

Abstract
Chk1 plays a key role in the DNA replication checkpoint and in preserving genomic integrity. Previous studies have shown that reduced Chk1 function leads to defects in the checkpoint response and is closely associated with tumorigenesis. Here, we report that glucose deprivation caused the degradation of Chk1 protein without perturbing cell cycle progression. The induction of Chk1 degradation in response to glucose deprivation was observed in various cancer cell lines and in normal human fibroblasts. Therefore, it appears to be a universal phenomenon in mammalian cells. A specific proteasome inhibitor blocked glucose deprivation-induced Chk1 degradation. Ubiquitination of Chk1 was detected, indicating that the proteasome-ubiquitin pathway mediates Chk1 degradation upon glucose deprivation. Mechanistic studies have demonstrated that ATR-dependent phosphorylation of Chk1 at the Ser317 and Ser345 sites is not required, suggesting that the molecular mechanism for Chk1 degradation upon glucose deprivation is distinct from genotoxic stress-induced degradation. Under conditions of glucose deprivation, the cells manifested a defective checkpoint response to replication stress, camptothecin or hydroxyurea. The forced expression of Myc-Chk1 partially rescued the defective response to the replication block upon glucose deprivation. Taken together, our results indicate that glucose deprivation induces ubiquitin-mediated Chk1 degradation and defective checkpoint responses, implying its potential role in genomic instability and tumor development.
Keywords: Chk1, glucose deprivation, protein stability, ubiquitin-proteasome pathway, genomic instability
1. Introduction
Faithful DNA replication and segregation are essential steps for normal cell growth and the maintenance of genomic integrity. Mammalian cells have conserved checkpoint pathways to ensure accurate DNA replication and to monitor DNA damage [1]. The ATM-Chk2 and ATR-Chk1 pathways represent major checkpoints that respond to a stalled replication fork or DNA damage and regulate various cellular responses, including cell cycle arrest, DNA repair, and apoptosis [2]. Inactivation of checkpoint pathways or suppression of important checkpoint regulators, including Chk1 and Chk2, frequently results in tumorigenesis or induces malignant tumor progression [2–4].
Chk1 was first identified in fission yeast as an essential kinase involved in DNA damage-induced cell cycle arrest [5]. Subsequent studies revealed that Chk1 also plays a critical role in the cellular response to genotoxic stresses in other organisms, including Xenopus and the mouse as well as in human cells [6]. Studies also revealed that Chk1 function is essential for maintaining DNA replication checkpoints in unperturbed cell cycles. Fission yeast cells lacking Chk1 progressed to mitosis with unreplicated DNA [7, 8]. Mouse embryos and embryonic stem (ES) cells lacking Chk1 exhibited defective checkpoint responses to replication blocks [6, 9]. More importantly, a recent study demonstrated that Chk1 heterozygosity (Chk+/−) induced abnormal cell cycle coordination caused by defective DNA replication [10]. These studies have established that Chk1 plays a pivotal role in normal cell cycle control and that maintaining proper Chk1 levels is essential to DNA integrity and tumor suppression.
In response to replication blocks or DNA damage, Chk1 is activated mainly by the upstream PI-3 kinase, ATR, through phosphorylation at Ser317 and Ser345 [6, 11]. The activated Chk1 phosphorylates downstream targets cdc25A and cdc25C and thereby induces cell cycle arrest at the S and G2/M phases, respectively [2]. After DNA repair is completed, the checkpoint signaling must be terminated to return cells to a homeostatic state. Previous studies have shown that the protein phosphatases PP1, PP2A, and PPM1D/Wip1 dephosphorylate activated Chk1 and promote termination of the checkpoint response [12–14]. In addition to Chk1 deactivation through dephosphorylation, recent studies have shown that Chk1 is deactivated through proteolytic degradation upon genotoxic treatment and replication stress [15–19]. Interestingly, these studies revealed that the phosphorylation of Chk1 at Ser 317 and Ser345 not only promotes full activation of Chk1, but also induces degradation of Chk1 through the ubiquitin-proteasome pathway. These studies indicated that Chk1 function is strictly regulated through complicated activation and inactivation pathways that maintain proper cell proliferation and DNA integrity. Therefore, it is not surprising that abrogation of normal Chk1 function is a critical step for tumorigenesis or tumor progression [1, 2]. Given the essential role of Chk1 in normal cell growth and tumorigenesis, unveiling how Chk1 function is suppressed during tumorigenesis and tumor development is an important issue.
Glucose concentrations are strictly maintained at about 5 mM under physiological conditions, but glucose deprivation develops under conditions in which the blood supply is restricted such as in tissue ischemia and solid tumors [20, 21]. Previous studies have shown that glucose concentrations are decreased to less than 2 mM within solid tumor masses [20, 22, 23]. The tumor microenvironment has been implicated as an inducer of genomic instability and tumor progression [24–26]; however, the effect of glucose deprivation on checkpoint function is largely unknown. In this report, we describe our finding that Chk1 stability is regulated in a glucose concentration-dependent manner. Our data suggest that glucose deprivation induces Chk1 degradation through the ubiquitin-proteasome pathway and induces defective checkpoint responses in response to replication blocks.
2. Materials and methods
2. 1 Cell culture, glucose deprivation and reagents
A549, HEK293, U2OS, Hela, HepG2, and IMR90 cell lines were maintained in DMEM-high glucose media (Invitrogen) containing 4 mM glutamine and 10 % fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA). For glucose deprivation, cells were washed three times with DPBS and replaced with glucose-free DMEM (Invitrogen) containing 4 mM glutamine and 10 % dialyzed FBS. To expose cells to low glucose conditions, sterile glucose solution (0.2 g/ml) was added into the medium at the desired final concentration.
Cycloheximide (CHX), MG132, camptothecin (CPT), hydroxyurea (HU), and caffeine were purchased from Sigma-Aldrich Co. (St. Louis, MO). Wortmannin was purchased from Calbiochem (San Diego, CA).
