

Abstract
Dysregulated recruitment of leukocytes into the intestine is a characteristic feature of IBD. Several families of molecules regulate the influx of these cells into sites of inflammation within the gastrointestinal tract. Interference with molecules that mediate the formation of stable bonds (integrins) with their endothelial ligands has already shown efficacy in the clinics. Antibodies that target participant molecules have been approved by the US Federal Drug Administration for use in Crohn’s, multiple sclerosis (MS) (i.e. natalizumab) and psoriasis (i.e. efalizumab). A more recent additional family of drugs, which might also interfere with lymphocyte traffic (i.e. shingosine-1-phosphate receptor agonists: fingolimod) is in clinical use for MS and just recently entered the clinical trial stage for ulcerative colitis. In the present review we discuss basic aspects of clinically relevant molecules and compile the clinical studies that support the targeting of specific steps of the leukocyte adhesion cascade for therapeutic purposes in IBD.
Keywords: adhesion molecules, chemokines, Crohn’s disease, integrins, sphingosine1-phosphate, ulcerative colitis
Introduction
Immune-mediated chronic inflammatory diseases (e.g. rheumatoid arthritis (RA), psoriasis, multiple sclerosis, and inflammatory bowel disease (IBD)) are characterized by dysregulated leukocyte recruitment 1–3. Neutrophils, which are short-lived outside the circulation, migrate to sites of inflammation and undergo apoptosis while monocytes remain in inflamed tissues for several days, while other monocytes become permanent residents. By contrast, naïve T cells migrate to lymphoid organs where they encounter antigens, proliferate and acquire effector functions, along with a repertoire of adhesion molecules, cytokine and chemokine receptors that allow them to recognize specific vascular beds 4, 5. Effector lymphocytes then are able recirculate from the blood into tissues, followed by migration to lymphoid organs before returning to blood. Their ability to recirculate is essential for the perpetuation of chronic inflammatory processes, such as IBD. The molecules that mediate leukocyte traffic have therefore been the focus of investigation due to their potential as therapeutic targets.
The Leukocyte Adhesion Cascade
Leukocytes migrate into sites of inflammation across the walls of post-capillary venules by engaging specific molecules expressed on specialized endothelial cells. Endothelial molecules serve as mechanical anchors and confer tissue specificity to the recruitment process 6. These molecules mediate a sequential series of steps (i.e. capture, rolling, activation and firm adhesion) that allow leukocytes to escape the circulation and migrate to sites of inflammation. The major families of molecules involved in leukocyte recruitment include: selectins and their glycoprotein ligands, chemokines and their receptors, integrins and immunoglobulin-superfamily molecules.
Although interference with any of these steps might conceivably be of therapeutic value, only targeting the integrins, which mediate arrest and firm addition, have crossed from the bench into the clinics. Both α4β1 (VLA-4) and α4β7 integrins have been shown to mediate firm adhesion to their respective ligands Vascular Cell Adhesion Molecule (VCAM)-1 and Mucosal Addressin Cell Adhesion Molecule (MAdCAM)-1 7, 8.
Integrins
Integrins are cell-adhesion receptors expressed on leukocytes as heterodimeric transmembrane glycoproteins 9. They interact with molecular components of the extracellular matrix but also with ligands displayed by endothelial (and other) cells 10. Binding of integrins to their respective ligands is stable, results in arrest of leukocytes on the vessel wall and ensures migration of lymphocytes to sites of inflammation. Besides cell adhesion, integrins have been shown to play important roles in other cellular processes, such as antigen recognition, cell proliferation and survival 11. Consequently, integrins constitute integral components of diverse biological phenomena, including cellular development, immunological reactions, hemostasis and neoplastic transformation 10.
Integrins are heterodimeric molecules formed by the noncovalent association of two subunits, namely the large (α, molecular weight: 120–170 kDa) and the small (β, molecular weight: 90–100 kDa) subunits. As of today, 18 α and 8 β-subunits have been described, which combine to generate at least 24 different integrin heterodimers in vertebrates, 14 of which are detected in cells of the immune system (Figure 1) 11–14. The high number of possible combinations ensures the wide functional diversity of these molecules. Each subunit is a type I transmembrane glycoprotein consisting of a large extracellular domain, a single pass transmembrane domain which induces intracellular signaling via binding to cytoskeleton 15 and a short cytoplasmic tail (with the notable exception of β4 integrin) 13. Extracellular domains contain the site for ligand binding, which is dependent upon the presence of divalent cations (Ca2+, Mg2+, and Mn2+).
Figure 1.

The integrins are αβ heterodimers that share common subunits that bind distinct ligands. Illustrated in color are the integrins that have been targeted for the treatment of IBD. RGD: arginine-glycine-aspartic acid sequence found in some integrin ligands. C3bi: Complement 3b inactivated
The type of the β-subunit that is present in each heterodimer defines discrete subtypes of integrins, which display unique patterns of structure, tissue-specificity and function. According to this functional scheme, the β2, α4 and β7 families of integrins play the most prominent roles during immunological and inflammatory conditions, as they are leukocyte-specific10, 16. Further diversity results from the fact that different types of leukocytes display unique patterns of integrin expression. Indeed, lymphocytes, macrophages, and polymorphonuclear cells all express distinct sets of β integrins which differ between subclasses and also between the resting state and activated states 17.
Integrin ligands – adhesion molecules of the immunoglobulin superfamily
Integrins that are displayed on the surface of circulating leukocytes associate with adhesion molecules that are expressed on endothelial cells, directing the migration of leukocytes from the blood to sites with inflammation. The endothelial ligands for integrins are members of the immunoglobulin superfamily. Proteins of this class share structural and genetic features with immunoglobulin molecules; they contain at least one immunoglobulin domain, comprising of two β-pleated sheets held together by a disulfide bond (Figure 2). These immunoglobulins play pivotal roles in IBD as they are major mediators of the aberrant trafficking of lymphocytes and other immune cells to the inflamed mucosal sites. In addition they may also participate in the pathogenesis of extraintestinal inflammatory reactions that are a frequent manifestation of IBD. Among the several members of the immunoglobulin superfamily, the following have established pathogenetic roles in IBD: intercellular adhesion molecule-1 (ICAM-1) or CD54, Vascular Cell Adhesion Molecule-1 (VCAM-1) or CD106, and Mucosal Addressin Cell Adhesion Molecule-1 (MAdCAM-1).
Figure 2.
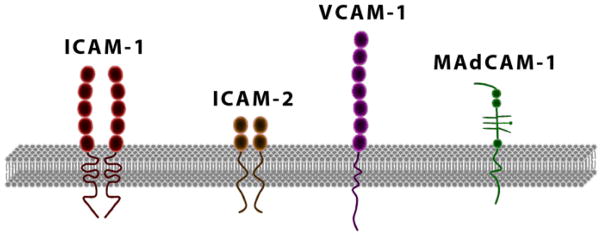
Immunoglobulin superfamily. Members of this family contain two to seven immunoglobulin domains and serve as ligands for leukocyte integrins. ICAM-1 and ICAM-2 exist as homodimers on the cell surface.
β2 Integrins (CD18)
The β2 integrins are also known as the “leukocyte” integrins because they are exclusively expressed on bone marrow-derived cells. Upon cellular activation, a process termed inside-out signaling or integrin activation is initiated whereby the extracellular region undergoes modulation that results in enhancement of ligand-binding capacity 13, 16. Integrin activation is triggered by the engagement of various membrane receptors such as the binding of a chemokine or other chemoattractant to its heptahelical G-protein coupled receptor (GPCR) on the leukocyte surface 18, 19. This triggering event is followed by inside-out signaling within milliseconds 13.
