

SUMMARY
Phagocytosis controls CNS homeostasis by facilitating the removal of unwanted cellular debris. Accordingly, impairments in different receptors or proteins involved in phagocytosis result in enhanced inflammation and neurodegeneration. While various studies have identified extrinsic factors that modulate phagocytosis in health and disease, key intracellular regulators are less understood. Here we show that the autophagy protein beclin 1 is required for efficient phagocytosis in vitro and in mouse brains. Furthermore, we show that beclin 1-mediated impairments in phagocytosis are associated with dysfunctional recruitment of retromer to phagosomal membranes, reduced retromer levels, and impaired recycling of phagocytic receptors CD36 and Trem2. Interestingly, microglia isolated from human Alzheimer’s disease (AD) brains show significantly reduced beclin 1 and retromer protein levels. These findings position beclin 1 as a link between autophagy, retromer trafficking, and receptor-mediated phagocytosis and provide insight into mechanisms by which phagocytosis is regulated and how it may become impaired in AD.
INTRODUCTION
Microglia are the immune cells of the brain. They constantly survey the brain for abnormalities and are quickly activated upon encountering tissue damage or injury (Nimmerjahn et al., 2005). Following activation, microglia become capable of numerous functions depending on the stimuli in the surrounding environment. One such function is phagocytosis, which facilitates brain homeostasis via the clearance of cellular debris and possibly the pruning of synapses (Lucin and Wyss-Coray, 2009; Nimmerjahn et al., 2005; Paolicelli et al., 2011). In addition to general maintenance roles, recent genome-wide association studies also suggest that microglial phagocytic receptors may have a critical role in Alzheimer’s disease (AD). Indeed, rare variants in the phagocytic receptor TREM2 triple the risk of developing AD and represent one of the strongest known risk factors (Guerreiro et al., 2013; Jonsson et al., 2013). In mice, genetic defects in different receptors or proteins involved in phagocytosis result in neurodegeneration (Kaifu et al., 2003; Lu et al., 1999; Lu and Lemke, 2001) and may be responsible for increased amyloidosis in mouse models of AD (Wyss-Coray et al., 2002). Conversely, driving microglial activation towards a more phagocytic phenotype reduces Aβ pathology in mouse models of AD (Heneka et al., 2012). These studies highlight the importance of phagocytosis in brain homeostasis and suggest that identifying key regulators of phagocytosis may represent a therapeutic target for the treatment of neurological disease. While various studies have identified extrinsic factors that modulate phagocytosis in health and disease (Lucin and Wyss-Coray, 2009), key intracellular regulators are much less understood.
Beclin 1 represents an intriguing target that may act to regulate phagocytic receptor function in health and disease. Indeed, beclin 1 is actively involved in protein degradation, host defense, and in mouse models of Alzheimer’s and Parkinson’s disease has a critical role in mitigating amyloidosis and neurodegeneration (Levine et al., 2011; Pickford et al., 2008; Spencer et al., 2009). While beclin 1 is classically associated with autophagy, a major protein degradation pathway, studies now suggest that beclin 1 may also have alternative functions independent of autophagy. This is suggested by studies showing that genetic deletion of beclin 1 results in lethality at embryonic day 7.5–8.5 (Qu et al., 2003; Yue et al., 2003), while genetic deletion of various downstream autophagy proteins results in postnatal lethality (Komatsu et al., 2005; Kuma et al., 2004). What these additional functions of beclin 1 might be is not entirely clear. However, given that apoptotic cell clearance is impaired during mouse embryonic development when beclin 1 is knocked-out (Qu et al., 2007), one possibility is a role in additional clearance processes such as phagocytosis. To that end, recent studies reveal that beclin 1 rapidly associates with phagosomes (Berger et al., 2010; Sanjuan et al., 2007) and receptor complexes at the cell surface (Berger et al., 2010; Yue et al., 2002) in the absence of autophagosomes. Whether beclin 1 has an essential role in receptor-mediated phagocytosis is unknown. Furthermore, whether microglial beclin 1 is dysfunctional during neurological disease and how this dysfunction may impair phagocytosis of disease relevant substrates also remains unexplored.
Here, we identify a novel role for microglial beclin 1 in receptor-mediated phagocytosis. Beclin 1, together with its phosphatidylinositol 3-kinase (PI3K) binding partner, Vps34, accomplish this by regulating the retromer complex, which is involved in sorting cellular components to the lysosome or recycling the components back to defined compartments (e.g., the cell surface). Consequently, genetic reduction of beclin 1 results in reduced retromer levels, phagocytic receptor recycling, and phagocytosis of latex beads and Aβ. Importantly, beclin 1 and retromer are reduced in microglia isolated from postmortem human AD brains. Together these findings suggest that similar mechanisms may be impaired in AD, possibly rendering microglia less efficient at phagocytosing Aβ or any other potentially toxic debris whose uptake depends on receptor-mediated phagocytosis.
RESULTS
Reduced beclin 1 impairs phagocytosis
To determine whether beclin 1 has a role in phagocytosis, we reduced its expression in BV2 microglial cells with lentivirus encoding beclin 1 shRNA (beclin 1 knockdown; KD) and assayed for microglial uptake of latex beads. Using this lentiviral approach, which allowed us to reduce beclin 1 expression by ~80% (Figure 1A), we find that reducing microglial beclin 1 levels significantly impaired the phagocytosis of fluorescent latex beads as determined by flow cytometry (Figure 1B – C). This effect was not exclusive to BV2 cells as N9 cells, another mouse microglial cell line, and C6 astrocyte cells showed a similar phagocytic defect when beclin 1 was reduced (Figure S1A–B). Importantly, phagocytosis was “rescued” in BV2 cells by recovering beclin 1 levels with a lentivirus encoding mouse beclin 1 (Figure 1D–E), demonstrating the specificity of the beclin 1 shRNA knockdown approach. Interestingly, reduced expression of Atg5, a protein critical for autophagy downstream of beclin 1, did not alter phagocytosis (Figure S1C–D), suggesting that beclin 1 may regulate phagocytosis through alternative pathways.
Figure 1.

Reduced beclin 1 impairs microglial phagocytosis. (A) BV2 microglia cells were infected with lentivirus encoding for luciferase shRNA (control) or beclin 1 shRNA (KD). Cell lysates were analyzed by western blot. Infected cells were provided fluorescent latex beads and phagocytosis was analyzed by flow cytometry (B–C). Data are expressed as phagocytic index, which is the percent of cells that phagocytose beads. (D) BV2 cells receiving beclin 1 shRNA lentivirus were transduced with lentivirus encoding for mouse beclin 1 (mBeclin 1) and cell lysates were analyzed by western blot. (E) Recovering beclin 1 levels was sufficient to improve phagocytosis of latex beads. Results were compared by an unpaired Student’s t test (C) or a one-way ANOVA with a Tukey’s post-test (E) and are representative of at least 3 independent experiments (n=3 per group). Values are mean ± SEM. *** p<0.001.
