

Abstract
Phase II clinical trials aim to identify promising experimental regimens for further testing in phase III trials. In this review article, we focus on phase II designs for initial predictive biomarker validation to determine if a drug should be developed for an unselected patient population or for a biomarker-defined patient subset only. Several prospective designs for biomarker-directed therapy have been proposed, differing primarily in the study population, or randomization scheme, or both. The design choice is driven by scientific rationale, marker prevalence, strength of preliminary evidence, assay performance, and turn-around times for marker assessment. The enrichment design is most appropriate when compelling preliminary evidence suggests treatment benefit in only certain marker-defined subgroups, the all-comers design is useful when preliminary evidence regarding treatment effects in marker subgroups is unclear, and adaptive designs have the most potential in the setting of multiple treatment options and multiple marker-defined subgroups. We recently proposed a 2-stage phase II design that has the option for direct assignment (i.e., stop randomization and assign all patients to the experimental arm in Stage 2) based on interim analysis (IA) results. This design recognizes the need for randomization but also acknowledges the possibility of promising but inconclusive results after pre-planned IA. Simulation studies demonstrated that the direct assignment-option design has minimal power loss, marginal increase in type I error rates, and reasonable robustness to population shift effects. Systematic evaluation and implementation of these design strategies in the phase II setting is essential for accelerating the clinical validation of biomarker guided-therapy.
Keywords: All-comers design, adaptive design, biomarker, direct assignment, enrichment design, interim analysis
Introduction
The Biomarkers Definitions Working Group defined a biomarker to be “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [1]. The term “biomarker” in oncology refers to a broad range of markers, and includes a range of measures derived from tumor tissues, whole blood, plasma, serum, bone marrow, or urine. From the perspective of clinical utility, biomarkers can be classified into three categories: prognostic biomarkers, predictive biomarkers, and surrogate endpoints, with the recognition that some biomarkers may fall into more than one category. Prognostic and predictive biomarkers focus on individual patient risk-classification and treatment selection respectively, whereas biomarkers used as surrogate endpoints aid in the evaluation of the efficacy of a new treatment. It is critical to realize that the ultimate intended usage of a biomarker determines its definition and the required validation methods. A prognostic biomarker predicts the natural history of the disease process in a given individual, and thus aids in the decision of whether a patient needs an intensive and possibly toxic treatment as opposed to no treatment or standard therapy [2]. A predictive biomarker predicts whether an individual patient will respond to a particular therapy or not, and hence its clinical utility is in allowing for individualized therapy. On the other hand, a surrogate endpoint replaces the primary clinical outcome (i.e., endpoint) and informs the efficacy of a new treatment with greater cost-effectiveness than the primary outcome (such as overall survival) at the population level [3]. We focus our discussion in this article on predictive biomarkers.
One key purpose of incorporating biomarkers into phase II studies of molecularly targeted agents is to determine if the new drug should be developed for an unselected patient population or for a biomarker-defined patient subset only. A single arm two-stage design such as the one proposed by Pusztai et al. [4] could be used to determine if a drug is likely to have a certain level of activity in unselected patients, and if activity is below the level of interest, whether a particular patient selection method can enrich the responding population to meet the targeted level of activity in the selected group. This tandem two-step phase II trial design assumes that a drug has completed phase I evaluation and a dose was selected for phase II testing; that at least one, but preferably more, putative predictive markers are available, but that the response rate in unselected patients is unknown. Even for such an initial validation, there is a growing consensus that randomization is important so as to ensure unbiased estimation of the treatment effect [5, 7]. Most single-arm designs conduct comparisons against historical controls, which may be inaccurate given changes in patient population based on biologic subsetting and/or evolution in imaging technologies. McShane et al. [5] show through simulations how misleading the results of a single-arm phase II trial in a selected patient population can be when the benchmark estimate is from prior trials of “unselected” patients and thus inappropriate for the enriched study population. In contrast, a randomized controlled trial (RCT) includes a control arm for comparison, thereby assuring that patients who are treated with the agent for whom the marker is purported to be predictive are comparable to those who are not. RCTs are essential for making the distinction between a prognostic and predictive marker, as well as to isolate any causal effect of the marker on therapeutic efficacy from the multitude of other factors that may influence the decision to treat or not to treat a patient [6]. In the setting of phase II trials, RCTs also provide the opportunity to simultaneously assess multiple promising therapies (and multiple possible markers) for a given disease. Freidlin et al. [7] propose guidelines for the design of randomized phase II trials with biomarkers that can inform the design of the subsequent phase III trial. Specifically, the results from the biomarker driven phase II trial (if the treatment is found promising in the phase II setting) can lead to three possible phase III trial decisions: enrichment/targeted design, biomarker stratified (marker by treatment interaction) design, or an unselected design [7].