β2 integrins/CD18 associate with different α chain ligands to generate four β2-heterodimers (Figure 1). Their cellular distribution varies between the different heterodimers and some such as CD11c are now commonly seen as markers of monocyte lineage cells. Critical information for the functional importance of β2 integrins has been gained from the study of the clinical and immunological phenotype of mice or humans bearing mutations in the respective genes. In particular, lymphocytes and neutrophils mainly express CD11a/CD18 (LFA-1). LFA-1 binds to ICAM-1 and ICAM-2 expressed on endothelial cells and mediates migration, antigen presentation and cell proliferation 20, 21. The existence of a null mutation in the gene encoding the LFA-1 α chain results to defects in lymphocyte migration to secondary lymphoid organs 22. CD11b/CD18 (Mac-1) is also expressed on lymphocytes; nevertheless its main cellular localization is on monocytes, granulocytes and some NK cells. In phagocytes, Mac-1 is necessary for respiratory burst and certain forms of phagocytosis 23, 24. The other two β2-heterodimers are not expressed on lymphocytes; instead CD11c/CD18 (p150, p95) is detected on monocytes and dendritic cells and CD11d/CD18 predominantly on macrophages and granulocytes in the red pulp of the spleen 25. Recent genetic studies have clearly elucidated an important role of CD18 integrins in innate immunity. Indeed, humans 26 and mice 21, bearing null or hypomorphic mutations in the Itgb2 gene (which encodes for the β2 (CD18) integrin subunit]) result in the genetic disorder leukocyte adhesion deficiency-1 (LAD-1) 26. This condition is characterized by recurrent bacterial infections. The pathophysiological disturbance that underlies the clinical phenotype is ineffective recruitment of granulocytes in response to bacterial invasion 26.
As mentioned above, ICAM-1 is a very important ligand for integrins LFA-1 and Mac-1 27. LFA-1/ICAM-1 interaction is dominant for the trafficking of inflammatory cells, whereas Mac-1 may associate with various ligands 28. ICAM-1 is constitutively expressed on endothelial cells. Nevertheless, its expression is highly upregulated when endothelial cells are stimulated by pro-inflammatory signals, including cytokines, such as IL-1α and IFN-γ 29. These cytokines are integral components of the mucosal milieu in IBD and may be responsible for the increased adhesiveness for integrins that characterizes the inflamed gut 30. In fact, mucosal upregulation of ICAM-1 has been reported in both CD and UC, whereas, in a recent study, response to treatment with anti-TNF agents correlated to decreases in mucosal ICAM-1 expression 31.
Therapeutic targeting of CD18 integrin/ICAM-1 pathway in IBD
Efalizumab in CD
Efalizumab (RAPTIVA) is a humanized monoclonal antibody (IgG1) that targets the integrin αL subunit (CD11a) of LFA-1 32. As a result, LFA-1 expressing lymphocytes are prevented from interacting with ICAM-1-expressing endothelial cells. This, in turn, results in compromised trafficking and defective activation of T-cells. Efalizumab was approved in 2003 for the treatment of plaque psoriasis 33. In 2011 the results of a clinical trial of efalizumab administration in treatment-refractory, moderate to severe CD (Crohn’s Disease Activity Index [CDAI] score 220–450) CD were published 34. Efalizumab was administered on a weekly schedule of subcutaneous 1 mg/kg injection for 8 weeks. The primary endpoint of clinical response (≥70 points decrease in the CDAI score) at 8 weeks was reached by 67% of patients. Clinical remission was achieved in 40% of cases and there was a significant increase in the mean Inflammatory Bowel Disease Questionnaire (IBDQ) score from 124 to 168 (P<0.001). Clinical response typically occurred during the first weeks of treatment (before week 4). There were no serious adverse events during the follow-up period (16 weeks). The most frequent adverse effect was headache that usually occurred after the first 3 doses and lasted for 1–2 days post-injection. In April 2009 efalizumab was voluntary withdrawn from the US market, due to the report of 3 cases of progressive multifocal leukoencephalopathy (PML) in patients with psoriasis.
Alicaforsen in IBD
Alicaforsen (ISIS2302) is a 20 base pair antisense oligonucleotide that inhibits the translation of ICAM-1 mRNA, leading to reduced expression of ICAM-1. The efficacy of this compound was originally studied in CD in various therapeutic protocols, including intravenous and subcutaneous administration. Analysis of these studies led to the conclusion that ICAM-1 is ineffective for the treatment of CD 35.
The efficacy of local alicaforsen delivery by enema was subsequently studied in patients with active, left-sided UC. Bioavailability studies did show that following rectal enema the concentration of alicaforsen at the colonic mucosa was more than 100-fold higher when compared to the maximum concentrations obtained in plasma 36. The efficacy of the compound was studied in a randomized, double-blind, both placebo and active-controlled multicenter studies, with mesalazine enema as the active comparator 37, 38. Patients with mild to moderate active, left-sided colitis were administered daily enemas of different doses of alicaforsen, mesalazine or placebo for 6 weeks. The major conclusions from these findings were the favorable safety profile and the durability of response to alicaforsen, which exceeded that of mesalazine.
α4 Integrins (CD49d) and their ligands: α4β1/VCAM-1 and α4β7/MAdCAM-1
There are two integrins that contain an α4 subunit, namely the α4β1 and the α4β7. The α4β1 homodimer is also known as very late antigen-4 (VLA-4, CD49d/CD29) 39, 40. VLA-4 binds to its ligand VCAM-1 (CD106), and is chiefly responsible for lymphocyte and monocyte adhesion to vascular endothelium. The steady state-expression of VCAM-1 on the endothelium is very low/undetectable. Nevertheless, under inflammatory or other stimulatory conditions VCAM-1 expression is upregulated. VCAM-1 is not exclusively expressed on endothelial cells. Studies have reported detection of this molecule on epithelial cells, dendritic cells (DCs), Kupffer cells and on smooth muscle cells within atherosclerotic lesions 41–43. Of particular importance is the fact that VLA-4/VCAM-1 system regulates the trafficking of lymphocytes to the central nervous system. VCAM-1 is expressed at very low levels in CNS microvessels at the healthy state; nevertheless its expression is significantly induced under inflammatory conditions. This was shown in the murine model of experimental autoimmune encephalitis and further confirmed in patients with multiple sclerosis 44. These findings may have important implications for the pathogenesis of PML, a devastating adverse effect of anti-integrin-based therapies.
The other α4 integrin, α4β7 is critically involved in gut-homing. Indeed, α4β7 expression is upregulated on plasma cells that are positive for the mucosa-associated immunoglobulin IgA and in memory/activated CD4+ subsets that re-circulate to the gut (defined as, α4β7high memory T cells) 45. This is in contrast to the immunophenotype displayed by non-mucosa associated memory T-lymphocytes. In this population, the expression of β7 integrins is lacking and replaced by upregulation of α4β7, which binds to VCAM-1 and directs T-cells to non-mucosal sites 46, 47. It has been recognized in recent years that the α4β7-mediated gut tropism of memory/activated gut-homing T cells (but also of and circulating B cells) is induced by retinoic acid, which originates from intestinal DCs 48, 49.
The ligand for α4β7 is mucosal addressin cell adhesion molecule-1 (MAdCAM-1) 50–52 which is selectively expressed by high endothelial venules of Peyer’s patches and gut associated lymphoid tissue 53. The α4β7/MAdCAM-1 interaction is pivotal for the homing of lymphocytes to Peyer’s patches. MAdCAM-1 may also be involved in L-selectin-mediated rolling of inflammatory cells54. Therefore, MAdCAM-1 may be the common critical factor through which L-selectin and α4β7 integrin synergize for lymphocyte homing to the intestine 7, 55.
Recent evidence shows increased numbers of intestinal mucosal vessels that stain positive for MAdCAM-1 patients with CD or UC 51. In addition, animal models of intestinal inflammation, such as the colitic IL-10 KO mice overexpress mucosal MAdCAM-1 56. This upregulation may be the result of the proinflammatory milieu that predominates at the inflamed mucosa. TNF-α and IL-1 are both abundant in areas of active CD or UC and have been shown to induce upregulation of MAdCAM-1 expression in the intestine, colon and MLN 56, 57. Very interestingly, during active IBD, MAdCAM-1 expression is detected in extra-intestinal sites, such as the joints, eyes, skin and liver58. As these organs are frequently affected in patients with IBD, the aberrant expression of a gut-homing molecule in these tissues may attract pathogenic cells and induce extra-intestinal inflammation. In addition to IBD, MAdCAM-1 expression is upregulated on inflamed venules in several other chronic inflammatory conditions as those occurring in diabetes, primary sclerosing cholangitis, and cirrhosis 59.
