

Abstract
Increases in the serum concentrations of parathyroid hormone (PTH), fibroblast growth factor 23 (FGF-23), and ultimately phosphate and decreases in 1,25-dihydroxyvitamin D are thought to play a central role in the progressive nature of kidney disease and the development of cardiovascular disease in patients with CKD. The initial changes in PTH and FGF-23 are adaptive to maintain the serum concentration of phosphate and the phosphate load within defined levels by increasing the urinary excretion of phosphate. Less well appreciated is the unanticipated finding that the absorption of phosphate from the gastrointestinal tract is not down-regulated in CKD. This maladaptive response maintains higher levels of phosphate absorption thereby contributing to the phosphate burden. Moreover, in response to a low phosphate diet, as is often prescribed to such patients, gut phosphate absorption may be enhanced, undermining the potential beneficial effects of this intervention. Given the poor response to limiting phosphate intake and the use of phosphate binders, we suggest that research efforts be oriented toward a better understanding of the factors that affect the absorption of phosphate in the gastrointestinal tract and the development of agents that directly inhibit the phosphate transporters in the small intestine and/or their associated binding proteins.
Index words: Gastrointestinal absorption of phosphate in CKD, Phosphate burden, Treatment strategies in CKD
Background
The purpose of this brief review is to highlight how the maladaptive responses of the gastrointestinal tract contribute to the progressive nature of kidney disease and the development of cardiovascular disease. These processes may also undermine some of the therapeutic strategies used in the management of patients with chronic kidney disease (CKD). There is a strong association between the presence of kidney disease and the development of an accelerated form of vascular disease characterized by alterations in both endothelial and vascular smooth muscle cells; the latter resulting in the development of arterial medial calcification (1,2). The presence of hypertension, abnormalities in lipid metabolism, and glucose intolerance in this patient population appears to provide only a partial explanation for the high prevalence of cardiovascular disease, and the presence of other non-traditional risk factors has been proposed. Attempts to link the progressive nature of kidney disease and the development of cardiovascular disease have begun to shift research efforts from the study of individual metabolic pathways that may be inappropriately regulated to other adaptations. A major emphasis of these investigations relates to abnormalities in phosphate balance, and the possibility that increases in the body burden of inorganic phosphate and ultimately hyperphosphatemia is the primary abnormality linking the progression of CKD to the development of cardiovascular disease (2,3,4).
Case Vignette
A 38-year-old hypertensive man with end-stage renal disease due to malignant hypertension returns to the dialysis unit after a recent hospitalization for acute chest pain. During the hospitalization, a non-enhanced ECG-gated spiral CT of the heart revealed previously unappreciated coronary calcification. Exercise stress test was unremarkable. Pertinent laboratory values included a serum calcium of 9.5 mg/dL, phosphate 7.0 mg/dL, 25-hydroxyvitamin D 30 ng/mL, intact PTH 900 pg/mL, and an alkaline phosphatase reading less than 1.5 times the reference value. Further review of the laboratory trends over the 12 months on hemodialysis identified that the hyperphosphatemia was never adequately controlled despite administration of phosphate binders and education about adhering to a low phosphate diet.
