including Ifng, CD122, and Cxr3i (52, 54), NBB transfusion mediated upregulation of Tbx21 may lead to the alteration of autoimmune responses distinct from those caused by the deletion of Tbx21 in both innate and adaptive immune systems (37, 52-53).
Low level of microchimerism is not unexpected in un-irradiated mice, similar to that found after blood transfusion in humans (30). Although this may be related to the fact that only <10% of the NBB expressed MHC class antigen, H-2Kb and these cells may not significantly expand in un-irradiated hosts. Alternatively, detection of CD45 variant expressed by C57BL/6 derived NBB may allow better determination of the level of chimerism in NOD recipients. Nevertheless, chimerism was detected in a majority (4/7) of NBB transfused mice by RT-PCR but it did not strictly correlate with the level of protection against diabetes. These data are consistent with previous studies indicating that induction of chimerism after bone marrow or HSC transfusion is highly variable in NOD mice (6-8, 12) and low levels of allogeneic but not syngeneic hematopoietic chimerism were sufficient to abrogate diabetes (7, 12, 29). Bone marrow transfusion induced mixed chimerism has been suggested to reinstate clonal deletion of autoreactive (beta cell-reactive) T cells (6). While it is difficult to track the diabetogenic CD4+ T cells by tetramer staining without in vitro expansion that may introduce bias, tetramers that bind to diabetogenic CD8+ T cells recognizing the IGRP214-223 peptide (55) may allow ex vivo determination of the modulation of a subset of diabetogenic T cells in protected mice.
In conclusion, the data presented herein demonstrate the utility of allogeneic NBB, functionally equivalent to umbilical cord blood, to ameliorate autoimmune diabetes in NOD mice. This model system may prove to be useful for the delineation of the mechanisms involved in abrogation of diabetes by allogeneic neonatal stem cell therapy in the absence of recipient preconditioning.
Acknowledgments
Mei Ling Chen and Andy Hall are acknowledged for assistance with confocal imaging and preparation of histological slides, respectively. Lisa Joseph is acknowledged for help with gel electrophoresis.
Footnotes
This work was supported by the Department of Surgery and by The National Institutes of Health Grant RO1 AI058190 to B.S.P.
Presented in abstract form at the Key Stone Symposia, Snowbird, UT, April 6-11, 2008.
Abbreviations used in this paper: GVHD, graft vs host disease; HSC, hematopoietic stem cells; NBB, newborn blood; Sca-1, stem cell antigen-1; T1D, type 1 diabetes.
Authorship
SJ conceived, designed and conducted animal experiments, histological analysis, confocal imaging, flow cytometry, ELISA, real-time RT-PCR, and wrote the paper. TP and VP performed real-time RT-PCR experiments. SA and AJ performed RT-PCR. RG performed ELISA and evaluated histology. SK evaluated histology and RS assisted in animal experiments and performed ELISA. DR, BP, and MH supported the work, participated in discussions, and critically read the paper.
conflict of interest statements
The authors declare no competing financial interests.
References
- 1.Eisenbarth GS. Update in type 1 diabetes. J Clin Endocrinol Metab. 2007;92:2403–2407. doi: 10.1210/jc.2007-0339. [DOI] [PubMed] [Google Scholar]
- 2.Leiter EH. Nonobese diabetic mice and the genetics of diabetes susceptibility. Curr Diab Rep. 2005;5:141–148. doi: 10.1007/s11892-005-0042-z. [DOI] [PubMed] [Google Scholar]
- 3.Ikehara S, Ohtsuki H, Good RA, Asamoto H, Nakamura T, Sekita K, Muso E, Tochino Y, Ida T, Kuzuya H, et al. Prevention of type I diabetes in nonobese diabetic mice by allogenic bone marrow transplantation. Proc Natl Acad Sci U S A. 1985;82:7743–7747. doi: 10.1073/pnas.82.22.7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LaFace DM, Peck AB. Reciprocal allogeneic bone marrow transplantation between NOD mice and diabetes-nonsusceptible mice associated with transfer and prevention of autoimmune diabetes. Diabetes. 1989;38:894–901. doi: 10.2337/diab.38.7.894. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Kaufman CL, Ildstad ST. Allogeneic chimerism induces donor-specific tolerance to simultaneous islet allografts in nonobese diabetic mice. Surgery. 1995;118:192–197. doi: 10.1016/s0039-6060(05)80323-x. [DOI] [PubMed] [Google Scholar]