Therapeutic targeting of α4 integrins/ligands in IBD
Natalizumab in CD
Natalizumab is a recombinant humanized IgG4κ monoclonal antibody that binds to α4-integrin. It was produced in murine myeloma cells and humanized by engrafting the complementarity-determining regions of a murine anti-α4 antibody (AN100226m) to a human immunoglobulin IgG4 framework. This resulted in a 95/5 % human /murine protein composition of natalizumab, with a molecular weight of 149 kD. Natalizumab is marketed under the brand name of Tysabri (formerly Antegren).
By targeting the α4 subunit of integrins, natalizumab blocks both α4β1/VCAM-1 and α4β7/MAdCAM-1 interactions. These properties were demonstrated in in vitro studies, as natalizumab effectively prevented adhesion of human Jurkat cells that expressed α4β1 to purified recombinant VCAM-1 and of RPMI-8866 cells that expressed α4β7 to recombinant MAdCAM-1. These data were complemented by in vivo studies in guinea pigs with experimental allergic encephalomyelitis (EAE). This model is mediated by T-lymphocytes that infiltrate regions of the central nervous system via α4β1/VCAM-1-mediated migration. Natalizumab administration did not allow leukocytes from crossing the blood-brain barrier and both prevented the development of neurological manifestations as well as reversed established disease 60. In all, these results provided a direct proof for the efficacy of natalizumab as an anti-adhesion drug. Pre-clinical studies were also performed in tamarins with IBD and provided evidence for an anti-inflammatory effect of α4 blockade in experimental intestinal inflammation61, 62. These pre-clinical studies were followed by a multicenter study of natalizumab in patients with active multiple sclerosis and a small phase I study in 26 healthy male volunteers, which showed that a single 3-mg/kg intravenous dose was safe and well tolerated. These data set the background for clinical studies of natalizumab in patients with IBD.
In a first study by Gordon et al., 30 patients with mild to moderate, active CD (CDAI >151 and <450) were blindly randomized to receive a single, 3-mg/kg infusion of natalizumab (n=18) or placebo (n=12) (n=12)63. The primary outcome was the change in CDAI at week 2 after infusion and the presence of clinical remission as defined by a CDAI<150. Among secondary outcomes were Inflammatory Bowel Disease Questionnaire (IBDQ) score, concentration of C-reactive protein (CRP) in the serum, and peripheral blood T-cells and B-cells counts. At week 2, the CDAI decreased significantly from baseline after infusion of natalizumab (mean 45 points) but not placebo (mean 11 points). The number of patients achieving clinical remission at week 2 was 7/18 (39%) and 1/12 (8%) for the natalizumab and control groups, respectively. Nevertheless, comparisons between natalizumab and placebo-treated patients regarding clinical response and remission did not reach statistical significance. At 4 weeks, patients treated with natalizumab experienced significant improvement of IBDQ scores, and decreases in the levels of inflammatory markers (CRP, ESR). Rescue therapies were needed by 4/12 (33%) of the placebo-treated patients in contrast to only 2/18 (11%) patients in the natalizumab group. Significant increases in circulating B and T lymphocytes were detected 1, 2, and 4 weeks after drug administration indicating that natalizumab interrupted lymphocyte trafficking. There were no significant differences in the frequency of adverse events between natalizumab and placebo-treated patients, the most common being headache, CD exacerbation, and abdominal pain. The overall modest results achieved in the study by Gordon may be attributed to the study protocol that included a single infusion of natalizumab. It was later found that this administration resulted in a satisfactory mean serum concentration of natalizumab by week 2, whereas at week 4, drug levels were lower, and potentially suboptimal according to the results of leukocyte saturation studies.
A second, double-blinded randomized study on natalizumab in CD was performed by Gosh et al.64 Patients (n=248) with moderate to severe CD (CDAI >220 and <450) were recruited from 35 centers. Patients were randomized to receive two infusions 4 weeks apart (week 0 and week 4) according to four different treatment regimens: wk-0: placebo, wk-4: placebo; wk-0: 3 mg/kg natalizumab, wk-4: placebo; wk-0: 3 mg/kg natalizumab, wk-4: 3 mg/kg natalizumab; or wk-0: 6 mg/kg* natalizumab, wk-4: 6 mg/kg* natalizumab. The primary outcome was the proportion of patients in remission (CDAI<150) by week 6 which was not reached as there were no significant differences between the rates of remission between the group that received 2 infusions of 6 mg/kg* natalizumab and the placebo group. In contrast, there were significantly more patients in remission in the 6 mg/kg* group both at 4 weeks (29% vs. 14% in the placebo group, P=0.028) and at 8 weeks (43% vs 16%, P=<0.001). In addition, patients receiving 3 mg/kg* of natalizumab had a significant higher chance of being in remission by week 4 (29% vs. 14%, P=0.027, week 6 (44% vs. 27%, P=0.03), week 8 (41% vs. 16%, P<0.001) and week 12 (42% vs. 27% in the placebo group, P=0.042). In addition, administration of 3 or 6 mg/kg* of natalizumab resulted in significant differences on several secondary outcomes of the study, including higher percentages of patients achieving clinical response by week 4, 6, 8, and 12, significant improvements in mean IBDQ scores at week 6, as well as significant decline of the baseline serum CRP levels at week 6 (P<0.05 vs. placebo treated). No significant differences in study outcomes were seen between the groups receiving 3 or 6 mg/kg* of natalizumab. Similar to the previous study, there were increased numbers of lymphocytes in the systemic circulation, a finding compatible with a natalizumab-mediated inhibition of lymphocytic extravasation due to inhibition of α4 integrin blockade.
The ENACT study was the first to include both induction and maintenance arms for the administration of natalizumab to patients with CD65. In addition, a fixed dose of 300mg of natalizumab was given to patients instead of body weight based adjustments. The study was conducted in 142 centers around the world and patients with moderate to severe CD (CDAI >220 and <450) were recruited. In the induction arm, patients received 3 infusions of 300 mg natalizumab (n=724) or placebo (n=181) at weeks 0, 4, and 8. The primary outcome of the study was clinical response at week 10, which was defined as a decrease in CDAI score of at least 70 points, whereas clinical remission (CDAI <150) was a secondary endpoint. At week 10 the response and remission rates were 56% and 37% for the natalizumab group and 49% and 30% for placebo-treated patients, respectively, the differences not being statistically significant (P=0.05 for response and P=0.12 for remission, respectively). However, when patients with elevated CRP at baseline were analyzed separately, significant differences were found.
In the maintenance phase of the study, primary responders of the induction arm (n=399) were randomly assigned to receive natalizumab or placebo every 4 weeks through week 56. Analysis of the data showed that significantly more patients on continuous natalizumab treatment remained in clinical response as compared with the placebo group (61% vs. 28%, P<0.001). In addition the time to loss of response was longer in the natalizumab group.
A final study (ENCORE trial) was published in 2005 by Targan et al.66 Only patients with objective evidence of active inflammation, as indicated by elevated CRP were included. Patients with moderate to severe CD were enrolled in 112 centers from around the world. Participants were randomized 1:1 to receive natalizumab 300mg on week 0-4-8 or placebo infusions. The primary outcome was clinical response (reduction of CDAI by 70 points from baseline) at week 8 that was maintained through week 12. The primary endpoint was met by 124/259 (48%) of patients receiving natalizumab and 81/250 (32%) of placebo-treated patients. The difference was significant (P<0.001). The secondary endpoint (clinical remission) was also significantly higher in the natalizumab group (26% vs. 16%, P=0.002). At week 12, the rates for clinical response and remission were 60% and 38% for the natalizumab group and 44% and 25% for the placebo group (both statistically significant). In addition, the median time to response was shorter for the natalizumab group (31 days) as compared with the placebo-treated group (51 days). Other secondary endpoints included significant improvements in IBDQ scores and consistent decreases in CRP concentrations in natalizumab treated patients. The rates of adverse events (including serious) did not differ between active drug and placebo groups.