Along with changes in overall phagocytosis, flow cytometry scattergrams also suggested that phagocytic efficiency was impaired in beclin 1-deficient BV2 cells, as indicated by the loss of highly phagocytic cell populations (See Figure 1B). Subsequent flow cytometry analysis revealed that while significantly more beclin 1-deficient cells phagocytosed a single bead, they were less competent at phagocytosing 3–10 beads (Figure 2A) when compared with control shRNA transduced BV2 cells. Impaired phagocytic efficiency by beclin 1-deficient BV2 cells could also be rescued by recovering beclin 1 levels (Figure 2B). To confirm these findings in primary cells, we next isolated microglia from beclin 1 heterozygous knockout mice (beclin 1+/−) (Figure 2C), which show a 40–50% reduction in beclin 1 levels (Pickford et al., 2008; Qu et al., 2003). In agreement with our lentiviral approach, beclin 1+/− microglia also showed impairments in phagocytic efficiency when analyzed by flow cytometry (Figure 2D). To determine if impaired phagocytic efficiency in beclin 1-deficient cells resulted from beads stalling at the cell surface or from a disruption in the kinetics of phagocytosis we used microscopy and live-cell imaging. We observed that while beads were initially phagocytosed at a similar rate (Figure S2A), beclin 1-deficient BV2 cells were less able to phagocytose subsequent beads (Figure S2B, Figure 2E). Quantification of cell migration confirmed that beclin 1-deficient cells have a similar migratory capacity as control cells, indicating that impaired movement is not responsible for phagocytic deficits (Figure 2F). Instead, our data suggest that beclin 1 deficiency impairs the ability of cells to phagocytose subsequent beads beyond the initial phagocytic event (see Figure 2G for representative live-cell images) resulting in overall reduced phagocytic uptake (Figure S2C).
Figure 2.

Reduced beclin 1 impairs microglial phagocytic efficiency. BV2 microglia cells were infected with lentivirus encoding for luciferase shRNA (control) or beclin 1 shRNA (KD). (A) Phagocytosis of fluorescent latex beads was assayed by flow cytometry and analysis of flow cytometry histograms determined the number of BV2 cells phagocytosing relative quantities of beads. (B) Recovering beclin 1 levels with a lentivirus encoding for mouse beclin 1 (mBeclin 1) was sufficient to rescue phagocytic efficiency as measured by flow cytometry. (C) Primary microglia were isolated from beclin 1+/− (Becn1+/−) or wildtype (WT) mice and labeled with fluorescently tagged antibodies against CD11b or isotype control antibodies. The percent of CD11b+ cells in the microglial population was determined by flow cytometry relative to the cells receiving isotype control antibody. The microglial population was determined to be ~90% pure. (D) The average number of beads phagocytosed by Becn1+/− and WT primary microglia was quantified by flow cytometry. (E) Using live-cell imaging the number of beads phagocytosed over time was monitored. (F) Cell motility was also monitored by live-cell imaging. (G) Shown are representative images from live-cell imaging at various times after the administration of beads. To better visualize the beads an outline is provided around each bead. Scale bar, 15 µm. For further analysis of bead uptake please see Figure S2. Results were compared by a two-way ANOVA with a Bonferroni post-test (A,E), a one-way ANOVA with a Tukey’s post-test (B), or an unpaired Student’s t test (D,F) and are representative of at least 3 independent experiments (A–B; n=3 per group, D; n=4–6 per group, E–F; n=10–16 per group). Values are mean ± SEM. * p<0.05, ** p<0.01, *** p<0.001.
Reduced beclin 1 disrupts retromer-mediated recycling of phagocytic receptors
Phagocytosis is initiated by numerous receptors that recognize molecular structures on extracellular substrates. Upon binding and internalization of substrates, phagocytic receptors are recycled back to the cell surface to be used again. Accordingly, disruptions in phagocytic receptor recycling have dramatic consequences on phagocytic efficiency (Chen et al., 2010). To determine if reduced phagocytic efficiency seen in beclin 1-deficient cells is due to changes in phagocytic receptor dynamics we used an established receptor recycling assay (Mitchell et al., 2004) (Figure 3B). Indeed, beclin 1-deficient BV2 cells showed a prominent reduction in recycling of the phagocytic receptor CD36 (Figure 3C–D), a class B scavenger receptor involved in phagocytosing a wide range of substrates, including Aβ (El Khoury et al., 2003) and latex beads (Figure 3A). Primary microglia obtained from beclin 1+/− mice also showed a similar deficit in CD36 recycling (Figure 3E–F). Importantly, flow cytometric analysis demonstrated that beclin 1 shRNA did not affect baseline cell surface expression of CD36 in the absence of ligand (Figure 3G). Additionally, because the phagocytic receptor Trem2 has been reported to recycle (Prada et al., 2006) and is a risk factor for AD (Guerreiro et al., 2013; Jonsson et al., 2013), we investigated Trem2 recycling in beclin 1-deficient BV2 cells. Trem2 recycling was impaired in beclin 1-deficient BV2 cells as was observed with CD36 (Figure 3H). Taken together, these data suggest that beclin 1 has a function in phagocytic receptor recycling.
Figure 3.

Reduced beclin 1 diminishes phagocytic receptor recycling and retromer localization. (A) To determine whether CD36 contributes to phagocytosis of latex beads, wildtype BV2 cells were cultured with latex beads in the presence or absence of CD36 neutralizing antibody or IgG control antibody. Phagocytosis was quantified by flow cytometry. (B–D) Using an established receptor recycling assay, CD36 receptor recycling was analyzed in BV2 cells infected with lentivirus encoding luciferase shRNA (control) or beclin 1 shRNA (KD). Scale bar, 100 µm. (E-F) CD36 receptor recycling was analyzed in Becn1+/− and WT primary microglia. The cell highlighted within the dashed box (E) is shown at a higher magnification in the insert. Scale bar, 100 ⌠m. (G) Using flow cytometry, surface expression of CD36 was determined in the absence of ligand for BV2 cells infected with beclin 1 shRNA or control lentivirus. (H) Trem2 receptor recycling was analyzed in BV2 cells infected with beclin 1 shRNA or control lentivirus. All bars are mean ± SEM; mean differences were compared by a one-way ANOVA with a Tukey’s post-test (A) or an unpaired Student’s t test (D,F–H). (A,D,G; n=3 per group, F,H; n=4–6 per group). *p<0.05, **p<0.01, n.s., not significant.
Beclin 1 and its binding partner, Vps34, regulate retromer localization
While receptor recycling is regulated by numerous mechanisms, a recent study in C. elegans showed that clearance of apoptotic cell corpses by the phagocytic receptor CED-1 was dependent on receptor recycling via the retromer complex (Chen et al., 2010). This conserved structure is responsible for endosome-to-Golgi retrograde transport of membrane proteins and in mammals consists of the subunits Vps26, Vps29, Vps35, and sorting nexins (Bonifacino and Hurley, 2008). To determine whether beclin 1 might regulate the retromer complex, we knocked down beclin 1 expression using a lentivirus encoding beclin 1 shRNA and subsequently investigated whether the retromer complex was affected by beclin 1 knock down. Remarkably, beclin 1 knock down resulted in a prominent reduction of the retromer complex (Figure 4A–B). To test whether diminished retromer expression impairs CD36 recycling, we knocked down Vps35 levels in BV2 cells using lentivirus encoding Vps35 shRNA (Figure 4C) and analyzed CD36 recycling. Reducing Vps35 significantly impaired CD36 recycling (Figure 4D). Furthermore, reducing Vps35 also resulted in a concomitant reduction in phagocytic efficiency (Figure 4E). Because previous studies have demonstrated that retromer dysfunction can be reversed by enhancing Vps35 levels (MacLeod et al., 2013), we rescued Vps35 levels in beclin 1-deficient BV2 cells. Rescuing Vps35 resulted in enhanced CD36 recycling (Figure 4G) and phagocytosis (Figure 4H). Together these data suggest that beclin 1 regulates the retromer complex, which is required to maintain phagocytic receptor recycling and phagocytosis.