Pathogenesis
In the early stages of CKD, the plasma concentration of phosphate is maintained within the normal range by increases in the serum concentration of parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF-23), which serve to increase the urinary excretion of phosphate, and decreases in the serum concentration of 1,25-dihydroxyvitamin D (1,25(OH)2D), which decreases phosphate absorption from the gastrointestinal tract. In more advanced stages of kidney disease, the changes in PTH, FGF-23, and 1,25(OH)2D are no longer sufficient and hyperphosphatemia ensues (Figure 1). These four parameters (called mineral bone disorder) collectively and/or individually, correlate with the development of progressive kidney disease and cardiovascular disease (5,6,7). Moreover, there is a pathophysiologic basis for the effects of elevated levels of PTH, FGF-23, and phosphate and decreased levels of 1,25(OH)2D on cardiovascular structure and function. Vitamin D deficiency is associated with activation of renin, increases in kidney and systemic levels of angiotensin II, and the development of left ventricular hypertrophy (8,9). PTH has been found to activate ossification pathways that recruit vascular smooth muscles cells to function as adult mesenchymal stem cells and result in vascular calcification (10,11). FGF-23 has been shown to cause hypertrophy of isolated rat cardiac myocytes, as well as left ventricular hypertrophy when injected into normal animals; an effect attenuated by a FGF-23 receptor blocker (12,13). Elevations in phosphate concentration have been demonstrated to increase proliferation of both endothelial and vascular smooth muscle cells (14,15). Thus, both clinical association data as well as results from animal and cell studies support the hypothesis that the mineral bone disorder resulting from CKD contributes to the development of cardiovascular disease (Figure 2). While the progressive decline in kidney function ultimately limits the urinary excretion of phosphate despite the stimulation of two powerful phosphaturic hormones, namely PTH and FGF-23, we propose that the failure to down-regulate the absorption of phosphate from the gastrointestinal tract sustains hormone secretion, thus impairing hydroxylation of 25-hydroxyvitamin D, increasing the phosphate burden, and resulting in hyperphosphatemia.
Figure 1.

A propose schema of the development of mineral bone disorder in patients with CKD. A diseased kidney is unable to excrete (UPV) absorbed dietary phosphate, resulting in increased serum inorganic phosphate (Pi). This triggers an increase in parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF-23), both of which inhibit renal tubular phosphate absorption, and increase phosphate excretion. In addition, FGF-23 inhibits intestinal phosphate absorption, helping to mitigate the increase in the phosphate burden. Abbreviation: GI, gastrointestinal.
Figure 2.

The pathogenesis of cardiovascular disease caused by mineral bone disorder in CKD. Elevated phosphorus and PTH result in vascular calcification through effects on vascular smooth muscle cells (SMC) and endothelial cells (EC). This, in turn, leads to vascular stiffness and left ventricular hypertrophy (LVH). Elevated fibroblast growth factor 23 (FGF-23) lowers 1,25-dihydroxyvitamin D (1,25(OH)2D) level by inhibiting 1α-hydroxylase and activating 24-hydroxylase, the elimination pathway for 25-hydroxyvitamin D (25(OH)D). Vitamin D deficiency results in increased renin and hypertension which, in turn, results in LVH. FGF-23 itself can also cause LVH. Abbreviations: HTN, hypertension; Pi, inorganic phosphate.
In individuals with normal kidney function, the absorption of dietary phosphate is balanced by relatively rapid adjustments in the urinary excretion to maintain tissue, cell, and serum phosphate concentrations (collectively called the phosphate burden) within a defined and narrow range. The rate limiting step in intestinal phosphate absorption is the sodium-dependent phosphate transporter 2b (Npt2b, or, in humans, NPT2b [which is encoded by the SLC34A2 gene]) located in the apical membrane of small intestinal cells (Figure 3) (16). From studies in conditional Npt2b knockout mice, it is estimated that 50% or more of phosphate absorption is mediated by the Npt2b transporter (17). A small percent of phosphate transport in the gut is likely absorbed by the Pit1 transporter, while the remaining transport is believed to occur via a passive paracellular route.
Figure 3.