Taken together, the aforementioned studies provided important evidence for the efficacy of natalizumab as a remission-inductive and maintenance therapy for CD.
Natalizumab in UC
In addition to the reviewed studies in CD, the efficacy of natalizumab was also tested in a small group of patients with UC63. Ten patients with active disease (Powell-Tuck activity score >4, median baseline score=10) received a single 3 mg/kg natalizumab infusion. There were significant decreases in the median Powell-Tuck score at 2 weeks (primary endpoint, decrease of 7.5 points) but also at 4 weeks post-infusion (decrease of 6). Patients also achieved significant improvements in quality of life scores by week 4. Further evaluation of the efficacy of natalizumab in UC in a randomized, placebo-control trial has not been reported yet and under the TOUCH program implemented after the reports of PML, patients with UC may not receive this drug in the United States.
AJM300 in CD
AJM300 is an oral compound that acts as an antagonist of α4 integrins. Several studies have reported the efficacy of this small molecule in animal models of IBD (i.e. TNBS, DSS, and adoptive transfer); nevertheless all have been presented in abstract form and associated full manuscripts are yet to be published. Similarly the results of a randomized, double-blind, placebo controlled trial in Japanese patients with active CD was presented during DDW 2009 by Takazoe et al. (DDW2009, A-181, presentation #S1066). In this trial seventy-one patients with active CD (CDAI>150 and elevated CRP) received placebo or one of 4 doses of AJM300: AJM300 40 mg TID, 120 mg TID, or 240 mg TID orally for 8 weeks. AJM300 was safe and well tolerated. The study did not meet the primary (decrease in CDAI at week 4) or secondary (decrease in CDAI by 70 points at week 4) endpoints as the differences between AJM300- and placebo-treated patients were not significantly different. However, patients with a CDAI≥200 at week 0, had a significant decrease of CDAI (41.5 ± 57.5 in the 120 mg group, P=0.0485, 41.6 ± 94.1 in the 240 mg group); in addition, there was a significant decrease in CRP level but only in the 240 mg group (1.87 mg/dL at week 0 to 0.96 mg/dL at week 8, P=0.022). Further evaluation of the efficacy of AJM300 in IBD is required.
Vedolizumab in IBD
An antibody that binds to a combinatorial epitope on α4β7 integrin (i.e. vedolizumab, MLN002) has been tested in patients with IBD. The topic is discussed elsewhere in this issue (see Parikh and Danese).
PF-00547659 (anti-MAdCAM-1) in UC
PF-00547659 is a fully human IgG2 antibody against MAdCAM-1. Its functional characteristics and pharmacological properties have been extensively characterized67. In particular, its ability to bind to human MAdCAM-1 and block adhesion of α4β7-expressing leukocytes was demonstrated both in in vitro systems and in animal models. In 2011, the results of the first human study utilizing PF-00547659 were reported in a population of patients with UC68. Eighty patients with active UC (defined as a total Mayo score ≥6, endoscopic subscore ≥2) were randomized in a double-blind way to receive either placebo or one of multiple PF-00547659 regimens (single intravenous infusions: 0.03, 0.1, 0.3, 1.0 or 10 mg/kg or a single subcutaneous injection of 3.0 mg/kg or multiple intravenous infusions: 0.1, 0.3 or 3.0 mg/kg 4 weeks apart or multiple subcutaneous infusions: 4 weeks apart 0.3 or 1.0 mg/kg). Clinical response was defined as the proportion of patients with ≥3-point reduction and 30% improvement in total Mayo score, and ≥1-point decrease in rectal bleeding subscore or absolute rectal bleeding score of 0 or1 and was achieved by 52% in PF-00547659-treated patients (vs. 32% in the placebo group) and 42% (vs. 21%) at week 4 and 12, respectively. Remission was defined as the proportion of patients with total Mayo score ≤2 points with no individual subscore exceeding 1 point, and was reached by 13% of PF-00547659-treated patients (vs. 11%) and 22% (vs. 0%) at week 4 and 12, respectively. Endoscopic response was defined as ≥1-point improvement in the Mayo endoscopic subscore and was achieved by 50% of PF-00547659-treated patients (vs. 26%) and 42% (vs. 29%) at week 4 and 12, respectively. These results were corroborated by objective measures of response such as the decrease in fecal calprotectin levels at week 4 (PF-00547659: 63%; placebo: 8%). There were no drug-related side effects in the study during the follow-up period. Further studies will be required to elucidate the clinical applicability of this novel anti-MAdCAM-1 antibody in patients with IBD.
αEβ7 Integrin (CD103)
Integrin αEβ7 (CD103) is a heterodimer composed by the alpha E chain (molecular weight: 175kDa) which only pairs with the β7 chain. The ligand for αEβ7 has been identified as E-cadherin that is expressed by epithelial cells 69. The adhesion of αEβ7 T cells to epithelial E-cadherin is promoted by the interaction between epithelial CCL25 and T-cell CCR9. The gut selectivity of this molecule is exemplified by the fact that αEβ7 was originally identified by a monoclonal antibody that recognizes lymphocytes in intestinal tissue (i.e. human mucosal lymphocyte antigen-1, HML-1). A minority (2%) of peripheral blood lymphocytes expressαEβ7. In sharp contrast, more than 90% of intraepithelial lymphocytes (IEL) express this marker, which is also present on fractions of DCs and lymphocytes at effector sites in the intestinal lamina propria70. In fact, the subpopulation of DCs that express αEβ7 may be responsible for imprinting gut tropism to T cells 71, 72. Furthermore, it has been recently demonstrated that the expression of αEβ7 defines also a unique population of regulatory cells 72, 73. The expression of αEβ7 by different leukocyte subsets might have implications for the targeting of this molecule in IBD.
Therapeutic targeting of β7 integrins in IBD
Etrolizumab in UC
Etrolizumab is a humanized IgG1 monoclonal antibody that is directed against the β7 integrin. Therefore, etrolizumab targets both the αEβ7 and the α4β7 integrins and blocks interactions to their respective ligands, MAdCAM-1 and E-cadherin. Pre-clinical studies showed that etrolizumab effectively inhibits migration of T-cells to mucosal sites, without affecting their homing to non-mucosal tissue 74. The results from a randomized, phase I study on the use of etrolizumab (PRO145223) in moderate to severe UC were recently reported 75. The study had two components. In the single ascending dose protocol, patients received once either placebo or one of 4 ascending doses of etrolizumab (0.3, 1.0, 3.0, 10 mg/kg intravenous, or 3.0 mg/kg subcutaneous). In the multipledose (MD) stage, patients received etrolizumab every for weeks for 3 cycles in 4 different regimens (0.5 mg/kg SC, 1.5 mg/kg SC,3.0 mg/kg SC or 4.0 mg/kg IV.) or placebo. The safety, pharmacokinetics, and clinical response (as estimated by Mayo Clinic Score calculations) were examined at various time-points. This study showed that etrolizumab was well tolerated and safe. Serious adverse effects included exacerbation of UC and impaired wound healing in two patients who underwent colectomy. The most common side effect was headache. There was a decrease in ‘availability” of β7 receptors on target CD4+ lymphocytes, providing evidence that etrolizumab administration decreases the number of gut homing lymphocytes and compromises lymphocytic migration to the gut. The duration of β7 receptors was dose-dependent. Clinical response (≥3 points and 30% reduction from baseline in Mayo Clinic Score, and ≥1 point decrease in rectal bleeding subscore or absolute rectal bleeding score of 0 or 1) was achieved by 12/18 etrolizumab-treated patients. Clinical remission (Mayo Clinic Score ≤2 with no individual subscore>1) was achieved in 3/18. Nevertheless, similar trends were seen in the placebo group and no conclusion on efficacy could be drawn.