The crucial component of RCTs is, of course, randomization. However, a controversial yet pertinent question is whether it is necessary to randomize throughout the entire duration of a trial. This might be relevant in the case, for example, where preliminary evidence for a new experimental agent is promising in a biomarker defined cohort, but not sufficiently compelling to pursue a non-randomized trial. In such cases, one might consider an alternative phase II design approach that starts as a RCT, but allows for a switch to direct assignment (i.e. all patients receive the experimental agent) after a pre-specified interim analysis. In this paper, we review the design strategies for initial predictive marker validation (i.e., phase II setting), including a recently proposed Phase II design that includes an option for direct assignment to the experimental treatment when there is promising but not definitive evidence of a treatment benefit at the end of an initial, randomized stage of the trial [8, 9].
Overview of prospective phase II designs for initial predictive biomarker validation
Phase II clinical trials are designed primarily to identify promising experimental regimens that are then tested further in definitive phase III trials. Here, we focus on phase II trial designs for initial predictive biomarker validation to determine if a drug should be developed for an unselected patient population or for a biomarker-defined patient subset only. Trial designs in this setting can be classified under enrichment, all-comers, adaptive, and the direct assignment option design categories elaborated below [6, 10]. Table 1 provides an overview of these designs, which are described in detail below.
Table 1.
Definitions and Criteria of Phase II biomarker driven designs
| Design Definitions | Design Criteria |
|---|---|
| Enrichment Designs: screen all patients for the marker, but only randomize those with certain molecular features. These evaluate treatment in the marker-defined subgroup only |
|
| All-Comers Designs: screen all patients for the marker and randomize patients with a valid marker result with analysis plans to test the treatment by marker interaction effect |
|
| Adaptive Designs: are a class of designs that adapt the design parameters during the course of the trial based on accumulated data. Direct Assignment Option |
|
| Designs: have the option for direct assignment (i.e., stop randomization and assign all patients to the experimental arm) based on pre-specified interim analysis (IA). |
|
Enrichment Designs
An enrichment design screens patients for the presence (or absence) of a biomarker profile, and then includes in the trial only patients who have (or do not have) the profile [11]. The goal of these designs is to understand the safety, tolerability and clinical benefit of the treatment within the patient subgroup determined by a specific marker status. This design is based on the paradigm that not all patients will benefit from the study treatment under consideration, but rather that the benefit will be restricted to a biomarker defined subgroup of patients. This design has gained considerable importance in the setting of targeted therapies which are most effective in particular biomarker subgroups and not effective in the general population, such as the investigation of anti-EGFR therapies in lung or colon cancer where the population is restricted to only include EGFR mutant lung cancer or KRAS wild type colon cancer patients. N0923 (Clinicaltrials.gov identifier (CT.gov id): NCT01017601) is an example of a phase II trial following an enrichment design strategy. This is a randomized double-blinded phase II study of a replication-competent picornavirus versus matching placebo, after standard platinum-containing cytroreductive induction chemotherapy in patients with extensive stage small cell lung cancer with a neuroendocrine histology as per presence of ≥1 neuroendocrine marker (synaptophysin, chromogranin and CD56) [12]. In addition, the marker prevalence also plays a role in the use of enrichment designs in the phase II setting.
All-comers (stratified by marker status) Designs
In this design, all patients meeting the eligibility criteria are entered [6]. The eligibility criteria may include the ability to provide adequate tissue, but do not include exhibiting a specific biomarker result or status of a biomarker characteristic [6]. The fundamental difference between this design and the common RCT is that only patients with a valid marker result are eligible and randomized. The sample size requirement for treatment-by-marker interaction design is based on a pre-specified analysis plan. A separate evaluation of the treatment effect can be tested in the different marker defined subgroups, or a preliminary test of interaction can be carried out first. Different sequential analysis plans can also be implemented [6, 13]. For example, when a preliminary test of interaction is not significant at a pre-specified significance level, then the treatment arms can be compared in the overall population (ignoring the biomarker status). If the interaction is significant, then the experimental treatment can be compared to the control arm within the strata determined by the marker. Such an elaborate sample size planning is usually more appropriate in a phase III setting, but a scaled down version can be utilized in larger randomized phase II trials with related type I and II error rates.
