

Abstract

An efficient method for the synthesis of fused heteroaromatic dihydrosiloles via Ni-catalyzed hydrosilylation/intramolecular Ir-catalyzed dehydrogenative coupling of the Si-H bond with the heteroaromatic C-H bond has been developed. The method is efficient for both electron-deficient and electron-rich heterocycles. It exhibits high functional group tolerance and good regioselectivity. Fused heteroaromatic dihydrosiloles can be smoothly halogenated and then oxidized or arylated. Application of these transformations allows obtaining highly functionalized heteroaromatic structures. A gram-scale synthesis of dihydropyridinosilole has also been accomplished using reduced amounts of Ni- and Ir-catalysts.
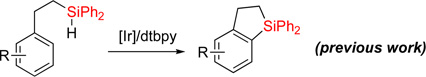
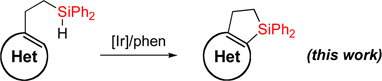
Transition metal-catalyzed C-H silylation reactions1 of aromatic and heteroaromatic systems serve as powerful tools for functionalization of these molecules.2–10 In recent years, a number of methodologies employing Ru,3 Ir,4 Rh,5 Pt,6 Pd,7 Re,8 and Lewis acid9 catalysis, have been developed for inter- and intramolecular dehydrogenative Si-H / C-H coupling reactions. Yet reports on analogous heteroaromatic C-H coupling are exceedingly rare.10 Recently, we reported a one-pot procedure for the efficient synthesis of dihydrobenzosiloles via hydrosilylation of styrenes with diphenylsilane followed by dehydrogenative cyclization (eq 1).11 However, the heteroaromatic analogues of dihydrobenzosiloles are virtually unknown.12 Given the importance of heteroaromatic molecules in various fields, herein we wish to report an efficient method for dehydrogenative Si-H / C-H cyclization of heteroaromatic molecules including pyridine, pyrrole, furan, and thiophene (eq 2). In this work, we also show that resulted fused heteroaromatic dihydrosiloles can be readily transformed into valuable highly functionalized building blocks.
 |
(1) |
 |
(2) |
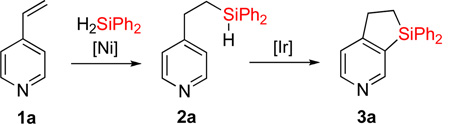
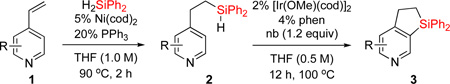
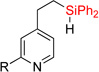
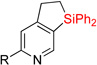
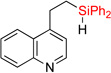
First, we attempted to apply the previously developed conditions11 for the synthesis of dihydropyridinosilole 3a from 4-vinylpyridine (eq 3). However, hydrosilylation of vinylpyridine 1a with diphenylsilane in the presence of NiBr2•(PPh3)2 produced traces of 2a. Use of Ni(cod)2/PPh3 combination was more effective in this transformation to give 2a in high yield. A subsiquent one-pot addition of [Ir(cod)(OMe)]2, dtbpy, and norbornene however did not provide any sufficient amounts of dihydropyridinosilole 3a. Optimization of the dehydrogenative cyclization of 2a revealed13 that use of 2% [Ir(cod)OMe]2 and 4% 1,10-phenantroline gives good yield of dihydropyridinosilole 3a (Table 1, entry 1). We also found that the presence of PPh3, which is a requisite ligand for the hydrosilylation reaction, inhibits the cyclization step. Hence, hydrosilylation/dehydrogenative coupling sequence was performed in a two-step sequence. Notably, both reactions can be easily scaled up to a gram-scale synthesis of dihydropyridinosilole 3a, which can be accomplished using lower amounts of Ni- and Ir-catalysts (Table 1, entry 2).
 |
(3) |
Table 1.