PML: a major complication of anti-integrin antibodies
The widespread use of anti-integrin monoclonal antibodies (natalizumab, efalizumab) for the treatment of IBD, multiple sclerosis or psoriasis has been hampered by the occurrence of a rare but potentially fatal complication, progressive multifocal leukoencephalopathy (PML)76. This condition is the result of reactivation of a polyoma virus, which is designated JC virus. The risk for developing PML after treatment with natalizumab has been estimated to approximately 1:1000 for patients treated for more than 2 years. Efalizumab has also been associated with cases of PML in patients with psoriasis. Up to 2009, 4 cases were described within a cohort of 6000 patients that had received efalizumab for psoriasis. Development of PML has result in the voluntary withdrawal of natalizumab from the market in February 2005 (it returned in July 2006) and of efalizumab in 2009. PML appears to be a true drug-effect as neither MS, CD nor psoriasis have been associated with PML per se. Currently, natalizubab (TYSABRI) is available in the US only through a restricted distribution program, called the TOUCH Prescribing Program.
The pathogenesis of PML in patients receiving natalizumab is largely unknown. Nevertheless, it may be primarily associated with the binding of α4β1 integrin by natalizumab. This may result in the blockade of migration of JCV-specific lymphocytes to the central nervous system, including cytolytic lymphocytes, as the latter have been related to increased survival from PML 77. Alternative pathogenetic mechanisms may also participate such as mobilization of JC-infected pre-B-cells from the bone marrow due to α4β1 blockade 78. In any case, if α4β1 blockade is mainly responsible for PML development it should be expected that selective blockade of α4β7 will avoid this complication. Indeed, there have been no cases of PML in patients treated with the specific anti-α4β7 antibody vedolizumab.
It is not clear whether PML is a ‘class” adverse effect. Cases of PML have been reported in patients receiving rituximab also 79. Rituximab is an anti-CD20 monoclonal antibody that primarily targets B-cells. Nevertheless, a causal association between this drug and PML cannot be directly established since the conditions for which rituximab was administered (lymphoproliferative disorders, systemic lupus erythematosous and rheumatoid arthritis) may inherently increase the risk for developing PML. The frequency of PML in patients who are negative for JC virus (around 50% of patients) is near zero, thus it is possible that anti-integrin antibodies might be much more safely used in seronegative patients.
Chemokines as therapeutic targets
Chemokines are small proteins originally named for their role in leukocyte chemotaxis: chemo/tactic cyto/kine. They are classified based on the relative position of cysteine residues (i.e. C, CC, CXC, CX3C) and bind to G-protein coupled chemokine receptors (CCR). CCL25 is produced by small intestinal epithelial cells, and serves as a homing beacon for the homeostatic recruitment of lymphocyte subpopulations (e.g., IgA antibody secreting cells, CD8αα and T cells) to the small intestine 80–85. The description of the restricted small intestinal expression of the chemokine CCL25/TECK 86 was exciting, as this expression pattern provided molecular evidence for the potential dichotomization of intestinal trafficking into distinct small- and large-intestinal compartments 87, 88. Dichotomization of homing to small and large intestine for the first time allowed us to understand a subset of patient with Crohn’s develop disease strictly localized to small bowel. Patients with small intestinal CD have an increased number of CCR9+ T cells increased in peripheral blood 89. It has also been shown that CCL25 is induced aberrantly in the chronically-inflamed hepatic microvasculature of patients with primary sclerosing cholangitis (PSC), a chronic immune-mediated disease of the biliary tree frequently associated with IBD 90. These findings imply that the role of CCL25/CCR9 may not be limited to homeostatic recruitment, but rather that this chemokine/receptor pair also participates in chronic inflammatory trafficking.
Traficet EN in CD
GSK-1605786 (Traficet-EN) is a small molecule that was developed as a selective antagonist of CCR9. Its mechanism of action relates to blockade of CCL25/CCR9 interaction, which is said to result in prevention of B- and T-cell trafficking to the intestine. An obvious theoretical advantage of this compound, besides its oral administration, is that migration of lymphocytes to non-intestinal tissues is theoretically unaffected, ameliorating therefore the risk for infectious complications, including PML. There has been one clinical trial of GSK-1605786 in CD (PROTECT-1 trial), which included an induction and a maintenance arm. So far, the results of this study have been presented only in abstract form. In the Protect-1 induction trial, 436 patients with active CD (CDAI>250 and <450) were randomized to receive one of three doses of Traficet-EN or placebo for 12 weeks. Clinical response (defined as the number of patients with decrease of ≥70 points on the CDAI) was 61% with Traficet-EN vs. 40% with placebo. In the maintenance trial, 241 responders from the induction phase were re-randomized to either Traficet-EN 250 mg twice daily (n=146) or placebo (n=95) for 36 weeks. At the end of the follow-up there was a significant difference in remission rates between patients on Traficet-EN (47%) in comparison with those who received placebo (31%) (P=0.01). This was associated with significant increases in the rates of corticosteroid-free remission and normal CRP levels. Traficet-EN was safe and well-tolerated with no increase in serious adverse events. Several phase III clinical trials are currently underway. Of interest is that we cannot find published evidence that this chemokine axis plays a role in traffic to the colon, yet patients with CD regardless of their disease localization (e.g. ileitis, colitis, ileocolitis) are being included. Results from these studies will define the applicability and efficacy of this attractive novel treatment for patients with CD.
BMS-936557 in UC
BMS-936557 is a fully human antibody that binds to the chemokine IP-10 (CXCL10), therefore blocking its interaction with its receptor CXCR3. Recently, the efficacy of BMS-936557 in moderate to severe UC was reported. This was a randomized, double-blind, placebo-control, phase II study which was carried out in 54 centers in 8 countries. Patients with active UC were administered BMS-936557 (10 mg/kg) or placebo at weeks 0, 2, 4, and 6. The primary endpoint was rate of clinical response at day 57 (≥3 points and 30% decrease in Mayo score, with ≥1 point decrease in rectal bleeding score or absolute rectal bleeding score ≤1). The primary endpoint was not met as the clinical response rate at day 57 was 52.7% (BMS-936557) vs. 35.2% (placebo) (P=0.083). The secondary endpoints of clinical remission and mucosal healing were also not met. Nevertheless, when patients with higher steady-state through concentration of BMS-936557 were analyzed, this population had a significantly increased clinical response (87.5% vs. 37%, P<0.001) and histological improvement (73% vs. 41%, P=0.004). More studies will be required to evaluate the efficacy of this compound in UC as well as the importance of achieving optimal drug levels.
Future Directions
Targeting sphingosine-1-phosphate receptors in IBD
Shingosine-1-phosphate (S1P) is a bioactive sphingolipid that regulates an array of physiological processes, including lymphocyte traffic. T cells express G-protein coupled receptors (GPCRs) that bind S1P and lymphocytes rely on S1P gradients to recirculate. Fingolimod (FTY720, a prototype S1P receptor agonist) is derived from myriocin, a fungal derivative used in Chinese medicine. Like natalizumab, fingolimod is effective in MS and already approved by the FDA for that indication under the brand name Gylenia. Fingolimod binds to four of the five S1P receptors (S1P1, 3,4,5). Due to this lack of specificity its bradycardic effect on heart rate has already resulted in 1 death 91. Refinement of the drug, to minimize their effect on heart rate is desirable. A more specific S1P1 agonist (RPC 1063) has been successfully tested in animal models of IBD92 and is currently being tested in patients with UC (http://www.clinicaltrials.gov/ct2/show/NCT01647516).
