

Abstract
The liver kinase B1 (LKB1) tumour suppressor functions as a master regulator of growth, metabolism and survival in cells, which is frequently mutated in sporadic human non-small cell lung and cervical cancers. LKB1 functions as a key upstream activator of the AMP-activated protein kinase (AMPK), a central metabolic switch found in all eukaryotes that govern glucose and lipid metabolism and autophagy in response to alterations in nutrients and intracellular energy levels. The LKB1/AMPK signalling pathway suppresses mammalian target of rapamycin complex 1 (mTORC1), an essential regulator of cell growth in all eukaryotes that is deregulated in a majority of human cancers. LKB1 inactivation in cancer leads to both tumorigenesis and metabolic deregulation through the AMPK and mTORC1-signalling axis and there remain critical challenges to elucidate the direct role LKB1 inactivation plays in driving aberrant metabolism and tumour growth. This review addresses past and current efforts to delineate the molecular mechanisms fueling metabolic deregulation and tumorigenesis following LKB1 inactivation as well as translational promise of therapeutic strategies aimed at targeting LKB1-deficient tumors.
Keywords: AMP-activated protein kinase, fluoro-deoxyglucose positron emission tomography, liver kinase B1, mammalian target of rapamycin complex 1, therapeutics, Warburg
INTRODUCTION
The liver kinase B1 (LKB1) tumour suppressor (LKB1 also known as serine/threonine kinase 11) that functions as a master regulator of growth, metabolism and survival in cells. LKB1 was originally identified as a tumour suppressor gene located on chromosome 19p13 where loss or mutation of this gene is responsible for the inherited cancer disorder Peutz-Jeghers Syndrome (PJS).[1] Importantly, LKB1 is frequently mutated in ~30% of sporadic human non-small cell lung cancer (NSCLC) and 20% of cervical cancers.[2,3,4] A critical link directly connecting LKB1 to cell metabolism and cancer came from the discovery that LKB1 was the key upstream activator of the AMP-activated protein kinase (AMPK),[5,6,7,8] a central metabolic switch found in all eukaryotes that governs glucose and lipid metabolism and autophagy in response to alterations in nutrients and intracellular energy levels. In addition, LKB1 functions as a negative regulator of mammalian target of rapamycin complex 1 (mTORC1), an essential integrator of nutrient and growth factor signals that controls cell growth in all eukaryotes and is deregulated in a majority of human cancers.[9] Nearly 90 years ago Otto Warburg first described aerobic glycolysis in tumour cells and renewed interest in the Warburg effect has propelled metabolism to the forefront of cancer biology.[10] LKB1 inactivation in cancer leads to both tumorigenesis and metabolic deregulation through the AMPK and mTORC1-signalling axis and there remain critical challenges to elucidate the direct role LKB1 inactivation plays in driving aberrant metabolism and tumour growth. This review will address past and current efforts to delineate the molecular mechanisms driving metabolic deregulation and tumorigenesis following LKB1 inactivation as well as therapeutic strategies to target LKB1 regulated pathways and their translational promise.
LKB1 IS A MASTER KINASE REGULATING METABOLISM AND GROWTH THROUGH AMPK AND MTOR SIGNALLING
LKB1 regulation of growth and metabolism through AMPK and mTOR signalling
The activated LKB1 kinase exists as a trimeric complex with the pseudo kinase STE20-related adaptor-α STRADα) and scaffolding protein mouse protein 25α,[11,12,13] where it regulates metabolism through the direct phosphorylation and activation of AMPK along with 12 other closely related AMPK-like kinases whose functions span a broad spectrum of biology including regulation of metabolism, growth, polarity and cell survival.[14,15] AMPK is the only LKB1 substrate that is activated under low adenosine triphosphate (ATP) conditions following nutrient deprivation or hypoxia and functions as a cellular rheostat maintaining energy homeostasis. AMPK exists as a heterotrimeric complex composed of a catalytic subunit (AMPKα1, α2) and two regulatory subunits (AMPKβ1, β2 and AMPKγ1, γ2, γ3). AMPK is activated upon the direct binding of adenosine diphosphate (ADP) or adenosine monophosphate (AMP) to the γ subunit where AMPK undergoes a conformational change leading to the phosphorylation of Thr172 on the activation loop of the α subunit.[16,17,18] LKB1 was found to be the key upstream activating kinase of AMPK,[6,7,8] however, AMPK is activated as weel by calmodulin-dependent protein kinase kinase β in response to calcium flux and potentially TAK1 in certain cellular contexts.[19,20,21]