Scope of Electron-deficient Heterocycles in Hydrosilylation - Dehydrogenative Cyclizationa
 | ||||
|---|---|---|---|---|
| entry | intermediate | 2, yieldb | product | 3, yieldb |
 |
 |
|||
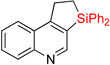
| 1 | 2a: R = H | 84% | 3a: R = H | 83% |
| 2c | 2a: R = H | 83% | 3a: R = H | 74–82% |
| 3 | 2b: R = Me | 86% | 3b: R = Me | 87% |
| 4 | 2c: R = F | 86% | 3c: R = F | 68% (5:1) |
| 5 | 2d: R = Cl | 61% | 3d: R = Cl | 64% |
 |
 |
|||
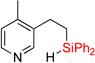
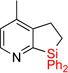
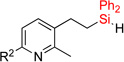
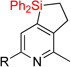
| 6 | 2e | 95% | 3e | traces |
 |
 |
|||
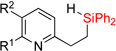
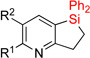
| 7 | 2f: R = H | 93% | 3f: R = H | 79% |
| 8 | 2g: R = Me | 90% | 3g: R = Me | 86% |
| 9 | 2h: R = Cl | 83% | 3h: R = Cl | 75% |
 |
 |
|||
| 10 | 2i: R1 = H, R2 = H | dec. | 3i: R1 = H, R2 = H | N/A |
| 11 | 2j: R1 = Me, R2 = H | 48%1 | 3j: R1 = Me, R2 = H | 36% |
| 12 | 2k: R1 = Me, R2 = F | 76%d | 3k: R1 = Me, R2 = F | 73% |
| 13 | 2l: R1 = OMe, R2 = H | 67% | 3l: R1 = OMe, R2 = H | 72% |
| 14 | 2m: R1 = CF3, R2 = H | 76% | 3m: R1 = CF3, R2 = H | 77% |
 |
 |
|||
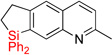
| 15 | 2n | 97% | 3n | 74% |
 |
||||
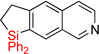
| 16 | 2o | 96% | 3o | 78% |
 |
||||
| 17 | 2p | 90% | 3p | 74%e |
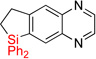 |
||||
| 18 | 2q | 94% | 3q | 56%e |
Hydrosilylation reaction conditions: 1 (1.0 mmol), H2SiPh2 (1.02 mmol), Ni(cod)2 (5 mol %), PPh3 (20 mol %), and THF (1.0 mL) were stirred at 90 °C for 2 h under nitrogen. Dehydrogenative coupling reaction conditions: 2 (0.5 mmol), [Ir(cod)OMe]2 (2 mol %) and 1,10-phenantroline (4 mol %), norbornene (1,2 equiv), and THF (1.0 mL) were stirred at 100 °C for 12 h under nitrogen.
Isolated yield.
Reaction was performed on 10 mmol scale using 2.5 mol % Ni-catalyst and 0.25–0.5 mol % Ir-catalyst.
10 mol % Ni(cod)2 was used.
48 h at 100 °C.
Having optimized conditions for dehydrogenative cyclization in hand, we examined the scope of the hydrosilylation – dehydrogenative cyclization reaction starting with 4-vinylpyridines bearing a substituent at the C2 position of the ring (Table 1, entries 3–5). It was found that both electron-donating and electron-withdrawing groups were tolerable at this position leading to dehydrogenative Si-H / C-H coupling reaction at less hindered site. Hydrosilylation of 4-methyl-3-vinylpyridine worked well (entry 6). However, attempts on cyclization step resulted in traces of cyclized product only, probably due to inhibition of Ir catalyst by its complexation with pyridine nitrogen atom. On the other hand, 2-methyl-3-vinylpyridine and its derivatives were converted into the cyclization products in good yield (entries 7–9). 2-vinylpyridine underwent smooth hydrosilylation reaction, as judged by GC/MS analysis of the crude reaction mixture, however it decomposed upon purification into 2-ethylpyridine (entry 10). This result can be explained by intramolecular pyridine nitrogen-assisted hydrolysis of the hydrosilylation product 2i. Hence, next, we examined reactions of more sterically hindered substrates possessing a substituent at C6 position of the pyridine ring. Thus, the hydrosilylation of 6-methyl-2-vinyl-pyridine lead to the corresponding hydrosilylation product 2j in moderate yield (entry 11). Subsequent dehydrogenative cyclization reaction of 2j allowed to obtain 3j in low yield. Introduction of fluorine at C5 position of 6-methyl-2-vinylpyridine, gave improved results (entry 12). Employment of substrates possessing MeO- and CF3- groups at C6 position of 2-vinylpyridine gave comparable good yields for both steps (entries 13–14). Having explored dehydrogenative cyclization of pyridine substrates with Si-tether at different positions, we turned our attention to other electron-defficient heterocycles. It was found that, quinoline, isoquinoline, and quinoxaline systems provided high yields of hydrosilylation reaction and good yields for a subsequent cyclization step (entries 15–18).
