

Abstract
Homogeneous transition metal–catalyzed reactions are indispensable to all facets of modern chemical synthesis. It is thus difficult to imagine that for much of the early 20th century, the reactivity and selectivity of all known homogeneous metal catalysts paled in comparison to their heterogeneous and biological counterparts. In the intervening decades, advances in ligand design bridged this divide, such that today some of the most demanding bond-forming events are mediated by ligand-supported homogeneous metal species. While ligand design has propelled many areas of homogeneous catalysis, in the field of Pd(II)-catalyzed C–H functionalization, suitable ligand scaffolds are lacking, which has hampered the development of broadly practical transformations based on C–H functionalization logic. In this review, we offer an account of our research employing three ligand scaffolds, mono-N-protected amino acids, 2,6-disubstituted pyridines, and 2,2′-bipyridines, to address challenges posed by several synthetically versatile substrate classes. Drawing on this work, we discuss principles of ligand design, such as the need to match a ligand to a particular substrate class, and how ligand traits such as tunability and modularity can be advantageous in reaction discovery.
1. Introduction
1.1 The Role of Ligand Design in Transition Metal Catalysis
The importance of ligands in modern homogeneous transition metal catalysis cannot be overstated. Because ligand coordination changes the structure and reactivity of a metal catalyst, it inherently changes the activation energy of elementary steps in a given catalytic process. This change is manifested in the kinetic reactivity,1 which can broaden the effective substrate scope of the reaction. Moreover, ligands can influence the selectivity (i.e., enantioselectivity, diastereoselectivity, regioselectivity, and chemoselectivity) in transformations where more than one product is produced, improve the solubility of metal catalysts in organic solvents, and extend catalyst lifetimes by suppressing metal catalyst degradation pathways. For instance, in cases where precipitation of metal nanoparticles is an irreversible catalyst deactivation pathway, strongly binding organic ligands can protect two molecules of catalyst from contacting one another and precipitating. Finally, as a consequence of these previous points, the ligand can affect the operational properties of the reaction: the compatibility with air and/or moisture, the reaction temperature, pressure, etc. In essence, ligands are chemists’ “hands” for influencing the bond-making and -breaking processes that occur at metal centers, and arguably, in the past several decades, ligand design has been the principal force driving the improvement of known catalytic reactions and the discovery of new ones.2
During the past century, simple, commonly available metal salts (e.g., CuCl, AuCl3, and Pd(OAc)2) have been applied extensively in catalysis, leading to the discovery of a myriad of different reactions, several of which have become mainstays in organic synthesis. This flurry of research in catalytic organometallic chemistry set the stage for the design of catalysts with improved reactivity and selectivity, such that desired products could be synthesized with high levels of purity and minimal amounts of waste. Because improved catalytic performance hinges on the properties of ancillary ligands, new ligand scaffolds have been vigorously pursued in all subfields of homogeneous metal catalysis.
Much of the search for new ligands has taken place in the context of research into catalytic asymmetric reactions, where in the absence of chiral information, the reaction will proceed via one of two pathways of equal energy to give a one-to-one mixture of enantiomeric products. By using a chiral nonracemic ligand, however, the reaction can be rendered enantioselective, leading to the predominant production of one enantiomer. Prominent early examples include the use of dialkyltartrate ligands in the Sharpless asymmetric epoxidation3,4 and the development of chiral diphosphine ligands for asymmetric hydrogenation, such as DIOP,5 DiPAMP,6 chiraphos,7 and BINAP8 by Kagan, Knowles, Bosnich, and Noyori, respectively. Ligand design has played an equally important role in developing enhanced kinetic reactivity for catalytic transformations that otherwise exhibit prohibitively slow reaction rates (or a complete lack of reactivity all together). Notable examples include the popularization of triphenylphosphine as a ligand in the 1960s, particularly in Wilkinson’s seminal applications to rhodium-catalyzed hydroformylation9 and hydrogenation,10 and more recently, the advent of dialkylaryl- and trialkylphosphines and N-heterocyclic carbenes as ligands in ruthenium-catalyzed olefin metathesis11,12 and palladium-catalyzed cross-coupling reactions.13–17 Moreover, there are also important cases where the search for ligands for stereoinduction led to the identification of more reactive catalysts and vice versa.18,19
1.2 “Privileged” Ligands in Transition Metal Catalysis
During the past half-century of research in asymmetric transition metal catalysis, a small collection of “privileged” ligand scaffolds have emerged, a term first coined by Yoon and Jacobsen.20 These ligand scaffolds have been found to be useful in many different types of asymmetric transformations, even among those with few or no mechanistic similarities (1–9, Figure 1). Privileged chiral ligands have been used extensively for reaction development in organic chemistry, and their application accounts for a high percentage of new asymmetric reactions developed each year.
Figure 1.
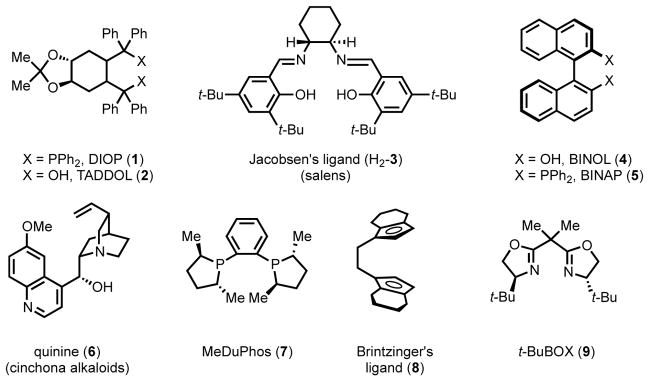
“Privileged” chiral ligands for asymmetric transition metal catalysis.20
As discussed above, in parallel to the progress in asymmetric catalysis, homogeneous catalytic transformations that do not induce chirality have been extensively studied. These reactions have required catalysts with high kinetic reactivity to ensure broad substrate scope, short reaction times, and favorable product distributions, and here too, ligand development has played a central role. In non-stereoselective reactions, there exist achiral “privileged” ligands, which have been utilized ubiquitously to enable a variety of different catalytic reactions, all with a myriad of distinct elementary steps (Figure 2). These include triaryl- and trialkylphosphines (10–13),21–24 biaryl(dialkyl)phosphines (14–19),25,26 diphosphines (20–25),27,28 “pincer” ligands (26–28),29,30 N-heterocyclic carbenes (29–33),16,31,32 cyclopentadienides (34–36),33–38 acetylacetonates (37–38),39 diamines (40–42),39–41 bipyridines (43–45),39,41–43 and pyridines (46–48).41
Figure 2.
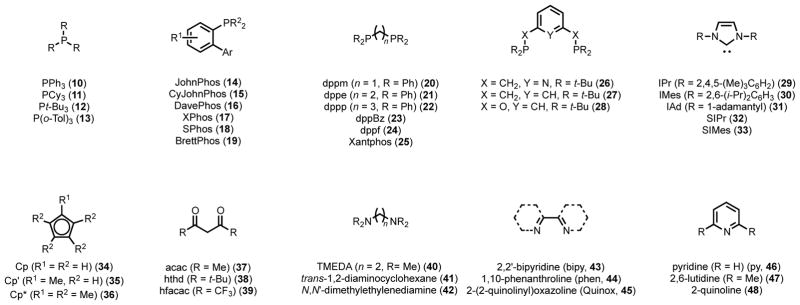
“Privileged” achiral ligands for high kinetic reactivity in transition metal catalysis.44
1.3 Matching a Substrate Class to a Metal Catalyst and Identifying a Suitable Ligand Scaffold
Though privileged ligand scaffolds are remarkably general in their utility across reaction types and substrate classes, they are still not universally applicable. One common feature of ligand-controlled metal-catalyzed reactions is that the catalytic system must be designed to match the properties of the substrate. Each individual class of substrates possesses a characteristic set of properties that will affect its approach to and interaction with the metal center. The course of the reaction is dictated by the intrinsic properties of the metal center (oxidation state, electron count, coordination geometry, etc.) and its established redox activity patterns. Together the properties of the substrate and metal then inform the selection and/or design of appropriate ligand scaffolds. Important substrate considerations include the steric and electronic properties (particularly those proximate to the reactive functional group), any existing stereochemistry, the presence or absence of other potentially reactive functional groups, and the presence or absence of Lewis basic chelating functional groups (directing groups).45,46 For example, consider asymmetric olefin hydrogenation (Scheme 1a). For substrates containing a chelating functional group, such as α,β- unsaturated carboxylic acid 49, the presence of an open binding site on the metal for carboxylate coordination is desirable. With the C2-symmetric Noyori [Ru(BINAP)(OAc)2] catalyst, the substrate can smoothly displace the acetate, without compromising catalyst activity, and subsequent directed asymmetric hydrogenation proceeds with excellent levels of stereoinduction.47 In contrast, in the Pfaltz non-directed asymmetric hydrogenation of olefins (51), a Crabtree-type catalyst48 bearing a non-C2-symmetric P,N-phosphite–pyridine ligand 52 and a non-coordinating counterion, BArF, is highly effective in the absence of a chelating functional group, relying on weak coordinative interactions for orienting the substrate.49,50 Similarly, for asymmetric olefin epoxidation (Scheme 1b), the Sharpless system3,4,51 is highly effective for allylic alcohols (54), and the Jacobsen system works well for internal (Z)-olefins not containing directing groups (57).52,53 Although the net transformations appear similar in both cases, the properties of the substrate dictate the selection of a metal, the mechanistic manifold, and ultimately the ligand used for stereoinduction.
Scheme 1.