During periods of energetic stress AMPK activation results in the activation of catabolic pathways and the concomitant inhibition of anabolic metabolism, which serve to restore energy homeostasis. Acute activation of AMPK regulates cellular metabolism through activation of AMPK substrates including the 6-phosphofructo-2-kinase pathway, acetyl coenzyme A carboxylase, 3-hydroxy-3-methyl coenzyme A reductase, which rapidly lead to the activation of glucose metabolism and fatty acid synthesis.[22,23,24,25] Long-term activation of AMPK results in the phosphorylation and activation of transcriptional factors involved in adaptive reprogramming of cellular metabolism. For complete and detailed reviews of AMPK's role in regulation of metabolism refer to reviews by Mihaylova and Shaw 2011 and Hardie et al., 2012.[26,27]
The LKB1/AMPK signalling pathway is a negative regulator of the mTORC1 signalling complex [Figure 1], a central integrator of nutrient and growth factor inputs that controls growth and the metabolic landscape of cells in all eukaryotes and is deregulated in a majority of human cancers,[28,29] mTOR is found in two biochemically and functionally discrete signalling complexes,[30] mTOR complex 1 includes raptor, which acts as a scaffold to recruit downstream substrates such as translation initiation factor 4E-binding protein 1 (4E-BP1) and ribosomal S6 kinase (p70S6K1) that contribute to mTORC1-dependent regulation of protein translation.[31] In contrast, mTOR complex 2 contains the rictor subunit and is neither sensitive to nutrients nor acutely inhibited by rapamycin,[9] mTORC1 controls the translation of a number of cell growth regulators, including cyclin D1, hypoxia inducible factor 1 α (HIF-1 α) and c-Myc, which in turn promote processes including cell cycle progression, cell growth, metabolism and angiogenesis, all of which can become deregulated during tumorigenesis.[9] AMPK inhibits mTORC1 through the direct phosphorylation of both tuberous sclerosis complex 2 (TSC2) and the mTORC1 scaffolding protein raptor.[32,33,34,35] TSC2 inhibits mTORC1 indirectly via regulation of the small guanine triphosphate hydrolase (GTPase) Rheb, such that loss of TSC1 or TSC2 leads to hyperactivation of mTORC1,[36] while phosphorylation of raptor was shown to be required for down regulation of mTORC1 and efficient G2/M cell cycle arrest following AMPK activation,[35] [Figure 1].
Figure 1.
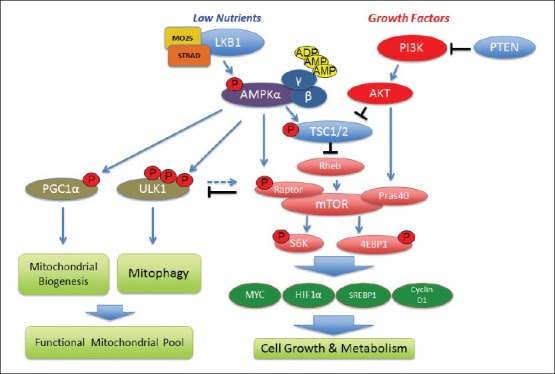
Schematic representation of the Liver Kinase B1 dependent regulation of growth, metabolism and mitochondrial homeostasis through AMP-activated protein kinase (AMPK) and mammalian target of rapamycin complex 1 (mTORC1) signalling pathways. Both tumour suppressors (in blue) and oncogenes (in red) regulate mTORC1 and are frequently mutated in cancer. mTORC1 effectors S6K and 4E-binding protein 1 and mTORC1-regulated transcription factors such as sterol regulatory element-binding protein 1 (SREBP1) and HIF1α, cMYC and Cyclin D1 (in green) are involved in cell growth, lipid and glucose metabolism and angiogenesis. AMPK suppresses mTORC1 through activation of tuberous sclerosis complex 2 and raptor and regulates mitochondrial homeostasis through peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1 α) and Unc51 like kinase 1
LKB1 INACTIVATION LEADS TO TUMORIGENESIS AND ALTERED METABOLISM
LKB1 is a frequently mutated tumour suppressor gene
Mutations in LKB1 lead to an inherited cancer syndrome known as PJS collectively belong to a group of syndromes known as phakmatoses in which patients develop benign hamartomas and are predisposed to a number of other malignancies, including breast, ovarian, endometrial and pancreatic tumors,[37] many of which have been studied in specific LKB1 mouse models [Table 1]. This increased cancer risk in PJS patients prompted the discovery that LKB1 is frequently mutated in lung and cervical cancer patients, which has intensified the focus to understanding the molecular basis of LKB1 mediated carcinogenesis.[2,4] LKB1 mutations are most prevalent in male smokers and frequently co-mutate with the Kirsten rat sarcoma viral oncogene homolog (KRAS) and p53 genes, which may in part explain the low frequency of lung cancer in PJS patients as cigarette smoke likely compounds the mutational spectrum in NSCLC, however, definitive studies directly linking cigarette smoke to LKB1 mutation have yet to be performed. Importantly, LKB1 inactivation may be an early event in carcinogenesis as LKB1 mutations have been found in early as well as late stage primary lung tumors.[38] A recent study examining organ specific deletion of LKB1 during the mouse development found that LKB1 inactivation alone lead to growth of pancreatic lesions ex vivo.[39] Studies of LKB1 inactivation in genetically engineered mouse models (GEMMs) of disease further support the idea that loss of LKB1 may in fact be an early event in carcinogenesis sufficient to drive tumour growth. GEMMs targeting LKB1 inactivation in a broad spectrum of tissue accurately mimic human tumors commonly found to bear LKB1 mutations [Table 1].