The hybrid designs are a class of all-comers designs where only a certain subgroup of patients based on their marker status are randomized between treatments, whereas patients in the other marker defined subgroups are assigned the standard of care treatment(s) [6]. This phase II design is an appropriate choice when there is compelling evidence demonstrating the efficacy of a certain treatment(s) for a marker defined subgroup, thereby making it unethical to randomize patients with that particular marker status to other treatment options. However, unlike the enrichment design strategy, all patients regardless of the marker status are enrolled, and followed. This provides the possibility for future testing for other potential prognostic markers. Sequential testing strategy designs and marker based strategy designs also fall under the all-comers designs, but are more appropriate in the setting of definitive predictive marker validation, i.e., phase III setting. One example of a marker based strategy design in the phase II setting is the Phase II Randomized Trial Utilizing Geriatric Assessment in treatment allocation of patients >70 years with Advanced Non-Small Cell Lung Cancer (Alliance A081203, under development) [Figure 1]. Patients will complete the geriatric assessment (GA) survey and classified as high or low-risk based on a model score. Patients will then be randomized, where in the marker independent arm, patients will be treated based on the physician’s choice of treatment, and in the marker based arm, high risk patients will be treated with single agent chemotherapy (based on histology), and low-risk patients will be treated with doublets (again, based on histology). The trial will include an analysis midway to ascertain rate of concordance between the oncologist preference and GA guided therapy to ascertain the overlap between the marker based and the marker independent treatment arms.
Figure 1. Example of a Biomarker strategy Phase II design.

*Stratification factors: ECOG performance status (0,1 versus 2); Age (≤75 versus > 75)
Adaptive Designs
A number of innovative adaptive designs have been proposed to validate putative predictive biomarkers in a phase III setting such as the adaptive accrual based on interim analysis design and the biomarker adaptive threshold design [14, 15]. We focus our discussion on the class of adaptive designs in the phase II setting where a variety of marker signatures and drugs can be tested under one umbrella protocol. In these designs, the success of the drug-biomarker subgroup is assessed in an ongoing manner which allows either the randomization ratio to be altered in order to place more patients on the most promising arm(s), and/or the under-performing drugs and/or the biomarker subgroups are eliminated midway through the trial [10]. Key requirements for adaptive designs include: 1) a rapid and reliable endpoint, which can be somewhat challenging in the oncology setting where time to event endpoints or endpoints that involve following a patient’s status for a predetermined time period (such as the progression status at 2 years), and 2) real time access to all clinical and biologic data, which can be a daunting task in multicenter trials at the current time, but may not be a rate-limiting step in the future with state of the art electronic data capture systems, and mobile device platforms. Other words of caution when using outcome based adaptive randomization include the potential for major imbalances among treatment arms, which in turn affects the statistical power of the trial; complicated statistical inference as the treatment assignments and the outcomes are correlated; potential accrual bias; and the resources (both in terms of manpower and cost) required to build and run these trials compared to conventional trials [16]. The articles by Korn and Freidlin [17] and by Berry [18] provide a point-counterpoint discussion revolving around the utility and limitations of adaptive (that alter the randomization ratio as the trial progresses) designs.
Examples of phase II trials that have utilized or are utilizing an outcome adaptive randomization design strategy are I-SPY 2 (investigation of serial studies to predict therapeutic response with imaging and molecular analysis 2; CT.gov id: NCT01042379) and BATTLE (biomarker-integrated approaches of targeted therapy of lung cancer elimination trial) [19, 20]. I-SPY 2 is an ongoing neo-adjuvant phase II trial in breast cancer that is designed to compare the efficacy of standard therapy to that of novel drugs in combination with chemotherapy. The goal is to identify improved treatment regimens for subsets on the basis of molecular characteristics (biomarker signatures) of their disease. All drugs will be evaluated within the biomarker-defined signature groups. Regimens that have a high Bayesian predicted probability of being successful in a phase III trial are moved forward to Phase III testing within sub-populations corresponding to the most promising biomarker signature(s). Regimens that have a low probability of efficacy for all biomarker signature subgroups will be dropped from further development [19].