Figure 4.

Beclin 1 regulates the retromer complex, which controls CD36 recycling. (A-B) BV2 cells were infected with a lentivirus encoding for luciferase shRNA (control) or beclin 1 shRNA (KD) and cell lysates were analyzed by western blot to detect levels of the retromer complex. (C) BV2 cells were infected with a lentivirus encoding for Vps35 shRNA (Vps35 KD) or luciferase shRNA (Control) and cell lysates were probed by western blot. CD36 receptor recycling (D) and phagocytic efficiency (E) were then analyzed in BV2 cells infected with Vps35 shRNA or control lentivirus. (F) To confirm our Vps35 rescue approach, HEK cells were infected with either control lentivirus, beclin 1 shRNA lentivirus (Becn KD) and transfected with a control RFP plasmid (Ctrl), or beclin 1 shRNA lentivirus and transfected with a Vps35-T2A-RFP plasmid (Vps35) and cell lysates were analyzed by western blot. CD36 receptor recycling (G) and phagocytosis (H) was impaired in BV2 cells infected with beclin 1 shRNA lentivirus and transfected with control plasmid when compared to BV2 cells infected with beclin 1 shRNA lentivirus and transfected with Vps35 plasmid. Phagocytic analysis by flow cytometry was investigated in cellular populations expressing RFP. All bars are mean ± SEM and are representative of at least two independent experiments (n=3 per group). Mean differences were compared by an unpaired Student’s t test. *p<0.05, **p<0.01, ***p<0.001.
To further explore this connection, we first tested whether beclin 1 might regulate retromer via a direct interaction. This proved unlikely as we were unable to coimmunoprecipitate beclin 1 and Vps35 (Data not shown). Moreover, a recent study using mass spectrometry screens to identify putative binding partners of mammalian beclin 1 and other known autophagy proteins that complex with beclin 1 did not reveal any binding partners exclusive to the retromer complex (Behrends et al., 2010). Therefore, we next tested whether beclin 1 may have a role in the recruitment of retromer.
Retromer is typically recruited to vesicles via its sorting nexin subunits. These subunits contain a Phox homology domain capable of binding to phosphatidylinositol 3-phosphate (PI3P) present on target membranes (Burda et al., 2002). Interestingly, phosphatidylinositol is converted to PI3P primarily by the PI3-kinase, Vps34, which is known to form a complex with beclin 1 and regulate autophagy and apoptosis (Funderburk et al., 2010). If the beclin 1/Vps34 complex were responsible for recruiting retromer in microglia, we speculated that reducing beclin 1 would diminish Vps34 levels. Consistent with our previous observations in neurons (Jaeger et al., 2010), beclin 1 knockdown in BV2 cells also resulted in a prominent reduction in Vps34 (Figure 5A). To begin to establish whether PI3P has a role in recruiting retromer in BV2 cells, we next tested whether Vps35 co-localized with vesicles containing PI3P. To visualize PI3P we utilized a reporter construct expressing an RFP fusion protein containing a 2xFYVE PI3P binding domain. Using this approach, we observed co-localization of PI3P and Vps35 in BV2 cells (Figure 5B). To determine the kinetics by which PI3P and Vps35 localize to vesicles we utilized live-cell imaging. To best monitor the recruitment of PI3P and Vps35 to clearly defined vesicles we provided latex beads to BV2 cells expressing either the PI3P reporter construct or a Vps35-RFP fusion construct. These cells allowed us to track the uptake of latex beads into clearly defined phagosomes. Given that the phagocytic receptor CD36 is involved in part in phagocytosing latex beads (Figure 3A), and that beclin 1 and Vps35 play a role in recycling CD36 (See Figure 3&Figure 4), the phagosomal membrane surrounding latex beads provides an excellent substrate to monitor PI3P and Vps35 recruitment. Using this paradigm, we find that PI3P is rapidly generated at the phagosomal membrane within 10mins and is followed by the recruitment of Vps35 around 20mins (Figure 5C).
Figure 5.

Reduced beclin 1 impairs Vps34-mediated recruitment of retromer. (A) Cell lysates from BV2 cells infected with lentivirus encoding for luciferase shRNA (control) or beclin 1 shRNA (KD) were analyzed by western blot. (B) BV2 cells transfected with a phosphatidylinositol 3-phosphate (PI3P) reporter (2xFYVE-RFP) were immunofluorescently labeled for Vps35 and co-localization was visualized by confocal microscopy. Arrows indicate vesicles with co-localization. Scale bar, 20µm. (C) Live-cell imaging of BV2 cells transfected with 2xFYVE-RFP or Vps35-RFP were used to monitor the kinetics by which PI3P and Vps35 localize to the phagosomal membrane of internalized 6µm latex beads. BV2 cells transfected with either 2xFYVE-RFP or Vps35-RFP were subsequently infected with beclin 1 or control shRNA lentivirus and the phagosomal localization of PI3P (D) and Vps35 (E) was similarly monitored by live-cell imaging after the internalization of 6µm latex beads. The X symbols in figures D and E represent the absence of localization in a 90min live-cell imaging session. Please see Movies S1–4 for representative examples of phagosomal localization. (F) BV2 cells transfected with Vps35-RFP were treated with 10mM of the PI3-kinase inhibitor 3-methyladenine (3MA) or PBS (control) for 3hrs and the localization of Vps35 was visualized. (G) Cell lysates from BV2 cells treated with 5mM 3MA for 48hrs were probed by western blot to detect levels of the retromer complex. (H) The average number of beads phagocytosed by BV2 cells receiving 10mM 3-MA or PBS (control) was quantified by flow cytometry. Values are mean ± SEM and are representative of at least two independent experiments (C–E; n=25–31 beads per group, H; n=3 per group).
To investigate the role of beclin 1 in these events, we knocked down beclin 1 and monitored PI3P generation and Vps35 recruitment. In agreement with beclin 1 deficiency causing diminished Vps34 levels (See Figure 5A), reducing beclin 1 also impaired the generation of PI3P at the phagosomal membrane (Figure 5D; Movie S1–2) and the recruitment of Vps35 to the phagosomal membrane (Figure 5E; Movie S3–4). Importantly, when Vps34 was inhibited with the PI3K inhibitor 3-methyladenine, which selectively inhibits Vps34 kinase activity (Miller et al., 2011), we observed mis-localization of Vps35 (Figure 5F) and reduced retromer complex levels (Figure 5G), similar to what was seen in beclin 1 knockdown cells. Treatment with 3-methyladenine also resulted in a significant impairment in phagocytic efficiency that was similar to what was seen in beclin 1 knockdown cells (Figure 5H). Given the role of PI3P in regulating phagosomal maturation (Vieira et al., 2001), we tested whether beclin 1-deficient BV2 cells show impairments in phagosomal maturation. Indeed, beclin 1-deficient BV2 cells also demonstrated impairments in phagosomal maturation (Figure S3). Together, these data indicate that beclin 1 works in collaboration with Vps34 to generate PI3P at phagosomal membranes, which then allows for the recruitment of the retromer complex. When this process is disrupted, phagocytic receptor recycling, phagocytic efficiency, and phagosomal maturation are impaired.