Table 1.
LKB1 mutations in human disease and modeling Lkb1 inactivation in cancer using genetically engineered mouse models (GEMMs)

LKB1-AMPK regulation of the Warburg effect in vivo
Deletion of LKB1 in human cancer cell lines and murine embryonic fibroblasts (MEFs) and gastrointestinal hamartomas from LKB1 +/- mice resulted in hyperactivation mTORC1.[34,40] Loss of function studies of LKB1 or AMPK in fibroblasts as well as epithelium from gastrointestinal hamartomas from Peutz-Jeghers patients or LKB1 +/- mice also show increased expression results in increased HIF-1 α and its targets Glucose transporter 1 (GLUT1) and hexokinase in a rapamycin-reversible manner,[41] suggesting that HIF-1 α may be a relevant target downstream of LKB1-deficiency in PJS. 18F-Fluoro-deoxyglucose positron emission tomography (18FDG-PET) imaging studies on LKB1 +/- mice showed that their gastrointestinal hamartomas have increased maximum standard uptake values (SUVmax) correlating with the increased GLUT1 expression and suggest that loss of LKB1 drives glycolytic metabolism.[41]
Recent studies in murine T and B-cells and hematopoietic stems cells (HSCs) have demonstrated the LKB1-AMPK pathway is critical to metabolism and survival.[42,43,44,45] A recent loss of function study of AMPKα1 in the Eμ-Myc murine lymphoma model and in fibroblasts confirmed the role of the LKB1-AMPK pathway in suppressing the Warburg effect as deletion of AMPKα1 resulted in increased mTORC1 and HIF1 α dependent glucose metabolism. This is the first study to identify AMPK as a potential tumour suppressor gene as inactivation of AMPKα1 -/- resulted in accelerated tumour growth of Eμ-Myc lymphomas.[46] While both T and B cells only have one copy of AMPKα1,[47] the majority of human and murine epithelial tissues expresses both α1 and α2 genes and it will be important to identify the tumour suppressor functions of both the α1 and α2 genes in various tissues. Studies of LKB1 inactivation in NSCLC likewise revealed increased mTORC1 signalling and metabolic deregulation that correlated with elevated GLUT1 expression and increased SUVmax following 18FDG-PET imaging in both human and murine lung tumors suggesting that increased glucose metabolism is a conserved phenotype in LKB1 -/- tumors.[48,49]
LKB1 mediated regulation of mitochondrial homeostasis
Recent work in HSCs and NSCLC has shed light on LKB1 role as a regulator of mitochondrial function and aerobic metabolism, where LKB1 -/- murine HSCs show mitochondrial defects including increased mitochondrial content and reduced mitochondrial membrane function.[43,44,45] It was recently discovered that AMPK directly phosphorylates the Unc51 like kinase (ULK1), a key upstream regulator of autophagy and mitochondrial turnover known as mitophagy.[50] AMPK -/- and ULK1 -/- MEFs displayed mitochondrial defects similar to LKB1 -/- HSCs and a loss of mitophagy resulting in the accumulation of defective mitochondria. Analysis of LKB1 -/- NSCLC tumour lines revealed mitochondrial defects and inactivation of the AMPK-ULK1 mitophagy signalling pathway as well as reduced oxidative phosphorylation.[48] These studies suggest that LKB1 inactivation and the resultant loss of AMPK-ULK1 regulated mitophagy may lead to aberrant mitochondrial pools compromising aerobic respiration in tumour cells. As both mitophagy and biogenesis are regulated by AMPK through peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1 α) and ULK1, the accumulation of defective mitochondria following LKB1 loss appears to reduce the respiratory capacity in tumour cells and may be another mechanism by which LKB1 inactivation drives glycolytic metabolism. Additionally, Myc overexpressing tumour cells were shown to rely on the LKB1 substrate AMPK-related protein kinase 5 (ARK5, also known as NUAK1) and the AMPK-mTORC1 signalling axis to maintain glutamine metabolism during energy stress and readily underwent apoptosis following glucose starvation or inhibition of ARK5 and mTORC1.[51] Future metabolomics studies investigating the integration of respiration, glycolysis and glutaminolysis will shed light to the global metabolic rewiring that occurs following LKB1 loss.