The BATTLE trial is completed, and used an outcome based adaptive randomization design for assigning patients to treatment choices based on multiple biomarker profiles in NSCLC. Patients had their tumors tested for 11 different biomarkers, and were subsequently categorized into one of five biomarker subgroups, and then randomized to one of four treatment choices. The first 97 patients were assigned using a balanced randomization to one of the four treatments equally. Subsequent patients were adaptively randomized, where the randomization rate was proportional to the marginal posterior eight week disease control rate. The results from the BATTLE trial showed, as hypothesized, that each drug works best for patients with a specific molecular profile [21].
A biomarker-adaptive parallel Simon two-stage design has been proposed for the evaluation of a targeted agent that is assumed to have different activity in subgroups defined by biomarker positive versus negative [22]. The design assumes that the biomarker is pre-specified, and starts with two parallel two-stage designs, one in each of the biomarker-positive and biomarker-negative groups, and then switches to an adaptive parallel design [refer [22] for full details].
Direct Assignment Option Design
Colton [23, 24] first proposed several designs that involved directly assigning patients to one of two treatment arms. He considered a cost function approach to clinical trial design for comparing two treatments, whereby the choice of design parameters was driven by minimization of the cost associated with treating patients. In his class of designs, the second stage always was a direct assignment. The direct assignment design that was recently proposed by An et al. [8] is a two-stage design (i.e. screen all patients for marker status, but only enroll and randomize a particular marker subgroup, e.g. marker-positive or marker-negative for a binary marker) that may stop early for futility or efficacy. The design can be implemented using a 2-stage strategy, i.e., halting accrual to assess efficacy outcomes, or using an interim analysis strategy, whereby accrual is not halted while the efficacy analysis from the first stage of patients is underway. As with any clinical trial, the decision to suspend or not suspend accrual to an ongoing trial awaiting an interim analysis depends on several factors, such as rapidity of accrual, endpoint data availability etc. For this reason, we use the term stage and interim analysis interchangeably.
In its simplest form, if the trial does not stop early for efficacy or futility after Stage I, then in Stage II the trial can continue in one of two ways: 1) continue with randomization as in Stage I; or 2) switch to “direct assignment,” where all patients are given the experimental treatment. A data-derived decision is made at interim analysis based on Stage I data regarding whether or not to direct assign in Stage II. The decision for direct assignment is based on observing promising, but not definitive, results indicating treatment benefit in Stage I [Figure 2]. Table 2 includes the simulation study results that demonstrate that this design strategy results in minimal power loss and marginal increase in type I error rates. See An et al. [8] for further details on the operating characteristics of this design. In the setting of Phase II trials, many have argued that a single interim look may be inadequate, and that multiple looks improves both the statistical and ethical properties of the design [25, 26]. Extensions to the direct assignment design have recently been proposed to either look earlier by shifting the timing of the single look or by incorporating two looks with different decisions rules at each look [9].
Figure 2. Direct Assignment option Design with a single pre-planned interim analysis for one biomarker defined subgroup.
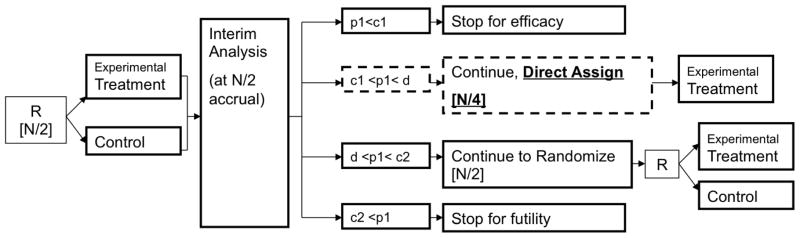
Square brackets [] indicate number of patients enrolled at the given stage. R: randomize; N: total number of patients allocated at start of trial; p1: p-value based on interim analysis patient data; c1, c2, and d: O’Brien-Fleming stopping boundaries (d is the overall trial efficacy boundary)
Table 2.