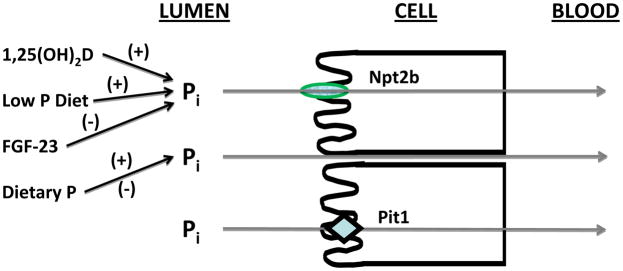
Pathways for phosphate absorption in the small intestine. In addition to passive paracellular transport, two transporters, sodium-dependent phosphate transporter 2b (Npt2b) and Pit1, account for 50% or more of phosphate absorption. Npt2b is down-regulated by fibroblast growth factor 23 (FGF-23), and up-regulated by 1,25-dihydroxyvitamin D (1,25(OH)2D) and a low phosphate (P) diet. Little is known about the regulation of the Pit1 transporter at the present time. Abbreviation: Pi, inorganic phosphate.
Npt2b is a unique gene product that differs significantly from the related kidney transporters Npt2a and Npt2c (18). The abundance of Npt2b is regulated by hormones such as vitamin D, which increases, and FGF-23, which decreases, the expression of Npt2b in the apical membrane of small intestinal cells and phosphate absorption in the gut (19,20,21). The effect of FGF-23 on the apical membrane expression of Npt2b has been associated with FGF-23-mediated decreases in 1,25(OH)2D levels. The regulation of Npt2b, however, is likely more complex. For example, FGF-23 administration had no effect on Npt2b expression in Npt2a knockout, Npt2c knockout, or Npt2a/Npt2c double knockout mice, although FGF-23 decreased the elevated 1,25(OH)2D levels in all these mutant mice (20). These findings provide evidence that intestinal Npt2b protein levels may be regulated by FGF-23 independent of changes in 1,25(OH)2D, and that there is cross talk between the renal Npt2a and Npt2c transporters and the gastrointestinal Npt2b transporter.
It is theorized that there are additional but as yet unknown factors that are important in linking the control of phosphate transport in the kidney and gastrointestinal tract, and that these factors may be operative in other circumstances such as the adaption to a low phosphate diet (20). When the kidney fails, however, the balance between gastrointestinal tract absorption and urinary excretion is impaired, and increases in the plasma and tissue concentrations of phosphate ensue. It is now well established that the fractional excretion of phosphate in the urine is increased in CKD; an early adaptive response that maintains near normal serum concentrations of phosphate until the level of kidney function is less than 10% of normal (21,22). It would have been reasonable to predict that an adaptive response to decrease phosphate absorption from the gastrointestinal tract would also be evident. Surprisingly, however, the absorption of phosphate from the gut has been found to be near-normal in patients with end stage renal disease and in animal models of CKD (21,22,23,24). In work published in the early 1990s, Loghman-Adham and colleagues demonstrated normal rates of sodium-dependent phosphate transport in brush border membranes from the intestine of rats that had undergone 5/6 nephrectomy, a model of CKD, one week prior to study (21,22). In these same studies, phosphate transport in kidney brush border membranes was significantly lower in the CKD animals. The experimental techniques used in these studies measures the component of phosphate absorption mediated by the sodium-dependent phosphate transporters. Gut perfusion studies by Marks et al found that phosphate transport in the gastrointestinal tract was not different between control animals and animals who had undergone a 5/6 nephrectomy 5 weeks before study (23). The perfusion technique measures both the facilitated as well as the paracellular transport of phosphate. When considered together, CKD did not affect either the facilitated or passive paracellular pathways of phosphate absorption in the small intestine of animals with CKD. Small intestine perfusion studies by Fordtran and co-workers showed only a modest decrease in phosphate transport in dialysis patients as compared to controls (24). When treated with vitamin D, the difference between controls and dialysis patients was no longer evident. In all of these studies, it would be predicted that FGF-23 levels were increased. We would suggest, therefore, that the inhibitory effect of FGF-23 on gastrointestinal phosphate absorption is blunted in CKD, and that the failure of the gastrointestinal tract to decrease phosphate absorption plays a critical role in the increase in the body burden of phosphate and the development of hyperphosphatemia.