THERAPEUTICALLY TARGETING THE LKB1-AMPK SIGNALLING PATHWAY IN CANCER
Targeting mTOR signalling in LKB1-deficient tumors
Preclinical studies examining rapamycin in Lkb1 deficient murine models of PJS, lung and endometrial cancers have yielded mixed results. Spontaneously arising hamartomas in Lkb1-/- mice responded well.[41,52] Translation of these rapamycin studies to treat phakmatoses in clinical trials has proven successful in TSC and Lymphangioleiomyomatosis patients,[53,54] while early phase clinical trial testing rapalogs in PJS patients have suffered from low enrollment. Rapamycin as a single agent has been shown to potently inhibit growth and viability of endometrial carcinomas, ovaductal neoplasias and papillary bladder tumors in Lkb1-/- and Lkb1-/-; Pten-/- GEMMs,[55,56,57] however, in KrasG12D driven LKB1 deficient GEMMs of lung cancer rapamycin failed to induce a therapeutic response.[58] Clinical trials assessing rapalogs on advanced stage tumors have seen variable results and much of the resistance of advanced tumors to rapamycin may be due to mTORC2-AKT mediated reactivation of mTORC1 and rapalogs have performed poorly in clinical trials as tumour frequently become resistant.[59] It will be important to identify the appropriate genetic and molecular context in advanced tumors that dictate either sensitivity or resistance to rapamycin and rapalogs.
The development of next generation mTOR inhibitors that target the kinase domain of mTOR or dual mTOR/PI3K may in fact induce a greater therapeutic response with targeted cytotoxicity to mTOR dependent tumors.[60,61,62] Treatment of KRAS driven, PI3KCA mutant murine lung tumors with the dual PI3K/mTOR inhibitor BEZ235 resulted in inhibition of both PI3K and mTOR signalling and regression of primary lung tumors. Importantly, inhibition of the PI3K/mTOR pathway, lead to a dramatic reduction of 18FDG uptake suggesting a robust inhibition of glucose metabolism.[63] A recent study that examined the mTOR kinase inhibitor INK128 found this drug to dramatically inhibit mTORC1 and mTORC2 signalling in both human and murine PTEN -/- prostate tumour models that resulted in tumour regression and inhibition of epithelial-mesenchymal transition through down regulation of the mTOR-mediated translatome.[64] The predicted effectiveness of targeted mTOR kinase inhibitors against LKB1 deficient tumors of different tissues remains to be tested.
Using AMPK agonists as cancer therapeutics
AMPK's negative regulation of mTORC1 [Figure 1] and its ability to both acutely and durably rewire cellular metabolism suggest that AMPK activating drugs may be useful as cancer therapeutics.[65] One of the most commonly used AMPK agonist is the biguanide metformin (Glucophage), which is the most widely used type 2 diabetes drug in the world and taken by approximately 120 million patients daily. Several retrospective studies revealed a strong correlation between reduced cancer risk and mortality in diabetic patients taking metformin,[66,67,68] agreeing with early studies showing that biguanides suppressed naturally arising tumors in both transgenic and carcinogen treated rodent cancer models.[69,70] Metformin and its more potent analog phenformin induce energy stress through inhibition of complex I of the mitochondrial respiratory chain, resulting in elevated intracellular ADP/AMP levels and AMPK activation.[71] Biguanides are thought exert their anti-cancer effects on tumour cells in part through activation of the AMPK pathway, but in reality these drugs work through a broad spectrum of mechanisms that extend beyond the AMPK pathway solely.[72]
AMPK agonists such as metformin, phenformin, AICAR, 2-deoxyglucose and thiazolidinediones have been shown to inhibit the growth of a wide variety of tumour cells in culture and xenografts in AMPK-dependent and independent manners,[73] for a detailed review please see reference.[74] Other widely used metabolic stress agents and AMPK activators include the natural product berberine found in green tea and salicylic acid (aspirin) found in most bathroom medicine cabinets. Recently, aspirin was identified as a direct AMPK activator,[75] and strikingly, colorectal cancer patients with PI3KCA mutations who took aspirin showed a significant reduction in mortality.[76] A separate study found increased AMPK activation and reduced mTORC1 signalling in colorectal cancer biopsies samples from patients taking aspirin daily,[77] suggesting that inhibition of PI3K/mTOR signalling via aspirin mediated activation of AMPK may lie at the heart of such striking results.