Operating Characteristics of the direct assignment option design vs. a Balanced randomized design based on 6000 simulations, and assuming stage I analysis at 50% accrual
| Response Rate Ratio (treatment versus control) | α= 0.10 | α= 0.20 | |||
|---|---|---|---|---|---|
|
| |||||
| Balanced Randomized Design | Design with Direct Assignment Option | Balanced Randomized Design | Design with Direct Assignment Option | ||
| Power (%) | 2.00 | 80.6 | 79.3 | 79.3 | 78.0 |
| 2.25 | 90.5 | 89.0 | 88.0 | 86.3 | |
| 2.50 | 96.5 | 95.5 | 94.5 | 93.2 | |
| 3.00 | 99.7 | 99.3 | 99.0 | 98.7 | |
|
| |||||
| Type I Error Rate (%) | - | 10.4 | 11.5 | 19.7 | 21.8 |
[1-β = 0.80; Response rate in the control arm = 0.20, Total planned sample size: 101 (α = 0.10); 65 (α =0.20)]
We emphasize that this design, unlike those proposed by Colton [23, 24], does not necessarily always switch to direct assignment in Stage II. Only when there is convincing though not definitive evidence from Stage I does the trial design switch to direct assignment. In the absence of such evidence, the trial continues with randomization in Stage II. As such, the direct assignment option provides an “extended confirmation phase” as an alternative to stopping the trial early for efficacy, which may help to avoid possibly prematurely launching into a Phase III trial. This design strategy can be incorporated within all-comers and enrichment biomarker based designs or implemented as a phase II biomarker independent trial. In the spectrum of designs proposed to date, with adaptive designs on the one end and fixed balanced randomized designs on the other, the direct assignment option design provides a possible middle ground, with likely clinical appeal.
Discussion
Key considerations for the choice of a biomarker driven design in a Phase II setting
Table 3 lists some of the key considerations when deciding between enrichment versus all-comers versus adaptive (including the direct assignment option) designs in a Phase II setting [10, 12]. The four main components include the marker prevalence, strength of the preliminary evidence, the assay reliability and validity, and turn-around times for marker assessment. We discuss these in more details below.
Table 3.
Criteria for choice of phase II design for initial validation of predictive marker
| Criteria | Design
|
|||
|---|---|---|---|---|
| Enrichment | All-Comers | Direct assignment option | Adaptive | |
| Preliminary Evidence | ||||
| 1. Strongly suggest benefit in marker defined subgroups. | Optimal | Not Recommended | Appropriate (with an early single IA, or two IA with option for direct at both IA) | Appropriate (assess multiple treatments/biomarker subgroups) |
| 2. Uncertain about benefit in overall population versus marker defined subgroups | Not Recommended | Appropriate | Appropriate (direct assignment option within the biomarker positive and negative cohorts) | Appropriate (learn and adapt as the trial proceeds) |
|
| ||||
| Assay Performance | ||||
| 1. Excellent (high concordance between local and central testing; commercially available kits; well established marker cutpoint etc.) | Required | Appropriate | Required | Required |
| 2. Questionable | Not Recommended | Appropriate | Not Applicable | Not Applicable |
|
| ||||
| Turnaround Times | ||||
| 1. Rapid (2–3 days; without causing delay in the start of therapy) | Optimal | Optimal | Optimal | Optimal |
| 2. Slow to Modest (one week or more) | Not Recommended | Appropriate (retrospective marker subgroup assessment) | Appropriate in some cases | Appropriate in some cases |
|
| ||||
| Marker Prevalence | ||||
| 1. Low (< 20%) | Optimal | Not Recommended | Appropriate (with an early single IA, or two IA with option for direct at both IA) | Appropriate |
| 2. Moderate (20%–50%) | Appropriate | Appropriate (stratified by marker status) | Appropriate, with 2 IA with direct assignment option only at the second IA | Appropriate |
| 3. High (> 50%) | Appropriate | Appropriate | Appropriate | Appropriate |
Enrichment designs are clearly appropriate when there is compelling preliminary evidence to suggest benefit of a treatment only in a marker defined subgroup(s), and/or when the marker prevalence is low (<10–20%). Under these situations, it is not feasible to use an all-comers strategy as the treatment effect in the overall population will be diluted, thus requiring a prohibitively large sample size. For enrichment designs, it is also essential to have an established assay with good performance and short turn-around times for marker assessment. Traditional designs that enroll a general (i.e. unselected) patient population often fail to identify promising targeted agents, since any subgroup treatment effect is diluted. A direct assignment option design with a single early interim analysis or two interim analyses with option for direct at both analysis [Figure 3] are other potential options in this setting. This is an enrichment strategy, but where the randomization to the control could be stopped based on the interim analysis results. Potential advantages of the direct assignment enrichment design strategy, compared to the enrichment design, include accrual savings and treating proportionally more patients with active versus control treatment, while maintaining desirable statistical properties.