Another factor that affects the absorption of phosphate in the gastrointestinal tract is the dietary intake of phosphate. In response to a low phosphate diet, there is an increase in the fractional absorption of phosphate in the gut associated with recruitment of additional phosphate transporters to the apical membrane of the cells of the small intestine (19). The effect of CKD on the adaptive response to a low phosphate diet has not been extensively studied. There was an increase observed in the uptake of phosphate in small intestinal brush border membrane vesicles from CKD rats adapted to a low phosphate diet in the studies of Loghman-Adham, but an absence of adaptation in the studies of Marks et al (22,23).
Recent Advances
Therapeutic approaches to treatment of the mineral bone disorder of patient with advanced kidney disease involve restriction of the dietary phosphate intake and the use of phosphate binders. As part of the strategy to limit the dietary phosphate intake, it is often suggested that the protein content of the diet be reduced. While seemingly logical, aspects of this approach are of potential concern. First, modest limitation of phosphate intake is unlikely to be effective given that phosphate absorption in the gut is not down-regulated in CKD patients (21,22,23,24). More severe restriction of protein raises the potential concern of inducing negative protein balance in these patients. It has been suggested that this latter concern might be obviated, at least in part, by increased education of health care providers about the relation between the protein and phosphorus content of food, the enhanced phosphate content of some processed foods, and the obstacles to patient compliance related to the absent or obscure labeling of the phosphate content of foods (25) (Box 1).
Box 1. Suitability of foods for CKD patients, sorted by available phosphate content.
|
Second, in response to a low phosphate diet, there is up-regulation of the NPT2b transporter and an increase in the fractional absorption of phosphate in the small intestine, at least in normal subjects (21). While there is conflicting evidence that patients and animals with CKD can up-regulate phosphate absorption from the gastrointestinal tract in response to restriction of the dietary phosphate intake, any up-regulation would tend to negate the potential beneficial effects of a low phosphate diet (22,23). Finally, in animals and humans adapted to a low phosphate diet, the acute ingestion of a high phosphate meal is associated with a rapid and significant increase in total phosphate absorption and a significant rise in the serum concentration of phosphate (26,27). In patients whose eating patterns are irregular and whose compliance to dietary restrictions is intermittent, it is easy to envision circumstances where patients adapted to a low phosphate diet would have periodic surges in the serum phosphate concentration as a consequence of dietary indiscretion. The clinical consequences of relatively sudden but significant rises in the serum phosphate concentration, however, remain to be determined.
The second approach to managing the phosphate burden and hyperphosphatemia in patients with CKD is the use of phosphate binders. Phosphate binders are recommended when there is a clear elevation in the serum concentration of phosphate. In the early stages of kidney disease, values in the normal range are sustained at a cost of elevated levels of PTH and FGF-23. This “tradeoff” may contribute to continuing fibrosis of the kidney and arteriosclerosis. As in the case vignette, use of phosphate binders has not proven to be uniformly effective. It is possible, although as yet not studied, that phosphate binders result in up-regulation of the gastrointestinal absorption of phosphate in a manner analogous to limiting the dietary intake of phosphate. A recently published study showed no change in FGF-23 levels and a paradoxical increase in vascular calcification in patients with moderate to advanced CKD treated with a variety of phosphate binders for up to 9 months (28). It should be noted that in this study, increased calcification was mainly seen in patients treated with calcium containing phosphate binders. In accord with this observation, other studies have shown that administration of some non-calcium based phosphate binders to CKD patients was associated with decreases in the serum concentration of FGF-23. In this regard, Eto and co-workers have demonstrated that nicotinamide inhibits intestinal phosphate reabsorption that is associated with a decrease in the abundance of Npt2b in the small intestine of rats (29,30,31,32).