Application of a diverse array of catalyst/ligand structures to asymmetric transformations of olefins. (a) Asymmetric hydrogenation of olefins by Noyori47,54 and Pfaltz.49 (b) Asymmetric epoxidation of olefins by Sharpless3,4 and Jacobsen.53 (c) Ligand structures.
In non-stereoselective catalysis, tailoring ligand properties to match the substrate is similarly important. For instance in Pd-catalyzed Suzuki–Miyaura55 cross-coupling reactions, unactivated aryl chlorides were traditionally an unreactive class of substrates. Ligands that are commonly used with aryl iodides, -bromides, and -triflates, such as triphenylphosphine, BINAP, and dppf are generally unreactive with electron-neutral or -rich aryl chlorides, such as 59,13 presumably because oxidative addition of a C(sp2)–Cl bond to a Pd(0) center is kinetically unfavorable.56 This challenge can be surmounted, however, through the application of sterically demanding, electron-rich phosphine ligands such as DavePhos (16)25 and P(t-Bu)3 (12) (Scheme 2).23 The σ-donating character of these ligands increases the nucleophilicity of the Pd(0) center which lowers the activation energy for oxidative addition. The steric bulk is thought to facilitate reductive elimination and promote formation of the catalytically active 1:1 Pd(0):ligand complex in solution.13 Through the selection and design of appropriate ligands, a new class of widely available substrates was rendered reactive in this and other related cross-coupling chemistries.
Scheme 2.
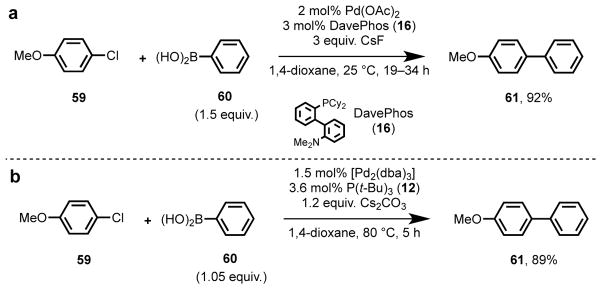
Application of sterically bulky, electron-rich phosphine ligands in the Suzuki–Miyaura cross-coupling of unactivated arylchlorides with boronic acids, as reported independently by the groups of (a) Buchwald25 and (b) Fu.23
1.4 Modifiability and Modularity of Ligand Scaffolds
Because it is difficult to predict a priori the potentially dramatic influence that subtle changes to a ligand’s structure will have on the metal center as it cycles through many elementary steps of a catalytic process;57,58 one key to the success of a ligand scaffold is that it can be readily modified. Multiples sites of variation both near and distal to the metal allow for the steric and electronic properties to be finely tuned to meet the energetic requirements of the reaction. For instance, the BINAP/BINOL scaffold has been adapted through modification at nearly every conceivable position since its introduction in asymmetric catalysis8,59–62 and has continued to play a central role in reaction discovery (Figure 3). In the early stages of reaction discovery and development, catalyst modifiability is particularly important, because many of the crucial reaction parameters are typically poorly understood. As evidence of the importance of modifiability, it is becoming increasingly common for commercial suppliers to offer ligand kits, containing multiple structural variants on a single ligand scaffold.
Figure 3.
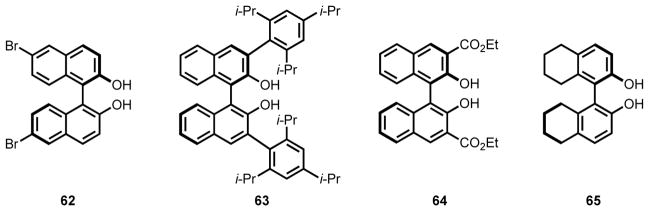
Selected examples of commercially available BINOL derivatives.
For similar reasons, many widely used ligand scaffolds are modular, meaning that they can be synthesized in a small number of steps from readily available components, allowing for a library of ligand candidates to be synthesized expediently.63 For instance, chiral phosphine–oxazoline (PHOX, 66) ligands pioneered by Pfaltz,64 Helmchen,65 and Williams,66 possess a non-symmetrical P,N-donor system,67 with one soft atom (P) and one hard atom (N) (Figure 4). Their unique structure has proven to be critical for challenging asymmetric metal-catalyzed transformations, particularly asymmetric allylic substitution. A major practical aspect that makes this class of ligands convenient to use is the modular synthesis, where an entire family of ligand candidates can be synthesized from widely available commercial chiral building blocks.68,69
Figure 4.
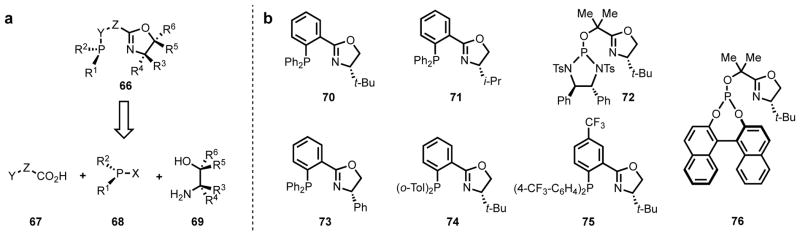
(a) Modular synthesis of chiral PHOX-type ligands.70 (b) Representative PHOX ligands.71
Tailoring ligand properties for a desired metal-catalyzed transformation requires careful consideration of the key features of the substrates class as well as the mechanism of each elementary step in the would-be catalytic process. Ligands that have a rich history of success in metal-catalyzed reactions (those that are considered privileged) are often useful starting points in reaction discovery and development. In this vein modifiability and modularity are key in providing libraries of ligands for medium- and high-throughput screening campaigns. As discussed below, many of the key advancements in our own laboratory have hinged upon ligand design, so the considerations outlined above are ones that we have returned to repeatedly as our research program in Pd(II)-catalyzed C–H functionalization has matured.
In the ensuing section, we discuss some of our general goals and sources of inspiration, and outline how ligand development plays a key role in the realization of our objectives. We then go on to describe the challenges in identifying effective ligand scaffolds for C(sp2)–H and C(sp3)–H functionalization using catalytic Pd(II). Drawing primarily from our own results, we present three stories about three different ligand scaffolds and the substrate classes with which they were designed to promote selective C–H functionalization. We show how ligand design can enable enantioselective C–H activation, and also serve to enhance reactivity with achiral substrates, thereby dramatically broadening substrate scope. We further discuss the need to match a target substrate and ligand to enable a productive interplay at the metal center as a prerequisite to efficient catalysis. In recounting these examples, we illustrate the principles articulated above and reflect on the lessons learned from our work.
2. Overview of Our Research Program in Pd(II)-Catalyzed C–H Functionalization
2.1 Philosophy and Guidelines for Reaction Development
While there remain numerous challenges in organic synthesis that can be addressed through transition metal catalysis, a primary focus of our own research program has been the creation of novel and strategic retrosynthetic disconnections72–75 that allow C–H bonds to be viewed as dormant functional groups that can be converted to a desired C–C or C–X bond at any stage of a synthesis.76,77 To make this approach broadly useful for synthetic applications, we have adhered to the following guidelines in developing C–H functionalization reactions:
Substrates: We utilize substrate classes that are readily available in both bulk quantity and structural diversity, and which can lead to a variety of complex synthetic targets upon C–H functionalization. Newly uncovered modes of reactivity can then be extended to feedstock chemicals, including those that contain weakly coordinating functional groups which can be used to facilitate C–H cleavage.77 Overall, appropriate selection of target substrate classes can further improve both the economy78 and sustainability79 of organic synthesis.
Products: Equally important is that C–H activation and subsequent functionalization takes place at a strategic position and installs a desirable functional group so that the product represents a commonly encountered structural motif, such as a β-functionalized carboxylic acid, amino acid, or biaryl. Establishing reliable routes to these substructures facilitates synthesis planning that incorporates C–H activation transformations as key disconnections. When combined with the selection of simple starting materials, drastic increases in molecular complexity can be achieved using this approach (Scheme 3).
Reactivity: We preferentially target catalytic manifolds that allow flexibility in terms of the scope of nucleophiles, electrophiles, and/or radical species as reacting partners with C–H bonds. Versatility in the functionalization step allows the [C–M] bond resulting from C–H cleavage to be efficiently mapped into any carbon–carbon or carbon–heteroatom bond (Scheme 4).
Catalysts/Ligands: For the reasons outlined above, ligand-controlled C–H functionalization reactions represent an especially fertile ground for discovering new modes of reactivity and for gleaning fundamental insights into chemical reactivity (Scheme 5). We view ligand development as the long-term solution for improving catalyst performance: reactivity (TON, TOF, and substrate scope), selectivity (site-selectivity, chemoselectivity, and stereoselectivity), and operating conditions (reaction time, temperature, and tolerance for air and moisture).
Operational Convenience: In order for novel C–H functionalization reactions to be embraced by the synthetic community, the reactions must be operationally simple and robust. In this respect, substrate, coupling partners, catalysts, and ligands that are commercially available or easily accessible are preferable. Reactions that do not require special equipment and can be set up on the benchtop without rigorous exclusion of air or moisture are similarly desirable.
Scheme 3.

Novel retrosynthetic disconnections enabled by our methodology: routes to (a) the kinamycins80 and (b) (+)-lithospermic acid.81
Scheme 4.

Diverse Pd(II)-catalyzed C–H functionalization of (a) benzoic acids82–86 and (b) benzoic acid–derived N-aryl amides87–89,90 reported by our group.
Scheme 5.
Enantioselective Pd(II)-catalyzed C–H activation using a chiral ligand.
In line with these goals, much of our research has focused on C–H functionalization reactions catalyzed by Pd(II).77,91–95 One of the attractive aspects of this approach is the established, versatile reactivity of [R–Pd(II)] (R = aryl or alkyl) intermediates. In the context of Pd(0)-catalyzed aryl halide functionalization, it has been well-established that [R–Pd(II)] intermediates can effectively couple with a range of carbon- and heteroatom-based nucleophiles via Pd(0)/Pd(II) catalysis. Thus, C–H cleavage with Pd(II), which is generally believed to be redox neutral with concomitant loss of HX (X = halide, acetate, etc.), can be viewed as an alternative and complimentary entry point to this same collection of bond-forming reactions. Key to the achieving this type of general reactivity is the development of effective ligands to promote C–H cleavage,76 which is typically the rate-limiting and selectivity-determining step in Pd(II)-catalyzed C–H functionalization reactions. It is also our intention that fundamental studies on the interplay between Pd(II), substrate, and ligand could lend useful information to the development of C–H functionalization reactions catalyzed by other metals, especially Cu(II),96 Rh(III),97 and Ru(II),98 which can potentially perform similar redox-chemistry to Pd(II).99
2.2 The Need for Effective Ligand Scaffolds
During the past 10 years that we have worked in this area, an impediment to our research has been the lack of ligand scaffolds for accelerating C–H cleavage with Pd(II).100 We have consistently resorted to the simplest Pd(II) salts available (Pd(OAc)2, Pd(TFA)2, PdI2, etc.), and with these basic catalysts, many of our goals, including the development of methodology for functionalizing bulk chemicals and for achieving site- and stereoselectivity in the C–H cleavage step, remained elusive. Thus, the lack of suitable ligand scaffolds represents a major roadblock in harnessing the full potential of Pd(II)-catalyzed C–H functionalization. Indeed, this need for ligands is at the heart of commonly cited problems in the field, like the inability to control positional selectivity of functionalized arenes101 or more generally, the inability to selectively functionalize one of the many inequivalent C–H bonds present in organic small molecules.
We became keenly aware of these shortcomings when developing methodology for stereoselective C–H activation which would require an efficient chiral [Pd(II)–ligand]* catalyst (Section 3.1).93 As we began to move from a chiral auxiliary approach to a chiral catalyst approach, we confronted a long-standing challenge in the field: uncovering a chiral metal catalyst for enantioselective C–H activation. Our motivation for investigating enantioselectivity was rooted in the hypothesis that a chiral ligand effective for stereoinduction is likely to be involved in the C–H cleavage transition state. Involvement in the C–H cleavage step suggests that this ligand scaffold, or a closely related scaffold, could ultimately be used for the interrelated problems of rate acceleration and site-selectivity.
3. Identification of Ligand Scaffolds for Pd(II)-Catalyzed C–H Functionalization
3.1 Considerations for Ligand Design
When considering potential ancillary ligand scaffolds for Pd(II)-catalyzed C–H functionalization, there are many interrelated challenges and complications to keep in mind:
Assembling a 1:1 ligand:substrate coordination structure. The ligand could outcompete the substrate for coordination to the Pd(II) center or vice versa. Ligands by their very nature contain coordinating functional groups (either in the form of non-bonding lone pair electrons or bonding π-systems). By contrast, unactivated C–H bonds are paraffin in nature, meaning that they are non-polar and have low inherent affinity for cationic metal centers. Achieving high effective molarity of the substrate in the presence of a ligand is thus difficult, particularly with simple hydrocarbon substrates, like toluene or hexane. In directed C–H activation, on the other hand, many traditional directing groups (pyridines, oxazolines, oximes, etc.),92,102,103 could inhibit ligand coordination prior to C–H cleavage or could render the C–H cleavage step so facile that the ligand would not play a dominant role in influencing reactivity and/or selectivity.
Promoting the C–H cleavage step. Under different mechanistic paradigms for C–H cleavage, the electronic requirements of the Pd(II) center, and thus the supporting ligand, are different. For instance in C(aryl)–H cleavage along a electrophilic palladation mechanism,104 there is a need to maintain relatively electrophilic Pd(II) center. In a concerted metalation/deprotonation (CMD) mechanism,105–108 a more nucleophilic Pd(II) center can be effective. The operative mechanism for C–H cleavage typically depends on a combination of factors, including the substrate, the reaction conditions, and also the ligand(s).76
Compatibility with the other steps in the catalytic cycle and the reaction conditions. Different Pd(II)-mediated C–H functionalization reactions proceed via different elementary steps, and each poses challenges in ligand engineering. For example, some steps may require ligand dissociation to provide a vacant coordination site, while others could require the ligand to be resistant to reductive elimination from high-valent intermediates. Additionally, under the reaction conditions, the ligand could react with the substrate, the coupling partner, or the Pd(II) catalyst along any of a number of possible degradation pathways.
Modifiability and modularity. Because it is unlikely that a single ligand will be adequate for each combination of substrate and reaction partner, it is advantageous to pursue multiple distinct ligand scaffolds in parallel. In addition to developing multiple complementary ligand scaffolds, utilizing modifiable and modular ligand scaffolds can be advantageous for fine-tuning a ligand to a specific combination of substrate and reaction type.
These general considerations and design guidelines provide a conceptual framework for ligand development in Pd(II)-catalyzed C–H functionalization. Naturally, as our research program has progressed, our theoretical understanding of ligand design principles has evolved as a result of new insights from experimental work. Owing to the inherent unpredictability of ligand discovery, there is also an element of empiricism and serendipity to this research area, which is why it is important to think carefully but still be adventurous in exploring old and new ligand architectures.
Below we present three case studies from our research group about three ligand scaffolds, mono-N-protected amino acids, 2,6-disubstitued pyridines, and 2,2′-bipyridines, which we have found to be effective for Pd(II)-catalyzed C–H functionalization (Figure 6). Of the three ligand scaffolds, we have studied mono-N-protected amino acids in the most depth to date; we thus provide a more comprehensive discussion of this work and shorter synopses of studies done with the other two ligand scaffolds. We describe early findings that led to identification of these ligand scaffolds, and examine how the principles outlined above are at play in each case.
Figure 6.
Ligand scaffolds discussed in this section.
3.2. Mono-N-Protected Amino Acid Ligands
3.2.1. Background
Drawn to the richness of mechanistic questions and potential synthetic applications of C–H activation chemistry, we established our research program in 2002 at the University of Cambridge to investigate stereoinduction in the palladation of prochiral C–H bonds, which we viewed as fundamentally important to the understanding of C–H cleavage by Pd(II) centers.93 To gain structural information concerning the transition state of directed palladation reactions, we focused on reactions that would proceed through well-defined intermediates that could be characterized. Inspired by the important role that removable chiral auxiliaries play in modern organic synthesis, our first approach centered on the use of a chiral oxazoline group109–111 that could be readily installed onto aliphatic carboxylic acids. Following this strategy, we found high levels of stereoinduction (>99:1 d.r.) could be achieved in the diastereoselective C–H iodination and acetoxylation of prochiral α,α-gem-dimethyl groups, α,α-gem-diphenyl groups, and cyclopropanes along a Pd(II)/Pd(IV) catalytic cycle (Scheme 6a).91,112–114
Scheme 6.

(a) Disastereoselective C(sp3)–H iodination of gem-dimethyl groups using a removable chiral oxazoline auxiliary. (b) General catalytic cycle for C–H iodination via Pd(II)/Pd(IV) catalysis. (c) Model for diastereocontrol, including proposed transition state structure.
tert-Butyl-substituted oxazoline 110 proved to be remarkably effective in directing C(sp3)–H activation, even under mild conditions (including temperatures as low as 0 °C). We have sought to understand the root cause of this reactivity through structural characterization both in solution and in the solid state. Further studies to elucidate the transition state structure via computational studies have also been performed in collaboration with the Houk group (Scheme 6b).115 We have characterized several trinuclear palladium intermediates (e.g., 113 and 119), performed calculations that revealed the involvement of monomeric precursor 115 as the reactive intermediate, and modeled various possible transition states for the diastereoselective C–H cleavage step (116). This work has confirmed the importance of conformation and geometry in the square planar pre-transition state coordination structure 115, in which the target C–H bond is approximately co-planar to the acetate anion. This arrangement minimizes the dihedral angle between the C–H bond and the Pd(II)–OAc, which is critical during the CMD transition state.
3.2.2 Enantioselective C–H Activation
As this work was progressing, we took the next conceptual step forward and questioned whether we could develop a chiral ligand capable of achieving similar levels of stereoinduction in the absence of a chiral auxiliary. To that end, we used diphenyl-2-pyridylmethane 120 as our pilot substrate and developed efficient non-stereoselective conditions to achieve Pd(II)-catalyzed C–H/R–BXn cross-coupling (Schemes 7 and 8).92 In the long term, we hoped to use this reaction system as a model for developing a broad range of enantioselective carbon–carbon and carbon–heteroatom bond-forming reactions, in which C–H cleavage could be directed by a diverse collection of functional groups.
Scheme 7.
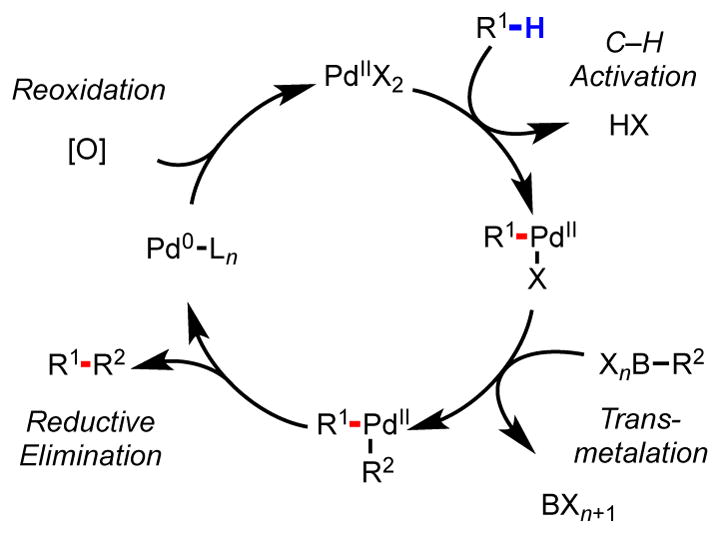
General catalytic cycle for C–H/R–BXn cross-coupling via Pd(II)/Pd(0) catalysis.
Scheme 8.