- 6.Sykes M, Nikolic B. Treatment of severe autoimmune disease by stem-cell transplantation. Nature. 2005;435:620–627. doi: 10.1038/nature03728. [DOI] [PubMed] [Google Scholar]
- 7.Beilhack GF, Scheffold YC, Weissman IL, Taylor C, Jerabek L, Burge MJ, Masek MA, Shizuru JA. Purified allogeneic hematopoietic stem cell transplantation blocks diabetes pathogenesis in NOD mice. Diabetes. 2003;52:59–68. doi: 10.2337/diabetes.52.1.59. [DOI] [PubMed] [Google Scholar]
- 8.Serreze DV, Osborne MA, Chen YG, Chapman HD, Pearson T, Brehm MA, Greiner DL. Partial versus full allogeneic hemopoietic chimerization is a preferential means to inhibit type 1 diabetes as the latter induces generalized immunosuppression. J Immunol. 2006;177:6675–6684. doi: 10.4049/jimmunol.177.10.6675. [DOI] [PubMed] [Google Scholar]
- 9.Madrigal JA, Cohen SB, Gluckman E, Charron DJ. Does cord blood transplantation result in lower graft-versus-host disease? It takes more than two to tango. Hum Immunol. 1997;56:1–5. doi: 10.1016/s0198-8859(97)00125-0. [DOI] [PubMed] [Google Scholar]
- 10.Haller MJ, Viener HL, Wasserfall C, Brusko T, Atkinson MA, Schatz DA. Autologous umbilical cord blood infusion for type 1 diabetes. Exp Hematol. 2008;36:710–705. doi: 10.1016/j.exphem.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lampeter EF, Homberg M, Quabeck K, Schaefer UW, Wernet P, Bertrams J, Grosse-Wilde H, Gries FA, Kolb H. Transfer of insulin-dependant diabetes between HLA-identical siblings by bone marrow transplantation. Lancet. 1993;341:1243–1244. doi: 10.1016/0140-6736(93)91148-f. [DOI] [PubMed] [Google Scholar]
- 12.Kaminitz A, Mizrahi K, Yaniv I, Farkas DL, Stein J, Askenasy N. Low levels of allogeneic but not syngeneic hematopoietic chimerism reverse autoimmune insulitis in prediabetic NOD mice. J Autoimmun. 2009;33:83–91. doi: 10.1016/j.jaut.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 13.de La Selle V, Gluckman E, Bruley-Rosset M. Newborn blood can engraft adult mice without inducing graft-versus-host disease across non H-2 antigens. Blood. 1996;87:3977–3983. [PubMed] [Google Scholar]
- 14.de La Selle V, Gluckman E, Bruley-Rosset M. Graft-versus-host disease and graft-versus-leukemia effect in mice grafted with peripheral newborn blood. Blood. 1998;92:3968–3975. [PubMed] [Google Scholar]
- 15.Harrison DE, Astle CM. Short-and long-term multilineage repopulating hematopoietic stem cells in late fetal and newborn mice: Models for human umbilical cord blood. Blood. 1997;90:174–181. [PubMed] [Google Scholar]
- 16.Hofer MJ, Finger C, Pagenstecher A. Molecular genotyping of the murine H-2K MHC class I allele. J Immunol Methods. 2005;302:168–171. doi: 10.1016/j.jim.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Arcellana-Panlilio MY, Schultz GA. Temporal and spatial expression of major histocompatibility complex class I H-2k in the early mouse embryo. Biol Reprod. 1994;51:169–183. doi: 10.1095/biolreprod51.2.169. [DOI] [PubMed] [Google Scholar]
- 18.Jayaraman S, Luo Y, Dorf ME. Tolerance induction in T helper (Th1) cells by thymic macrophages. J Immunol. 1992;148:2672–2281. [PubMed] [Google Scholar]
- 19.Overbergh L, Giulietti A, Valckx D, Decallonne R, Bouillon R, Mathieu C. The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J Biomol Tech. 2003;14:33–43. [PMC free article] [PubMed] [Google Scholar]