Reduced microglial beclin 1 impairs Aβ phagocytosis
Given that beclin 1 deficiency impairs the recycling of CD36, a known Aβ phagocytic receptor, we next asked whether phagocytosis of Aβ is impaired in beclin 1-deficient BV2 cells. To evaluate Aβ phagocytosis, we used a previously described ex-vivo brain slice assay (Bard et al., 2000) (Figure 6A), where microglial cells are added to unfixed brain slices from aged, plaque-depositing APP transgenic mice. Microglia then surround and engulf Aβ (Figure 6B) and its removal can be measured immunohistochemically or by ELISA. While control BV2 cells efficiently cleared Aβ deposits from these brain sections, beclin 1-deficient cells were significantly less efficient at clearing the aggregates in both the hippocampus (Figure 6C – D) and cortex (Figure 6E). These histological findings were supported by independent measurements of Aβ content in brain slices by ELISA (Figure 6F – G). Moreover, primary microglia isolated from beclin 1+/− mice showed similar impairments in Aβ phagocytosis when compared to wildtype littermates (Figure S4A–B). Importantly, Aβ phagocytosis by beclin 1-deficient BV2 cells could be rescued by recovering beclin 1 levels (Figure S4C–D).
Figure 6.

Reduced microglial beclin 1 impairs Aβ phagocytosis. (A) BV2 cells were infected with beclin 1 shRNA (KD) or luciferase shRNA (control) lentivirus and cultured on 10µm thick freshly cut, unfixed brain slices from APP transgenic mice. After 24hrs the slices were processed for Aβ histology or ELISA. (B) Confocal microscopy confirmed that BV2 cells cultured on APP brain slices contained Aβ after 24hrs of culture. Transduced BV2 cells were visualized with GFP and Aβ was visualized with 3D6. Scale bar, 10µm. (C) Sections were then probed for Aβ and Aβ load was quantified in the hippocampus (D) and cortex (E). Scale bar, 200µm. Aβ was also extracted from replicate slices and total Aβ (F) and Aβ1–42 (G) were measured by ELISA. Please see Figure S4 for analysis with primary microglia. All bars are mean ± SEM. Mean differences were compared by a one-way ANOVA with a Tukey’s post-test (D,E) or an unpaired Student’s t test (F,G) and are representative of at least two independent experiments (n=6–10 per group). *p<0.05.
To determine whether beclin 1 has a role in the removal of extracellular Aβ in vivo we used beclin 1+/− mice and injected fibrillar Aβ into the frontal cortex (Figure 7A). In agreement with our ex-vivo phagocytosis assay, the removal of Aβ was significantly impaired in beclin 1-deficient mice, resulting in twice as much Aβ remaining in the brains of beclin 1+/− mice compared with wildtype littermates (Figure 7B – C). We then tested whether reduced phagocytic capacity could explain impairments in the removal of Aβ in vivo by injecting pH-sensitive beads, which fluoresce when internalized by microglial cells (Insert of Figure 7D), into the frontal cortex of beclin 1+/− mice or wildtype littermates. Quantification of fluorescence as an indicator of phagocytosed beads showed that beclin 1+/− mice phagocytosed almost 4-fold fewer beads than wildtype mice (Figure 7D–E). Because the fluorescence read-out of our pH-sensitive beads could be confounded by changes in phagosomal or lysosomal pH, we tested whether reduced beclin 1 levels might affect phagosomal or lysosomal pH. To do this, we isolated primary microglia from beclin 1+/− mice and analyzed lysosomal pH in these cells using LysoSensor Yellow/Blue dextran (Lee et al., 2010) (Figure 7F). In addition, we analyzed phagosomal and lysosomal pH using FITC conjugated beads (Figure S5). Phagosomal and lysosomal pH in beclin 1-deficient cells were not significantly different than control cells (Figure 7F; Figure S5), suggesting that the diminished fluorescent signal in beclin 1+/− mice injected with pH-sensitive beads is likely due to reduced uptake of the pH-sensitive beads. Together, these ex vivo and in vivo studies support our cell culture experiments and demonstrate that beclin 1 is necessary for efficient phagocytosis.
Figure 7.

Beclin 1-deficient mice demonstrate impaired extracellular Aβ clearance and phagocytosis. (A) To determine whether beclin 1+/− mice (Becn1+/−) demonstrate deficits in extracellular Aβ phagocytosis and removal, fibrillar Aβ1–42 or pH-sensitive beads were stereotaxically injected into the frontal cortex. Brain image was obtained from Allen Brain Atlas. (B-C) Residual Aβ was quantified histologically 48hrs after injection. Scale bar, 500µm. (D-E) Fluorescence from pH-sensitive beads, which was confirmed with BV2 cells in vitro (See Insert), was quantified from confocal images 48hrs after injection. Scale bar, 200µm. (F) Primary microglia isolated from beclin 1+/− mice were analyzed by LysoSensor Yellow/Blue dextran to determine relative lysosomal pH levels. For additional measures of phagosomal and lysosomal pH please see Figure S5. Results were analyzed by an unpaired Student’s t test (n=9–11 (C,E) or n=4 (F) per group). All values are mean ± SEM. *p<0.05; n.s., non significant.
Microglia from human AD brains exhibit reduced beclin 1 and retromer
While our studies point to a key role for beclin 1 in regulating retromer function and phagocytosis in vitro and in vivo (using mouse models), we next asked whether our findings might be clinically relevant for human disease. Indeed, previous studies have demonstrated that beclin 1 protein levels are reduced in AD brain lysates (Crews et al., 2010; Jaeger et al., 2010; Pickford et al., 2008) and that retromer mRNA is selectively decreased in entorhinal cortex vs. the dentate gyrus of AD patients (Small et al., 2005). However beclin 1 and retromer levels in AD microglia is unknown. To determine whether beclin 1 and retromer are reduced in AD brains and in microglia in particular, we analyzed brain lysates from the midfrontal gyrus and we isolated microglia from superior and middle frontal gyri of postmortem AD and non-demented control brains. We discovered that Vps35 levels were reduced by roughly half in brain homogenates from beclin 1+/− mice and AD patients (Figure S6). Importantly, we find that microglia obtained from AD brains have prominently reduced levels of beclin 1 and VPS35 (Figure 8A – B) when compared to non-demented controls. However, neither beclin 1 nor Vps35 levels were reduced in brain lysates from plaque depositing 16-month-old APP transgenic mice (Figure S7). Collectively, these findings suggest that beclin 1 is reduced in microglia within AD brains and that this deficiency is not the result of amyloid accumulation alone but may have other causes.
Figure 8.