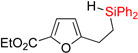
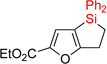
Next, we examined the possibility of using substrates possessing electron-rich heterocycles in this hydrosilylation – dehydrogenative coupling reaction sequence (Table 2). We were pleased to find that furan, thiophene, and pyrrole systems worked well in both reactions producing the corresponding cyclization products in good yields (entries 1–3). In the case of pyrrole with Si-tether at the C4 position, the cyclization selectively occured at the C3 position (entry 3). Expectedly, benzothiophene with tether at C3 position cyclized at C2 site (entry 4). Cyclization of fused heteroaromatic systems at phenyl ring were efficient, as well. Thus, employment of benzothiophene with tether at the C5 resulted in cyclization product at the C6 position as a major regioisomer (entry 5). Similarly, indole and N-Ts indazole with alkylsilyl tether at C5 gave the C6 cyclization products as major regioisomers (entries 6–7). On the other hand, N-Me indazole with Si-tether at C5 cyclized at the C6 exclusively (entry 8).
Table 2.
Scope of Electron-rich Heterocycles in Hydrosilylation - Dehydrogenative Cyclizationa
 | ||||
|---|---|---|---|---|
| entry | intermediate | 2, yieldb | product | 3, yieldb |
 |
 |
|||
| 1 | 2aa | 93% | 3aa | 88% |
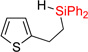 |
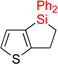 |
|||
| 2 | 2bb | 70% | 3bb | 67% |
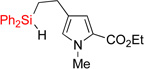 |
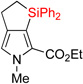 |
|||
| 3 | 2cc | 91% | 3cc | 84% |
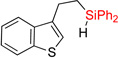 |
 |
|||
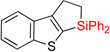
| 4 | 2dd | 94% | 3dd | 63% |
 |
||||
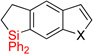
| 5 | 2ee: X = S | 92% | 3ee: X = S | 61% (10:1) |
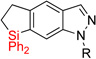
| 6 | 2ff: X =NMe | 89% | 3ff: X =NMe | 94% (10:1) |
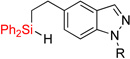 |
 |
|||
| 7 | 2gg: R = Ts | 85% | 3gg: R = Ts | 52% (7:1) |
| 8 | 2hh: R = Me | 84% | 3hh: R = Me | 59% |
Hydrosilylation reaction conditions: 1 (1.0 mmol), H2SiPh2 (1.02 mmol), Ni(cod)2 (5 mol %), PPh3 (20 mol %), and THF (1.0 mL) were stirred at 90 °C for 2 h under nitrogen. Dehydrogenative coupling reaction conditions: 2 (0.5 mmol), [Ir(cod)OMe]2 (2 mol %) and 1,10-phenantroline (4 mol %), norbornene (1,2 equiv), and THF (1.0 mL) were stirred at 100 °C for 12 h under nitrogen.
Isolated yield.