- 20.Fina D, Sarra M, Fantini MC, Rizzo A, Caruso R, Caprioli F, Stolfi C, Cardolini I, Dottori M, Boirivant M, Pallone F, Macdonald TT, Monteleone G. Regulation of gut inflammation and Th17 cell response by interleukin-21. Gasteroenterology. 2008;134:1038–1048. doi: 10.1053/j.gastro.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 21.Oko L, Aduddell-Swope B, Willils D, Hamor R, Coons TA, Hjelle B, Schountz T. Profiling helper T cell subset gene expression in deer mice. BMC Immunol. 2006;7:18. doi: 10.1186/1471-2172-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serre K, Mohr E, Toellner KM, Cunningham AF, Granjeaud S, Bird R, MacLennan IC. Molecular differences between the divergent responses of ovalbumin-specific CD4 T cells to alum-precipitated ovalbumin compared to ovalbumin expressed by Salmonella. Mol Immunol. 2008;45:3558–3566. doi: 10.1016/j.molimm.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 24.Goodison S, Urquidi V, Tarin D. CD44 cell adhesion molecules. Mol Pathol. 1999;52:189–196. doi: 10.1136/mp.52.4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nadri S, Soleimani M, Hosseni RH, Massumi M, Atashi A, Izadpanah R. An efficient method of isolation of murine bone marrow mesenchymal stem cells. Int J Dev Biol. 2007;51:723–29. doi: 10.1387/ijdb.072352ns. [DOI] [PubMed] [Google Scholar]
- 26.Holmes C, Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells. 2007;25:1339–1347. doi: 10.1634/stemcells.2006-0644. [DOI] [PubMed] [Google Scholar]
- 27.Ikehara S, Kawamura M, Takao F, Inaba M, Yasumizu R, Than S, Hisha H, Sugiura K, Koide Y, Yoshida TO, et al. Organ-specific and systemic autoimmune diseases originate from defects in hematopoietic stem cells. Proc Natl Acad Sci U S A. 1990;87:8341–8344. doi: 10.1073/pnas.87.21.8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.André I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease: Lessons from diabetes models. Proc Natl Acad Sci USA. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zorina TD, Subbotin VM, Bertera S, Alexander AM, Haluszczak C, Gambrell B, Bottino R, Styche AJ, Trucco M. Recovery of endogenous beta cell function in the NOD model of autoimmune diabetes. Stem cells. 2003;21:377–388. doi: 10.1634/stemcells.21-4-377. [DOI] [PubMed] [Google Scholar]
- 30.Utter GH, Reed WF, Lee TH, Busch MP. Transfusion-associated microchimerism. Vox Sang. 2007;93:188–195. doi: 10.1111/j.1423-0410.2007.00954.x. [DOI] [PubMed] [Google Scholar]
- 31.Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, McDevitt HO. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med. 1994;180:995–1004. doi: 10.1084/jem.180.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pakala SV, Chivetta M, Kelly CB, Katz JD. In autoimmune diabetes the transition from benign to pernicious insulitis requires an islet cell response to tumor necrosis factor alpha. J Exp Med. 1999;189:1053–1062. doi: 10.1084/jem.189.7.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee LF, Xu B, Michie SA, Beilhack GF, Warganich T, Turley S, McDevitt HO. The role of TNF-alpha in the pathogenesis of type 1 diabetes in the nonobese diabetic mouse: analysis of dendritic cell maturation. Proc Natl Acad Sci U S A. 2005;102:15995–16000. doi: 10.1073/pnas.0508122102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He J, Haskins K. Pathogenicity of T helper 2 T-cell clones from T-cell receptor transgenic non-obese diabetic mice is determined by tumor necrosis factor-alpha. Immunology. 2007;123:108–117. doi: 10.1111/j.1365-2567.2007.02715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oikawa Y, Shimada A, Kasuga A, Morimoto J, Osaki T, Tahara H, Miyazaki T, Tashiro F, Yamato E, Miyazaki J, Saruta T. Systemic administration of IL-18 promotes diabetes development in young nonobese diabetic mice. J Immunol. 2003;171:5865–5875. doi: 10.4049/jimmunol.171.11.5865. [DOI] [PubMed] [Google Scholar]