(a) Racemic route to dimeric palladacycle 122 from prochiral starting material 120. (b) Initial hypothesis for achieving stereoinduction in the C–H cleavage step through use of a chiral carboxylate ligand.116
Following the isolation and characterization of cyclopalladated dimer 122, we reasoned that prior to the C–H cleavage step, the putative pre-transition state coordination structure 124 offered the opportunity to replace the achiral non-bridging acetate ligands on the Pd(II) center with chiral carboxylates. In this manner, we hypothesized, stereoselective C–H cleavage could take place at one of the two prochiral phenyl groups. Although this line of thinking is not entirely accurate based on our current understanding, it nonetheless served as an invaluable starting point to probe enantioselective C–H activation.
To develop a catalytic enantioselective reaction, we first surveyed a variety of chiral acid ligands94g in an attempt to induce stereoselectivity in the Pd(II)-catalyzed enantioselective C–H activation of 125 (Table 1).117 We were encouraged by modest enantioselectivity (<25%) using this approach, and we reasoned that a more rigid backbone structure would limit the conformational degrees of freedom that the ligand possessed. For this reason, we prepared chiral cyclopropane amino acids 136–139, and found that the ee values could be improved to >40%, while maintaining good yield (Table 2). Consistent with expectations, enantiomers 136 and 138 led to product mixtures with opposite enantioselectivity values. Surprisingly, pseudo-C2-symmetric ligand 139 led to nearly racemic mixture. These data, in conjunction with earlier work in the literature characterizing amino acid–bound palladacycles118 suggested that during the C–H cleavage step, the ligand was bound in a bidentate fashion with the N–H bond possibly remaining intact, inducing chirality at the nitrogen atom. In this way, chiral information could be effectively geared from the α-carbon atom of the ligand to the nitrogen atom, which would rationalize why 139, which has two α-substituents of similar steric bulk, was ineffective.
Table 1.
Early attempts to use chiral carboxylate ligands in the catalytic enantioselective C(sp2)–H activation of 125.117

| |||
|---|---|---|---|
| Entry | Ligand | Yield (%)a | ee (%) b |
| 1 |
 128 |
62 | 5 |
| 2 |
 Boc-Pro-OH, 129 |
75 | 20 |
| 3 |
 Cbz-Pro-OH, 130 |
65 | 20 |
| 4 |
 131 |
87 | 6 |
| 5c,d |
 132 |
25 | 0 |
| 6 |
 133 |
78 | 20 |
| 7 |
 134 |
63 | 15 |
| 8b,e |
 135 |
9 | 21 |
Isolated yield.
ee determined by HPLC using a chiral stationary phase.
Yield determined by 1H NMR.
10 mol% Ligand, 85 °C.
15 mol% Ligand, 85 °C.
Table 2.
Catalytic enantioselective C(sp2)–H activation of 125 with chiral cyclopropane amino acid ligands.116




Isolated yield.
ee determined by HPLC using a chiral stationary phase.
With a refined mechanistic understanding, we returned to optimizing the ligand scaffold by maximizing the steric difference between the two α-substituents. To our delight, we found that simple, commercially available Boc-protected amino acids119 gave high levels of stereoinduction (Entries 1–7, Table 3). Among the Boc-protected amino acids examined, leucine (Entry 7) proved to be the optimal backbone structure. In an effort to improve the yield, we then fine-tuned the alkyl substituent on the carbamoyl protecting group. By introducing a (−)-menthyl group, we were able to improve the yield while maintaining the stereoselectivity (Entry 11).
Table 3.
Catalytic enantioselective C(sp2)–H activation of 125 with mono-N-protected amino acid ligands.116

| |||
|---|---|---|---|
| Entry | Ligand | Yield (%)a | ee (%) b,c |
| 1 |
 Boc-tert-Leu-OH, 140 |
60 | 52 |
| 2 |
 Boc-Val-OH, 141 |
69 | 70 |
| 3 |
 Boc-Abu-OH, 142 |
47 | 85 |
| 4 |
 Boc-Ala-OH, 143 |
60 | 80 |
| 5 |
 Boc-Ser-OH, 144 |
65 | 88 |
| 6 |
 Boc-Phe-OH, 145 |
65 | 88 |
| 7 |
 Boc-Leu-OH, 146 |
63 | 90 |
| 8 |
 MeCO2-Leu-OH, 147 |
88 | 79 |
| 9 |
 TcBoc-Leu-OH, 148 |
89 | 85 |
| 10 |
 (+)-Men-Leu-OH, 149 |
87 | 85 |
| 11 |
 (−)-Men-Leu-OH, 150 |
91 | 87 |
Isolated yield.
ee determined by HPLC using a chiral stationary phase.
Absolute configuration of 127 determined by single-crystal X-ray diffraction.120
Importantly we were also able to extend this concept to enantioselective C(sp3)–H activation using pyridyl substrates containing gem-dimethyl groups (Scheme 9). In this case, one of the rigid cyclopropane amino acid ligands from above, 136, was found to be optimal. Although, the alkylated product was only obtained in modest yield and ee, it served as crucial proof of concept that through future advances in ligand design, synthetically useful levels of stereoinduction could be achieved in enantioselective C(sp3)–H activation.
Scheme 9.
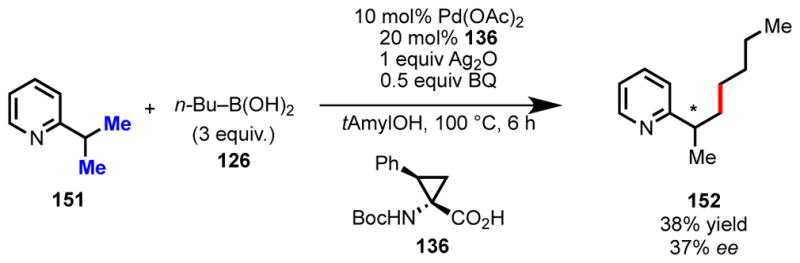
Catalytic enantioselective C(sp3)–H activation of 151.116
These experiments led us to elucidate how the chiral mono-N-protected amino acid ligand coordinates with the Pd(II) center and to propose a stereomodel as a basis for further development of amino acid–based ligands to control C–H activation (Scheme 10).116,120 Through NMR studies and inorganic synthesis of putative coordinating structures, we believe that the mono-N-protected amino acid ligand coordinates to Pd(II) with high affinity via a bidentate binding mode (153). Upon complexation, the nitrogen atom becomes stereogenic, and the sterically encumbered protecting group is forced trans to the bulky alkyl substituent through a “gearing” effect. The pyridine of the substrate coordinates trans to the carbamoyl moiety of the ligand, situating the substrate’s ortho-C–H bonds in close proximity to the acetate group. Computational studies performed in collaboration with the Musaev group suggest that at this stage, activation of the N–H bond by internal acetate takes place, opening a coordination site at the Pd(II) center.120 Concerted metalation/deprotonation via external base delivers the palladacycle with concomitant formation of a new stereocenter at the methine carbon atom. Consistent with our experimental results, pre-transition state coordination structure 155 and transition state 156, which lead to 157, containing a newly formed (R)-stereocenter, are computed to be lower in energy that 159 and 160, which lead to 161, containing a newly formed (S) stereocenter. The precise steric and electronic effects of the amino acid ligand and its influence on the relative energies of key intermediates 154, 155, 158, and 159 and transition states 156 and 160 are topics of continuing investigation. After enantioselective C–H activation, palladacycle 157 then reacts with the boronic acid via a transmetalation/reductive elimination sequence, releasing of Pd(0), which is reoxidized by Ag(I) and reenters the catalytic cycle (Scheme 7).
Scheme 10.
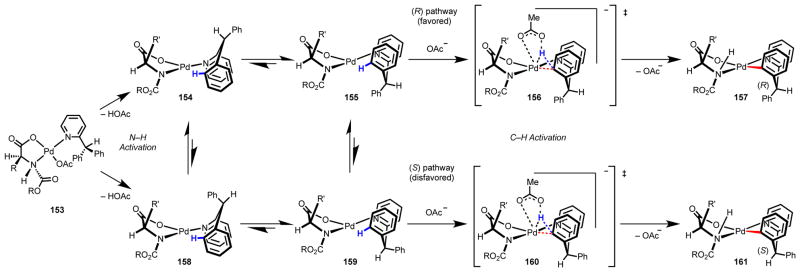
Working stereomodel, as supported by computational studies.116,120–122
Our next objectives were (1) to broaden this chemistry to substrate- and product classes more relevant to organic synthesis and (2) to test the viability of other catalytic manifolds for C–C and C–heteroatom bond formation. Drawing on our extensive experience using carboxylic acid–containing substrates (see Scheme 4a),77 we questioned whether we could employ mono-N-protected amino acid ligands to induce enantioselectivity in a Pd(II)-catalyzed aerobic C–H olefination reaction of phenylacetic acids that we had recently discovered in our laboratory.123 Consistent with our philosophy for reaction development, we envisioned at the outset that the carboxylate directing group82,86,124–127 would be versatile for further functionalization, and in this way, we would have a unique method of constructing enantioenriched chiral building blocks containing densely substituted stereocenters. Gratifyingly, exposing sodium 2,2-diphenylpropanoate (162) to our parent reaction conditions in the presence of Boc-Ile-OH (164) led to the product in high yield and with high ee (Scheme 11).123 Interestingly, the combination of the pre-formed sodium salt and KHCO3 as the base were critical; alternative combinations led to lower yield and/or ee.
Scheme 11.
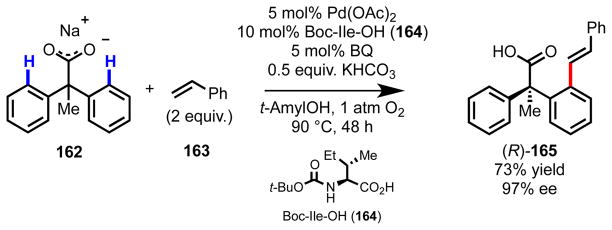
Enantioselective C(sp2)–H olefination of diphenylacetic acid derivative 162.121
We were able to confirm the absolute configuration of one product through single crystal X-ray diffraction and assign the other products by analogy, including (R)-165. The observation that this set of products has the same absolute configuration as (R)-127 in Table 3 is consistent with the notion that the two mechanisms of stereoinduction are analogous, with the sodium carboxylate acting like the 2-pyridyl group as an L-type neutral donor during the enantioselective cyclometalation step. Importantly, this work also established the compatibility of the amino acid ligand scaffold with other elementary steps in catalysis: namely, olefin association, 1,2-migratory insertion, and β-hydride elimination (Scheme 12).
Scheme 12.