Finally we explored a synthetic usefulness of this method by transforming the obtained fused heteroaromatic dihydrosiloles into valuable heterocylic building blocks (Scheme 1). Thus, dihydropyridinosilole 3a upon treatement with tBuOOH/KH and TBAF14 can be oxidized into 4-(2-hydroxyethyl)pyridin-3-ol derivative 4 in 85% yield. Upon reaction with N-halosuccinimides and AgF, C(Het)-Si bond of dihydropyridinosilole 3a can be cleaved selectively over the C(Ph)-Si bond to produce 3-halopyridinefluorosilanes 5a–c in good yields. The Sigroup in 5a–c can be further oxidized to 6a–c, or replaced with aryl-15 group to form 7a,b in good yields. Compound 7b can undergo efficient intramolecular direct arylation with formation of tricyclic dihydrobenzoquinoline 8. Moreover, upon reaction with LiAlH4,16 3-chloropyridinefluorosilane 5a was converted to hydrosilane 2a’, which was cyclized into 3a’ under the Ir-catalyzed dehydrogenative cyclization reaction conditions. Subsequent dihydrosilole ring opening with NIS and AgF, and oxidation of the Si-group in the resulted 3-chloro-5-iodopyridinefluorosilane 5d using tBuOOH/KH and TBAF, led to the highly functionalized 2-(3-chloro-5-iodopyridin-4-yl)ethanol 9.
Scheme 1.

Further Modifications of Dihydropyridinosilole
In summary, we developed an efficient method for the synthesis of fused heteroaromatic dihydrosiloles via the Ni-catalyzed hydrosilylation of heteroaromatic styrenes, followed by the Ir-catalyzed dehydrogenative Si-H / C-H coupling sequence. This method proved to be very effective for elecron-defficient and electron-rich heterocycles. These newly formed fused heteroaromatic dihydrosiloles can be further transformed into valuable heterocyclic building blocks, posessing halogen-, hydroxyl-, and aryl- functionalities.
Supplementary Material
Acknowledgment
We thank the National Institutes of Health (GM-64444) and (1P50 GM-086145) for financial support of this work. The Mass Spectrometry Lab at the University of Illinois at Urbana-Champaign (UIUC) is greatfully acknowledged for acquisition of HRMS data.
Footnotes
Supporting Information Available Detailed experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org..
References
- 1.For recent reviews on C-H silylation reactions, see: Kakiuchi F, Chatani N. Adv. Synth. Catal. 2003;345:1077. Hartwig JF. Acc. Chem. Res. 2012;45:864. doi: 10.1021/ar200206a. Kuhl N, Hopkinson MN, Wencel-Delord J, Glorius F. Angew. Chem. Int. Ed. 2012;51:10236. doi: 10.1002/anie.201203269.
- 2.For pioneering works on transition metal-catalyzed silylation reactions, see: Gustavson WA, Epstein PS, Curtis MD. Organometallics. 1982;1:884. Sakakura T, Tokunaga Y, Sodeyama T, Tanaka M. Chem. Lett. 1987:2375. Ishikawa M, Okazaki S, Naka A, Sakamoto H. Organometallics. 1992;11:4135. Uchimaru Y, El Sayed AMM, Tanaka M. Organometallics. 1993;12:2065. Ishikawa M, Naka A, Ohshita J. Organometallics. 1993;12:4987.
- 3.For recent examples on Ru-catalyzed C-H silylation reactions, see: Kakiuchi F, Igi K, Matsumoto M, Chatani N, Murai S. Chem. Lett. 2001:422. Kakiuchi F, Igi K, Matsumoto M, Hayamizu T, Chatani N, Murai S. Chem. Lett. 2002:396. Kakiuchi F, Matsumoto M, Tsuchiya K, Igi K, Hayamizu T, Chatani N, Murai S. J. Organomet. Chem. 2003;686:134. Kakiuchi F, Tsuchiya K, Matsumoto M, Mizushima E, Chatani N. J. Am. Chem. Soc. 2004;126:12792. doi: 10.1021/ja047040d. Shima T, Hou Z. Chem. Lett. 2008:298. Ihara H, Suginome M. J. Am. Chem. Soc. 2009;131:7502. doi: 10.1021/ja902314v. Ihara H, Ueda A, Suginome M. Chem. Lett. 2011;40:916. Sakurai T, Matsuoka Y, Hanataka T, Fukuyama N, Namikoshi T, Watanabe S, Murata M. Chem. Lett. 2012;41:374. Mita T, Michigami K, Sato Y. Org. Lett. 2012;14:3462. doi: 10.1021/ol301431d.