2.2 Western blot analyses and antibodies
Cells harvested after treatment were lysed in RIPA buffer and subjected to western blot analyses, as previously described [27]. Antibodies for Chk1 (G-4), ubiquitin (P4D1), and Myc (9E10) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for Chk2 (Clone-7) were obtained from Upstate (Billerica, MA). Antibodies for phospho-Chk1 (S317), phospho-Chk1 (S345), phospho-Chk2 (T68), and ATR were obtained from Cell Signaling Technology Inc. (Danvers, MA). Actin levels were monitored as internal loading controls using anti-actin (Sigma) antibodies. The western blot analyses were repeated three times. Band intensities were quantified by densitometry using AlphaEaseFC 4.0 software (Alpha Innotech). Relative band intensities were calculated based on the band intensity of untreated samples after normalization to the actin signal.
2.3 Synchronizations and Cell cycle analyses
To synchronize A549 cells to G1/S boarder, sub-confluent cells were treated with 2 mM thymidine for 16 h and released by washing three times with pre-warmed PBS. After grown for 10 h in complete medium, 2 mM thymidine was added again and the cells were cultured for an additional 16 h. Cells were washed with PBS and released into complete medium containing indicated concentration of glucose. To synchronize at G0 phase, A549 cells were cultured in the medium containing 0.1% FBS for 72 h and released into serum-enriched media.
To determine the cell cycle distribution, 1 × 106 cells were seeded into a 100 mm dish. After glucose deprivation treatments, cells were trypsinized at various time points and fixed with 70% ethanol. Subsequently, cells were stained with propidium iodide (PI), and analyzed by flow cytometry using EPICS XL cytometer and WINCYCLE software (Beckman Coulter Inc.). A total of 10,000 events were analyzed for each sample, and the experiment was repeated at least twice.
2.4 Semi-quantitative RT-PCR analyses
For semi-quantitative reverse transcriptase (RT)-PCR analyses, total RNA was isolated using TRIZOL reagent (Invitrogen) according to the manufacturer’s instructions. The cDNA was obtained using Moloney murine leukemia virus reverse transcriptase (MMLV-RT) and oligo-dT primers (Promega, Madison, WI). PCR was carried out with Accupower PCR premix (Bioneer Co., Taejon, Korea) using cDNA as template. PCR products were separated by 2 % agarose gel electrophoresis. PCR conditions were as follows: one cycle at 95 °C for 5 min, followed by 25 cycles at 95 °C for 30 sec, 60 °C for 30 sec, and 72 °C for 30 sec. The primers employed in RT-PCR were as follows: Chk1 forward primer 5′-CTTTGGCTTGGCAACAGT-3′, Chk1 reverse primer 5′-CCAGTCAGAATACTCCTG-3′, Chk2 forward primer 5′-GCGCCTGAAGTTCTTGTTTC-3′, Chk2 reverse primer 5′-GCCTTTGGATCCACTACCAA-3′, actin forward primer 5′-CAAGAGATGGCCACGGCTGCT-3′, and actin reverse primer 5′-TCCTTCTGCATCCTGTCGGCA-3′. Experiments were repeated at least three times.
2.5 Plasmids, transfection and establishment of cell lines
An expression plasmid encoding Flag-Chk1 was kindly provided by Dr. Yolanda Sanchez (University of Cincinnati, Cincinnati, OH). The HA-tagged ubiquitin expression plasmid (HA-pCS2-FA-Ub) has been previously described [28]. Expression plasmids encoding Myc-tagged wild-type, S317A, and S345A Chk1 were kindly provided by Dr. Helen Piwnica-Worms (Washington University, St. Louis, MO). DNA transfections were performed using Lipofectin reagent (Invitrogen) according to the manufacturer’s instructions. To generate HEK293 cells stably expressing Myc-Chk1 wild type, S317A, and S345 mutants, the pcDNA3 control vector or Myc-Chk1 plasmid were transfected and cells were selected with 500 μg/ml of neomycin for 2 weeks. After selection, resistant cells were pooled and the expression of Myc-Chk1 was confirmed by western blotting.
2.6 Ex vivo ubiquitination assays
Chk1 ex vivo ubiquitination assays were performed as previously described [28]. Briefly, A549 or HEK293 cells were transfected with 10 μg of expression plasmid for Flag-tagged Chk1 and HA-ubiquitin. After 24 h, the cells were exposed to glucose deprivation together with MG132 (10 μM) or DMSO vehicle for 16 h, lysed with sodium dodecyl sulfate (SDS) buffer, and boiled for 5 min. The extracts were diluted 10-fold with buffer A containing proteasome inhibitors and 10 mM N-ethylmaleimide (Sigma) and then immunoprecipitated with anti-Flag or anti-Myc antibodies. The immunoprecipitated protein was immunoblotted with anti-ubiquitin antibodies. Experiments were repeated at least three times.
2.7 RNA interference
An siRNA pool specific for ATR (ON-TARGETplus SMARTpool Human ATR, L-003202000005) and control siRNA (ON-TARGETplus Not-targeting siRNA, D-0018100105) were obtained from Dharmacon (Lafayette, CO). To knockdown ATR expression, A549 cells (4 × 105) were seeded into a 60-mm dish and siRNA was transfected using DharmaFECT1 transfection reagent (Dharmacon) according to the manufacturer’s instructions.
2.8 Fluorescence micronucleus analysis
Cells were grown on cover slides and fixed with 3 % paraformaldehyde for 10 min at room temperature. Cells were then permeabilized with 0.2 % Triton X-100 and stained with DAPI (4,6-diamino-2-phenylindole, Sigma). The nucleus was examined at 200× magnification using a fluorescence microscope (Olympus BX50F, Olympus Optical Co. Ltd, Tokyo). After capturing nuclear images by CCD camera (Olympus DP70), 200 to 500 cells for each sample in several fields were examined and the number of micronucleated cells or multinucleated cells was counted.