General catalytic cycle for C–H olefination via Pd(II)/Pd(0) catalysis.
More recently, we targeted another important and underutilized class of starting materials, cyclopropanecarboxylic acids. Encouraged by the high levels of diastereoselectivity that we previously observed in cyclopropane C–H iodination using our chiral oxazoline auxiliary112 and by our preliminary results using amino acid ligands for enantioselective C(sp3)–H activation of gem-dimethyl groups (Scheme 9),116 we reasoned that we could develop a catalytic enantioselective method for cyclopropane C–H functionalization.
We viewed this as a potentially powerful and direct means of synthesizing enantiopure cis chiral cyclopropanes, which are challenging to access using other methods. Due to the partial π-character of the C–C bonding system in cyclopropanes (Walsh orbital analysis), the cleavage of cyclopropyl C–H bonds is known to be more facile than that of completely unactivated C(sp3)–H bonds, and one would expect the pre-transition state coordination structure to be rigid, conceivably allowing C–H activation to take place under mild conditions. To facilitate C(sp3)–H cleavage we installed an acidic N-aryl amide directing group (see Scheme 4b),128,129 enabling Pd(II)-catalyzed C–H/R–BX2 cross-coupling at temperatures as low as 40 °C.130 Following optimization of a non-stereoselective version of the reaction, we systematically examined a structurally diverse array of amino acid ligands for stereoinduction (Table 4). In our early screening studies, we found that of a wide range of protecting groups examined, the TcBoc group was most successful, likely due to the large steric bulk and moderate electron-withdrawing character of the Me2(Cl3C)C– moiety. Of the backbones tested, those that had aromatic rings like phenylalanine, tyrosine, and tryptophan were superior. Within the TcBoc-protected series, TcBoc-Phe-OH (173) proved to be optimal (Entry 6). Increasing the steric demands of the alkyl substituents on the TcBoc group and tuning of the electronic properties on the arene led to the identification of ligand 179 (Entry 12). Though the yield was low, this problem could be remedied by adding the reagents consecutively in two batches, giving up to 81% yield, 91% ee for the model reaction of 166 to 168. This reaction was found to be compatible with aryl-, vinyl-, and alkylboron reagents and could tolerate a number of different substituents at the α-position.
Table 4.
Catalytic enantioselective C(sp3)–H activation of cyclopropanecarboxylic acid derivative 166 with mono-N-protected amino acid ligands.116

| |||
|---|---|---|---|
| Entry | Ligand | Yield (%) a | ee (%) b |
| 1 |
 TcBoc-Ala-OH, 169 |
55 | 80 |
| 2 |
 TcBoc-Val-OH, 170 |
42 | 75 |
| 3 |
 TcBoc-Leu-OH, 148 |
47 | 78 |
| 4 |
 Tc-Boc-PhG-OH, 171 |
47 | 55 |
| 5 |
 TcBoc-MePhe-OH, 172 |
31 | 33 |
| 6 |
 TcBoc-Phe-OH, 173 |
47 | 85 |
| 7 |
 174 |
51 | 88 |
| 8 |
 175 |
44 | 91 |
| 9 |
 176 |
50 | 90 |
| 10 |
 177 |
48 | 90 |
| 11 |
 178 |
40 | 88 |
| 12 |
 179 |
47 | 93 |
Yield determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard.
ee determined by HPLC using a chiral stationary phase.
3.2.3. Position-Selective C–H Activation
In light of mounting evidence that the mono-N-protected amino acid ligands were intimately involved in the C–H cleavage step of the reactions described above, we then examined the use of these ligands for a conceptually related challenge, controlling the positional selectivity in the C–H functionalization of substituted arenes.
While exploring the substrate scope of non-stereoselective Pd(II)-catalyzed ortho-C–H olefination of phenylacetic acids123 we became interested in the functionalization of 2-(3-methoxy-5-methylphenyl)acetic acid 180 because of its potential as a synthon in natural products synthesis. When we attempted the reaction under our parent reaction conditions, we observed an intractable 1.4:1 mixture of the two possible products, 182-A and 182-B in 68% conversion. Upon surveying a collection of ligands, we ultimately found that the application of For-Ile-OH (189) furnished 182-A and 183-B in a 20:1 ratio without a substantial decrease in conversion (45%) (Entry 12). Simply increasing the catalyst loading to 7% lifted the conversion to 75% with identical levels of selectivity.
Though a precise mechanistic rationalization of this change in selectivity remains to be determined, one plausible explanation is that the ligand causes a change in the mechanism of C–H cleavage. In the absence of ligand, electron-rich and electron-neutral phenylacetic acids are highly reactive, while substrates with electron-withdrawing groups are unreactive, a pattern consistent with an electrophilic palladation mechanism for C–H cleavage.123 In the presence of certain amino acid ligands, electron-deficient phenylacetic acids are preferentially reactive, which is more consistent with a CMD mechanism (vide infra).76 It is possible that the drastic change in product distribution is a reflection of the difference in relative rates under the two mechanistic regimes. An alternative explanation is that the ligand could merely be enhancing the steric demands around the metal center, favoring reaction away from the bulkier methyl group.131,132
3.2.4. Ligand-Accelerated C–H Activation
The ability of mono-N-protected amino acids to control enantioselectivity and positional selectivity serves as evidence that the ligand is coordinated to Pd(II) during the C–H cleavage event and is influencing the corresponding transition state energies. Accordingly, we wondered whether these ligands could promote C–H activation for otherwise unreactive substrates.76,123,133 In our C–H olefination of phenylacetic acids,123 we had found that electron-poor substrates gave low yields even after extended reaction times (up to 48 h). For instance, 2-(trifluoromethyl)phenylacetic acid 190 was found to give <15% yield of 191 after 48 h under our standard reaction conditions. We began investigating the effect of mono-N-protected amino acids (Table 6), and in order to measure the kinetic reactivity of the various ligands, we assayed the conversion at 20 min and 2 h. Of the Boc-protected amino acids tested (Entries 2–6), valine was found to be the best backbone (Entry 6), giving 46% conversion after 20 min and nearly quantitative conversion after 2 h. Optimizing the structure of the protecting group revealed that acetyl was the most reactive (Entries 7–10). We then re-optimized the backbone structure using acetyl as the protecting group, leading to the discovery that Ac-Ile-OH (188) gave the highest kinetic reactivity for this transformation (Entry 12).
Table 6.
Ligand-accelerated C–H olefination of 2-(trifluoromethyl)phenylacetic acid 190 using mono-N-protected amino ligands.76

| |||
|---|---|---|---|
| Entry | Ligand | 20 min | 2 h |
| 1 | --- | <5c | 7c |
| 2 |
 Boc-Ala-OH, 143 |
28 | 88 |
| 3 |
 Boc-tert-Leu-OH, 140 |
38 | 98 |
| 4 |
 Boc-lle-OH, 164 |
37 | 98 |
| 5 |
 Boc-Leu-OH, 146 |
37 | 92 |
| 6 |
 Boc-Val-OH, 141 |
46c | 98c (96) |
| 7 |
 Me2OC-Val-OH, 192 |
21 | n.d.d |
| 8 |
 Piv-Val-OH, 193 |
2 | n.d.d |
| 9 |
 For-Val-OH, 194 |
31 | n.d.d |
| 10 |
 Ac-Val-OH, 195 |
57 | >99 |
| 11 |
 Ac-Leu-OH, 196 |
55 | >99 |
| 12 |
 Ac-lle-OH, 188 |
72 c | >99 (96) |
The conversion was determined by 1H NMR of the crude reaction mixture.
Isolated yield shown in parentheses.
Average of three trials.
n.d. = not determined.
With this new ligand-accelerated catalytic system, the substrate scope could be dramatically expanded to include electron-poor phenylacetic acids bearing CF3 or NO2 substituents, and it gave improved activity with electron-rich and -neutral substrates.76,133 By employing amino acid ligands, hydrocinnamic acids, which contain a more distal carboxylate directing group (via a seven-membered palladacycle) were also found to be reactive in C–H olefination for the first time (Scheme 13).123,133 Using Ac-Ile-OH (188) as the ligand, exceptionally high catalytic activity was observed with phenylacetic acid substrates. With 190, for example, catalyst loadings as low as 0.2 mol% could be used to give >90% conversion (>450 TON), which is among the most efficient aerobic C–H olefination reactions reported to date.134,135 Due to the stability of the catalyst, the reaction can also be run with air (1 atm), rather than O2 (1 atm), as the sole terminal oxidant, albeit with extended reaction times (87% conv. of 190 to 191 after 48 h with 1 mol% catalyst). This transformation can readily be applied in practical settings, for instance in the direct functionalization of NSAIDs (e.g., ibuprofen, ketoprofen, and naproxen). Moreover, it has been used as the key convergent step in the synthesis of (+)-lithospermic acid (see Scheme 3b for retrosynthesis).81
Scheme 13.
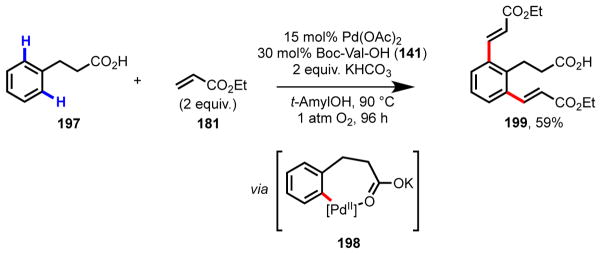
Ligand-promoted diolefination of hydrocinnamic acid (197). The mono-olefinated product (35% conv.) was also observed by 1H NMR, but was not isolated.133
Mechanistic studies performed in our laboratory76 and in collaboration with the Blackmond group136 suggest that the observed rate enhancement in the presence of amino acid ligand is due to acceleration of the C–H cleavage step, possibly due to a change in the mechanism of C–H cleavage from electrophilic palladation (in the absence of ligand) to a CMD pathway (with ligand), as described previously.120 Through kinetic studies, off-cycle catalyst reservoirs from olefin- and product binding to the active Pd(II) catalyst have been identified, which decrease the reaction rate but may protect against catalyst deactivation.136
In recent work, we were able to transpose the ligand-enhanced reactivity of phenylacetic acids to the first example of ligand-accelerated Pd(II)-catalyzed C(sp2)–H/Ar–BXn cross-coupling. By using mono-N-protected amino acid ligands in conjunction with Ag2CO3 as a stoichiometric reoxidant, we developed a new protocol for the functionalization of phenylacetic acid substrates that offers shorter reaction times, improved substrate scope, and higher yields compared to our previous method.13 Importantly, this work established that mono-N-protected amino acids are compatible with the transmetalation, reductive elimination, and reoxidation steps in ligand-accelerated C–H/R–M cross-coupling.
In addition to carboxylic acid substrates, we have found that other key classes of widely available, synthetically useful starting materials can undergo Pd(II)-mediated C–H functionalization promoted by amino acid ligands. This catalytic system has proven to be instrumental for substrates containing weakly coordinating functional groups, where in the absence of a highly reactive catalyst, cyclopalladation is sluggish. Contemporaneous to our initial studies in ligand-accelerated catalysis with phenylacetic acids, we also began exploring the effects of amino acid ligands in alcohol-directed reactions.137–139 We viewed aryl- and alkylalcohols as ideal starting materials in C–H functionalization because of their abundance and prevalence in desirable product motifs.140–143 With the exception of examples using phenol-type directing groups,144,145 hydroxy-directed C–H functionalization using Pd catalysts had not been reported prior to our work. Through careful optimization of the reaction conditions, we were able to achieve ortho-C–H olefination137 and carbonylation139 of phenethyl alcohols (Scheme 14). The use of (+)-Men-Leu-OH as a supporting ligand was critical for obtaining synthetically useful yields; in both the transformations depicted in Scheme 14, <40% yield was observed in the absence of ligand. For both reactions, selection of a non-coordinating solvent was key due to the weak coordinative affinity of –OH for Pd(II) centers in comparison to traditional directing groups. As part of a collaboration with Pfizer, we applied amino acid ligands to achieve high reactivity in the ortho-C–H olefination directed by sulfonamides,146 a privileged pharmocophore in medicinal chemistry (Scheme 15a).147 With this result as the entry point, we developed a collection of categorically distinct, divergent C–H functionalization reactions for synthesizing analog libraries of drugs like Celebrex. With ether directing groups,148 another class of prevalent, weakly coordinating functionality, we were able to develop an ortho-C–H olefination reactions in the presence of Ac-Gly-OH and hexafluoro-2-propanol (HFIP) (Scheme 15b).149
Scheme 14.