Inhibiting tumour cell growth and proliferation through AMPK activation in LKB1-/- tumour cells can also be achieved by AMPK agonists that work independently of LKB1. The small molecule Abbott A769662 is an allosteric activator of AMPK that can directly activate AMPK independently of LKB1, however, the exact region of the AMPK complex to which A769662 binds is unknown.[78,79] A769662 showed in vivo activation of AMPK and anti-tumour efficacy in a Pten +/- model of lymphoma.[80] Selective killing of LKB1 -/- tumour cells can also be achieved by exploiting metabolic and oxidative stress pathways that become deregulated upon inactivation of the LKB1/AMPK pathway. Tumour cells lacking LKB1 are hypersensitive to apoptosis in culture following treatment with energy stress inducing agents, presumably originating from an inability to restore ATP levels due to AMPK deficiency.[8] We have recently shown that LKB1 loss selectively sensitizes human and murine models of NSCLC to phenformin through metabolic and oxidative stress,[48] [Figure 2]. Our results agree with recent studies that showed AMPK mediates cell survival not only through maintaining cellular ATP but also by restoring nicotinamide adenine dinucleotide phosphate (NADPH) levels required to quench reactive oxygen species that accumulate during periods of metabolic stress.[83] Expanding the use of phenformin beyond LKB1 deficient lung tumors, a recent study found that phenformin significantly inhibited growth of estrogen receptor positive and triple negative breast cancer cells in xenograft models.[81] Phenformin was withdrawn from clinical use in the late 1970s due to the occurance of lactic acidosis in a subset of diabetics on the drug,[82] however, it may find modern use as an anti-cancer agent as the dosing and duration of its use for cancer would be quite different from its use for diabetes.[84] Given the known pharmacokinetics and widespread long-term clinical use of biguanides, the potential for metabolic therapies to be repurposed as chemotherapies to target LKB1 deficient tumors warrants further attention.
Figure 2.
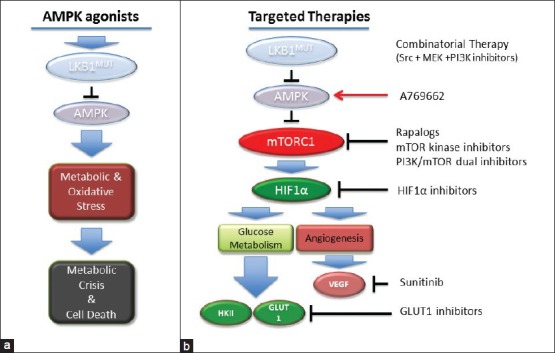
Therapeutically targeting the AMP-activated protein kinase (AMPK) and mammalian target of rapamycin complex (mTORC1) signaling pathways in liver kinase B1 (LKB1) -/- tumors. Schematic representation of therapeutic strategies targeting AMPK and the mTORC1 signaling pathway following LKB1 inactivation. (a) A representation of AMPK agonists that mimic cellular energy stress creating metabolic and oxidative stress that results in tumor cell death. (b) A representation of therapies targeting critical upstream and downstream effectors of mTORC1 in LKB1 -/- tumors
Translational outlook – the promise and the reality
Before LKB1 -/- tumors can ever be successfully treated with targeted or metabolic therapies it must become common practice to perform genetic screening for LKB1 mutations in tumour types with a high frequency of LKB1 mutations. The elevated glycolytic metabolism of LKB1-deficient tumors in patients should allow these tumors to be readily imaged by 18FDG-PET. 18FDG uptake need not only be viewed as a surrogate end stage biomarker but as a diagnostic tool providing details to tumour metabolism and therapeutic response following treatment.[48,49] AMPK activating drugs and metabolic therapies, like most targeted therapeutics, are expected to be most effective against tumors of specific genotypes. The relative success of biguanides in preclinical studies must be taken cautiously as many studies are performed at supra-physiological concentrations in cell culture and rodent cancer models that will never be achieved in man.[84] Therefore, careful studies of biguanides and other AMPK agonists at physiologically relevant concentrations are needed to understand their true potential. Given the multitude of deregulated effectors following LKB1 inactivation, it is likely that a combination of therapies will be required. Combining PI3K and MEK inhibitors with the Src inhibitor dasatinib in murine KrasG12D; Lkb1 -/- (KL) GEMMs induced a potent tumour response in non-small cell lung tumors and to a lesser degree in melanomas.[85,86] The tyrosine kinase and VEGF receptor inhibitor sunitinib decrease tumor burden, increased tumor necrosis and prolonged survival in KL GEMMs.[89] The elevated expression of both HIF1 α and GLUT1 in LKB1 -/- tumors suggests both these proteins may be likely candidates for targeted therapies such as the small molecule inhibitor of GLUT1, STF31, which was recently identified for the treatment of renal cell carcinoma,[87,88] [Figure 2]. The advent of recent phase I/II clinical trials combining metformin and chemotherapies for metastatic breast cancer (Clinical Trial NCT01310231) along with the completion of recent clinical trials testing rapalogs and next generation PI3K/mTOR inhibitors should provide valuable insight into the effectiveness and future of these drugs as realistic therapies for the treatment of LKB1 deficient cancer.[59] Achieving personalized treatments will require defining which oncogenic genotypes sensitize tumors to AMPK activating or mTOR inhibiting drug treatments and is an important goal for future studies.