Conclusions and implications for future anti-adhesion strategies
During the past decade, the success of anti-TNF-α strategies has revolutionized the treatment of IBD 93. Yet, only about 70% of patients respond to this therapy, and another 20% lose response after a year, driving the continued search for other therapeutic strategies 93, 94. Interference with leukocyte recirculation to the intestine by targeting specific molecules involved with leukocyte traffic has resulted in the development of agents that have advanced into clinical use (e.g. Natalizumab (Biogen/Elan Pharmaceuticals, www.tysabri.com), vedolizumab (Millenium Pharmaceuticals), RPC 1063 (Receptos)) have been either approved or are being evaluated in CD and UC 95. Several other molecules that target these pathways are currently being evaluated. Despite their progression from the bench to bedside 64, 96, much remains to be learned regarding their fundamental mechanism of action. This enhanced understanding may allow us to optimize these therapies64, minimize risks and potentially expand their use to other chronic inflammatory conditions which share self-perpetuating and dysregulated lymphocyte recruitment as a basic pathogenic mechanism.
References
- 1.Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001;345:340–350. doi: 10.1056/NEJM200108023450506. [DOI] [PubMed] [Google Scholar]
- 2.Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why and where it occurs. Nat Med. 2001;7:899–905. doi: 10.1038/90935. [DOI] [PubMed] [Google Scholar]
- 3.O’Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol. 2002;2:37–45. doi: 10.1038/nri702. [DOI] [PubMed] [Google Scholar]
- 4.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 5.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 6.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 7.Bargatze RF, Jutila MA, Butcher EC. Distinct roles of L-selectin and integrins alpha 4 beta 7 and LFA-1 in lymphocyte homing to Peyer’s patch-HEV in situ: the multistep model confirmed and refined. Immunity. 1995;3:99–108. doi: 10.1016/1074-7613(95)90162-0. [DOI] [PubMed] [Google Scholar]
- 8.Rose DM, Han J, Ginsberg MH. Alpha4 integrins and the immune response. Immunol Rev. 2002;186:118–124. doi: 10.1034/j.1600-065x.2002.18611.x. [DOI] [PubMed] [Google Scholar]
- 9.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 10.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 11.Pribila JT, Quale AC, Mueller KL, Shimizu Y. Integrins and T cell-mediated immunity. Annu Rev Immunol. 2004;22:157–180. doi: 10.1146/annurev.immunol.22.012703.104649. [DOI] [PubMed] [Google Scholar]
- 12.Lacy-Hulbert A, Smith AM, Tissire H, Barry M, Crowley D, Bronson RT, Roes JT, Savill JS, Hynes RO. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc Natl Acad Sci U S A. 2007;104:15823–15828. doi: 10.1073/pnas.0707421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, Wang Y, Bernstein X, Huang X, Reichardt LF, Bluestone JA, Sheppard D. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–365. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5:546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 16.Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006;119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. The leukocyte integrins. J Biol Chem. 2000;275:23409–23412. doi: 10.1074/jbc.R000004200. [DOI] [PubMed] [Google Scholar]
- 18.Alon R, Grabovsky V, Feigelson S. Chemokine induction of integrin adhesiveness on rolling and arrested leukocytes local signaling events or global stepwise activation? Microcirculation. 2003;10:297–311. doi: 10.1038/sj.mn.7800195. [DOI] [PubMed] [Google Scholar]
- 19.Ley K. Arrest chemokines. Microcirculation. 2003;10:289–295. doi: 10.1038/sj.mn.7800194. [DOI] [PubMed] [Google Scholar]
- 20.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813–819. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 21.Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, Smith CW, Montgomery CA, Rich S, Beaudet AL. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. 1998;188:119–131. doi: 10.1084/jem.188.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmits R, Kundig TM, Baker DM, Shumaker G, Simard JJ, Duncan G, Wakeham A, Shahinian A, van der Heiden A, Bachmann MF, Ohashi PS, Mak TW, Hickstein DD. LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J Exp Med. 1996;183:1415–1426. doi: 10.1084/jem.183.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross GD. Regulation of the adhesion versus cytotoxic functions of the Mac-1/CR3/alphaMbeta2-integrin glycoprotein. Crit Rev Immunol. 2000;20:197–222. [PubMed] [Google Scholar]
- 24.Zen K, Cui LB, Zhang CY, Liu Y. Critical role of mac-1 sialyl lewis x moieties in regulating neutrophil degranulation and transmigration. J Mol Biol. 2007;374:54–63. doi: 10.1016/j.jmb.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Van der Vieren M, Le Trong H, Wood CL, Moore PF, St John T, Staunton DE, Gallatin WM. A novel leukointegrin, alpha d beta 2, binds preferentially to ICAM-3. Immunity. 1995;3:683–690. doi: 10.1016/1074-7613(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 26.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–194. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 27.Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- 28.Shang XZ, Issekutz AC. Contribution of CD11a/CD18, CD11b/CD18, ICAM-1 (CD54) and -2 (CD102) to human monocyte migration through endothelium and connective tissue fibroblast barriers. Eur J Immunol. 1998;28:1970–1979. doi: 10.1002/(SICI)1521-4141(199806)28:06<1970::AID-IMMU1970>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 29.Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1) J Immunol. 1986;137:245–254. [PubMed] [Google Scholar]
- 30.Van Assche G, Rutgeerts P. Physiological basis for novel drug therapies used to treat the inflammatory bowel diseases. I Immunology and therapeutic potential of antiadhesion molecule therapy in inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2005;288:G169–174. doi: 10.1152/ajpgi.00423.2004. [DOI] [PubMed] [Google Scholar]
- 31.Baert FJ, D’Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, Geboes K, Rutgeerts PJ. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology. 1999;116:22–28. doi: 10.1016/s0016-5085(99)70224-6. [DOI] [PubMed] [Google Scholar]
- 32.Werther WA, Gonzalez TN, O’Connor SJ, McCabe S, Chan B, Hotaling T, Champe M, Fox JA, Jardieu PM, Berman PW, Presta LG. Humanization of an anti-lymphocyte function-associated antigen (LFA)-1 monoclonal antibody and reengineering of the humanized antibody for binding to rhesus LFA-1. J Immunol. 1996;157:4986–4995. [PubMed] [Google Scholar]
- 33.Simmons DL. Anti-adhesion therapies. Curr Opin Pharmacol. 2005;5:398–404. doi: 10.1016/j.coph.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 34.James DG, Seo da H, Chen J, Vemulapalli C, Stone CD. Efalizumab, a human monoclonal anti-CD11a antibody, in the treatment of moderate to severe Crohn’s Disease: an open-label pilot study. Dig Dis Sci. 2011;56:1806–1810. doi: 10.1007/s10620-010-1525-6. [DOI] [PubMed] [Google Scholar]