- 4.For recent examples on Ir-catalyzed C-H silylation reactions, see: Ishiyama T, Sato K, Nishio Y, Miyaura N. Angew. Chem. Int. Ed. 2003;42:5346. doi: 10.1002/anie.200352399. Saiki T, Nishio Y, Ishiyama T, Miyaura N. Organometallics. 2006;25:6068. Simmons EM, Hartwig JF. J. Am. Chem. Soc. 2010;132:17092. doi: 10.1021/ja1086547. Mita T, Michigami K, Sato Y. Org. Lett. 2012;14:3462. doi: 10.1021/ol301431d.
- 5.For recent examples on Rh-catalyzed C-H silylation reactions, see: Tobisu M, Ano Y, Chatani N. Chem. Asian J. 2008;3:1585. doi: 10.1002/asia.200800090. Ureshino T, Yoshida T, Kuninobu Y, Takai K. J. Am. Chem. Soc. 2010;132:14324. doi: 10.1021/ja107698p. Tobisu M, Hasegawa J, Kita Y, Kinuta H, Chatani N. Chem. Commun. 2012:11437. doi: 10.1039/c2cc36601k. Kuninobu Y, Nakahara T, Takeshima H, Takai K. Org. Lett. 2013;15:426. doi: 10.1021/ol303353m. Kuninobu Y, Yamauchi K, Tamura N, Seiki T, Takai K. Angew. Chem. Int. Ed. 2013;52:1520. doi: 10.1002/anie.201207723.
- 6.For recent examples on Pt-catalyzed C-H silylation reactions, see: Williams NA, Uchimaru Y, Tanaka M. J. Chem. Soc. Chem. Commun. 1995:1129. Tsukada N, Hartwig JF. J. Am. Chem. Soc. 2005;127:5022. doi: 10.1021/ja050612p. Fukuyama N, Wada J-i, Watanabe S, Masuda Y, Murata M. Chem. Lett. 2007;36:910.
- 7.For example on Pd-catalyzed allylic C-H silylation, see: Larsson JM, Zhao TSN, Szabó KJ. Org. Lett. 2011;13:1888. doi: 10.1021/ol200445b.
- 8.For example on Re-catalyzed C-H silylation, see: Jiang Y, Blacque O, Fox T, Frech CM, Berke H. Chem. Eur. J. 2009;15:2121. doi: 10.1002/chem.200802019.
- 9.For Lewis acid-catalyzed C-H silylation examples, see: Furukawa S, Kobayashi J, Kawashima T. J. Am. Chem. Soc. 2009;131:14192. doi: 10.1021/ja906566r. Oyamada J, Nishiura M, Hou Z. Angew. Chem. Int. Ed. 2011;50:10720. doi: 10.1002/anie.201105636. For Lewis base-catalyzed C-H silylation example, see: Fedorov A, Toutov AA, Swisher NA, Grubbs RH. Chem. Sci. 2013;4:1640.
- 10.For heteroaromatic C-H silylation examples, see: Ishiyama T, Sato K, Nishio Y, Saiki T, Miyaura N. Chem. Commun. 2005:5065. doi: 10.1039/b511171d. Lu B, Falck JR. Angew. Chem. Int. Ed. 2008;47:7508. doi: 10.1002/anie.200802456. Klare HF, Oestreich M, Ito J-i, Nishiyama H, Ohki Y, Tatsumi K. J. Am. Chem. Soc. 2011;133:3312. doi: 10.1021/ja111483r.
- 11.Kuznetsov A, Gevorgyan V. Org. Lett. 2012;14:914. doi: 10.1021/ol203428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamao K, Nakamura K, Ishii H, Yamaguchi S, Shiro M. J. Am. Chem. Soc. 1996;118:12469. [Google Scholar]
- 13.See Supporting Information for details.
- 14.Jones GR, Landais Y. Tetrahedron. 1996;52:7599. [Google Scholar]
- 15.Beaulleu L-PB, Delvos LB, Charette AB. Org. Lett. 2010;12:1348. doi: 10.1021/ol1002863. [DOI] [PubMed] [Google Scholar]
- 16.Cheng AH-B, Lee ME, Rouss P, Jones PR. Organometallics. 1985;4:581. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.