3. Results
3.1 Glucose deprivation cause a reduction in Chk1protein levels
Over the course of our studies on Chk1 function, we observed that Chk1 levels were significantly reduced under low glucose conditions. To investigate the effect of glucose concentration on Chk1 function, we first examined Chk1 protein levels in A549 cells that had been exposed to various concentrations of glucose for 12 h. We found that the amount of Chk1 was significantly decreased when cells were cultured in medium containing less than 2 mM glucose compared to normal physiological glucose levels (5 mM), but high glucose concentrations (10 mM) did not influence Chk1 levels (Fig. 1A). A time-course study revealed that the amount of Chk1 was reduced to less than 10 % by 12 h after glucose deprivation (0 mM) (Fig. 1B). Conversely, levels of Chk2, another important checkpoint kinase, were not changed under any of the glucose concentration conditions tested. Semi-quantitative RT-PCR results showed that Chk1 mRNA levels were unchanged following glucose deprivation (0 mM) (Fig. 1C), suggesting that the reduction in Chk1 upon glucose deprivation is regulated by a posttranscriptional mechanism. A similar reduction in Chk1 protein levels, without changes in Chk1 mRNA levels, following glucose deprivation (0 mM) was also observed in HEK293 cells (Supplementary Fig. S1A and B). The reduction in levels of Chk1 upon glucose deprivation was restored by re-addition of glucose (Fig. 1D), suggesting that Chk1 protein levels are reversibly regulated by glucose concentration.
Fig. 1. Chk1 protein levels are regulated by glucose concentration.
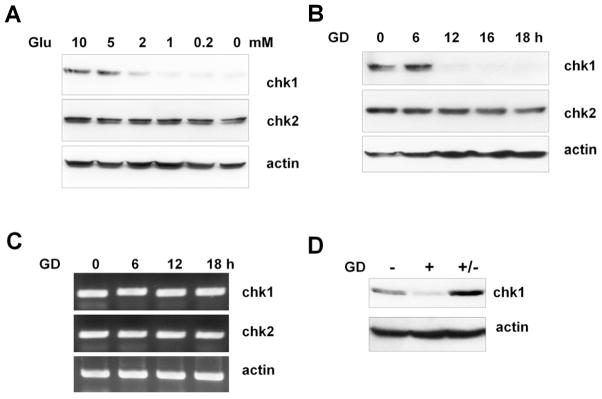
(A) A549 cells were cultured for 12 h in media containing various concentrations of glucose (Glu) and harvested. Cell lysates were subjected to western blotting using anti-Chk1, anti-Chk2 and anti-actin antibodies. B–D. A549 cells were exposed to glucose deprivation (GD, 0 mM) conditions and harvested at the indicated time points for western blotting (B), and RT-PCR (C). RT-PCR analyses were performed as described in the Materials and Methods. (D) A549 cells were cultured for 12 h in 10 mM glucose (−) or 0 mM glucose (+). A549 cells exposed to glucose deprivation were then changed to high glucose (10 mM) media and subsequently cultured for an additional 24 h. Cell lysates were subjected to western blotting. The data shown are representative of three independent experiments.
3.2 Chk2 levels are decreased by glucose deprivation independently of an effect on cell cycle
Chk1 levels are regulated in a cell cycle-dependent manner [29, 30]. Because glucose deprivation often induces cell cycle arrest and cell death, we examined whether Chk1 reduction following glucose deprivation results from altered cell cycle distribution or cell death. Cell cycle analyses after glucose deprivation (0 mM) showed no significant changes in A549 cells until at least 18 h after glucose deprivation (Supplementary Fig. S2). HEK293 cells also showed no changes to the cell cycle profile upon glucose deprivation (0 mM) until 18 h (Supplementary Fig. S1C). Cell proliferation assays employing A549 cells exposed to various glucose concentrations (from 10 mM to 0 mM) showed that cell death was observed only when cells were cultured with glucose concentrations below 0.2 mM. In addition, the cells proliferated continuously, albeit growth rate is little bit decreased, at glucose concentrations over 1 mM and showed no signs of cell death (Supplementary Fig. S3A). However, in 1 mM and 2 mM glucose condition, decreased Chk1 protein levels were evident (Supplementary Fig. S3B).
To avoid possible complications of glucose deprivation on cell cycle progression, we synchronized A549 cells at the G1/S border by double thymidine block and released the cells into medium containing high glucose (10 mM) or glucose deprivation conditions (1 mM). Whereas cell cycle propagation after release from thymidine block was not affected by glucose deprivation (Supplementary Fig. S4A), Chk1 levels were significantly reduced in cells exposed to glucose deprivation conditions (Fig. 2A). Next, A549 cells were synchronized by serum deprivation for 72 h. Consistent with a previous study [29], Chk1 protein was hardly detectable after serum starvation and increased 16 h after serum stimulation (Fig. 2B). While cell cycle propagation was not affected (Supplementary Fig. S4B), glucose deprivation inhibited the increase in Chk1 level after serum stimulation (Fig. 2B). Using these collective data, we concluded that the reduction of Chk1 protein observed following glucose deprivation is not the result of either cell cycle arrest or cell death.
Fig. 2. Reduced Chk1 in response to glucose deprivation is cell cycle-independent.
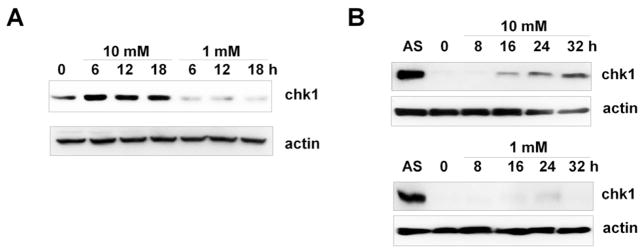
(A) A549 cells were synchronized by double thymidine block and released into media containing high glucose (10 mM) or glucose deprived (1 mM), as described in the Materials and Methods. Cell lysates were prepared at the indicated time points and subjected to western blotting using anti-Chk1 and anti-actin antibodies. (B) A549 cells were synchronized by culturing in 0.1% FBS medium for 72 h and subsequently restimulated with 10 % FBS containing high glucose (10 mM) or low glucose (1 mM). Cell lysates were prepared at the indicated time points and subjected to western blotting using the indicated antibodies. AS denotes asynchronous cells.
3.3 Reduced levels of Chk1 in response to glucose deprivation is a universal phenomenon
To further confirm that glucose deprivation induced Chk1 degradation and to avoid possible artifacts of using specific cell lines, we examined Chk1 protein expression after glucose deprivation in various cell lines. As shown in Fig. 2, the amount of Chk1 protein was also significantly decreased after 12 h of glucose deprivation (0 mM) in all cancer cell lines examined, including HEK293, U2OS, Hela, and HepG2 cells. Similarly, reduced levels of Chk1 following glucose deprivation were also observed in normal lung epithelial IMR90 cells. These results suggest that Chk1 degradation in response to glucose deprivation is a universal phenomenon.