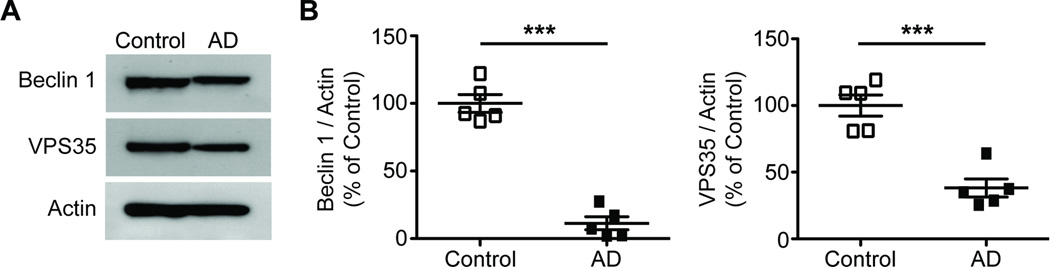
Microglia isolated from human AD brains show reduced levels of beclin 1 and VPS35. Human microglia isolated from the superior and middle frontal gyri of postmortem AD or non-demented control patients were lysed, the detergent soluble protein fraction was probed by western blot (A), and protein levels were quantified relative to actin (B). Results were analyzed by an unpaired Student’s t test (n=5 per group). All values are mean ± SEM ***p<0.001.
DISCUSSION
In this study, we define a novel function for the autophagy protein beclin 1 in regulating phagocytosis and phagocytic receptor recycling. Importantly, our observations appear to be clinically relevant as microglia isolated from human AD postmortem brains showed reduced levels of beclin 1 and VPS35. These findings open the possibility that reduced microglial beclin 1 levels in AD patients impairs phagocytic capacity when compared to microglia from healthy controls (See Figure S8). Independent studies are in line with this notion and show that microglia in mouse models of AD are inefficient at phagocytosing and clearing Aβ (Meyer-Luehmann et al., 2008). Whether “healthy” microglia are active participants in controlling Aβ levels remains controversial (Grathwohl et al., 2009). In spite of this uncertainty, numerous studies show that activating microglia with either lipopolysaccharide (Herber et al., 2004), genetic manipulation (Heneka et al., 2012; Liu et al., 2010; Town et al., 2008; Wyss-Coray et al., 2001), or following Aβ vaccination (Bard et al., 2000) is sufficient to promote removal of Aβ in vivo. Emerging evidence supports the idea that microglial phagocytosis may have important roles in AD progression. This is indicated by genetic studies showing that variants in the phagocytic receptor TREM2 triple the risk for AD (Guerreiro et al., 2013; Jonsson et al., 2013). While the mechanisms underlying this risk are unclear, it is postulated that microglia may play an active role in removing Aβ, apoptotic cellular or synaptic debris and in the process prevent the activation of rampant inflammation during AD. Taken together, microglial phagocytosis may have multiple functions in the healthy and diseased brain, which help to prevent amyloid accumulation and clear cellular debris. Given that we find general defects in phagocytosis when beclin 1 is reduced (i.e., with latex beads and Aβ), it is possible that recovering beclin 1 and phagocytic receptor recycling levels may be necessary for promoting optimal and sustained phagocytosis of disease-relevant substrates.
Additionally, our findings may also provide insight into phagocytic effectiveness beyond AD. For example, pathogens, including HSV-1 and γ-herpes viruses, encode factors that directly antagonize beclin 1 (Ku et al., 2008; Orvedahl et al., 2007). This may represent a strategy to impair phagocytosis and prevent viral clearance. Although inhibiting beclin 1 is likely to affect various cellular processes that could influence substrate clearance, including autophagy and potentially phagosomal maturation (which has been described for phagosomes containing apoptotic cells, entotic cells, and bacteria (Berger et al., 2010; Florey et al., 2011; Ma et al., 2012; Martinez et al., 2011)), our studies further reveal that inhibiting beclin 1 may also cause impairments upstream at the receptor level to disrupt phagocytic efficiency. One way that inhibiting beclin 1 might disrupt phagocytic efficiency is by impairing phagocytic receptor recycling as our studies on CD36 and Trem2 recycling indicate.
If the mechanisms described here for CD36 and Trem2 recycling are used more widely, it is tempting to speculate that beclin 1 deficiency might also result in dysfunctional turnover and availability of other membrane receptors. Notably, receptors for various growth factors or NMDA are dysregulated in AD (Ikonomovic et al., 1999; Moloney et al., 2010; Tesseur et al., 2006). Additionally, beclin 1 has been shown to be associated with several surface receptors, including delta 2 glutamate receptors (Yue et al., 2002) and bacterial SLAM receptors (Berger et al., 2010; Ma et al., 2012). It is currently unclear if beclin 1 is involved in the regulation and trafficking of these or any other receptors. Intriguingly, studies in C. elegans reveal a conserved role for beclin 1 in regulating Wnt receptor recycling. Indeed, C. elegans expressing a mutant form of BEC-1, the C. elegans ortholog of beclin 1, display defective recycling of the Wnt receptor MIG-14/Wntless, a receptor that is classically recycled by the retromer complex. Moreover, BEC-1 mutants exhibit reduced levels of PI3P and the retromer subunit RME-8 (Ruck et al., 2011). Our findings are in line with these observations and together they support the possibility that mechanisms described herein may be applicable to other receptors that utilize retromer-mediated recycling.
Given that retromer is involved in the sorting of various disease-relevant proteins, beclin 1-mediated impairments in retromer sorting could also play an important role in neurodegenerative disease. For example, both APP and β-Secretase are dependent on retromer trafficking (Andersen et al., 2005; Finan et al., 2011; Wen et al., 2011). Additionally, the retromer-binding receptor SorLA is reduced in AD and may disrupt APP trafficking and subsequent processing (Andersen et al., 2005). Most recently, genetic mutations in VPS35 have been linked to autosomal dominant Parkinson’s disease in two independent studies (Vilarino-Guell et al., 2011; Zimprich et al., 2011), suggesting retromer-mediated impairments in receptor recycling or protein sorting might also play an important role in Parkinson’s disease. It remains to be shown whether these mutations or other retromer abnormalities lead to impaired receptor recycling or phagocytosis. It is also unknown why beclin 1 is changed in AD and in microglia. Nevertheless, if beclin 1 proves to be a critical upstream regulator of retromer function in humans, restoring proper beclin 1 expression may have beneficial effects on sustaining various retromer-mediated processes in conditions where beclin 1 is disrupted or reduced. In particular, restoring beclin 1 expression in AD may represent a therapeutic approach for enhancing phagocytic efficiency and removal of Aβ aggregates.
EXPERIMENTAL PROCEDURES
Mice
T41 APP transgenic mice (mThy-1-hAPP751V171I, KM670/671NL) and beclin 1+/− mice have been described previously (Pickford et al., 2008; Qu et al., 2003). Beclin 1+/− mice were crossed with heterozygous T41 transgenic mice. All lines were maintained on a C57BL/6 genetic background. Brains were harvested from mice anesthetized with 400 mg/kg chloral hydrate (Sigma-Aldrich) and transcardially perfused with 0.9% saline. Brains were then dissected, and 1 hemibrain was fixed for 24 h in 4% paraformaldehyde and cryoprotected in 30% sucrose. Serial coronal sections (30 or 50µm) were cut with a freezing microtome (Leica) and stored in cryoprotective medium. When possible, the other hemibrain was frozen immediately at −80°C for additional analyses. All animal procedures were conducted with approval of the animal care and use committees of the Veterans Administration Palo Alto Health Care System.