Figure 3. Direct Assignment option Design with 2 planned interim analyses (IA), with an option for direct assignment at each analysis for one biomarker defined subgroup.

Fut: futility; eff: efficacy; dir: direct; rand: randomize
c1, c2, c3, c4 (=c2), c5 (=c7), c6, c7: O’Brien-Fleming stopping boundaries
An all-comers design is appropriate when 1) the preliminary evidence is unclear and the marker prevalence is high (≥50%) and/or 2) the assay performance is not well established (i.e., no established cutpoint for marker status definition) and/or 3) the turn-around time for marker assessment is long (more than a week for example in second or third line treatment settings. In most instances however, an all-comers design should incorporate a prospectively specified subgroup analyses of the treatment effect within biomarker defined subgroups. This is critical to ensure that the effect of the drug is tested both on the overall as well as prospectively defined subsets of patients so as to not incorrectly conclude that the drug is not effective, when it may be effective for a smaller subset of the population [6]. If the preliminary evidence for a new experimental agent is promising in only the biomarker positive subgroup, but is not sufficiently compelling or clear in the biomarker negative cohort, then a direct assignment option design could be considered for each of the positive and the negative biomarker cohorts.
As noted earlier, adaptive designs have the greatest potential when assessing multiple treatments and marker subsets where many questions are addressed, and not recommended in the context of two-armed trials, fixed sample size, and no biomarkers [18]. They do require established assays, and reasonable turn-around times for marker assessment, but can be applied to any marker prevalence scenario (low, moderate and high). Of course, a major consideration is the real time access to outcome data for the adaptation to be informative, in contrast to the direct assignment option design, which although incorporates a simple adaptation, requires the same infrastructure as conventional designs.
In cases where the prevalence of the marker in question is moderate (say between 20%–50%), and the preliminary evidence is not compelling, two possible strategies are as follows:
-
Strategy 1:
First, perform a single arm enrichment trial (pilot) as proof of concept that the treatment likely has a major effect within the marker subgroup;
-
Second, based on data from the pilot trial, perform an all-comers Phase II (randomized) trial, using either:
an adaptive design where the relationship between markers to treatment success is assessed in an ongoing manner, or
a trial stratified by marker status, with the primary hypothesis defined within the marker subgroup hypothesized to derive the most benefit. Accrue sufficient patients to the other subgroup(s) to demonstrate lack of benefit.
Strategy 2:Instead of a 2-step process, perform a direct assignment option design with 2 interim analyses, where the option for direct assignment is only possible at the second IA. This is a modification to the design proposed in Figure 3 [9].
Summary.
The wealth of opportunities in cancer drug development mandates intelligent clinical trial design. Incorporating biomarkers in the design of phase II studies of molecularly targeted agents informs the Phase III trial design strategy, thus assuring optimal use of limited phase III financial and patient resources.
Acknowledgments
Mandrekar and Sargent: National Cancer Institute Grants No CA-15083 (Mayo Clinic Cancer Center) and CA-25224 (North Central Cancer Treatment Group)
Footnotes
CONFLICTS OF INTEREST: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 2.Mandrekar SJ, Sargent DJ. Clinical Trial Designs for Biomarker Evaluation. Personalized Medicine in Oncology. (in press) [Google Scholar]
- 3.Buyse M, Sargent DJ, Grothey A, et al. Biomarkers and surrogate end points--the challenge of statistical validation. Nat Rev Clin Oncol. 2010;7:309–17. doi: 10.1038/nrclinonc.2010.43. [DOI] [PubMed] [Google Scholar]
- 4.Pusztai L, Anderson K, Hess KR. Pharmacogenomic predictor discovery in phase II clinical trials for breast cancer. Clin Cancer Res. 2007;13:6080–6. doi: 10.1158/1078-0432.CCR-07-0809. [DOI] [PubMed] [Google Scholar]