3.4 Chk1 protein stability is decreased upon glucose deprivation
Next, to investigate the molecular mechanisms by which glucose deprivation causes reduction in Chk1 levels, we examined the half-life of the Chk1 protein after glucose deprivation. To examine whether Chk1 protein stability is modulated by glucose deprivation, we measured Chk1 turnover rates under high glucose (10 mM) or glucose deprivation conditions (0 mM) in the presence of a protein synthesis inhibitor, cycloheximide. Consistent with a previous report [19], we found that Chk1 has a half-life of approximately 4 h in 10 mM glucose (Fig. 3). However, the half-life of Chk1 was decreased to less than 2 h under glucose deprivation conditions (0 mM). These results indicate that glucose deprivation-induced downregulation of Chk1 is due to degradation of Chk1 protein.
Fig. 3. Glucose deprivation leads to reduced Chk1 levels in various cell lines.

Various human cancer cell lines and the normal human lung epithelial cell line IMR90 were exposed to glucose deprivation (GD; 0 mM) and harvested at the indicated time points. Cell lysates were subjected to western blot analyses using anti-Chk1 and anti-actin antibodies. The western blot analyses were repeated three times.
3.5 Chk1 is degraded through the ubiquitin-proteasome pathway in response to glucose deprivation
Rapid protein degradation is usually mediated by the ubiquitin-proteasome pathway [31, 32]. To test whether Chk1 degradation is mediated by this pathway in response to glucose deprivation, we examined the effect of MG132, a potent proteasome inhibitor, on Chk1 degradation in A549 and HEK93 cells under glucose deprivation conditions. As shown in Fig. 4A and 4B, glucose deprivation-induced Chk1 degradation was completely abolished by MG132 treatment in A549 and HEK293 cells, respectively. In addition, MG132 treatment completely inhibited glucose deprivation-induced degradation of transiently expressed Flag-Chk1 (Fig. 4C).
Fig. 4. The half-life of Chk1 protein is decreased upon glucose deprivation.

A549 cells were cultured in either 10 mM or 0 mM glucose for 12 h. Cells were then treated with cycloheximide (CHX) (50 μg/ml) and harvested at the indicated time points. Cell lysates were subjected to western blotting using anti-Chk1 and anti-actin antibodies. Band intensities were quantified by densitometry, and the band intensity values were calculated relative to the band intensity of the 0 h sample after normalization to the actin signal. The band intensity of Chk1 from the 0 h sample was defined as 1.
Proteasome-dependent degradation usually requires ubiquitination of the substrate protein for it to be recognized by the 26S proteasome [33]. To examine whether Chk1 is ubiquitinated upon glucose deprivation, we performed ex vivo ubiquitination assays using Flag-Chk1. Following glucose deprivation (0 mM) in both A549 and HEK293 cells, immunoprecipitation with anti-Flag antibodies followed by immunoblot analyses with anti-ubiquitin antibodies revealed the presence of high-molecular-mass smears, which are a common characteristic of polyubiquitinated proteins (Fig. 5D). Together, these results suggest that Chk1 is ubiquitinated in response to glucose deprivation and degraded by the 26S proteasome.
Fig. 5. The ubiquitin-proteasome pathway mediates Chk1 degradation in response to glucose deprivation.
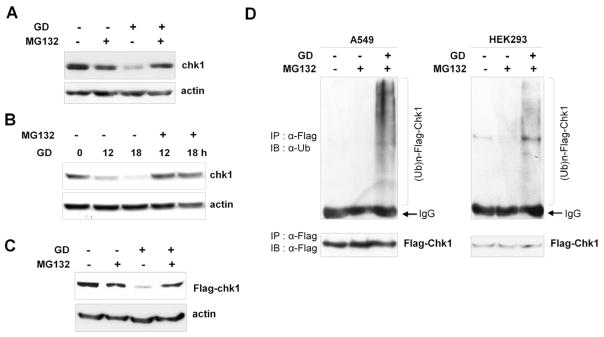
(A) A549 cells were cultured in either 10 mM glucose or glucose deprivation (GD) conditions (0 mM) in the presence or absence of MG132 (10 μM) for 16 h. Cell lysates were subjected to western blotting using anti-Chk1 and anti-actin antibodies. (B) HEK293 cells were cultured as in (A), and cell lysates were harvested at the indicated time points. Western blot analyses were performed using anti-Chk1 and anti-actin antibodies. (C) HEK293 cells were transfected with a Flag-Chk1 plasmid and cultured as in (A). Cell lysates were subjected to western blotting using anti-Chk1 and anti-actin antibodies. (D) A549 and HEK293 cells were transfected with Flag-Chk1 and HA-ubiquitin-expressing plasmids, and ex vivo ubiquitination assays were performed as described in the Materials and Methods under glucose deprivation (GD) conditions (0 mM). Cell lysates were immunoprecipitated with anti-Flag antibodies (M2) and immunoblotted with anti-ubiquitin antibodies. The band corresponding to the immunoglobulin G heavy chain that reacts with the secondary antibody is labeled ‘IgG’ in the figure. Immunoprecipitated Flag-Chk1 was also detected by immunoblotting with anti-Flag antibodies.
3.6 ATR-dependent phosphorylation at Ser317 and Ser345 is not required for glucose deprivation-induced Chk1 degradation
Recent studies have reported that the phosphorylation of Chk1 by ATR at Ser317 and Ser345 plays an important role in its ubiquitination and degradation following various genotoxic stresses and replication blocks [15, 18, 19]. To address whether ATR-mediated Chk1 phosphorylation is important for glucose deprivation-induced Chk1 degradation, we first examined Chk1 phosphorylation upon glucose deprivation (0 mM) using anti-phospho antibodies specific for Chk1 phosphorylated at Ser317 and Ser345. Because Chk1 is rapidly degraded upon glucose deprivation, protein lysate samples were prepared in the presence of MG132 to examine Chk1 phosphorylation. As shown in Fig. 6A, phosphorylation of Ser317 and Ser345 was hardly detected after glucose deprivation in contrast to phosphorylation after UV irradiation, which is a representative genotoxic stress that induces Chk1 phosphorylation [2].