Antibodies for immunohistochemistry and immunoblotting
The following antibodies were used: 3D6 (1:8000; Elan Pharmaceuticals), which was biotinylated using EZ-link NHS Biotin (Pierce Biotechnology); actin (diluted 1:5,000; Sigma-Aldrich); Atg5 (diluted 1:500; Novus Biologicals); Beclin 1 (diluted 1:500; BD Biosciences); CD36 (Abcam; [JC63.1] for receptor recycling assays and [FA6–152] for neutralization); CD68 (diluted 1:50; Serotec); EEA1 (Abcam); Iba-1 (diluted 1:2,500; Wako Bioproducts); Lamp1 (Abcam); NSE (LabVision); Rab5 (Sigma); Rab7 (Cell Signaling); Trem2 (R&D Systems); Vps26 (diluted 1:500; abcam); Vps29 (diluted 1:500; abcam); Vps34 (diluted 1:200; Invitrogen); and Vps35 (diluted 1:500; Abcam).
Viral infection
BV2 and N9 microglial cells were maintained in DMEM media containing 10% FBS. Cells were transduced with lentivirus (LV) containing an shRNA plasmid targeting mouse beclin 1 at nucleotides 405–423, which also expressed a co-poped GFP. LV targeting Atg5 contained an shRNA plasmid targeting nucleotides 302–320 of the mouse Atg5 transcript. LV targeting Vps35 consisted of a mixture of 3 separate LVs, each containing a 19–25 nucleotide shRNA sequence targeting a different region of the mouse Vps35 transcript. Control LV contained the same plasmid backbone, but expressed an shRNA sequence targeting luciferase. For beclin 1 rescue experiments, LV particles contained a plasmid encoding mouse beclin 1. Cells were infected in 96 well plates at a multiplicity of infection of 50 or 100 in the presence of polybrene (8 µg/ml). 18hrs after infection, virus-containing media was removed and replaced with normal maintenance media. 24hrs later cells were split into 24well plates. 24 hrs later (72hrs after infection), cells were used in phagocytosis and receptor recycling assays. At 72hrs posttransduction our lentiviral constructs were well tolerated, with nearly 95% viability as measured by trypan blue exclusion (data not shown). When used in ex-vivo phagocytosis assays, BV2 cells were transferred to 10cm dishes and grown until confluent. When possible, successful transduction was monitored by GFP expression and confirmed by western blot. Vps35 shRNA LV particles were purchased from Santa Cruz Biotechnology. All other LV plasmids were prepared as previously described (Marr et al., 2003) and generated by the Stanford Neuroscience Gene Vector and Virus Core.
Western blotting
All tissues or cells were lysed in RIPA buffer and total protein concentrations were determined with a BCA Protein Assay Kit (Thermo Scientific). 10–20 µg of total protein was loaded for each sample into pre-cast 4–12% bis-tris gels and run with MOPS buffer (Invitrogen). Gels were transferred onto PVDF membranes (Millipore). Antigen specific primary antibodies were incubated overnight at 4°C and detected with species-specific horseradish-peroxidase labeled secondary antibodies. An ECL Western Blotting Detection kit (GE Healthcare) was used to obtain a chemiluminescence signal, which was detected using Amersham Hyperfilm ECL (GE Healthcare). Band quantification was performed using ImageJ software (version 1.44; NIH). Bands of interest were normalized to actin or neuron specific enolase for a loading control.
Isolation of primary mouse microglia
Beclin 1+/− and wildtype littermate pups were sacrificed at postnatal day 6–8. Brains were dissected from the pups, the meninges were removed, and the cortex was isolated. Cortical tissue was then dissociated into single cells using the Neural Tissue Dissociation Kit P (Miltenyi Biotec). Cells were washed with Hank’s Balanced Salt Solution and resuspended in PBS containing 0.5% bovine serum albumin and 2mM EDTA (MACS buffer). To positively select for microglia, 10µL of CD11b MicroBeads (Miltenyi Biotec) were added per 107 total cells. Cells were then washed with MACS buffer and applied to a LS separation column placed in a magnetic holder (Miltenyi Biotec). Unlabeled cells were allowed to pass through the column while labeled cells remained in the magnetic field. After washing the column with MACS buffer, the column was removed from the magnetic field and the labeled cells were flushed from the column with MACS buffer. The resultant cell population, which was confirmed by flow cytometry to be ~90% pure for microglia (Figure 2C), was then cultured in DMEM:F12 with 10% FBS for at least three days before being used for functional studies.
Phagocytosis assay
BV2 cells were plated in 24 well plates at a density of 30,000 cells per well in DMEM with 10% FBS. Cells were cultured at 37°C for 1hr. 6µm latex beads (internally dyed with the fluorophore Flash Red; Polysciences, Inc.) were pre-opsonized in 50% FBS and PBS. BV2 cell media was then replaced with DMEM alone and pre-opsonized beads were added to the cells at a concentration of 10 beads per cell. BV2 cells and beads were incubated at 37°C for 1hr and were subsequently transferred to 5mL polystyrene Fluorescence-activated cell sorting (FACS) tubes with the aid of 0.25% trypsin. Cells were centrifuged at 1,200rpm for 5mins and washed twice with cold FACS buffer, consisting of 1% FBS and 0.02% sodium azide in PBS. Cells were resuspended in FACS buffer and analyzed by flow cytometry, as described below.
Flow cytometry
All flow cytometry experiments were performed on an LSR-II FACS machine (BDbioscience). For phagocytosis assays, events were thresholded for size as to exclude the visualization of free latex beads. At least 2,500 events were recorded for each sample. CD36 surface expression was assayed on fixed un-permeablized cells using a phycoerythrin-tagged CD36 antibody (BioLegend). At least 25,000 events were recorded for each sample. Analysis was performed using FlowJo (version 9.2; Tree Star Inc.).
Live-cell phagocytosis imaging
BV2 cells were transduced with lentivirus encoding either beclin 1 shRNA or luciferase shRNA control. Both lentiviruses also expressed a co-poped GFP to allow for the visualization of infected cells. 48hrs after lentiviral transduction, beclin 1 shRNA and Luciferase shRNA control cells were plated in separate chambers of 2-well chamber slides and incubated for 1 hr at 37°C in DMEM with 5ng/mL GM-CSF. Polystyrene beads (Polysciences, Inc.) pre-opsonized in 50% FBS were added at a concentration of 5 beads per cell immediately before imaging. Several fields from each well were imaged on a Zeiss Observer.Z1 inverted microscope. Cells were kept at 37°C and 5% CO2 with a Zeiss XL S1 stage incubator. Phase and GFP images were collected every 5 min for 2hrs using Axiovision 4.7.1 software (Zeiss) and exported as individual JPEG files. Individual frames were compiled and analyzed in ImageJ. Images from three independent experiments were blinded and only GFP-positive cells (n=15) in each condition were analyzed. The number of beads in each cell was counted for every frame. A bead was considered cell-associated when at least half of the bead was within the cell’s perimeter and stayed associated for the remainder of the movie. Cell movement was analyzed with ImageJ using the MTrackJ plug-in. Live cell imaging for phagosomal PI3P, phagosomal Vps35, or intracellular pH analysis with FITC beads was performed with a LSM 700 confocal microscope (Zeiss) using Zen 2010 software (Zeiss). In these studies, cells were kept at 37°C with 5% CO2 and imaged every 5mins for 30–90mins.