Ligand-promoted ortho-C–H functionalization of phenethyl alcohols: (a) olefination137 and (b) carbonylation.139 (c) General catalytic cycle C–H carbonylation via Pd(II)/Pd(0) catlaysis; Nu = generic nucleophile.
Scheme 15.
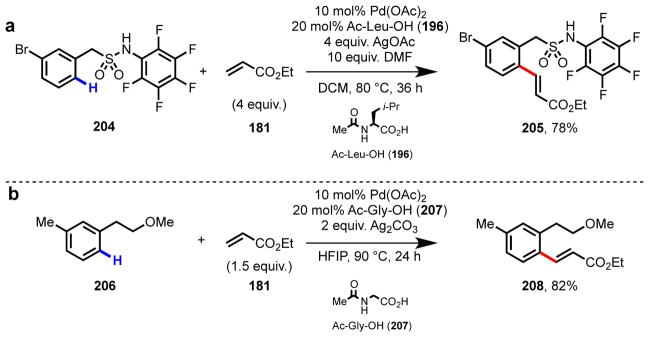
Ligand-promoted ortho-C–H olefination of (a) benzylsulfonamides (204)147 and (b) phenethylethers (206).149
Finally, we again turned to amino acid ligands during our efforts to develop a meta-selective Pd(II)-catalyzed C–H olefination reaction of hydrocinnamic acid derivatives (Scheme 16).150 Our reaction design centered on the use of an end-on template,125,126 which was inspired by gas-phase studies of Schwarz in which metal centers were observed to adopt a linear coordination geometry with nitriles, enabling activation of remote C–H bonds.151,152 To reduce this idea to practice, a simple nitrile-containing amide auxiliary was installed on a hydrocinnamic acid. End-on coordination of Pd(II) to the nitrile through the nitrogen atom situates the catalyst directly over the desired C–H bond. In this system the combination of Ac-Gly-OH (207) and HFIP were found to be critical for high activity in C–H activation.
Scheme 16.
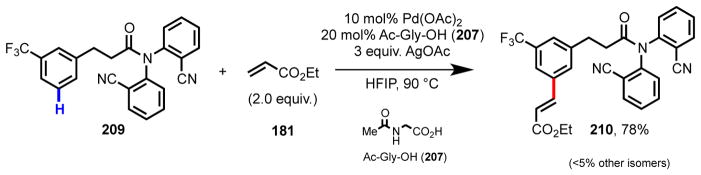
Ligand-promoted meta-C–H olefination of hydrocinnamic acid derivative 209 using an end-on template approach.150
3.2.5. Outlook
This collection of results, which we have gathered during the past five years, demonstrates that mono-N-protected amino acid ligands effect enantioselectivity, positional selectivity, and rate enhancement in Pd(II)-catalyzed C–H functionalization. It establishes that amino acid ligands are bound to the Pd(II) center during C–H cleavage and are capable of controlling the mechanism (and thus activation energy) of C–H cleavage. Amino acid ligands have been proven to control reactivity with many different substrate classes containing diverse directing groups, and they are compatible with a variety of elementary steps found in the catalytic cycles of Pd(II)-catalyzed C–H/R–BXn cross-coupling, C–H olefination, and C–H carbonylation.
An advantage of mono-N-protected amino acid ligands for directed Pd(II)-catalyzed C–H functionalization is the non-symmetrical N,O-mixed donor system, which desymmetrizes the remaining two sites on the square planar Pd(II) center, such that the directing group can favorably bind at one, and the C–H bond react at the other. Another benefit is their practical convenience; many are commercially available and others can easily be prepared through modular synthesis. They are air- and moisture-stable and simple to employ in screening libraries. Furthermore, the mono-N-protected amino acid ligand scaffold offers several sites for tuning, which was pivotal for us in adapting the steric and electronic properties of the ligand to the unique demands of each of the transformations depicted above. Indeed, much to our delight, other groups have already found success employing them to develop new Pd(II)-catalyzed C–H activation reactions.153–159 We further anticipate the use of amino acid ligands for metal catalysis will be applied to develop hybrid transition metal/enzyme catalysts for C–H activation.
3.3 2,6-Disubstituted Pyridine Ligands
3.3.1 Background
Simple, monofunctionalized arenes constitute a class of abundant chemicals with rich synthetic potential. An attractive approach to map these substrates onto more complex multi-substituted arene building blocks would be position-selective C–H functionalization to form new C–C bonds. Since the early work of Fujiwara and Moritani in the late 1960s (Scheme 17),160,161 C–H olefination of simple arenes has been extensively studied.134,135,162–168 Despite these efforts, several problems have persisted: (a) the need for high molar excess of arene substrates, which is often used as solvent (b) limited positional selectivity, (c) the need for high catalyst loadings and stoichiometric metal reoxidants, and (d) low reactivity with electron-poor substrates.169,170 Through extensive efforts during the past several years, methods for position-selective C–H olefination of electron-rich heterocycles171–173 and substrates containing ortho-directing groups125,174 have been established (Scheme 18). To realize the full synthetic utility of this approach, however, new strategies to promote reactivity and control selectivity are needed.
Scheme 17.
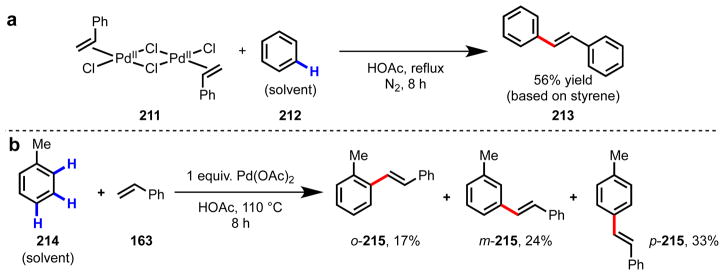
(a) Initial discovery of Pd(II)-mediated C–H olefination using stoichiometric complex 211 by Fujiwara and Moritani in 1967.160 (b) Distribution of positional isomers (ortho-, meta- and para215) in the stoichiometric Pd(II)-mediated C–H olefination of toluene 214.170
Scheme 18.
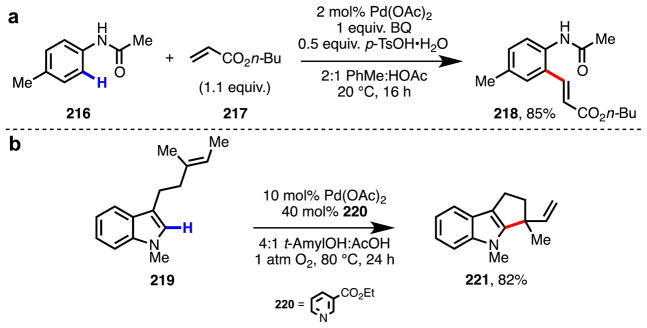
Representative examples of tactics for controlling positional selectivity in C–H olefination: (a) intermolecular ortho-C–H olefination of N-(p-tolyl)acetamide (216) by de Vries and van Leeuwen174 (b) intramolecular C–H olefination of N-methyl indole substrate 219 by Stoltz.173
We sought to address these problems through the design of an ancillary ligand that could accomplish the following goals: (1) enhance reactivity with electron-poor arenes, ideally using only 1 equiv of substrate, (2) favor selectivity at the meta position to offer complementary selectivity to directed methods, and (3) promote reoxidation with molecular O2134,135 at ambient pressure.
In order to realize these goals, there were several obstacles that warranted consideration at the outset. The first potential problem is that the arene π-systems of electron-deficient arenes are comparatively poor σ-donors, meaning that they may be unable to compete with the majority of L-type ligands for binding to Pd(II). An ideal ligand would be capable of tightly binding to Pd(II), yet would still allow the substrate to bind, even in low concentration. The second challenge, as alluded to above, is that it is generally believed that under standard reaction conditions, Pd(II)-mediated C–H olefination proceeds through an electrophilic palladation mechanism for C–H cleavage, in a similar manner to that of a Friedel–Crafts reactions. To achieve high reactivity with electron-poor arenes, the supporting ligand would need to increase greatly the electrophilicity of the Pd(II) catalyst or bias the C–H cleavage step towards a CMD mechanism. Third, to achieve meta selectivity, the catalyst would need to be able to recognize minor differences in the steric and electronic environments of the different C–H bonds, promoting selective C–H cleavage according to these subtle cues. Lastly, the ligand would need to be compatible with (and ideally would promote) the reoxidation step of Pd(0) to Pd(II) with O2.
3.3.2. meta-Selective C–H Activation of Electron-Deficient Arenes
To begin our investigation, we first attempted to develop conditions to effect Pd(II)-catalyzed C–H olefination of 1,3-bis(trifluoromethyl)benzene (222), a highly electron-deficient arene.175 Due to steric effects, we presumed that this substrate would give a single product in C–H olefination, which would allow us to focus on reactivity during optimization. Under standard aerobic Fujiwara–Moritani conditions, using Pd(OAc)2 as the catalyst and 222 as the solvent, we found the reaction gave only trace quantities of the desired product (Entry 1, Table 7).
Table 7.
Ligand-promoted C–H olefination of 1,3-bis(trifluoromethyl)benzene (222).175

| |||||
|---|---|---|---|---|---|
| Entry | Ligand | Yield (%)a | Entry | Ligand | Yield (%)b |
| 1 | --- | <2 | 6 |
 220 |
<5 |
| 2 |
 py, 46 |
<5 | 7 |
227 |
10 |
| 3 |
 47 |
<2 | 8 |
228 |
24 (E:Z = 7.8:1)b |
| 4 |
 225 |
<5 | 9 |
229 |
52 (E:Z = 5.9:1)b |
| 5 |
 226 |
<2 | |||
The yield was determined by 1H NMR of the crude reaction mixture using CH2Br2 as an internal standard.
The E:Z ratio was determined by 1H NMR of the crude reaction mixture.
To overcome the low reactivity and lack of catalytic turnover, we first turned to pyridine-based ligands, which have long been known to be beneficial in oxidative Pd(II)-catalyzed transformations, especially those under aerobic conditions.176–183 Seminal work by Ferreira and Stoltz established that an electron-deficient pyridine ligand, ethyl nicotinate, greatly improved the yield in an intramolecular C–H olefination reaction of an electron-rich nitrogen heterocycles under aerobic conditions (Scheme 18b).173 Other groups have also successfully employed pyridine ligands in C–H functionalization reactions along a Pd(II)/Pd(0) catalytic cycle.168,182,184–188 In our study, we first tested pyridine (py, 46) and 2,6-lutidine (47), but these ligands gave low yields (Entries 2 and 3). At this stage, we speculated that the pyridine ligands were bis-ligated to the Pd(II) center (e.g., Pd(II)(py)2OAc)2 and that displacement of a ligand with the electron-poor arene 222 was unfavorable, owing to the strength of the Pd(II)–N bond. We hypothesized that the Pd(II)–N bond could be destabilized by introducing steric bulk at the 2- and 6-positions of the pyridine ring or by substituting the pyridine ring with an electron-withdrawing group; however, 2,6-di-iso-propylpyridine (225), 2,6-di-tert-butylpyridine (226), and ethyl nicotinate (220) all gave low yields (Entries 5–7). With these ligands, formation of the corresponding Wacker olefin oxidation product and accompanying precipitation of palladium black (Pd(0)n) suggested that ligand binding was too weak.
We reasoned that what was needed was a pyridine-based ligand that would be strongly binding yet preferentially mono-ligating. Our breakthrough came in recognizing that we could achieve this combination of properties by minimizing steric bulk at the carbon atoms immediately adjacent to the nitrogen atom and introducing steric bulk at a more distal position. In this manner, the one molecule of ligand would be free to bind Pd(II), but coordination of a second ligand molecule trans to the first would be unfavorable because of steric clashing away from the metal center. In other words, the ligands would experience “mutual repulsion” when both bound to the Pd(II) center. To reduce this idea to practice, we prepared and tested ligands 227, 228, and 229, and found, much to our delight, that the yield could be improved to 10–52% (Entries 7–9). Longer and more branched side chains led to improved yield, with 229 proving to be optimal.
The substrate scope with electron-deficient arenes proved to be quite broad (selected examples shown in Table 8). The reaction was compatible with a variety of electron-withdrawing groups; trifluoromethyl and nitro substituents were well tolerated, as were esters and ketones. 1,3-Disubstituted arenes bearing two electron-withdrawing groups, reacted selectively at the 5 position (Entry 1). 1,4-Disubstituted arenes with one electron-releasing group and one electron-withdrawing group reacted meta- to the electron-withdrawing group (Entry 2). Monosubstituted electron-poor arenes reacted at the meta- and para- position with an m:p ratio typically around 4:1 (Entries 4–8). A variety of olefin coupling partners could be used, including those with substitution as the β-position.
Table 8.
Substrate scope of ligand-promoted C–H olefination of electron-poor arenes (selected examples).175
| Entrya | Product | Yield (%) b | m:p c | Time (h) |
|---|---|---|---|---|
| 1d |
 230 |
62 | --- | 36 |
| 2 |
 231 |
68 | --- | 24 |
| 3 |
 232 |
77 | --- | 24 |
| 4d |
 m/p-233 |
70 | 4.0:1 | 2 |
| 5 |
 m/p-234 |
72 | 3.5:1 | 20 |
| 6 |
 m/p-234 |
71 | 4.9:1 | 5 |
| 7 |
 m/p-236 |
80 | 3.5:1 | 56 |
| 8 |
 m/p-237 |
71 | 3.3:1 | 36 |
Unless otherwise specified the reaction conditions were as follows: alkene (0.6 mmol), arene (2 mL, 20–30 equiv), Pd(OAc)2 (10 mol%), 229 (20 mol%), Ac2O (1.0 equiv), O2 (1 atm), 90 °C.
Isolated yield.
The m:p ratio was determined by GC. Reference samples of the pure meta- and para-isomers were prepared independently via 0Mizoroki–Heck chemistry.
Ac2O (1.5 equiv).
As mentioned above, one limitation of traditional non-directed Fujiwara–Moritani reactions is the need to use the substrate as the solvent. This requirement is highly undesirable, and inherently limits the substrate scope to simple, low-melting arene starting materials. The high levels of reactivity promoted by 229 allowed us to reduce the arene substrate loading to 5 equiv relative to olefin using EtOAc as the solvent (see Scheme 19 for an example). These findings suggest that with future advances in ligand design, a catalytic system could be devised such that 1 equiv of arene starting material and exclusive selectivity is obtained for functionalization of a single C–H bond.
Scheme 19.
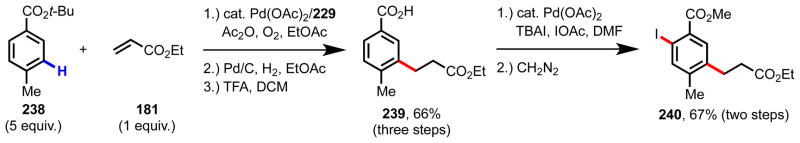
Sequential C–H functionalization route to tetra-substituted arene 240.175
In an effort to showcase the synthetic utility of this meta-selective transformation, we attempted to use it as the first step in a sequential C–H functionalization route133 to build up molecular complexity from tert-butyl 4-methylbenzoate 238, a commercially available benzoic acid derivative (Scheme 19). Following ligand-promoted C–H olefination, Pd/C-mediated hydrogenation, and tert-butyl ester deprotection, free acid 239 was obtained in 66% yield over the three steps. Next, we found that 239 could undergo directed ortho-C–H iodination82 to give differentially functionalized 1,2,4,5-tetrasubstituted arene 240 in 67% yield following conversion to the methyl ester to simplify purification.
To understand the mechanism of this reaction and the role of the 2,6-dialkyl pyridine ligand 229, we studied relevant Pd(II) intermediates by 1H NMR and X-ray crystallography (Scheme 20). By stirring 2 equiv ligand 229 and Pd(OAc)2 in hexanes, we were able to prepare Pd(229)2(OAc)2 complex 241, which we characterized by single-crystal X-ray diffraction. The structure shows that the two ligand molecules 229 are approximately co-planar and coordinated trans to one another. Consistent with our mutual repulsion hypothesis, the two sets of branching arms lie on opposite sides of the Pd–N–C2 plane (as expressed in the relative stereochemistry drawn for 241), and the Pd–N bond length is 0.05 Å longer than the Pd–N bond in Pd(Py)2(OAc)2•H2O.189 When we dissolved complex 241 in CDCl3 and monitored the solution by 1H NMR at 2 h and 12 h, we observed formation of a new species consistent with 243 along with free ligand 229. In contrast, when we performed the same experiments with the pyridine and 2,6-lutidine complexes, Pd(py)2(OAc)2 and Pd(47)(OAc)2, we observed that they were stable in CDCl3 for several days.
Scheme 20.
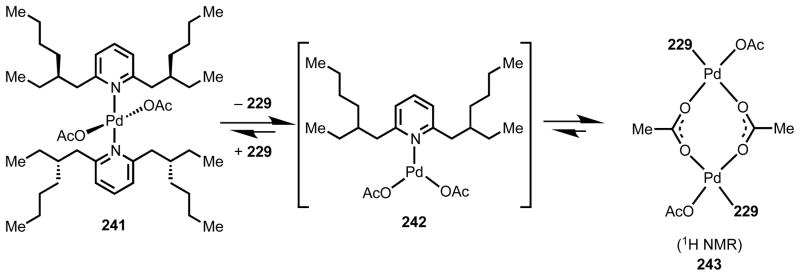
Equilibrium species observed based on structural and spectroscopic studies.
On the basis of these data, we propose that the exceptional reactivity observed with ligand 229 is due to its mutually repulsive properties. It is energetically unfavorable for two molecules of 229 to be bound at Pd(II) simultaneously, which creates an opportunity for the substrate π-system to coordinate to the metal. The high reactivity with electron-poor substrates is consistent with a CMD mechanism for the C–H cleavage step. Furthermore, in line with the precedents mentioned above, the pyridine-type 229 ligand could be effective in part because it stabilizes reduced Pd(0) species and facilitates reoxidation with O2. In terms of selectivity, the observed product distribution remains somewhat unclear. The lack of reactivity at the ortho position with monosubstituted arenes can be attributed to the steric bulk of the catalyst, which would force substituents on the arene away upon association to the catalyst. The precise origin of meta-selectivity over para-selectivity is possibly due to subtle differences in electronic density or C–H bond acidity between the two positions. Differences between the thermodynamic stability of the developing Pd(II)–C bonds formed via C–H cleavage at the different positions could also contribute to the observed selectivity patterns.
Computational studies performed by Zhang and coworkers found that a CMD mechanism was favorable for the C–H cleavage step in this system and supported the notion that steric interactions between the pyridine ligand and the substrate are dominant in influencing the positional selectivity, with electronic effects playing a minor effect.190 Their model also suggests that the ligand dissociates from the metal upon olefin binding, a step that would also be facilitated by a ligand with weaker Pd–N bond strength.
We have recently gone on to apply the mutually repulsive ligand design concept to address a different problem, methylene C(sp3)–H functionalization of aliphatic acid derivatives (Scheme 21a).191 Our goal was to expand the diverse reactivity that we have developed using our weakly coordinating N-aryl amide auxiliary to methylene C(sp3)–H functionalization. However, methylene C(sp3)–H cleavage is generally sluggish with Pd(II) catalysts in the absence of a strongly coordinating (typically bidentate directing group),192,193 Drawing on the lessons from above, we discovered an electron-rich, mutually repulsive 2-alkyoxy quinolone ligand 246, which dramatically promoted methylene C(sp3)–H arylation of 244 along a Pd(II)/Pd(IV) catalytic cycle (Scheme 21b).
Scheme 21.