Fig. 6. ATR-dependent phosphorylation of Chk1 at Ser317 and Ser345 is not required for glucose deprivation-induced degradation.

(A) A549 cells were exposed to glucose deprivation (GD; 0 mM) in the presence of MG132 (10 μM) and harvested at the indicated time points. Cell lysates were analyzed by western blotting using the indicated antibodies. A549 cell lysates harvested 2 h after UV irradiation (20 J/m2) were used as positive controls. (B) HEK293 cells stably expressing Myc-Chk1 wild type, S317A, or S345A were transfected with a plasmid containing HA-ubiquitin, and ex vivo ubiquitination assays were performed as described in Fig. 5D using anti-Myc and anti-ubiquitin antibodies. (C) HEK293 cells stably expressing Myc-Chk1 wild type, S317A, or S345A were cultured in 10 mM glucose or in glucose deprivation conditions (0 mM) for 16 h and harvested. Cell lysates were analyzed by western blotting using the indicated antibodies. * p>0.05 as determined by Student’s t-test. (D) A549 cells were exposed to glucose deprivation (GD; 0 mM) and treated with 5 mM of caffeine or 5 μM of wortmannin for 16 h. Cell lysates were prepared and subjected to western blotting using the indicated antibodies. (E) A549 cells were transfected with either negative control siRNA (NC) or siRNA against ATR (ATRi) as described in the Materials and Methods. After 48 h, cells were cultured in 10 mM glucose or in glucose deprivation conditions (0 mM) for 16 h and cell lysates were analyzed by western blotting using the indicated antibodies.
If the phosphorylation of the Ser317 and Ser345 sites is important for glucose deprivation-induced Chk1 degradation, then mutating these sites should inhibit Chk1 ubiquitination and degradation upon glucose deprivation. To test this possibility, we generated HEK293 cells stably expressing wild-type Myc-Chk1 or the S317A or S345A mutants [11] and examined glucose deprivation-induced ubiquitination. We found that both Chk1 S317A and S345A mutants were ubiquitinated in a manner similar to wild-type Chk1 (Fig. 6B). Moreover, both Myc-Chk1 S317A and S345A mutants were degraded upon glucose deprivation (0 mM) in a similar manner to wild-type Chk1. The statistical analyses of three independent experiments indicate that the extent of degradation was not statistically significant (p > 0.05) (Fig. 6C). These results indicate that Chk1 phosphorylation at Ser317 and Ser345 is not essential for glucose deprivation-dependent degradation.
Consistent with these data, co-treatment with caffeine and wortmannin, which are both potent chemical inhibitors of the ATR/ATM-Chk1/Chk2 pathway, failed to inhibit glucose deprivation-induced degradation of Chk1 (Fig. 6D). Moreover, specific suppression of ATR expression by siRNA did not affect Chk1 degradation upon glucose deprivation (0 mM) (Fig. 6E). These results indicate that the glucose deprivation-induced degradation of Chk1 is independent of ATR function. Thus, our data suggest that Chk1 is degraded through a mechanism distinct from genotoxic stress-induced Chk1 degradation.
3.7 Glucose deprivation induces a defective checkpoint response to DNA replication blocks
Previous studies have demonstrated that reduced Chk1 function induces a defect in cell cycle arrest and the checkpoint response upon replication arrest and DNA damage [9, 10]. Our results show that glucose deprivation results in decreased levels of Chk1 protein. Thus, we expected that the cells would fail to execute normal checkpoint responses to replication blocks in the glucose deprivation conditions. To test this possibility, we examined the cellular checkpoint response following replication blocks under both high glucose and glucose deprivation conditions. HEK293 cells were cultured in high glucose (10 mM) or glucose deprivation (1.5 mM) conditions for 12 h and subsequently treated with camptothecin (CPT) or hydroxyurea (HU) as a replication block. Cell cycle analyses showed that HEK293 cells were arrested at the G1/S phase in response to CPT and HU treatments, and these arrests were maintained until 48 h under high glucose conditions (10 mM), indicating that the replication checkpoint pathway was normally activated (Fig. 7A, Supplementary Table 1). Conversely, under glucose deprivation conditions (1.5 mM), a portion of cells failed to arrest at the G1/S phase at 24 h, and extensive cell death was observed at 48 h after both CPT and HU treatments (Fig. 7A and B), suggesting that the checkpoint response to the replication block was not properly working due to the reduced level of Chk1 protein. Western blot analyses confirmed the reduced level of Chk1 protein in cells exposed to 1.5 mM glucose and Chk1 activation levels were much lower than in the high glucose condition, whereas Chk2 activation was not affected by glucose deprivation (Fig. 7C and D). Previous studies have shown that a defective checkpoint response to genotoxic stress or the inactivation of checkpoint genes often results in the formation of abnormal nuclear structures such as micronuclei and multinuclei [34, 35]. Fluorescence microscopic analysis after staining cells with DAPI revealed that the proportion of HEK293 cells exhibited micronuclei or multinuclei was profoundly increased 72 h after HU treatment in cells exposed to the glucose deprivation conditions (1.5 mM) (Fig. 7E). These results further support the notion that glucose deprivation leads to deactivation of the replication checkpoint through ubiquitin-proteasome pathway mediated Chk1 degradation and, in turn, results in an abnormal checkpoint response to replication blocks.
Fig. 7. Glucose deprivation induces defective checkpoint responses to replication blocks.