Receptor recycling assay
Receptor recycling assays were performed as previously described (Mitchell et al., 2004). Briefly, BV2 cells were plated on poly-L-lysine coated glass coverslips in 24 well plates at a density of 70,000 cells per well. Cells were maintained in DMEM with 10% FBS for 48hrs. Cells were then incubated in DMEM with 10% donkey serum (the source of the secondary antibody) for 15mins at 37°C. Antibodies against CD36 (Abcam) or Trem2 (R&D Systems) were added to the cells in DMEM with 1% donkey serum for 1hr at 37°C. Cells were then acid washed with cold DMEM at pH 2.0. Cells were cultured in DMEM with 10% donkey serum for 1hr at 37°C and then provided fluorophore-conjugated secondary antibodies (Alexa Fluor 555 or Alexa Fluor 647 for Vps35 rescue experiments; Invitrogen) in 1% donkey serum for 1hr at 37°C. Cells were again acid washed with cold DMEM at pH 2.0 and washed with cold PBS. Cells were then fixed with 4% paraformaldehyde, washed with PBS, and mounted on glass slides using Prolong Gold (Invitrogen). Fluorescent signal from vesicles containing recycled receptors was thresholded and the area of fluorescent signal was determined by ImageJ. The area of fluorescent signal was then divided by the total number of cells present in the field to generate a measurement of fluorescent area per cell. For all experiments investigators were blinded with respect to the treatment condition.
Cloning of Vps35 plasmids
Mouse wild-type Vps35 cDNA was purchased (Origene) and the full-length cDNA was cloned into the ligase-free cloning site of the pPS-EF1-LCS-T2A-RFP lentiviral vector (System Biosciences), which co-expresses RFP. To generate a Vps35-RFP fusion construct, the T2A domain of the plasmid described above was deleted using a QuikChange site-directed mutagenesis kit (Agilent Technologies) with the following primer: CTGTTCGAGAGGGCAGAGGAGAATTCATGGCCCTTAGTAAGC.
Transfection of BV2 cells by electroporation
BV2 cells were transiently transfected by electroporation as previously described (Smale, 2010). Briefly, cells were resuspended in DMEM media containing 10% FBS at a concentration of 3.75 × 107 cells/mL 200 µl of cells and 20 µg of plasmid DNA were transferred to Gene Pulser cuvettes with a 0.4cm electrode gap (Bio-Rad). A 250V charge was applied to each cuvette using a Gene Pulser II electroporation system with a 950µF capacitor (Bio-Rad). Transfected cells were plated in DMEM media containing 10% FBS and utilized 48hrs later.
Ex-vivo Aβ phagocytosis assay
Ex-vivo Aβ phagocytosis assays were performed as previously described (Bard et al., 2000). Briefly, 18 month-old T41 transgenic mice were perfused with saline. Brains were removed and snap frozen in cooled isopentane. Frozen brains were cut into 10µm thick sagittal sections using a cryostat (Leica) and mounted onto poly-L-lysine coated glass coverslips. Brain sections were allowed to dry for at least 2hrs at room temperature and then washed with hybridoma serumfree media (H-SFM; Invitrogen) containing 1% FBS. Brain sections were then cultured with 5×105 BV2 cells in H-SFM containing 1% FBS for 18hrs at 37°C with 5% CO2 or 1.75×105 primary mouse microglial cells in H-SFM containing 1% FBS and 5ng/mL GM-CSF (R&D Systems) for 60hrs at 37°C with 5% CO2. Sections were then washed twice with PBS and either fixed with 4% paraformaldehyde for subsequent histology or placed in 6.25M Guanidinhydrochloride to extract Aβ for ELISAs (see below).
Aβ ELISA
Enzyme linked immunosorbent assays were performed using Meso Scale technology (Meso Scale Discovery). Multi-array 96 well plates (Meso Scale Discovery) were coated with capture antibody 21D12 for total Aβ (Aβ13–28) or antibody 21F12 for Aβ42 (Aβ33–42). Plates were washed and diluted samples and Aβ standards were added. Aβ was detected using biotinylated-3D6 antibody (Ab1–5) and SULFO-TAG streptavidin (Meso Scale Discovery). Plates were read on a Sector Imager 2400 (Meso Scale Discovery) and samples were normalized to Aβ standards. All Aβ antibodies were provided by Elan Pharmaceuticals.
In vivo phagocytosis assays
Fibrillar Aβ1–42 was prepared by incubating synthetic monomers, diluted to a stock concentration of 1µg/µL in PBS, overnight at 37°C. pH-sensitive beads were prepared by coupling 3µm latex beads with CypHer5E mono N-hydroxysuccinimide (NHS) ester (GE Healthcare). Beads were washed extensively and diluted in PBS to a stock concentration of 2×104 beads/µL. 8 week-old mice were then anesthetized with an inhaled isoflurane/oxygen mixture and 1 µL of Aβ or pH-sensitive beads were stereotaxically injected into the frontal cortex using the following coordinates from bregma: +1.9µm anterior, +1.5µm laterally, and a depth of 1µm with an injection speed of 0.2µL/min. 48hrs later mice were perfused as described above and brains were prepared for histological analysis.
Histology
Fixed sections were permeabilized with 0.1% Triton × −100 and 0.6% hydrogen peroxide. For Aβ histology, sections were blocked using a streptavidin and biotin blocking kit (Vector) and biotinylated 3D6 antibody was applied (1:8000) overnight at 4°C. Primary antibody labeling was revealed using ABC kit (Vector) with diaminobenzidine (DAB; Sigma-Aldrich). For fluorescent double-immunolabeling, Iba-1 and 3D6 primary antibodies were detected with fluorophoreconjugated secondary antibodies (Alexa Fluor 488 and 555, respectively; Invitrogen). For studies with beclin 1+/− mice, coronal sections were cut at 50µm for in vivo pH bead analysis using a freezing microtome (Leica). All sections with the appearance of a needle track were selected and pH-sensitive beads could be observed with confocal microscopy without histological procedures. Sections stained for Aβ were imaged with a CoolSNAP HQ camera (Photometrics) mounted on an Olympus I×71 microscope (Olympus America Inc.) using Spot Software (version 4.7; Diagnostic Instruments Inc.). For ex vivo phagocytosis sections, digitized images were thresholded for positive Aβ labeling and percent thresholded area was determined with Metamorph (Molecular Devices Corporation). For pH-sensitive bead analysis, 6µm thick optical z-stack slices were generated with Zeiss software (version 4.2) for all sections where beads were visible. The total area of bead fluorescence in all slices from sections with positive signal was determined with ImageJ. For all experiments investigators were blinded with respect to the genotype of the mice or treatment condition.
Isolation of human microglia
Microglia were isolated from postmortem AD-confirmed or non-demented, non-pathological control brains (See Table S1 for more details) as previously described (Lue et al., 2001). Cells were then cultured in DMEM media supplemented with 10% FBS for 10–14 days at 37°C with 7% CO2. Cells were isolated from culture flasks with 0.25% trypsin and manual separation using a cell scraper (Nunc) and prepared for flow cytometry or western blot analysis as described above. Previous studies show that these cultures are ~99% pure by demonstrating positive CD68 immunoreactivity and negative immunoreactivity for glial fibrillary acidic protein and galactocerebroside (Lue et al., 2001). Purity of microglial cultures was also confirmed in our studies using antibodies against CD11b (BDbiosciences) as a marker for myeloid-derived cells. Flow cytometric analysis confirmed that our cultures were greater than 80% positive for CD11b (Data not shown).