- 5.McShane LM, Hunsberger S, Adjei AA. Effective incorporation of biomarkers into phase II trials. Clin Cancer Res. 2009 Mar 15;15(6):1898–905. doi: 10.1158/1078-0432.CCR-08-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandrekar SJ, Sargent DJ. Clinical trial designs for predictive biomarker validation: theoretical considerations and practical challenges. J Clin Oncol. 2009 Aug 20;27(24):4027–34. doi: 10.1200/JCO.2009.22.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freidlin B, McShane LM, Polley MY, Korn EL. Randomized Phase II Trial Designs With Biomarkers. J Clin Oncol. 2012 Sep 10;30(26):3304–9. doi: 10.1200/JCO.2012.43.3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.An MW, Mandrekar SJ, Sargent DJ. A 2-Stage Phase II Design with Direct Assignment Option in Stage II for Initial Marker Validation. Clin Cancer Res. 2012 Aug 15;18(16):4225–33. doi: 10.1158/1078-0432.CCR-12-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mandrekar SJ, An MW, Sargent DJ. A phase II trial design with direct assignment option for initial marker validation. J Clin Oncol. 2012;30 doi: 10.1158/1078-0432.CCR-12-0686. (suppl 30; abstr 34) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandrekar SJ, Sargent DJ. All-comers versus enrichment design strategy in phase II trials. J Thorac Oncol. 2011 Apr;6(4):658–60. doi: 10.1097/JTO.0b013e31820e17cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon R, Maitournam A. Evaluating the efficiency of targeted designs for randomized clinical trials. Clin Cancer Res. 2004 Oct 15;10(20):6759–63. doi: 10.1158/1078-0432.CCR-04-0496. [DOI] [PubMed] [Google Scholar]
- 12.Mandrekar SJ, Sargent DJ. Design of clinical trials for biomarker research in oncology. Clin Investig (Lond) 2011 Dec;1(12):1629–36. doi: 10.4155/CLI.11.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Q, Mandrekar SJ, Sargent DJ. Predictive biomarkers in colorectal cancer: usage, validation, and design in clinical trials. Scand J Gastroenterol. 2012 Mar;47(3):356–62. doi: 10.3109/00365521.2012.640836. [DOI] [PubMed] [Google Scholar]
- 14.Wang SJ, O’Neill RT, Hung HM. Approaches to evaluation of treatment effect in randomized clinical trials with genomic subset. Pharm Stat. 2007;6:227–44. doi: 10.1002/pst.300. [DOI] [PubMed] [Google Scholar]
- 15.Jiang W, Freidlin B, Simon R. Biomarker-adaptive threshold design: a procedure for evaluating treatment with possible biomarker-defined subset effect. J Natl Cancer Inst. 2007;99:1036–43. doi: 10.1093/jnci/djm022. [DOI] [PubMed] [Google Scholar]
- 16.Buyse M. Limitations of Adaptive Clinical Trials. American Society of Clinical Oncology 2012 Educational Book. doi: 10.14694/EdBook_AM.2012.32.13. [DOI] [PubMed] [Google Scholar]
- 17.Korn EL, Freidlin B. Outcome--adaptive randomization: is it useful? J Clin Oncol. 2011 Feb 20;29(6):771–6. doi: 10.1200/JCO.2010.31.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berry DA. Adaptive clinical trials: the promise and the caution. J Clin Oncol. 2011 Feb 20;29(6):606–9. doi: 10.1200/JCO.2010.32.2685. [DOI] [PubMed] [Google Scholar]
- 19.Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther. 2009 Jul;86(1):97–100. doi: 10.1038/clpt.2009.68. [DOI] [PubMed] [Google Scholar]
- 20.Zhou X, Liu S, Kim ES. Bayesian adaptive design for targeted therapy development in lung cancer-a step towards personalized medicine. Clinical Trials. 2008;5:181–193. doi: 10.1177/1740774508091815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Jr, Tsao A, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011 Jun;1(1):44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones CL, Holmgren E. An adaptive Simon two-stage design for phase 2 studies of targeted therapies. Contemp Clin Trials. 2007;28:654–61. doi: 10.1016/j.cct.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Colton T. A model for selecting one of two medical treatments. JASA. 1963;58(302):388–400. [Google Scholar]
- 24.Colton T. A two-stage model for selecting one of two treatments. Biometrics. 1965;21(1):169–180. [Google Scholar]
- 25.Pocock SJ. Interim Analyses for Randomized Clinical Trials: The Group Sequential Approach. Biometrics. 1982;38(1):153–162. [PubMed] [Google Scholar]
- 26.Chang MN, Therneau TM, Weiand HS, Cha SS. Designs for Group Sequential Phase II Clinical Trials. Biometrics. 1987;43(4):865–875. [PubMed] [Google Scholar]