- 35.Yacyshyn B, Chey WY, Wedel MK, Yu RZ, Paul D, Chuang E. A randomized, double-masked, placebo-controlled study of alicaforsen, an antisense inhibitor of intercellular adhesion molecule 1, for the treatment of subjects with active Crohn’s disease. Clin Gastroenterol Hepatol. 2007;5:215–220. doi: 10.1016/j.cgh.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Miner PB, Jr, Geary RS, Matson J, Chuang E, Xia S, Baker BF, Wedel MK. Bioavailability and therapeutic activity of alicaforsen (ISIS 2302) administered as a rectal retention enema to subjects with active ulcerative colitis. Aliment Pharmacol Ther. 2006;23:1427–1434. doi: 10.1111/j.1365-2036.2006.02909.x. [DOI] [PubMed] [Google Scholar]
- 37.van Deventer SJ, Wedel MK, Baker BF, Xia S, Chuang E, Miner PB., Jr A phase II dose ranging, double-blind, placebo-controlled study of alicaforsen enema in subjects with acute exacerbation of mild to moderate left-sided ulcerative colitis. Aliment Pharmacol Ther. 2006;23:1415–1425. doi: 10.1111/j.1365-2036.2006.02910.x. [DOI] [PubMed] [Google Scholar]
- 38.Miner PB, Jr, Wedel MK, Xia S, Baker BF. Safety and efficacy of two dose formulations of alicaforsen enema compared with mesalazine enema for treatment of mild to moderate left-sided ulcerative colitis: a randomized, double-blind, active-controlled trial. Aliment Pharmacol Ther. 2006;23:1403–1413. doi: 10.1111/j.1365-2036.2006.02837.x. [DOI] [PubMed] [Google Scholar]
- 39.Hemler ME, Huang C, Schwarz L. The VLA protein family. Characterization of five distinct cell surface heterodimers each with a common 130,000 molecular weight beta subunit. J Biol Chem. 1987;262:3300–3309. [PubMed] [Google Scholar]
- 40.Hynes RO. Integrins: a family of cell surface receptors. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- 41.Cybulsky MI, Gimbrone MA., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 42.Manka DR, Wiegman P, Din S, Sanders JM, Green SA, Gimple LW, Ragosta M, Powers ER, Ley K, Sarembock IJ. Arterial injury increases expression of inflammatory adhesion molecules in the carotid arteries of apolipoprotein-E-deficient mice. J Vasc Res. 1999;36:372–378. doi: 10.1159/000025676. [DOI] [PubMed] [Google Scholar]
- 43.Rice GE, Munro JM, Bevilacqua MP. Inducible cell adhesion molecule 110 (INCAM-110) is an endothelial receptor for lymphocytes. A CD11/CD18-independent adhesion mechanism. J Exp Med. 1990;171:1369–1374. doi: 10.1084/jem.171.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee SJ, Benveniste EN. Adhesion molecule expression and regulation on cells of the central nervous system. J Neuroimmunol. 1999;98:77–88. doi: 10.1016/s0165-5728(99)00084-3. [DOI] [PubMed] [Google Scholar]
- 45.Schweighoffer T, Tanaka Y, Tidswell M, Erle DJ, Horgan KJ, Luce GE, Lazarovits AI, Buck D, Shaw S. Selective expression of integrin alpha 4 beta 7 on a subset of human CD4+ memory T cells with Hallmarks of gut-trophism. J Immunol. 1993;151:717–729. [PubMed] [Google Scholar]
- 46.Erle DJ, Briskin MJ, Butcher EC, Garcia-Pardo A, Lazarovits AI, Tidswell M. Expression and function of the MAdCAM-1 receptor, integrin alpha 4 beta 7, on human leukocytes. J Immunol. 1994;153:517–528. [PubMed] [Google Scholar]
- 47.Rott LS, Briskin MJ, Andrew DP, Berg EL, Butcher EC. A fundamental subdivision of circulating lymphocytes defined by adhesion to mucosal addressin cell adhesion molecule-1. Comparison with vascular cell adhesion molecule-1 and correlation with beta 7 integrins and memory differentiation. J Immunol. 1996;156:3727–3736. [PubMed] [Google Scholar]
- 48.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 49.Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, Rajewsky K, Adams DH, von Andrian UH. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 50.Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, Weissman IL, Hamann A, Butcher EC. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–195. doi: 10.1016/0092-8674(93)90305-a. [DOI] [PubMed] [Google Scholar]
- 51.Briskin M, Winsor-Hines D, Shyjan A, Cochran N, Bloom S, Wilson J, McEvoy LM, Butcher EC, Kassam N, Mackay CR, Newman W, Ringler DJ. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am J Pathol. 1997;151:97–110. [PMC free article] [PubMed] [Google Scholar]
- 52.Briskin MJ, McEvoy LM, Butcher EC. MAdCAM-1 has homology to immunoglobulin and mucin-like adhesion receptors and to IgA1. Nature. 1993;363:461–464. doi: 10.1038/363461a0. [DOI] [PubMed] [Google Scholar]
- 53.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 54.Berg EL, McEvoy LM, Berlin C, Bargatze RF, Butcher EC. L-selectin-mediated lymphocyte rolling on MAdCAM-1. Nature. 1993;366:695–698. doi: 10.1038/366695a0. [DOI] [PubMed] [Google Scholar]
- 55.Kunkel EJ, Ramos CL, Steeber DA, Muller W, Wagner N, Tedder TF, Ley K. The roles of L-selectin, beta 7 integrins, and P-selectin in leukocyte rolling and adhesion in high endothelial venules of Peyer’s patches. J Immunol. 1998;161:2449–2456. [PubMed] [Google Scholar]
- 56.Connor EM, Eppihimer MJ, Morise Z, Granger DN, Grisham MB. Expression of mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in acute and chronic inflammation. J Leukoc Biol. 1999;65:349–355. doi: 10.1002/jlb.65.3.349. [DOI] [PubMed] [Google Scholar]
- 57.Sikorski EE, Hallmann R, Berg EL, Butcher EC. The Peyer’s patch high endothelial receptor for lymphocytes, the mucosal vascular addressin, is induced on a murine endothelial cell line by tumor necrosis factor-alpha and IL-1. J Immunol. 1993;151:5239–5250. [PubMed] [Google Scholar]
- 58.Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol. 2006;6:244–251. doi: 10.1038/nri1784. [DOI] [PubMed] [Google Scholar]
- 59.Salmi M, Andrew DP, Butcher EC, Jalkanen S. Dual binding capacity of mucosal immunoblasts to mucosal and synovial endothelium in humans: dissection of the molecular mechanisms. J Exp Med. 1995;181:137–149. doi: 10.1084/jem.181.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 61.Hesterberg PE, Winsor-Hines D, Briskin MJ, Soler-Ferran D, Merrill C, Mackay CR, Newman W, Ringler DJ. Rapid resolution of chronic colitis in the cotton-top tamarin with an antibody to a gut-homing integrin alpha 4 beta 7. Gastroenterology. 1996;111:1373–1380. doi: 10.1053/gast.1996.v111.pm8898653. [DOI] [PubMed] [Google Scholar]
- 62.Picarella D, Hurlbut P, Rottman J, Shi X, Butcher E, Ringler DJ. Monoclonal antibodies specific for beta 7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) reduce inflammation in the colon of scid mice reconstituted with CD45RBhigh CD4+ T cells. J Immunol. 1997;158:2099–2106. [PubMed] [Google Scholar]
- 63.Gordon FH, Hamilton MI, Donoghue S, Greenlees C, Palmer T, Rowley-Jones D, Dhillon AP, Amlot PL, Pounder RE. A pilot study of treatment of active ulcerative colitis with natalizumab, a humanized monoclonal antibody to alpha-4 integrin. Aliment Pharmacol Ther. 2002;16:699–705. doi: 10.1046/j.1365-2036.2002.01205.x. [DOI] [PubMed] [Google Scholar]
- 64.Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask-Madsen J, Rutgeerts P, Vyhnalek P, Zadorova Z, Palmer T, Donoghue S. Natalizumab for active Crohn’s disease. N Engl J Med. 2003;348:24–32. doi: 10.1056/NEJMoa020732. [DOI] [PubMed] [Google Scholar]
- 65.Sandborn WJ, Colombel JF, Enns R, Feagan BG, Hanauer SB, Lawrance IC, Panaccione R, Sanders M, Schreiber S, Targan S, van Deventer S, Goldblum R, Despain D, Hogge GS, Rutgeerts P. Natalizumab induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2005;353:1912–1925. doi: 10.1056/NEJMoa043335. [DOI] [PubMed] [Google Scholar]