Human brain tissue
Brain tissues from confirmed AD and age-matched, non-demented, non-pathological controls (See Table S2 for more details) were obtained from ADRC at the University of California - San Diego, The Institute for Brain Aging and Dementia Tissue Repository at the University of California - Irvine, Stanford Brain Bank at Stanford University, and The L. J. Roberts Alzheimer’s Center at Banner Sun Health Research Institute in strict accordance with all ethical and institutional guidelines. Cortical mid-frontal gray matter tissues were cut from frozen tissue blocks and subjected to protein extraction using procedures described above. Microglia were isolated from cortical mid-frontal gray matter tissues at the Banner Sun Health Research Institute using fresh tissue obtained within an average postmortem delay interval of 3.11 ± 0.55 hrs.
Statistics
All statistical analyses were computed using Prism5 (GraphPad Software). Differences between treatment conditions were established using a student’s unpaired t-test (for two conditions), a one-way ANOVA with a Tukey’s post-test for multiple comparisons, or a two-way ANOVA with a Bonferroni post-test when comparing groups with multiple variables. p values of less than 0.05 were considered significant. Statistic details are indicated in the respective figure legends.
Supplementary Material
HIGHLIGHTS.
Reducing beclin 1 impairs the phagocytosis of latex beads or Aβ in vitro and in vivo.
Reducing beclin 1 impairs recycling of the phagocytic receptors CD36 and Trem2.
Reducing beclin 1 impairs the recruitment of retromer and retromer levels.
Beclin 1 and retromer are reduced in human AD brains including microglia.
ACKNOWLEDGMENTS
The authors thank Dr. Saul Villeda and Dr. Joseph Castellano for critical review of the manuscript, Zhaoqing Ding for assistance with flow cytometry, Dr. Manuel Buttini for assistance with the ex-vivo phagocytosis assay, Dr. Sergio Grinstein for providing the 2×FYVE-mRFP construct, the Stanford Virus Core (supported by NINDS P30 NS069375-01A1) for producing the lentiviruses used in this study, and Dr. Philipp Jaeger and Dr. Scott Small for helpful discussions of the manuscript. Funding for these studies was provided by the Department of Veterans Affairs (T.W.-C), the National Institutes of Health Institute on Aging (R01 AG030144, T.W.-C), a California Initiative for Regenerative Medicine Award (T.W.-C), The Larry L. Hillblom Foundation (K.M.L., T.W-C.), The John Douglas French Alzheimer’s Foundation (K.M.L.), a National Science Foundation pre-doctoral fellowship (K.I.M), and a Kirschstein NRSA predoctoral fellowship (1 F31 AG040877-01A1, K.I.M.). We are also grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of human microglia. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05–901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors report no conflicts of interest.
REFERENCES
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SB, Romero X, Ma C, Wang G, Faubion WA, Liao G, Compeer E, Keszei M, Rameh L, Wang N, et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat Immunol. 2010;11:920–927. doi: 10.1038/ni.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda P, Padilla SM, Sarkar S, Emr SD. Retromer function in endosome-to-Golgi retrograde transport is regulated by the yeast Vps34 PtdIns 3-kinase. J Cell Sci. 2002;115:3889–3900. doi: 10.1242/jcs.00090. [DOI] [PubMed] [Google Scholar]
- Chen D, Xiao H, Zhang K, Wang B, Gao Z, Jian Y, Qi X, Sun J, Miao L, Yang C. Retromer is required for apoptotic cell clearance by phagocytic receptor recycling. Science. 2010;327:1261–1264. doi: 10.1126/science.1184840. [DOI] [PubMed] [Google Scholar]
- Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan GM, Okada H, Kim TW. BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin. J Biol Chem. 2011;286:12602–12616. doi: 10.1074/jbc.M110.170217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13:1335–1343. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, et al. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2012 doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, Gordon MN. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Mizukami K, Warde D, Sheffield R, Hamilton R, Wenthold RJ, Armstrong DM. Distribution of glutamate receptor subunit NMDAR1 in the hippocampus of normal elderly and patients with Alzheimer’s disease. Exp Neurol. 1999;160:194–204. doi: 10.1006/exnr.1999.7196. [DOI] [PubMed] [Google Scholar]
- Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One. 2010;5:e11102. doi: 10.1371/journal.pone.0011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaifu T, Nakahara J, Inui M, Mishima K, Momiyama T, Kaji M, Sugahara A, Koito H, Ujike-Asai A, Nakamura A, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest. 2003;111:323–332. doi: 10.1172/JCI16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku B, Woo JS, Liang C, Lee KH, Hong HS, E X, Kim KS, Jung JU, Oh BH. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog. 2008;4:e25. doi: 10.1371/journal.ppat.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. 2010;30:17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Gore M, Zhang Q, Camenisch T, Boast S, Casagranda F, Lai C, Skinner MK, Klein R, Matsushima GK, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398:723–728. doi: 10.1038/19554. [DOI] [PubMed] [Google Scholar]
- Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–311. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer’s disease with human postmortem microglial cultures. Neurobiol Aging. 2001;22:945–956. doi: 10.1016/s0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- Ma C, Wang N, Detre C, Wang G, O’Keeffe M, Terhorst C. Receptor signaling lymphocyte-activation molecule family 1 (Slamf1) regulates membrane fusion and NADPH oxidase 2 (NOX2) activity by recruiting a Beclin-1/Vps34/ultraviolet radiation resistance-associated gene (UVRAG) complex. The Journal of biological chemistry. 2012;287:18359–18365. doi: 10.1074/jbc.M112.367060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, Verma IM, Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Tavshanjian B, Oleksy A, Perisic O, Houseman BT, Shokat KM, Williams RL. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science. 2011;327:1638–1642. doi: 10.1126/science.1184429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell H, Choudhury A, Pagano RE, Leof EB. Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Molecular biology of the cell. 2004;15:4166–4178. doi: 10.1091/mbc.E04-03-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31:224–243. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada I, Ongania GN, Buonsanti C, Panina-Bordignon P, Meldolesi J. Triggering receptor expressed in myeloid cells 2 (TREM2) trafficking in microglial cells: continuous shuttling to and from the plasma membrane regulated by cell stimulation. Neuroscience. 2006;140:1139–1148. doi: 10.1016/j.neuroscience.2006.03.058. [DOI] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Ruck A, Attonito J, Garces KT, Nunez L, Palmisano NJ, Rubel Z, Bai Z, Nguyen KC, Sun L, Grant BD, et al. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C. elegans. Autophagy. 2011;7:386–400. doi: 10.4161/auto.7.4.14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- Smale ST. Transfection by electroporation of RAW 264.7 macrophages. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.prot5374. pdb prot5374. [DOI] [PubMed] [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J Clin Invest. 2006;116:3060–3069. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. The Journal of cell biology. 2001;155:19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, Shen C, Jung JU, Xiong F, Lee DH, et al. VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J Cell Biol. 2011;195:765–779. doi: 10.1083/jcb.201105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu G, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-p1 promotes microglial amyloid-p clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci USA. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa M, Tsukuba T, Nishioku T, Okamoto Y, Okamoto K, Takii R, Terada Y, Nakayama KI, Kadowaki T, Yamamoto K. Cathepsin E deficiency induces a novel form of lysosomal storage disorder showing the accumulation of lysosomal membrane sialoglycoproteins and the elevation of lysosomal pH in macrophages. The Journal of biological chemistry. 2007;282:1851–1862. doi: 10.1074/jbc.M604143200. [DOI] [PubMed] [Google Scholar]
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–933. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.