(A) HEK293 cells were cultured in either 10 mM or 1.5 mM glucose for 12 h (0 h) and then treated with 100 nM camptothecin (CPT) or 1.5 mM of hydroxyurea (HU). Cells were harvested at the indicated time points, stained with PI, and analyzed by flow cytometry as described in the Materials and Methods. The data shown are representative of three independent experiments with similar results. (B) The percentage of cells in the sub-G1 phase was determined using WINCYCLE software and plotted as the mean value with standard deviation. * p<0.05; ** p<0.01 as determined by Student’s t-test. (C) Protein levels of Chk1 and actin were determined in HEK293 cells cultured as in (A) and harvested at the indicated time point using anti-Chk1 and anti-actin antibodies. (D) HEK293 cells were cultured as in (A) and harvested 2 h after CPT and HU treatment. Cell lysates were subjected to western blotting using anti-pS317 Chk1 (pChk1), anti-pT68 Chk1 (pChk2), and anti-actin antibodies (E) HEK293 cells cultured on cover slides were treated with HU under high glucose (10 mM) or glucose deprivation conditions (1.5 mM) as in (A). After 72 h, Cells were stained with DAPI and the cells with micronuclei or multinuclei were counted as described in the Materials and Methods. A representative image of each sample is shown (left panels). Arrows indicate micronuclei, and the arrowhead indicates a binucleated cell. Cells with micronuclei or multinuclei from the samples were plotted as the mean value in the right panel.
To further confirm that Chk1 degradation was responsible for the defective checkpoint response in the glucose deprivation condition, we examined whether Chk1 overexpression could ameliorate the defects in response to replication blocks. To this end, we used HEK293 cells stably expressing Myc-Chk1 or control vector. While the endogenous Chk1 level was remarkably reduced in the HEK293 vector control cells (vec), a considerable amount of Myc-Chk1 protein remained in the glucose deprivation condition (1.5 mM) in HEK293-Myc-Chk1 cells (Fig. 8A). Flow cytometry analysis showed that HU-induced cell death in the glucose deprivation condition (1.5 mM) was reduced in HEK293 Myc-Chk1 cells by about 30% (Fig. 7B). Moreover, the proportion of the cells exhibiting micronuclei or multinuclei after HU treatment was also decreased to about 50% (Fig. 7C), indicating that Chk1 overexpression can rescue the defective checkpoint response and cell death in glucose deprivation conditions. Taken together, these results suggest that glucose deprivation induces deactivation of the replication checkpoint through ubiquitin-proteasome pathway-mediated Chk1 degradation and, in turn, results in an abnormal checkpoint response to replication blocks.
Fig. 8. Overexpression of Myc-Chk1 rescues the defective checkpoint response to replication blocks under glucose deprivation conditions.
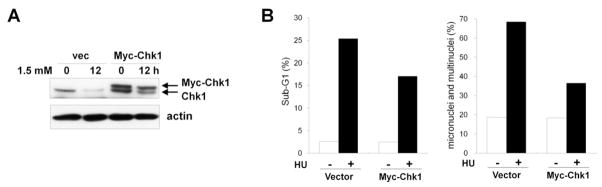
(A) HEK293 cells expressing Myc-Chk1 or control vector (vec) were cultured in glucose deprivation conditions (1.5 mM) for 12 h and harvested. Cell lysates were subjected to western blot analyses using anti-Chk1 and anti-actin antibodies. (B) HEK293 cells expressing Myc-Chk1 or control vector (vec) were cultured in 1.5 mM glucose for 12 h and treated with 1.5 mM of HU. Cells were harvested at 48 h to determine the sub-G1 fraction (left panel) and at 72 h to determine the proportion of cells harboring micronuclei or multinuclei (right panel) as described in Fig. 7.
4. Discussion
The present study reveals, for the first time, that Chk1 protein levels are regulated differently depending on the glucose concentration. First, we showed that the amount of Chk1 protein is significantly reduced in the presence of 2 mM or lower glucose as compared to physiological glucose concentrations (Fig. 1A). Cell cycle analyses and proliferation assay results demonstrated that Chk1 degradation occurs prior to cell cycle arrest upon glucose deprivation and at glucose concentrations that do not induce cell death (Supplementary Fig. S1, S2 and S3). Cell synchronization experiments further confirmed that glucose deprivation induces Chk1 degradation independently of its effect on cell cycle (Fig. 2).
The half-life of the Chk1 protein is dramatically decreased under conditions of glucose deprivation, whereas levels of mRNA expression are not changed (Fig. 1C and Supplementary Fig. S1). These findings indicate that the Chk1 protein is rapidly degraded in response to glucose deprivation. Consistent with endogenous Chk1, the ectopically expressed Flag-Chk1 and Myc-Chk1 proteins were also degraded in response to glucose deprivation (Fig. 5C and 6C). Further analyses using a proteasome inhibitor and ex vivo ubiquitination assays demonstrated that Chk1 was degraded through the ubiquitination-proteasome pathway (Fig. 5D). These results confirm that the Chk1 protein is specifically downregulated in response to glucose deprivation. The glucose deprivation-induced degradation of Chk1 was observed in all cell lines tested (Fig. 3), suggesting that glucose concentration-dependent control of Chk1 stability is a common regulatory mechanism.
Recent studies show that Chk1 is ubiquitinated and degraded through ATR-dependent phosphorylation at Ser317 and Ser345 upon various genotoxic treatments [15, 18, 19]. Initially, we hypothesized that ATR-dependent phosphorylation regulates Chk1 degradation upon glucose deprivation. However, our results indicate that glucose deprivation-induced Chk1 degradation is independent of ATR function. First, phosphorylation of Chk1 at Ser317 and Ser345 upon glucose deprivation was barely observable compared to phosphorylation of Chk1 upon UV irradiation (Fig. 6A). Second, the S317A and S345A Chk1 mutants are still ubiquitinated and degraded at a rate comparable to wild-type Chk1 upon glucose deprivation (Fig. 6B and C). Finally, glucose deprivation-induced Chk1 degradation was not abrogated in cells in which ATR had been knocked-down by RNA interference or inhibited by specific inhibitory compounds (Fig. 6D and E). These results indicate that Chk1 is ubiquitinated and degraded through a mechanism distinct from genotoxic treatment-induced Chk1 degradation.
Our results suggest that Chk1 is ubiquitinated and degraded through an ATR-independent pathway. In accordance with this notion, Rodriguez-Bravo et al. showed that Chk1 is able to activate and regulate cell cycle progression through an ATR-independent mechanism in the presence of stalled replication forks [36]. Identification of the E3 ubiquitin ligase responsible for Chk1 ubiquitination upon glucose deprivation would be helpful in understanding the underlying mechanism of glucose deprivation-induced degradation of Chk1. Several E3 ligases, such as Bmi1/Ring1A, RNF182, and cullin-containing E3 ubiquitin ligase complexes, were recently shown to be involved in protein degradation under glucose deprivation conditions [37–40]. We are currently working toward identifying the responsible E3 ubiquitin ligase. Further studies on these topics would provide a better understanding of the precise molecular mechanisms of glucose deprivation-induced degradation of Chk1.