- 66.Targan SR, Feagan BG, Fedorak RN, Lashner BA, Panaccione R, Present DH, Spehlmann ME, Rutgeerts PJ, Tulassay Z, Volfova M, Wolf DC, Hernandez C, Bornstein J, Sandborn WJ. Natalizumab for the treatment of active Crohn’s disease: results of the ENCORE Trial. Gastroenterology. 2007;132:1672–1683. doi: 10.1053/j.gastro.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 67.Pullen N, Molloy E, Carter D, Syntin P, Clemo F, Finco-Kent D, Reagan W, Zhao S, Kawabata T, Sreckovic S. Pharmacological characterization of PF-00547659, an anti-human MAdCAM monoclonal antibody. Br J Pharmacol. 2009;157:281–293. doi: 10.1111/j.1476-5381.2009.00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vermeire S, Ghosh S, Panes J, Dahlerup JF, Luegering A, Sirotiakova J, Strauch U, Burgess G, Spanton J, Martin SW, Niezychowski W. The mucosal addressin cell adhesion molecule antibody PF-00547,659 in ulcerative colitis: a randomised study. Gut. 2011;60:1068–1075. doi: 10.1136/gut.2010.226548. [DOI] [PubMed] [Google Scholar]
- 69.Kilshaw PJ. Alpha E beta 7. Mol Pathol. 1999;52:203–207. doi: 10.1136/mp.52.4.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cepek KL, Parker CM, Madara JL, Brenner MB. Integrin alpha E beta 7 mediates adhesion of T lymphocytes to epithelial cells. J Immunol. 1993;150:3459–3470. [PubMed] [Google Scholar]
- 71.Johansson-Lindbom B, Agace WW. Generation of gut-homing T cells and their localization to the small intestinal mucosa. Immunol Rev. 2007;215:226–242. doi: 10.1111/j.1600-065X.2006.00482.x. [DOI] [PubMed] [Google Scholar]
- 72.Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R, Agace WW. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med. 2005;202:1063–1073. doi: 10.1084/jem.20051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lehmann J, Huehn J, de la Rosa M, Maszyna F, Kretschmer U, Krenn V, Brunner M, Scheffold A, Hamann A. Expression of the integrin alpha Ebeta 7 identifies unique subsets of CD25+ as well as CD25- regulatory T cells. Proc Natl Acad Sci U S A. 2002;99:13031–13036. doi: 10.1073/pnas.192162899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stefanich EG, Danilenko DM, Wang H, O’Byrne S, Erickson R, Gelzleichter T, Hiraragi H, Chiu H, Ivelja S, Jeet S, Gadkari S, Hwang O, Fuh F, Looney C, Howell K, Albert V, Balazs M, Refino C, Fong S, Iyer S, Williams M. A humanized monoclonal antibody targeting the beta7 integrin selectively blocks intestinal homing of T lymphocytes. Br J Pharmacol. 2011;162:1855–1870. doi: 10.1111/j.1476-5381.2011.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rutgeerts PJ, Fedorak RN, Hommes DW, Sturm A, Baumgart DC, Bressler B, Schreiber S, Mansfield JC, Williams M, Tang M, Visich J, Wei X, Keir M, Luca D, Danilenko D, Egen J, O’Byrne S. A randomised phase I study of etrolizumab (rhuMAb beta7) in moderate to severe ulcerative colitis. Gut. 2012 doi: 10.1136/gutjnl-2011-301769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berger JR, Houff SA. Neurological infections: the year of PML and influenza. Lancet Neurol. 2010;9:14–17. doi: 10.1016/S1474-4422(09)70337-0. [DOI] [PubMed] [Google Scholar]
- 77.Du Pasquier RA, Kuroda MJ, Zheng Y, Jean-Jacques J, Letvin NL, Koralnik IJ. A prospective study demonstrates an association between JC virus-specific cytotoxic T lymphocytes and the early control of progressive multifocal leukoencephalopathy. Brain. 2004;127:1970–1978. doi: 10.1093/brain/awh215. [DOI] [PubMed] [Google Scholar]
- 78.Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology. 2008;71:1350–1354. doi: 10.1212/01.wnl.0000327671.91357.96. [DOI] [PubMed] [Google Scholar]
- 79.Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, Bennett CL. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 80.Bowman EP, Kuklin NA, Youngman KR, Lazarus NH, Kunkel EJ, Pan J, Greenberg HB, Butcher EC. The intestinal chemokine thymus-expressed chemokine (CCL25) attracts IgA antibody-secreting cells. J Exp Med. 2002;195:269–275. doi: 10.1084/jem.20010670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hieshima K, Kawasaki Y, Hanamoto H, Nakayama T, Nagakubo D, Kanamaru A, Yoshie O. CC chemokine ligands 25 and 28 play essential roles in intestinal extravasation of IgA antibody-secreting cells. J Immunol. 2004;173:3668–3675. doi: 10.4049/jimmunol.173.6.3668. [DOI] [PubMed] [Google Scholar]
- 82.Marsal J, Svensson M, Ericsson A, Iranpour AH, Carramolino L, Marquez G, Agace WW. Involvement of CCL25 (TECK) in the generation of the murine small-intestinal CD8alpha alpha+CD3+ intraepithelial lymphocyte compartment. Eur J Immunol. 2002;32:3488–3497. doi: 10.1002/1521-4141(200212)32:12<3488::AID-IMMU3488>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 83.Pabst O, Ohl L, Wendland M, Wurbel MA, Kremmer E, Malissen B, Forster R. Chemokine receptor CCR9 contributes to the localization of plasma cells to the small intestine. J Exp Med. 2004;199:411–416. doi: 10.1084/jem.20030996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 85.Staton TL, Habtezion A, Winslow MM, Sato T, Love PE, Butcher EC. CD8(+) recent thymic emigrants home to and efficiently repopulate the small intestine epithelium. Nat Immunol. 2006;7:482–488. doi: 10.1038/ni1319. [DOI] [PubMed] [Google Scholar]
- 86.Vicari AP, Figueroa DJ, Hedrick JA, Foster JS, Singh KP, Menon S, Copeland NG, Gilbert DJ, Jenkins NA, Bacon KB, Zlotnik A. TECK: a novel CC chemokine specifically expressed by thymic dendritic cells and potentially involved in T cell development. Immunity. 1997;7:291–301. doi: 10.1016/s1074-7613(00)80531-2. [DOI] [PubMed] [Google Scholar]
- 87.Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity. 2002;16:1–4. doi: 10.1016/s1074-7613(01)00261-8. [DOI] [PubMed] [Google Scholar]
- 88.Kunkel EJ, Campbell DJ, Butcher EC. Chemokines in lymphocyte trafficking and intestinal immunity. Microcirculation. 2003;10:313–323. doi: 10.1038/sj.mn.7800196. [DOI] [PubMed] [Google Scholar]
- 89.Papadakis KA, Prehn J, Moreno ST, Cheng L, Kouroumalis EA, Deem R, Breaverman T, Ponath PD, Andrew DP, Green PH, Hodge MR, Binder SW, Targan SR. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn’s disease. Gastroenterology. 2001;121:246–254. doi: 10.1053/gast.2001.27154. [DOI] [PubMed] [Google Scholar]
- 90.Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hubscher SG, Briskin M, Salmon M, Adams DH. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med. 2004;200:1511–1517. doi: 10.1084/jem.20041035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Espinosa PS, Berger JR. Delayed fingolimod-associated asystole. Mult Scler. 2011;17:1387–1389. doi: 10.1177/1352458511410344. [DOI] [PubMed] [Google Scholar]
- 92.Clemons BDH, Powell R, Martinborough E, Timony G, Peach R, Scott FL. A small molecule S1P1 receptor agonist with significant efficacy in animal models of inflammatory bowel disease. Gastroenterology DDW abstracts. 2012 [Google Scholar]
- 93.Sandborn WJ, Hanauer SB. Infliximab in the treatment of Crohn’s disease: a user’s guide for clinicians. Am J Gastroenterol. 2002;97:2962–2972. doi: 10.1111/j.1572-0241.2002.07093.x. [DOI] [PubMed] [Google Scholar]
- 94.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 95.Feagan BG, Greenberg GR, Wild G, Fedorak RN, Pare P, McDonald JW, Dube R, Cohen A, Steinhart AH, Landau S, Aguzzi RA, Fox IH, Vandervoort MK. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med. 2005;352:2499–2507. doi: 10.1056/NEJMoa042982. [DOI] [PubMed] [Google Scholar]
- 96.Podolsky DK, Lobb R, King N, Benjamin CD, Pepinsky B, Sehgal P, deBeaumont M. Attenuation of colitis in the cotton-top tamarin by anti-alpha 4 integrin monoclonal antibody. J Clin Invest. 1993;92:372–380. doi: 10.1172/JCI116575. [DOI] [PMC free article] [PubMed] [Google Scholar]