(a) Ligand-promoted methylene C(sp3)–H arylation of butanoic acid derivative 244.191 (b) General catalytic cycle for C–H arylation with aryl iodides via Pd(II)/Pd(IV) catalysis.
3.3.3. Outlook
Generally speaking, mutually repulsive 2,6-disubstituted pyridines represent a ligand scaffold that is carefully tailored for the steric and electronic demands of electron-poor arene substrates that do not contain directing groups. This class of substrates poses unique challenges in Pd(II)-mediated C–H cleavage that informed our ligand design criteria. A benefit of the ligand scaffold is that it has multiple tuning sites. In our case, we adjusted the steric properties to minimize steric crowding near nitrogen and maximizing steric clashing away from nitrogen. The ability to tune both adjacent to and remote from the binding atom were critical. In addition, modular synthesis allowed each of the ligand candidates to be synthesized in a single step, which was vital in the reaction discovery stage. The compatibility of this ligand scaffold with other elementary steps in other catalytic manifolds remains to be examined.
3.4. 2,2′-Bipyridine Ligands
3.4.1. Background
Ubiquitous in natural products, pharmaceutical agents, agrochemicals, organic materials, and ligands for coordination chemistry, pyridines (and related azines) represent one of the most common heterocyclic motifs. Owing to the potential synthetic utility of functionalized pyridine derivatives in many different realms of chemistry, metal-catalyzed position-selective pyridine C–H functionalization has been the topic of considerable interest in the past few years.194 However, owing to its tendency to adopt an N-bound coordination mode and its electron-poor nature, pyridine has proven to be a challenging substrate to use in these transformations.
Nonetheless, with metals other than Pd(II), some success has been achieved in position-selective C–H functionalization of pyridines.194 Representative strategies include the use of low-valent metals (e.g., Ru(0) and Rh(I)) for C2 functionalization facilitated by N-chelation,195–199 C3/C4-selective borylation using Ir(III) species,200–206 Ni(0)/Lewis acid catalysis for functionalization of the C2 or C4 positions,207–209 and utilization of ortho-directing groups on the pyridine ring with Ru(0) or Pd(0) catalysts.210–212
We were intrigued with the idea of developing position-selective pyridine C–H functionalization methodology using Pd(II) catalysts due to the potential versatility in the functionalization step. In terms of precedents, previously published approaches relied upon minimizing or eliminating the chelation strength of the nitrogen atom through substitution of the pyridine ring. For example, by protecting the nitrogen atom as an N-oxide213–217 or N-iminopyridinium ylide,218,219 C2 selectivity has been achieved with Pd(II), Pd(0), and Cu(I) catalysts (Scheme 22a). Additionally, the introduction of steric blocking groups and/or electron-withdrawing groups has been demonstrated to suppress N-chelation; for instance, C4-selective C–H functionalization of 2,3,5,6-tetrafluoropyridine has been achieved using a Pd(II) catalyst (Scheme 22b).220,221
Scheme 22.
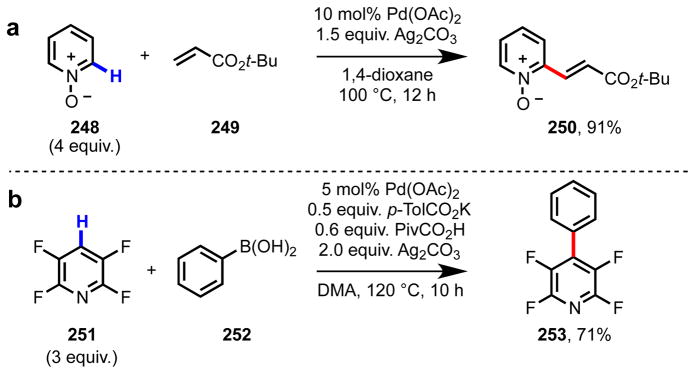
Literature precedents for pyridine C–H functionalization with Pd(II) catalysts: (a) C2-selective C–H olefination of pyridine N-oxide (248);214 (b) C4-selective C–H/R–B(OH)2 cross-coupling of 2,3,5,6-tetrafluoropyridine (251).220
Based on our success in obtaining reactivity with electron-deficient arenes, we hoped that we could draw on the lessons learned about the interplay between substrate and ligand at the Pd(II) center to design a ligand scaffold that would accomplish the following: (1) promote pyridine C–H bond cleavage, (2) functionalize the C3 position to offer complementary selectivity to other methodologies, and (3) maintain compatibility with a variety of coupling partners when applied to categorically distinct transformations.
In order to achieve these goals through ligand design, several factors were considered. The first major problem is that pyridine is a strongly binding ligand, tending to adopt an N-bound coordination mode with the catalyst, which situates the metal center distal from the target C–H bonds (Scheme 23). Pyridine substrate molecules could potentially outcompete the ancillary ligand for binding to the metal center. Second, the pyridine ring is electron-poor, which makes initial coordination of the π-system unfavorable and renders C–H cleavage via an electrophilic palladation mechanism untenable (similar to the situation described with electron-poor arenes in Section 3.2). Thus, the ligand, in addition to disfavoring the N-bound coordination mode, would need to promote C–H cleavage along a CMD pathway. Thirdly, achieving C3 selectivity would require that the catalyst be sensitive to the steric or electronic properties of the three different positions and would need to be resistant to N-coordination-assisted C2 functionalization. Fourthly, to accommodate many catalytic transformations along Pd(II)/Pd(0) and Pd(II)/Pd(IV) redox couples, the ligand must be compatible with a range of different elementary steps. For example in C–H arylation along a Pd(II)/Pd(IV) catalytic cycle, it should stabilize high-energy Pd(IV) intermediates and be recalcitrant to reductive elimination.
Scheme 23.
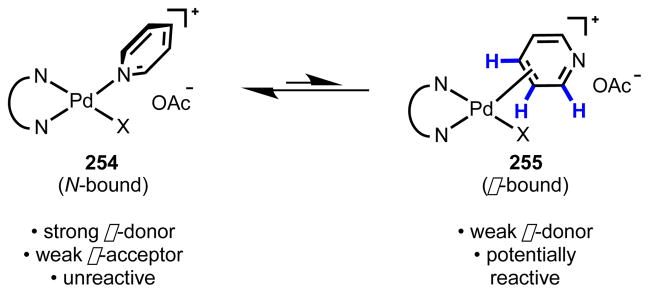
Postulated coordination equilibrium of pyridine with Pd(II): unproductive N-bound (left) and productive π-bound (right).
3.4.2. C3-Selective Pyridine C–H Functionalization
At the outset, we hypothesized that a strongly chelating bidentate ligand would be required to compete effectively for binding at the Pd(II) center and to disfavor N-bound coordination of the substrate. In particular, we surmised that N,N-bidentate ligands might prove to be effective in these pursuits. Bidentate N(sp2),N(sp2)-ligands, such as 2,2′-bipyridines, 2,2′-bis(2-oxazoline)s, 2-(2′-pyridyl)oxazoline, and 2-(2-quinolinyl)oxazolines, have a rich history in oxidative transformations with Pd(II), both in asymmetric and non-stereoselective reactions.42,181,222–229 In the context of Pd(II)-catalyzed C–H functionalization, Eberson probed the effects of pyridine- and 2,2′-bipyridine-type ligands in C(aryl)–H acetoxylation along a Pd(II)/Pd(IV) catalytic cycle, and found that use of 2,2′-bipyridine (bipy, 43) improved the reactivity and positional selectivity with substituted arenes.230–233 Fujiwara employed 1,10-phenanthroline (phen, 44) as a ligand in the C–H carbonylation of naphthalene along a Pd(II)/Pd(0) catalytic cycle, finding that it gave both improved conversion and better selectivity for the β-position.234 More recently, bidentate N(sp2),N(sp2)-ligands have been found to be beneficial in a variety of Pd(II)-catalyzed C–H functionalization transformations involving Pd(II)/Pd(0) and Pd(II)/Pd(IV) catalysis.43,142,148,235–245
We envisioned that by using a bidentate N,N-ligand, we could enhance dissociation of the N-bound substrate both sterically and electronically (Scheme 23). The ligand would potentially increase the steric demands of N-bound coordination due to unfavorable edge-to-edge or edge-to-face interactions and could also exert a kinetic trans-labilizing effect through electronic induction (vide infra). By promoting dissociation of the N-bound substrate, the catalyst would facilitate high effective concentration of reactive pyridine molecules around the Pd(II) center. The dissociated substrate would possess various orientations with respect to the Pd(II) catalyst, and under these conditions, C3-selective C–H cleavage could take place upon assembly of an intermediate with the appropriate orientation between the pyridine π-system and the Pd(II) center.
We began by studying Pd(II)-catalyzed C3-selective C–H olefination of pyridine (py, 46) (Table 9),246 with the long term aim of developing a wide range of C–C and C–heteroatom bond-forming reactions. We elected to use an excess of substrate to ensure high concentration around the Pd(II) catalyst. In the absence of an ancillary ligand, using ethyl acrylate (181) as the limiting reagent in the presence of Ag2CO3 (0.5 equiv) and Pd(OAc)2 (10 mol%) in DMF we observed 21% yield of 256 in a 5:1:1 C3:C2:C4 mixture (Entry 1). We reasoned that at such high concentration of pyridine in solution, it was likely that during the C–H cleavage step, at least two pyridine molecules were bound through nitrogen and were serving as ligands, suggesting that the pyridine moiety had approximately the appropriate donor strength for promoting the reaction. In accordance with our rationale for ligand design, we turned our attention to bidentate N,N-ligand scaffolds, surveying a broad range of different structures. We found that 2,2′-bipyridine ligands had a modestly positive effect on yield but gave substantially higher selectivity (Entries 2–6). Mixed N,N-donors like pyrox (Entry 7) and quinox (Entry 8) were ineffective, potentially because the oxazoline moiety is too electron-donating. 1,10-Phenanthroline-based ligands, which are less conformationally flexible and thus stronger binding, proved to be optimal in terms of combining high yield and selectivity (Entries 9 and 10).
Table 9.
Ligand-promoted C3-Selective C–H olefination of pyridine (py, 46).246