The results presented in this study suggest a possible link between glucose deprivation and cellular checkpoint function. It has been demonstrated that glucose deprivation conditions develop in solid tumors. Due to architectural and functional abnormalities in the capillary network, pronounced spatial heterogeneities in the metabolic milieu exist in a solid tumor mass, and thereby, a number of unique metabolic characters, such as hypoxia, low pH, and nutrient starvation including glucose deprivation, are created [20, 21]. Previous studies have shown that glucose concentrations in some parts of solid tumors are decreased to less than 1 mM [22, 23, 41]. Interestingly, it has been proposed that the tumor microenvironment itself may be a major cause of the genomic instability frequently observed in malignant cancers [25, 26]. Because these metabolic abnormalities appear at early stages of tumor development as a consequence of insufficient blood perfusion [42], microenvironmental parameters may influence cell cycle checkpoint control or DNA repair functions, which are essential for maintaining genomic integrity. In accordance with this notion, several recent studies have shown that factors within the tumor microenvironment, such as hypoxia and low pH, may contribute to genomic instability by increasing mutation frequencies and diminishing DNA repair activities [25, 43, 44]. In addition to hypoxia and low pH, glucose deprivation has also recently been implicated in the induction of high mutation rates [45, 46]. Consistent with this report, we observed that cells show defective checkpoint control under glucose deprivation conditions in response to replication blocks (Figure 6). Moreover, fluorescence microscopic observations after staining with DAPI revealed that cells exhibiting abnormal nuclear structures, such as micronuclei and multinuclei, were substantially increased after HU treatments under glucose deprivation (Figure. 6c and d). Abnormal nuclear structures, such as chromatin bridges, micronuclei, and multinuclei, are known to be typical phenotypes of chromosomal damage and genomic instability [34, 35]. Interestingly, Jardim et al. recently showed that reduced Chk1 function resulted in abnormal nuclear structures, including micronuclei and multinuclei [47]. Thus, it is possible to speculate that glucose deprivation could promote genomic instability, at least in part, through the degradation of Chk1 and subsequent abrogation of checkpoint control. However, whether glucose deprivation indeed induces spontaneous genomic instability and how glucose deprivation-induced Chk1 degradation may contribute to this process remain to be addressed in future studies.
In conclusion, this study reveals that Chk1 function is regulated by proteasome-mediated proteolysis under glucose deprivation conditions. Our results provide a possible explanation for how checkpoint defects and genomic instability are induced in tumor microenvironments. Although the exact mechanism by which glucose deprivation promotes the ubiquitination and degradation of Chk1 remains to be explored, further studies will provide valuable information for understanding the processes of tumor development, and they may offer novel strategies for cancer treatment.
Supplementary Material
Research highlights.
Chk1 protein levels are regulated differently depending on glucose concentrations.
Chk1 is degraded in response to glucose deprivation through the ubiquitin-proteasome pathway.
The molecular mechanism for Chk1 degradation upon glucose deprivation is distinct from genotoxic stress-induced degradation.
Cells showed defective checkpoint responses to replication blocks in glucose deprivation conditions.
Acknowledgments
We thank Dr. Yolanda Sanchez, and Dr. Helen Piwnica-Worms for providing expression plasmids. This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-2008-512-E00012) by a grant of the Korea Healthcare technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A090331), and by National Research Foundation of Korea grant funded by the Korea government (2009-0093197).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 2.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 3.Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286:2528–2531. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- 4.Bertoni F, Codegoni AM, Furlan D, Tibiletti MG, Capella C, Broggini M. CHK1 frameshift mutations in genetically unstable colorectal and endometrial cancers. Genes Chromosomes Cancer. 1999;26:176–180. [PubMed] [Google Scholar]
- 5.Walworth N, Davey S, Beach D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature. 1993;363:368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- 6.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 7.Boddy MN, Furnari B, Mondesert O, Russell P. Replication checkpoint enforced by kinases Cds1 and Chk1. Science. 1998;280:909–912. doi: 10.1126/science.280.5365.909. [DOI] [PubMed] [Google Scholar]
- 8.Zeng Y, Forbes KC, Wu Z, Moreno S, Piwnica-Worms H, Enoch T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature. 1998;395:507–510. doi: 10.1038/26766. [DOI] [PubMed] [Google Scholar]
- 9.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M, Nakayama K. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 10.Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 11.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.den Elzen NR, O’Connell MJ. Recovery from DNA damage checkpoint arrest by PP1-mediated inhibition of Chk1. Embo J. 2004;23:908–918. doi: 10.1038/sj.emboj.7600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leung-Pineda V, Ryan CE, Piwnica-Worms H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol. 2006;26:7529–7538. doi: 10.1128/MCB.00447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–1174. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collis SJ, Barber LJ, Clark AJ, Martin JS, Ward JD, Boulton SJ. HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat Cell Biol. 2007;9:391–401. doi: 10.1038/ncb1555. [DOI] [PubMed] [Google Scholar]
- 16.Feng JM, Zhu H, Zhang XW, Ding J, Miao ZH. Proteasome-dependent degradation of Chk1 kinase induced by the topoisomerase II inhibitor R16 contributes to its anticancer activity. Cancer Biol Ther. 2008;7 doi: 10.4161/cbt.7.11.6728. [DOI] [PubMed] [Google Scholar]