- 36.Miljkovic D, Cvetkovic I, Momcilovic M, Maksimovic-Ivanic D, Stosic-Grujicic S, Trajkovic V. Interleukin-17 stimulates inducible nitric oxide synthase-dependent toxicity in mouse beta cells. Cell Mol Life Sci. 2005;62:2658–2668. doi: 10.1007/s00018-005-5259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esensten JH, Lee MR, Glimcher LH, Bluestone JA. T-bet-deficient NOD mice are protected from diabetes due to defects in both T cell and innate immune system function. J Immunol. 2009;183:75–82. doi: 10.4049/jimmunol.0804154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 39.Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, Leeds KE, Du H, Wu B, Waterman ML, Zhu Z, Sen JM. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. 2009;10:992–999. doi: 10.1038/ni.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Couri CE, Oliveira MC, Stracieri AB, Moraes DA, Pieroni F, Barros GM, Madeira MI, Malmegrim KC, Foss-Freitas MC, Simões BP, Martinez EZ, Foss MC, Burt RK, Voltarelli JC. C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 mellitus. JAMA. 2009;301:1573–1579. doi: 10.1001/jama.2009.470. [DOI] [PubMed] [Google Scholar]
- 41.Fiorina P, Jurewicz M, Augello A, Vergani A, Dada S, La Rosa S, Selig M, Godwin J, Law K, Placidi C, Smith RN, Capella C, Rodig S, Adra CN, Atkinson M, Sayegh MH, Abdi R. Immunomodulatory function of bone marrow-derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol. 2009;183:993–1004. doi: 10.4049/jimmunol.0900803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonner-Weir S, Weir G. New sources of pancreatic β cells. Nat Biotechnol. 2005;23:857–861. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- 43.Bluestone JA, Tang Q, Sedwick CE. T regulatory cells in autoimmune diabetes: past challenges, future prospects. J Clin Immunol. 2008;28:677–684. doi: 10.1007/s10875-008-9242-z. [DOI] [PubMed] [Google Scholar]
- 44.Cheatem D, Ganesh BB, Gangi E, Vasu C, Prabhakar BS. Modulation of dendritic cells using granulocyte-macrophage colony-stimulating factor (GM-CSF) delays type 1 diabetes by enhancing CD4+CD25+ regulatory T cell function. Clin Immunol. 2009;131:260–270. doi: 10.1016/j.clim.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pakala SV, Kurrer MO, Katz JD. T helper 2 (Th2) T cells induce acute pancreatitis and diabetes in immune-compromised nonobese diabetic (NOD) mice. J Exp Med. 1997;186:299–306. doi: 10.1084/jem.186.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, Shapiro AM. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. 2009;58:1302–1311. doi: 10.2337/db08-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rapoport MJ, Jaramillo A, Zipris D, Lazarus AH, Serreze DV, Leiter EH, Cyopick P, Danska JS, Delovitch TL. Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J Exp Med. 1993;178:87–99. doi: 10.1084/jem.178.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol. 2009;39:216–224. doi: 10.1002/eji.200838475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bending D, De La Peña H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, Cooke A. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009;119:565–572. doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Southern C, Schulster D, Green IC. Inhibition of insulin secretion by interleukin-1 beta and tumour necrosis factor-alpha via an L-arginine-dependent nitric oxide generating mechanism. FEBS Lett. 1990;276:42–44. doi: 10.1016/0014-5793(90)80502-a. [DOI] [PubMed] [Google Scholar]
- 51.Corbett JA, McDaniel ML. Intraislet release of interleukin 1 inhibits beta cell function by inducing beta cell expression of inducible nitric oxide synthase. J Exp Med. 1995;181:559–568. doi: 10.1084/jem.181.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 53.Melanitou E, Liu E, Miao D, Yu L, Glimcher LH, Eisenbarth G. Absence of the T-bet gene coding for the Th1-related transcription factor does not affect diabetes-associated phenotypes in Balb/c mice. Ann N Y Acad Sci. 2003;1005:187–191. doi: 10.1196/annals.1288.024. [DOI] [PubMed] [Google Scholar]
- 54.Matsuda JL, George TC, Hagman J, Gapin L. Temporal dissection of T-bet functions. J Immunol. 2007;178:3457–3465. doi: 10.4049/jimmunol.178.6.3457. [DOI] [PubMed] [Google Scholar]
- 55.Trudeau JD, Kelly-Smith C, Verchere CB, Elliott JF, Dutz JP, Finegood DT, Santamaria P, Tan R. Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest. 2003;111:217–223. doi: 10.1172/JCI16409. [DOI] [PMC free article] [PubMed] [Google Scholar]