AUTHOR'S PROFILE
Dr. David B. Shackelford: Is an assistant professor in the Department of Pulmonary and Critical Care Medicine, David Geffen School of Medicine at University of California, Los Angeles, California, USA.
ACKNOWLEDGMENT
The author would like to acknowledge the UCLA David Geffen School of Medicine and the Department of Pulmonary and Critical Care Medicine for support of his research as well as Dr. Reuben J. Shaw for his years of valuable mentorship and training. Dr. Shackelford is funded by the NIH/CTSA UCLA-CTSI Translational Science Awards grant numbers KL2TR000122 and UL1TR000124 as well as the Bonnie J. Addario Lung Cancer Research Foundation and Kure-It Cancer Research Foundation.[106]
Footnotes
This article is available from: http://www.carcinogenesis.com/content/12/1/16
Source and Support: The author supported by the NIH/CTSA UCLA-CTSI Translational Science Award grant number KL2TR000122 and UL1TR000124
Conflict of Interest: None declared.
REFERENCES
- 1.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–7. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–62. [PubMed] [Google Scholar]
- 3.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wingo SN, Gallardo TD, Akbay EA, Liang MC, Contreras CM, Boren T, et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS One. 2009;4:e5137. doi: 10.1371/journal.pone.0005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci U S A. 2003;100:8839–43. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–8. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 8.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 11.Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, et al. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–72. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeqiraj E, Filippi BM, Deak M, Alessi DR, van Aalten DM. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 2009;326:1707–11. doi: 10.1126/science.1178377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, et al. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–14. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–43. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaleel M, McBride A, Lizcano JM, Deak M, Toth R, Morrice NA, et al. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 2005;579:1417–23. doi: 10.1016/j.febslet.2005.01.042. [DOI] [PubMed] [Google Scholar]
- 16.Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: Also regulated by ADP? Trends Biochem Sci. 2011;36:470–7. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–3. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–5. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- 19.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–6. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 22.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223:217–22. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 23.Sato R, Goldstein JL, Brown MS. Replacement of serine-871 of hamster 3-hydroxy-3-methylglutaryl-CoA reductase prevents phosphorylation by AMP-activated kinase and blocks inhibition of sterol synthesis induced by ATP depletion. Proc Natl Acad Sci U S A. 1993;90:9261–5. doi: 10.1073/pnas.90.20.9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- 25.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–55. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 26.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardie DG, Ross FA, Hawley SA. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex1. Mol Cell. 2010;39:171–83. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 31.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–80. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 32.Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–8. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 34.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, Manning BD. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem J. 2008;412:179–90. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007;26:7825–32. doi: 10.1038/sj.onc.1210594. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene. 2007;26:5911–8. doi: 10.1038/sj.onc.1210418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lo B, Strasser G, Sagolla M, Austin CD, Junttila M, Mellman I. Lkb1 regulates organogenesis and early oncogenesis along AMPK-dependent and-independent pathways. J Cell Biol. 2012;199:1117–30. doi: 10.1083/jcb.201208080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE, et al. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–7. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 41.Shackelford DB, Vasquez DS, Corbiel J, Wu S, Leblanc M, Wu CL, et al. mTOR-dependent regulation of polyposis and HIF-1 α mediated glucose metabolism in an LKB1 mouse model of peutz-jeghers syndrome. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0900465106. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. 2011;187:4187–98. doi: 10.4049/jimmunol.1100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–8. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–4. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–63. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–24. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamás P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+in T lymphocytes. J Exp Med. 2006;203:1665–70. doi: 10.1084/jem.20052469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–58. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–7. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L, Ulbrich J, Müller J, Wüstefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483:608–12. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- 52.Wei C, Amos CI, Zhang N, Wang X, Rashid A, Walker CL, et al. Suppression of Peutz-Jeghers polyposis by targeting mammalian target of rapamycin signaling. Clin Cancer Res. 2008;14:1167–71. doi: 10.1158/1078-0432.CCR-07-4007. [DOI] [PubMed] [Google Scholar]
- 53.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–51. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shorning BY, Griffiths D, Clarke AR. Lkb1 and Pten synergise to suppress mTOR-mediated tumorigenesis and epithelial-mesenchymal transition in the mouse bladder. PLoS One. 2011;6:e16209. doi: 10.1371/journal.pone.0016209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanwar PS, Kaneko-Tarui T, Zhang L, Tanaka Y, Crum CP, Teixeira JM. Stromal liver kinase B1 STK11 signaling loss induces oviductal adenomas and endometrial cancer by activating mammalian Target of Rapamycin Complex1. PLoS Genet. 2012;8:e1002906. doi: 10.1371/journal.pgen.1002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Contreras CM, Akbay EA, Gallardo TD, Haynie JM, Sharma S, Tagao O, et al. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Dis Model Mech. 2010;3:181–93. doi: 10.1242/dmm.004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang MC, Ma J, Chen L, Kozlowski P, Qin W, Li D, et al. TSC1 loss synergizes with KRAS activation in lung cancer development in the mouse and confers rapamycin sensitivity. Oncogene. 2010;29:1588–97. doi: 10.1038/onc.2009.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–41. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, et al. Targeted polypharmacology: Discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–9. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 63.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 66.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–8. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 68.Pollak M. Metformin and other biguanides in oncology: Advancing the research agenda. Cancer Prev Res (Phila) 2010;3:1060–5. doi: 10.1158/1940-6207.CAPR-10-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schneider MB, Matsuzaki H, Haorah J, Ulrich A, Standop J, Ding XZ, et al. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120:1263–70. doi: 10.1053/gast.2001.23258. [DOI] [PubMed] [Google Scholar]
- 70.Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685–93. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 71.Hardie DG. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology. 2006;131:973. doi: 10.1053/j.gastro.2006.07.032. author reply 974-5. [DOI] [PubMed] [Google Scholar]
- 72.Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, et al. Metformin: Multi-faceted protection against cancer. Oncotarget. 2011:96–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–22. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367:1596–606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Din FV, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K, et al. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012;142:1504–15. doi: 10.1053/j.gastro.2012.02.050. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–16. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 79.Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008;15:1220–30. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 80.Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–21. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 81.Appleyard MV, Murray KE, Coates PJ, Wullschleger S, Bray SE, Kernohan NM, et al. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br J Cancer. 2012;106:1117–22. doi: 10.1038/bjc.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–14. [PMC free article] [PubMed] [Google Scholar]