| |||
|---|---|---|---|
| Entry | Ligand | Yield (%)a | C3:C2:C4b |
| 1 | --- | 21 | 5:1:1 |
| 2 |
 bipy, 43 |
50 | 14:1:2 |
| 3 |
 257 |
29 | 5:1:1 |
| 4 |
 258 |
55 | 11:1:1 |
| 5 |
 DAF, 259 |
16 | 3:1:1 |
| 6 |
 DAF, 260 |
27 | 4:1:1 |
| 7 |
 pyrox, 261 |
11 | 6:4:1 |
| 8 |
 quinox, 45 |
7 | 5:1:1 |
| 9 |
 phen, 44 |
87 | 12:1:1 |
| 10 |
 bphen, 262 |
86 | 12:1:1 |
| 11 |
 267 |
6 | 1:1:1 |
| 12 |
 bcp, 268 |
9 | 4:4:1 |
The yield was determined by 1H NMR of the crude reaction mixture using CH2Br2 as an internal standard.
The C3:C2:C4 ratio was determined by 1H NMR of the crude reaction mixture.
Generally speaking, other methodology for C3- and C4-selective pyridine C–H functionalization has suffered from restricted substrate scope. We were thus delighted to find that under our optimized reaction conditions for C3-selective C–H olefination, a wide range of azines could be efficiently olefinated, generally with high selectivity for the C3 position (selected examples shown in Table 10). Both electron-releasing (Entries 2 and 3) and electron-withdrawing substituents (Entries 1 and 3) were tolerated on the pyridine ring, and quinoline was also found to be compatible (Entry 4). Several different functional groups were tolerated on the olefin as well (Entries 5–8). Under the optimal conditions, a large excess of substrate was required for high yield; however, the compatibility of the reaction with DMF as a solvent nonetheless allowed for the use of substrates that were solids at room temperature.
Table 10.
Substrate scope for ligand-promoted C3-Selective C–H olefination of pyridines (selected examples).246
| Entrya | Product | Yield (%) b | C3:C2:C4c |
|---|---|---|---|
| 1 |
 269 |
54 | --- |
| 2 |
 C3/C2/C4-270 |
61 | 10:2:1 |
| 3 |
C3/C2/C4-271 |
63 | 30:1:1 |
| 4 |
 C3/C2-272 |
44 | 3:1:0 |
| 5 |
 C3/C2/C4-256 |
73 | 12:1:1 |
| 6 |
 C2/C3/C4-273 |
68 | 7:1:1 |
| 7 |
 C2/C3/C4-274 |
57 | 14:1:1 |
| 8 |
 C2/C3/C4-275 |
45 | 30:3:1 |
Unless otherwise specified the reaction conditions were as follows: alkene (0.5 mmol), pyridine substrate (16 equiv), Pd(OAc)2 (10 mol%), phen (44) (13 mol%), Ag2CO3 (0.5 equiv), DMF (1 mL), air (1 atm), 140 °C, 12 h.
Isolated yield of C3-substitued product.
The C3:C2:C4 ratio was determined by 1H NMR of the crude product mixture prior to purification.
Having established high reactivity and C3-selectivty in C–H activation using the [Pd(II)-phen]-catalyst,247 we subsequently sought to test other coupling partners and catalytic manifolds with this system. In particular, owing to the importance of heterobiaryls in medicinal chemistry and agrochemistry, our next target was a C3-selective C–H arylation reaction with aryl iodides via Pd(II)/Pd(IV) catalysis. Starting with conditions similar to those we had developed for C–H olefination, we were ultimately able to devise a protocol for C–H arylation with aryl iodides and bromides.248 Through the use of Cs2CO3, which presumably regenerates a catalytically active Pd(II) species from PdI2 or PdI(OAc) we were able to avoid Ag(I) salts, which are often employed as halide scavengers in arylation reactions. To achieve high yields, we found that a large excess of pyridine substrate was required (generally used as solvent). The scope of the reaction was quite broad, tolerating a vast array of substituted aryl bromides and iodides,249 as well as a variety of azines (Table 11). Importantly, these results established the viability of this catalyst/substrate combination in supporting multiple coupling partners and elementary steps during the catalytic cycle.
Table 11.
Substrate scope for ligand-promoted C3-selective C–H arylation of pyridine (selected examples).248

| |||
|---|---|---|---|
| Entrya | Product | Yield (%) b | C3:C2:C4c |
| 1 |
 C3/C2-279 |
58 | 15:1:0 |
| 2 |
 C3/C2-280 |
65 | 3:1:0 |
| 3e |
 281 |
88 | --- |
| 4e |
 282 |
70 | --- |
| 5 |
 C3/C2/C4-283 |
82 | 28:1:1 |
| 6f |
 C2/C4-284 |
90 | 15:0:1 |
| 7 |
 C2/C3/C4-285 |
80 | --- |
| 8f |
 C2/C3-286 |
73 | 9:1:0 |
Unless otherwise specified the reaction conditions were as follows: aryl iodide (0.5 mmol), pyridine (3 mL), Pd(OAc)2 (5 mol%), phen (44) (15 mol%), Cs2CO3 (3.0 equiv), 140 °C, 48 h.
Isolated yield of C3-substitued product.
The C3:C2:C4 ratio was calculated from the isolated yields for all product isomers.
pyridine substrate (60 equiv) and DMF (mL).
The corresponding ArBr was used in place of the ArI.
To demonstrate the practicality of this transformation, we developed a concise route to the preclinical drug (±)-preclamol (290), a partial dopamine receptor agonist, using our C3-selective C–H arylation on gram-scale as the first step (Scheme 24). Following construction of the carbon skeleton, a three-step sequence of N-alkylation, hydrogenation, and deprotection yielded the final product.
Scheme 24.
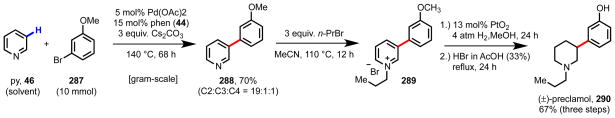
Expedient synthesis of (±)-preclamol using C3-selective C–H arylation.
While a detailed mechanistic model for these reactions remains the subject of active investigation in our laboratory, there are nonetheless several pertinent observations that we would like to highlight here to help guide future analysis. First, in both the pyridine C–H olefination and arylation reactions, large primary kinetic isotope effects were observed kH/kD ≥ 4.0, suggesting that a CMD mechanism, rather than an electrophilic palladation mechanism, may be operative. (At this stage, however, we cannot rule out the possibility of electrophilic palladation with rate-limiting deprotonation.) Moreover, with the C–H arylation reaction, we have observed that pyridine reacts at a higher rate than benzene, despite being far less electron-rich, which is consistent with a CMD mechanism. This observation is also potentially consistent with the hypothesis that initial N-coordination of pyridine increases the molarity of reactive substrate around Pd(II) to facilitate C–H cleavage.
Several relevant solid state structures had also been characterized by other groups prior to our work (Figure 7),189,250–255 analysis of which sheds some light on the role of the ligand in this reaction. Qualitatively, there seems to be more strain in the Pd(phen)(py2)(PF6)2 (293) and Pd(bipy)(py2)(PF6)2 (294) structures compared to the Pd(py)4(BF4)2 structure (292). In the Pd(py)4(BF4)2 structure, adjacent pyridine rings are all oriented face-to-face to one another and perpendicular to the Pd(II) square plane. In contrast, in the bipy- and phen-ligated structures, there are two enforced and sterically unfavorable edge-to-face interactions between the ligand and the pyridine substrate, which would potentially make pyridine dissociation more favorable. It should be noted, however, that across structures 291–294, the [Pd(II)–py] bond lengths remain nearly constant (all within 0.011 Å).
Figure 7.
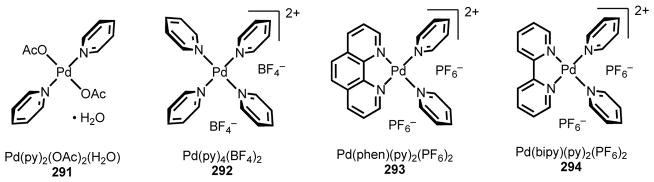
Relevant examples of Pd(II)(Py)nLn complexes that have been characterized by X-ray diffraction.189,250–252
Alongside steric considerations, the electronic influence of the ligand also warrants discussion. Generally speaking N- and N,N-ligands are known to be relatively weak trans-labilizing ligands (though still stronger than OAc−).41,256,257 In the case of our reaction system, however, because dissociation of the bidentate ligand will be prohibitively slow compared to dissociation of pyridine, the effective trans influence of the phen ligand on N-bound pyridine could still be substantial.
The solution dynamics of complexes of the type shown in Figure 8 have also been studied, and pyridine-dissociation mechanisms mediated by coordinating solvents and acetate anion have been reported.252,255 It would seem likely that these mechanism act in concert with the steric and electronic influences of the ligand in promoting the dissociation of N-bound pyridine to allow for population of the π-bound state at elevated temperatures (Scheme 23). Beyond its role in assisting the assembly of the active pre-transition state coordination structure, the ligand could have other beneficial roles in the catalytic cycle, such as promoting reoxidation of reduced Pd(0) species in the C–H olefination reaction, or stabilizing Pd(IV) species in the C–H arylation reaction.
The strong preference for C–H cleavage at the C3 position is not entirely clear at this time. The C3 position is expected to be the most electron-rich, so one possibility is that initial π-coordination and subsequent CMD is consequently more favorable. There are other explanations that are also consistent with the observed selectivity trends. Recent computational studies of C(heteroaryl)–H cleavage selectivity patterns via CMD with a related complex, (Ph)(PMe3)Pd(OAc), have been performed by Gorelsky using distortion–interaction analysis, and with pyridine, C3 selectivity was found to be favorable based on the proposed CMD mechanism.258 Ess and coworkers have studied the same system using density functional calculations, and have correlated the CMD transition state energy to the thermodynamic stability of the developing [Pd(II)–Ar], and have found that this too is in agreement with selectivity at the C3 position for pyridine.259 While these findings are intriguing, caution should be taken in extrapolating the data from this more electron-rich (Ph)(PMe3)Pd(OAc) system to the [Pd(II)–phen] catalyst used in our studies; further studies are needed to evaluate whether the same trends hold in both cases.
Recently, our group and others have gone on to apply this [Pd(II)–phen] catalytic system to effect selective C–H arylation of other classes of biologically active azaheterocycles, including pyrazoles and 1H-indazoles.260 Importantly, we were also able to demonstrate the practical utility of this procedure in a concise total synthesis of the nature product nigellidine hydrobromide.
3.4.3. Outlook
2,2′-Bipyridines, and N(sp2),N(sp2)-ligands more generally, have a rich history in Pd(II) catalysis and are now beginning to be used to address problems in Pd(II)-catalyzed C–H functionalization. One of the strengths of this ligand scaffold is that there are multiple tuning sites to adjust the steric and electron properties proximate and distal to the nitrogen donor atoms. Additionally, the ground state torsion angle (and thus the binding strength) of the biaryl backbone can further be adjusted. Each of the two donor sites can be tuned individually, offering the opportunity to desymmetrize the two remaining sites on the Pd(II) square plane. Many ligands within this class are commercially available and others can be prepared in a small number of steps through modular synthesis, which facilitates ligand screening and reaction optimization. This ligand scaffold was demonstrated to be compatible with the elementary steps of C–H olefination and C–H arylation along Pd(II)/Pd(0) and Pd(II)/Pd(IV) catalytic manifolds, respectively.
Despite being a strong chelator to Pd(II), this ligand scaffold still allows substrate coordination and subsequent C–H activation. In our case, the strong bidentate coordination seems to be key for facilitating the dissociation of N-bound pyridine, leading to the productive π-bound assembly. Through this interplay, high effective molarity of pyridine can be maintained around the catalyst, allowing high reactivity and selectivity to be achieved, despite the electron-poor nature of the arene. Further optimization of the catalyst to achieve high yield with 1 equiv of pyridine substrate relative to olefin is the next challenge that our group is pursuing.
4. Conclusions
Ligand development has been a principal driving force in the rapid maturation of the field of homogeneous catalysis during the past several decades. In both asymmetric and non-stereoselective reactions, privileged ligand scaffolds have played leading roles in the discovery of a wide range of catalytic reactions with little to no mechanistic similarity. Nonetheless, each combination of substrate, reaction type, and catalyst poses a unique set of requirements for the ligand (steric and electronic properties, denticity, etc.) that are difficult to predict from first principles. For this reason, ligand scaffolds that are tunable and modular are particularly useful because the ligand can rapidly be adjusted to meet the challenge at hand.
In our own research in Pd(II)-catalyzed C–H functionalization, we have made the discovery, development, and application of suitable ligands for controlling C–H cleavage a major priority. We view ligand development as the key to meeting our long term goals of engineering reactions to convert feedstock chemicals into value-added intermediates through selective and efficient C–H functionalization to form any desired C–C or C–heteroatom bond. In this review, we have discussed the challenges that we faced in trying to develop catalytic systems with high reactivity and selectivity with several distinct substrate classes, and we have examined the ligand design considerations at play in each reaction. Using mono-N-protected amino acid ligands, we were able to achieve enantioselectivity, positional selectivity, and rate enhancement with substrates containing directing groups. With 2,6-disubstituted pyridine ligands we were able to achieve meta-selective C–H olefination of electron-poor arenes under aerobic conditions and promote methylene C(sp3)–H functionalization of aliphatic acid derivatives. Finally with 2,2′-bipyridine ligands, we successfully developed C3-selective C–H olefination and arylation of pyridines and related azines. All three ligand classes have proven to be compatible with both Pd(II)/Pd(0) and Pd(II)/Pd(IV) catalysis. Across these case studies, our success in achieving unprecedented levels of reactivity and selectivity came by tailoring the ligand structure to the substrate properties, enabling a productive interplay at the Pd(II) center. In this work, identifying an optimal ligand typically stemmed from a dialogue between theory and empiricism, requiring robust ligand scaffolds that are compatible with a multitude of catalytic transformations and possess several sites for tuning.
Moving forward, ligand development will play an increasingly important role in realizing the untapped synthetic potential of Pd(II)-mediated C–H functionalization, and as a central challenge in the field, it continues to stimulate and inspire us.
Figure 5.
Aryl- and alkylcarboxylic acids, representative starting material classes that are abundant in quantity and rich in structural diversity
Table 5.
Position-selective C–H olefination of 180 using mono-N-protected amino ligands.