Despite attempts to limit the dietary intake of phosphate and the use of phosphate binders, it would appear that many dialysis patients fail to achieve the recommended serum concentration of phosphate (<5.5 mg/dl). What is even more abundantly clear is that only a small percent of dialysis patients achieve serum phosphate concentrations in the normal range (< 4.5 mg/dl) with present management protocols. Moreover, in patients with CKD, these same management strategies do not regularly normalize the abnormalities in the phosphate burden, or the serum concentrations of Vitamin D metabolites, PTH, and FGF-23.
Summary
In our view, treatment plans for the mineral bone disorder of CKD are in flux. Initial evaluation of strategies to decrease the concentrations or activities of PTH or FGF-23 has not been definitive. For example, recent studies suggest that inhibition of PTH with cinacalcet had only a small effect on all-cause mortality or first non-fatal cardiovascular events despite improved biochemical parameters in dialysis patients (33). Furthermore, blocking the effects of FGF-23 increased aortic calcification and decrease survival in a mouse model of CKD (34). From a number of perspectives, correction of deficiencies of 25-hydroxyvitamin D and 1,25(OH)2D would be a desired therapeutic goal. On the other hand, vitamin D also increases gut absorption of phosphate, thereby nullifying some of the more beneficial effects of vitamin D repletion.
Central to the hypothesis that the abnormalities in mineral and bone metabolism are causative factors in the progression of decreased kidney function and the development of cardiovascular disease is the continuing stimulus provided by the increase in the phosphate burden. Since there are no current strategies available to further increase the urinary excretion of phosphate in CKD patients, the major focus of current therapy is targeted to decrease the gastrointestinal absorption of phosphate. The clinical response to a low phosphate diet and phosphate binders has been sub-optimal, at best. The reasons for this limited response are likely multifactorial, and include poor patient compliance and poor understanding of the phosphate content of food. We would suggest, however, that the maladaptation of the gastrointestinal tract to low phosphate diets and, perhaps to phosphate binders in CKD patients, may contribute to the relative ineffectiveness of the current treatment protocols. We propose that other therapeutic strategies need to be explored and recommend that future research efforts be directed at understanding why there is failure to down-regulate gastrointestinal absorption of phosphate in the presence of decreased kidney function and to determine if gut transport of phosphate is up-regulated by a low phosphate diet and/or the use of phosphate binders in CKD patients. Phosphate absorption in the gastrointestinal tract has received limited attention, and additional experiments are required to understand the factors that regulate Npt2b and, perhaps, other phosphate transporters. Finally, there is a need to develop new therapies targeted to directly inhibit these transporter(s) and/or disrupt their association with transporter binding-proteins such as the sodium-hydrogen exchanger regulatory factor 1 (NHERF-1) (35). Schiavi et al recently reported studies in CKD mice in which the Npt2b gene was silenced, and demonstrated decreases in the serum concentrations of phosphate and FGF-23 (36). Moreover, treatment of these mice with sevelamer further decreased the serum concentration of phosphate. These studies would suggest that treatment strategies directed at inhibiting proteins involved in the facilitated absorption of phosphate in the gastrointestinal tract, with or without dietary phosphate restriction and the use of phosphate binders, might be of value in the treatment of patients with advancing kidney and cardiovascular disease.
Acknowledgments
Support: This work was supported, in whole or in part, by National Institutes of Health Grant DK55881 (to EJW) and by the Research Service, Department of Veterans Affairs (to EJW).
Footnotes
The authors recognize the contributions of Adaani E. Frost, M.D., Baylor College of Medicine.