- 83.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–5. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: Translational challenges. J Mol Endocrinol. 2012;48:R31–43. doi: 10.1530/JME-12-0007. [DOI] [PubMed] [Google Scholar]
- 85.Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–59. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu W, Monahan KB, Pfefferle AD, Shimamura T, Sorrentino J, Chan KT, et al. LKB1/STK11 inactivation leads to expansion of a prometastatic tumor subpopulation in melanoma. Cancer Cell. 2012;21:751–64. doi: 10.1016/j.ccr.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3:94ra70. doi: 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gandhi L, McNamara KL, Li D, Borgman CL, McDermott U, Brandstetter KA, et al. Sunitinib prolongs survival in genetically engineered mouse models of multistep lung carcinogenesis. Cancer Prev Res (Phila) 2009;2:330–7. doi: 10.1158/1940-6207.CAPR-08-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miyoshi H, Nakau M, Ishikawa TO, Seldin MF, Oshima M, Taketo MM. Gastrointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice. Cancer Res. 2002;62:2261–6. [PubMed] [Google Scholar]
- 91.Jishage K, Nezu J, Kawase Y, Iwata T, Watanabe M, Miyoshi A, et al. Role of Lkb1, the causative gene of Peutz-Jegher's syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci U S A. 2002;99:8903–8. doi: 10.1073/pnas.122254599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rossi DJ, Ylikorkala A, Korsisaari N, Salovaara R, Luukko K, Launonen V, et al. Induction of cyclooxygenase-2 in a mouse model of Peutz-Jeghers polyposis. Proc Natl Acad Sci U S A. 2002;99:12327–32. doi: 10.1073/pnas.192301399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Katajisto P, Vaahtomeri K, Ekman N, Ventelä E, Ristimäki A, Bardeesy N, et al. LKB1 signaling in mesenchymal cells required for suppression of gastrointestinal polyposis. Nat Genet. 2008;40:455–9. doi: 10.1038/ng.98. [DOI] [PubMed] [Google Scholar]
- 94.Nakau M, Miyoshi H, Seldin MF, Imamura M, Oshima M, Taketo MM. Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res. 2002;62:4549–53. [PubMed] [Google Scholar]
- 95.Wei C, Amos CI, Stephens LC, Campos I, Deng JM, Behringer RR, et al. Mutation of Lkb1 and p53 genes exert a cooperative effect on tumorigenesis. Cancer Res. 2005;65:11297–303. doi: 10.1158/0008-5472.CAN-05-0716. [DOI] [PubMed] [Google Scholar]
- 96.Takeda H, Miyoshi H, Kojima Y, Oshima M, Taketo MM. Accelerated onsets of gastric hamartomas and hepatic adenomas/carcinomas in Lkb1+/-p53-/-compound mutant mice. Oncogene. 2006;25:1816–20. doi: 10.1038/sj.onc.1209207. [DOI] [PubMed] [Google Scholar]
- 97.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–10. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 98.Cancer Genome Atlas Research Network. Hammerman et al., Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Contreras CM, Gurumurthy S, Haynie JM, Shirley LJ, Akbay EA, Wingo SN, et al. Loss of Lkb1 provokes highly invasive endometrial adenocarcinomas. Cancer Res. 2008;68:759–66. doi: 10.1158/0008-5472.CAN-07-5014. [DOI] [PubMed] [Google Scholar]
- 100.McCarthy A, Lord CJ, Savage K, Grigoriadis A, Smith DP, Weigelt B, et al. Conditional deletion of the Lkb1 gene in the mouse mammary gland induces tumour formation. J Pathol. 2009;219:306–16. doi: 10.1002/path.2599. [DOI] [PubMed] [Google Scholar]
- 101.Partanen JI, Tervonen TA, Myllynen M, Lind E, Imai M, Katajisto P, et al. Tumor suppressor function of Liver kinase B1 (Lkb1) is linked to regulation of epithelial integrity. Proc Natl Acad Sci U S A. 2012;109:E388–97. doi: 10.1073/pnas.1120421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pearson HB, McCarthy A, Collins CM, Ashworth A, Clarke AR. Lkb1 deficiency causes prostate neoplasia in the mouse. Cancer Res. 2008;68:2223–32. doi: 10.1158/0008-5472.CAN-07-5169. [DOI] [PubMed] [Google Scholar]
- 103.Hezel AF, Gurumurthy S, Granot Z, Swisa A, Chu GC, Bailey G, et al. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol Cell Biol. 2008;28:2414–25. doi: 10.1128/MCB.01621-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gurumurthy S, Hezel AF, Sahin E, Berger JH, Bosenberg MW, Bardeesy N. LKB1 deficiency sensitizes mice to carcinogen-induced tumorigenesis. Cancer Res. 2008;68:55–63. doi: 10.1158/0008-5472.CAN-07-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qiu W, Schönleben F, Thaker HM, Goggins M, Su GH. A novel mutation of STK11/LKB1 gene leads to the loss of cell growth inhibition in head and neck squamous cell carcinoma. Oncogene. 2006;25:2937–42. doi: 10.1038/sj.onc.1209325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morton JP, Jamieson NB, Karim SA, Athineos D, Ridgway RA, Nixon C, et al. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology. 2010;139:586–97. doi: 10.1053/j.gastro.2010.04.055. 597e1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]