- 17.Leung-Pineda V, Huh J, Piwnica-Worms H. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 2009;69:2630–2637. doi: 10.1158/0008-5472.CAN-08-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang YW, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, Manning G, Abraham RT, Hunter T. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell. 2009;35:442–453. doi: 10.1016/j.molcel.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 20.Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol. 2004;14:198–206. doi: 10.1016/j.semradonc.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 21.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–6465. [PubMed] [Google Scholar]
- 22.Gullino PM. Extracellular compartments of solid tumors. Vol. 3. Plenum Press; New York: 1975. [Google Scholar]
- 23.Mueller-Klieser W, Walenta S. Geographical mapping of metabolites in biological tissue with quantitative bioluminescence and single photon imaging. Histochem J. 1993;25:407–420. doi: 10.1007/BF00157805. [DOI] [PubMed] [Google Scholar]
- 24.Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509–4515. [PubMed] [Google Scholar]
- 25.Yuan J, Glazer PM. Mutagenesis induced by the tumor microenvironment. Mutat Res. 1998;400:439–446. doi: 10.1016/s0027-5107(98)00042-6. [DOI] [PubMed] [Google Scholar]
- 26.Rockwell S, Yuan J, Peretz S, Glazer PM. Genomic instability in cancer. Novartis Found Symp. 2001;240:133–142. doi: 10.1002/0470868716.ch9. discussion 142–151. [DOI] [PubMed] [Google Scholar]
- 27.Kim MA, Kim HJ, Brown AL, Lee MY, Bae YS, Park JI, Kwak JY, Chung JH, Yun J. Identification of novel substrates for human checkpoint kinase Chk1 and Chk2 through genome-wide screening using a consensus Chk phosphorylation motif. Exp Mol Med. 2007;39:205–212. doi: 10.1038/emm.2007.23. [DOI] [PubMed] [Google Scholar]
- 28.Yun J, Lee WH. Degradation of transcription repressor ZBRK1 through the ubiquitin-proteasome pathway relieves repression of Gadd45a upon DNA damage. Mol Cell Biol. 2003;23:7305–7314. doi: 10.1128/MCB.23.20.7305-7314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaneko YS, Watanabe N, Morisaki H, Akita H, Fujimoto A, Tominaga K, Terasawa M, Tachibana A, Ikeda K, Nakanishi M. Cell-cycle-dependent and ATM-independent expression of human Chk1 kinase. Oncogene. 1999;18:3673–3681. doi: 10.1038/sj.onc.1202706. [DOI] [PubMed] [Google Scholar]
- 30.Lukas C, Bartkova J, Latella L, Falck J, Mailand N, Schroeder T, Sehested M, Lukas J, Bartek J. DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res. 2001;61:4990–4993. [PubMed] [Google Scholar]
- 31.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 32.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 33.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 34.Fenech M. Cytokinesis-block micronucleus assay evolves into a “cytome” assay of chromosomal instability, mitotic dysfunction and cell death. Mutat Res. 2006;600:58–66. doi: 10.1016/j.mrfmmm.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 35.Iarmarcovai G, Bonassi S, Botta A, Baan RA, Orsiere T. Genetic polymorphisms and micronucleus formation: a review of the literature. Mutat Res. 2008;658:215–233. doi: 10.1016/j.mrrev.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Bravo V, Guaita-Esteruelas S, Florensa R, Bachs O, Agell N. Chk1- and claspin-dependent but ATR/ATM- and Rad17-independent DNA replication checkpoint response in HeLa cells. Cancer Res. 2006;66:8672–8679. doi: 10.1158/0008-5472.CAN-05-4443. [DOI] [PubMed] [Google Scholar]
- 37.Alchanati I, Teicher C, Cohen G, Shemesh V, Barr HM, Nakache P, Ben-Avraham D, Idelevich A, Angel I, Livnah N, Tuvia S, Reiss Y, Taglicht D, Erez O. The E3 ubiquitin-ligase Bmi1/Ring1A controls the proteasomal degradation of Top2alpha cleavage complex - a potentially new drug target. PLoS One. 2009;4:e8104. doi: 10.1371/journal.pone.0008104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu QY, Lei JX, Sikorska M, Liu R. A novel brain-enriched E3 ubiquitin ligase RNF182 is up regulated in the brains of Alzheimer’s patients and targets ATP6V0C for degradation. Mol Neurodegener. 2008;3:4. doi: 10.1186/1750-1326-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei S, Yang HC, Chuang HC, Yang J, Kulp SK, Lu PJ, Lai MD, Chen CS. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J Biol Chem. 2008;283:26759–26770. doi: 10.1074/jbc.M802160200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yun J, Kim YI, Tomida A, Choi CH. Regulation of DNA topoisomerase IIalpha stability by the ECV ubiquitin ligase complex. Biochem Biophys Res Commun. 2009;389:5–9. doi: 10.1016/j.bbrc.2009.08.066. [DOI] [PubMed] [Google Scholar]
- 41.Walenta S, Schroeder T, Mueller-Klieser W. Metabolic mapping with bioluminescence: basic and clinical relevance. Biomol Eng. 2002;18:249–262. doi: 10.1016/s1389-0344(01)00107-1. [DOI] [PubMed] [Google Scholar]
- 42.Moulder JE, Rockwell S. Tumor hypoxia: its impact on cancer therapy. Cancer Metastasis Rev. 1987;5:313–341. doi: 10.1007/BF00055376. [DOI] [PubMed] [Google Scholar]
- 43.Yuan J, Narayanan L, Rockwell S, Glazer PM. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res. 2000;60:4372–4376. [PubMed] [Google Scholar]
- 44.Mihaylova VT, Bindra RS, Yuan J, Campisi D, Narayanan L, Jensen R, Giordano F, Johnson RS, Rockwell S, Glazer PM. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol. 2003;23:3265–3273. doi: 10.1128/MCB.23.9.3265-3273.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papp-Szabo E, Josephy PD, Coomber BL. Microenvironmental influences on mutagenesis in mammary epithelial cells. Int J Cancer. 2005;116:679–685. doi: 10.1002/ijc.21088. [DOI] [PubMed] [Google Scholar]
- 46.Shahrzad S, Quayle L, Stone C, Plumb C, Shirasawa S, Rak JW, Coomber BL. Ischemia-induced K-ras mutations in human colorectal cancer cells: role of microenvironmental regulation of MSH2 expression. Cancer Res. 2005;65:8134–8141. doi: 10.1158/0008-5472.CAN-05-0713. [DOI] [PubMed] [Google Scholar]
- 47.Jardim MJ, Wang Q, Furumai R, Wakeman T, Goodman BK, Wang XF. Reduced ATR or Chk1 expression leads to chromosome instability and chemosensitization of mismatch repair-deficient colorectal cancer cells. Mol Biol Cell. 2009;20:3801–3809. doi: 10.1091/mbc.E09-04-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.