Financial Disclosure: Dr Suki has received speaking honoraria from Genzyme/Sanofi, Otsuka America, and Affymax/Takeda. The other authors declare that they have no relevant financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sarnak MJ, Levey AS. Cardiovascular disease and chronic renal disease: A new paradigm. Am J Kidney Dis. 2000;35:S117–S131. doi: 10.1016/s0272-6386(00)70239-3. [DOI] [PubMed] [Google Scholar]
- 2.Matsushita K, van der Velde M, Astor BC, Woodward M, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375:2073–2081. doi: 10.1016/S0140-6736(10)60674-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams ME. Chronic kidney disease/Bone and mineral metabolism: The imperfect storm. Seminars in Nephrology. 2009;29:97–104. doi: 10.1016/j.semnephrol.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Kiattisunthorn K, Moe SM. Chronic kidney disease-mineral bone disorder (CKD-MBD) IBMS Bonekey. 2010;7:447–457. [Google Scholar]
- 5.Mendoza JM, Isakova T, Ricardo AC, Xie H, Navaneethan SD, Anderson AH, et al. Fibroblast Growth Factor 23 and inflammation in CKD. Clin J Am Soc Nephrol. 2012;7:1155–1162. doi: 10.2215/CJN.13281211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation. 2005;112:2627–2633. doi: 10.1161/CIRCULATIONAHA.105.553198. [DOI] [PubMed] [Google Scholar]
- 7.Kestenbaum B, Sampson JN, Rudser KD, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol. 2005;16:520–528. doi: 10.1681/ASN.2004070602. [DOI] [PubMed] [Google Scholar]
- 8.Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1, 25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2012;110:229–238. doi: 10.1172/JCI15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forman JP, Williams JS, Fisher ND. Plasma 25-hydroxyvitamin D and regulation of the renin-angiotensin system in humans. Hypertension. 2010;55:1283–1288. doi: 10.1161/HYPERTENSIONAHA.109.148619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garfinkel A, Tintut Y, Petrasek D, Boström K, Demer LL. Pattern formation by vascular mesenchymal cells. Proc Natl Acad Sci. 2004;101:9247–9250. doi: 10.1073/pnas.0308436101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. 2010;7:528–536. doi: 10.1038/nrcardio.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faul C. Fibroblast growth factor 23 and the heart. Curr Opin Nephrol Hypertens. 2012;21:369–375. doi: 10.1097/MNH.0b013e32835422c4. [DOI] [PubMed] [Google Scholar]
- 14.Di Marco GS, König M, Stock C, et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013;83:213–222. doi: 10.1038/ki.2012.300. [DOI] [PubMed] [Google Scholar]
- 15.Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:10–17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 16.Radanovic T, Wagner CA, Murer H, Biber J. Regulation of intestinal phosphate transport. I. Segmental expression and adaptation to low-P(i) diet of the type IIb Na(+)-P(i) cotransporter in mouse small intestine. Amer J Physiol Gastrointest Liver Physiol. 2005;288:G496–G500. doi: 10.1152/ajpgi.00167.2004. [DOI] [PubMed] [Google Scholar]
- 17.Sabbagh Y, O’Brien SP, Song W, Boulanger JH, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20:2348–2358. doi: 10.1681/ASN.2009050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Werner A, Moore ML, Mantei N, Biber J, Semenza G, Murer H. Cloning and expression of cDNA for a Na/Pi cotransport system of kidney cortex. Proc Natl Acad Sci. 1991;88:9608–9612. doi: 10.1073/pnas.88.21.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams KB, DeLuca HF. Characterization of intestinal phosphate absorption using a novel in vivo method. Amer J Physiol Endocrinol Metab. 2007;292:E1917–E1921. doi: 10.1152/ajpendo.00654.2006. [DOI] [PubMed] [Google Scholar]
- 20.Tomoe Y, Segawa H, Shiozawa K, et al. Phosphaturic action of fibroblast growth factor 23 in Npt2 null mice. Amer J Physiol Renal Physiol. 2010;298:F1341–F1350. doi: 10.1152/ajprenal.00375.2009. [DOI] [PubMed] [Google Scholar]
- 21.Loghman-Adham M, Szczepanska-Konkel M, Dousa TP. Phosphate transport in brush border membranes from uremic rats. Response to phosphonoformic acid. J Am Soc Nephrol. 1992;6:253–1259. doi: 10.1681/ASN.V361253. [DOI] [PubMed] [Google Scholar]
- 22.Loghman-Adham M. Renal and intestinal Pi transport adaptation to low phosphorus diet in uremic rats. J Am Soc Nephrol. 1993;12:1930–1937. doi: 10.1681/ASN.V3121930. [DOI] [PubMed] [Google Scholar]
- 23.Marks J, Churchill LJ, Srai SK, et al. Intestinal phosphate absorption in a model of chronic renal failure. Kidney Int. 2007;72:166–73. doi: 10.1038/sj.ki.5002292. [DOI] [PubMed] [Google Scholar]
- 24.Davis GR, Zerwekh JE, Parker TF, Krejs GJ, Pak CY, Fordtran JS. Absorption of phosphate in the jejunum of patients with chronic renal failure before and after correction of vitamin D deficiency. Gastroenterology. 1983;85:908–916. [PubMed] [Google Scholar]
- 25.Gutierrez OM, Wolf M. Dietary phosphorus restriction in advanced chronic kidney disease: merit, challenges, and emerging strategies. Sem in Dialysi. 2010;23:401–406. doi: 10.1111/j.1525-139X.2010.00750.x. [DOI] [PubMed] [Google Scholar]
- 26.Giral H, Cranston D, Lanzano L, et al. Regulation of rat intestinal Na-dependent phosphate transporters by dietary phosphate. Am J Physiol Renal Physiol. 2009;297:F1466–F1475. doi: 10.1152/ajprenal.00279.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishida Y, Taketani Y, Yamanaka-Okumura H, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int. 2006;70:2141–2147. doi: 10.1038/sj.ki.5002000. [DOI] [PubMed] [Google Scholar]
- 28.Block GA, Wheeler DC, Persky MS, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. 2012;23:1407–1415. doi: 10.1681/ASN.2012030223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliveira RB, Cancela AL, Graciolli FG, et al. Early control of PTH and FGF23 in normophosphatemic CKD patients: A new target in CKD-MBD therapy? Clin J Am Soc Nephrol. 2010;5:286–291. doi: 10.2215/CJN.05420709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koiwa F, Kazama JJ, Tokumoto A, et al. Sevelamer hydrochloride and calcium bicarbonate reduce serum fibroblast growth factor 23 levels in dialysis patients. Ther Apher Dial. 2005;9:336–339. doi: 10.1111/j.1744-9987.2005.00293.x. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez-Parra E, Gonzalez-Casaus ML, Galán A, Martinez-Calero A, Navas V, Rodriguez M, Ortiz A. Lanthanum carbonate reduces FGF23 in chronic kidney disease stage 3 patients. Nephrol Dial Transplant. 2011;26:2567–2571. doi: 10.1093/ndt/gfr144. [DOI] [PubMed] [Google Scholar]
- 32.Eto N, Miyata Y, Ohno H, Yamashita T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol Dial Transplant. 2005;20:1378–1384. doi: 10.1093/ndt/gfh781. [DOI] [PubMed] [Google Scholar]
- 33.Chertow GM, Block GA, Correa-Rotter R, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;27;367:2482–2494. doi: 10.1056/NEJMoa1205624. [DOI] [PubMed] [Google Scholar]
- 34.Shalhoub V, Shatzen EM, Ward SC, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest. 2012;122:2543–2553. doi: 10.1172/JCI61405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giral H, Cranston D, Lanzano L, et al. NHE3 Regulatory Factor 1 (NHERF1) modulates intestinal sodium-dependent phosphate transporter (NaPi-2b) expression in apical microvilli. J Biol Chem. 2012;287:35047–35056. doi: 10.1074/jbc.M112.392415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schiavi SC, Tang W, Bracken C, et al. Npt2b Deletion attenuates hyperphosphatemia associated with CKD. J Am Soc Nephrol. 2012;23:1691–700. doi: 10.1681/ASN.2011121213. [DOI] [PMC free article] [PubMed] [Google Scholar]