| |||
|---|---|---|---|
| Entry | Ligand | Conv. (%)a | A:B a |
| 1 | --- | 68 | 1.4:1 |
| 2 |
 Boc-Thr(Bzl)-OH, 183 |
17 | 3:1 |
| 3 |
 Boc-Abu-OH, 142 |
17 | 5:1 |
| 4 |
 Boc-Val-OH, 141 |
23 | 6:1 |
| 5 |
 Boc-Leu-OH, 146 |
24 | 7:1 |
| 6 |
 Boc-lle-OH, 164 |
27 | 8:1 |
| 7 |
 Boc2-Leu-OH, 184 |
50 | 3:1 |
| 8 |
 H-Leu-OH, 185 |
16 | 6:1 |
| 9 |
 For-Leu-OH, 186 |
24 | 13:1 |
| 10 |
 Fmoc-lle-OH, 187 |
16 | 5:1 |
| 11 |
 Ac-lle-OH, 188 |
23 | 10:1 |
| 12 |
 For-lle-OH, 189 |
45 (75)b | 20:1 |
The conversion and A:B ratio were determined by 1H NMR of the crude reaction mixture.
Value in parenthesis represents the conversion using 7 mol% Pd(OAc)2 and 14 mol% For-Ile-OH under otherwise identical conditions.
Acknowledgments
This work was made possible by financial support from TSRI, the NSF (CHE-1011898), the NIH (NIGMS, 1 R01 GM084019-04; NIGMS 1 R01 GM102265-01), NSF under the CCI Center for Stereoselective C–H Functionalization (CHE-1205646), Bristol-Myers Squibb, Syngenta, Novartis, Pfizer, Amgen, and Eli Lilly. We are grateful to TSRI, the NSF GRFP, the NDSEG Fellowship program, and the Skaggs-Oxford Scholarship program for predoctoral fellowships to K.M.E. We acknowledge Peter S. Thuy-Boun (Yu lab, TSRI), Will R. Gutekunst (Baran lab, TSRI), Jae H. Chu (Mauro Lab, TSRI), and Matthew Tredwell (Gouverneur lab, Oxford) for generous assistance in editing this review and for thoughtful advice during its preparation. We also wish to recognize colleagues at University of Cambridge, Brandeis University, and TSRI for encouragement and inspiration over the years. Finally, and most importantly, we thank all present and former group members, whose tenacity and creativity has been at the heart of the research described here. TSRI Manuscript no. 21887.
Biography

Jin-Quan Yu received his B.Sc. in Chemistry from East China Normal University, M.Sc. from the Guangzhou Institute of Chemistry, and Ph.D. from the University of Cambridge (with Prof. J. B. Spencer). Following time as a Junior Research Fellow at Cambridge, he joined the laboratory of Prof. E. J. Corey at Harvard University as a postdoctoral fellow. He then began his independent career at Cambridge (2003–2004), before moving to Brandeis University (2004–2007), and finally to TSRI, where he is currently Frank and Bertha Hupp Professor of Chemistry.
References
- 1.Berrisford DJ, Bolm C, Sharpless KB. Angew Chem Int Ed. 1995;34:1059–1070. [Google Scholar]
- 2.As a point of clarification, ‘ligand’ can refer to any molecule that is coordinated to a metal catalyst (anionic donor, X-type; or neutral donor, L-type). Throughout the text we generally focus on ligands that possesses an element of design (i.e., a molecule that was independently synthesized for the express purpose of coordinating it to the metal to influence the reactivity), rather than common counterions (OAc, OTf, Cl, etc.) or solvent molecules (MeCN, THF, etc.), but those too can have a dramatic impact on the reactivity and/or selectivity of catalytic transformations.
- 3.Katsuki T, Sharpless KB. J Am Chem Soc. 1980;102:5974–5976. [Google Scholar]
- 4.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J Am Chem Soc. 1987;109:5765–5780. [Google Scholar]
- 5.Dang TP, Kagan HB. J Chem Soc D. 1971:481. [Google Scholar]
- 6.Vineyard BD, Knowles WS, Sabacky MJ, Bachman GL, Weinkauff DJ. J Am Chem Soc. 1977;99:5946–5952. [Google Scholar]
- 7.Fryzuk MD, Bosnich B. J Am Chem Soc. 1977;99:6262–6267. doi: 10.1021/ja00461a014. [DOI] [PubMed] [Google Scholar]
- 8.Miyashita A, Yasuda A, Takaya H, Toriumi K, Ito T, Souchi T, Noyori R. J Am Chem Soc. 1980;102:7932–7934. [Google Scholar]
- 9.Osborn JA, Wilkinson G, Young JF. Chem Commun. 1965:17. [Google Scholar]
- 10.Osborn JA, Jardine FH, Young JF, Wilkinson G. J Chem Soc A. 1966:1711–1732. [Google Scholar]
- 11.Trnka TM, Grubbs RH. Acc Chem Res. 2001;34:18–29. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- 12.Nolan SP, Clavier H. Chem Soc Rev. 2010;39:3305–3316. doi: 10.1039/b912410c. [DOI] [PubMed] [Google Scholar]
- 13.Littke AF, Fu GC. Angew Chem Int Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 14.Hartwig JF. Acc Chem Res. 2008;41:1534–1544. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrmann WA. Angew Chem Int Ed. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 17.Hillier AC, Grasa GA, Viciu MS, Lee HM, Yang C, Nolan SP. J Organomet Chem. 2002;653:69–82. [Google Scholar]
- 18.Jacobsen EN, Markó I, Mungall WS, Schröder G, Sharpless KB. J Am Chem Soc. 1988;110:1968–1970. [Google Scholar]
- 19.Andersson MA, Epple R, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:472–475. doi: 10.1002/1521-3773(20020201)41:3<472::aid-anie472>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 20.Yoon TP, Jacobsen EN. Science. 2003;299:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 21.Tolman CA. Chem Rev. 1977;77:313–348. [Google Scholar]
- 22.Nishiyama M, Yamamoto T, Koie Y. Tetrahedron Lett. 1998;39:617–620. [Google Scholar]
- 23.Littke AF, Fu GC. Angew Chem Int Ed. 1998;37:3387–3388. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3387::AID-ANIE3387>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 24.Hartwig JF, Kawatsura M, Hauck SI, Shaughnessy KH, Alcazar-Roman LM. J Org Chem. 1999;64:5575–5580. doi: 10.1021/jo990408i. [DOI] [PubMed] [Google Scholar]
- 25.Old DW, Wolfe JP, Buchwald SL. J Am Chem Soc. 1998;120:9722–9723. [Google Scholar]
- 26.Nieto-Oberhuber C, López S, Echavarren AM. J Am Chem Soc. 2005;127:6178–6179. doi: 10.1021/ja042257t. [DOI] [PubMed] [Google Scholar]
- 27.Hughes OR, Unruh JD. J Mol Catal. 1981;12:71–83. [Google Scholar]
- 28.Kranenburg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM, Goubitz K, Fraanje J. Organometallics. 1995;14:3081–3089. [Google Scholar]
- 29.Gupta M, Hagen C, Flesher RJ, Kaska WC, Jensen CM. Chem Commun. 1996:2083–2084. [Google Scholar]
- 30.Liu F, Pak EB, Singh B, Jensen CM, Goldman AS. J Am Chem Soc. 1999;121:4086–4087. [Google Scholar]
- 31.Nolan SP. Acc Chem Res. 2011;44:91–100. doi: 10.1021/ar1000764. [DOI] [PubMed] [Google Scholar]
- 32.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 33.Tajima Y, Kunioka E. J Org Chem. 1968;33:1689–1690. [Google Scholar]
- 34.Stern R, Sajus L. Tetrahedron Lett. 1968;9:6313–6314. [Google Scholar]
- 35.Miyake A, Kondo H. Angew Chem Int Ed. 1968;7:631–632. [Google Scholar]
- 36.Funk RL, Vollhardt KPC. J Am Chem Soc. 1977;99:5483–5484. doi: 10.1021/ja00458a044. [DOI] [PubMed] [Google Scholar]
- 37.Uto T, Shimizu M, Ueura K, Tsurugi H, Satoh T, Miura M. J Org Chem. 2007;73:298–300. doi: 10.1021/jo7022087. [DOI] [PubMed] [Google Scholar]
- 38.Hyster TK, Rovis T. Chem Sci. 2011;2:1606–1610. doi: 10.1039/C1SC00235J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054–3131. doi: 10.1021/cr8002505. [DOI] [PubMed] [Google Scholar]
- 40.Surry DS, Buchwald SL. Chem Sci. 2010;1:13–31. doi: 10.1039/C0SC00107D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Togni A, Venanzi LM. Angew Chem Int Ed. 1994;33:497–526. [Google Scholar]
- 42.Michel BW, Camelio AM, Cornell CN, Sigman MS. J Am Chem Soc. 2009;131:6076–6077. doi: 10.1021/ja901212h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell AN, White PB, Guzei IA, Stahl SS. J Am Chem Soc. 2010;132:15116–15119. doi: 10.1021/ja105829t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The examples included in this figure are not intended to constitute a comprehensive list. Additionally, it should also be noted that many of the ligands highlighted in Figure 1 are also known to give enhanced kinetic reactivity in asymmetric and non-stereoselective transformations.
- 45.Brown JM. Angew Chem Int Ed. 1987;26:190–203. [Google Scholar]
- 46.Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307–1370. [Google Scholar]
- 47.Ohta T, Takaya H, Kitamura M, Nagai K, Noyori R. J Org Chem. 1987;52:3174–3176. [Google Scholar]
- 48.Crabtree RH, Felkin H, Morris GE. J Organomet Chem. 1977;141:205–215. [Google Scholar]
- 49.Bell S, Wüstenberg B, Kaiser S, Menges F, Netscher T, Pfaltz A. Science. 2006;311:642–644. doi: 10.1126/science.1121977. [DOI] [PubMed] [Google Scholar]
- 50.Kaiser S, Smidt SP, Pfaltz A. Angew Chem Int Ed. 2006;45:5194–5197. doi: 10.1002/anie.200601529. [DOI] [PubMed] [Google Scholar]
- 51.Hanson RM, Sharpless KB. J Org Chem. 1986;51:1922–1925. [Google Scholar]
- 52.Zhang W, Loebach JL, Wilson SR, Jacobsen EN. J Am Chem Soc. 1990;112:2801–2803. [Google Scholar]
- 53.Jacobsen EN, Zhang W, Muci AR, Ecker JR, Deng L. J Am Chem Soc. 1991;113:7063–7064. [Google Scholar]
- 54.Noyori R. Angew Chem Int Ed. 2002;41:2008–2022. [PubMed] [Google Scholar]
- 55.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 56.Grushin VV, Alper H. Chem Rev. 1994;94:1047–1062. [Google Scholar]
- 57.Knowles WS. Acc Chem Res. 1983;16:106–112. [Google Scholar]
- 58.Reetz MT. Angew Chem Int Ed. 2008;47:2556–2588. doi: 10.1002/anie.200704327. [DOI] [PubMed] [Google Scholar]
- 59.Sakane S, Maruoka K, Yamamoto H. Tetrahedron Lett. 1985;26:5535–5538. [Google Scholar]
- 60.Sasai H, Suzuki T, Arai S, Arai T, Shibasaki M. J Am Chem Soc. 1992;114:4418–4420. [Google Scholar]
- 61.Keck GE, Tarbet KH, Geraci LS. J Am Chem Soc. 1993;115:8467–8468. [Google Scholar]
- 62.Hoveyda AH, Schrock RR. Chem Eur J. 2001;7:945–950. doi: 10.1002/1521-3765(20010302)7:5<945::aid-chem945>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 63.Harper KC, Sigman MS. Science. 2011;333:1875–1878. doi: 10.1126/science.1206997. [DOI] [PubMed] [Google Scholar]
- 64.von Matt P, Pfaltz A. Angew Chem Int Ed. 1993;32:566–568. [Google Scholar]
- 65.Sprinz J, Helmchen G. Tetrahedron Lett. 1993;34:1769–1772. [Google Scholar]
- 66.Dawson GJ, Frost CG, Williams JMJ, Coote SJ. Tetrahedron Lett. 1993;34:3149–3150. [Google Scholar]
- 67.Brown JM, Hulmes DI, Guiry PJ. Tetrahedron. 1994;50:4493–4506. [Google Scholar]
- 68.Helmchen G, Pfaltz A. Acc Chem Res. 2000;33:336–345. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- 69.Williams JMJ. Synlett. 1996:705–710. [Google Scholar]
- 70.Pfaltz A, Drury WJ., III Proc Natl Acad Sci U S A. 2004;101:5723–5726. doi: 10.1073/pnas.0307152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tani K, Behenna DC, McFadden RM, Stoltz BM. Org Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]
- 72.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. John Wiley & Sons; New York: 1995. [Google Scholar]
- 73.McMurray L, O’Hara F, Gaunt MJ. Chem Soc Rev. 2011;40:1885–1898. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- 74.Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- 75.Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 76.Engle KM, Wang DH, Yu JQ. J Am Chem Soc. 2010;132:14137–14151. doi: 10.1021/ja105044s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2012;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Newhouse T, Baran PS, Hoffmann RW. Chem Soc Rev. 2009;38:3010–3021. doi: 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li CJ, Trost BM. Proc Natl Acad Sci U S A. 2008;105:13197–13202. doi: 10.1073/pnas.0804348105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kimura S, Kobayashi S, Kumamoto T, Akagi A, Sato N, Ishikawa T. Helv Chim Acta. 2011;94:578–591. [Google Scholar]
- 81.Wang DH, Yu JQ. J Am Chem Soc. 2011;133:5767–5769. doi: 10.1021/ja2010225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mei TS, Giri R, Maugel N, Yu JQ. Angew Chem Int Ed. 2008;47:5215–5219. doi: 10.1002/anie.200705613. [DOI] [PubMed] [Google Scholar]
- 83.Zhang YH, Yu JQ. J Am Chem Soc. 2009;131:14654–14655. doi: 10.1021/ja907198n. [DOI] [PubMed] [Google Scholar]
- 84.Giri R, Yu JQ. J Am Chem Soc. 2008;130:14082–14083. doi: 10.1021/ja8063827. [DOI] [PubMed] [Google Scholar]
- 85.Zhang YH, Shi BF, Yu JQ. Angew Chem Int Ed. 2009;48:6097–6100. doi: 10.1002/anie.200902262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ. J Am Chem Soc. 2007;129:3510–3511. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]
- 87.Yoo EJ, Ma S, Mei TS, Chan KSL, Yu JQ. J Am Chem Soc. 2011;133:7652–7655. doi: 10.1021/ja202563w. [DOI] [PubMed] [Google Scholar]
- 88.Chan KSL, Wasa M, Wang X, Yu JQ. Angew Chem Int Ed. 2011;50:9081–9084. doi: 10.1002/anie.201102985. [DOI] [PubMed] [Google Scholar]
- 89.Dai HX, Yu JQ. J Am Chem Soc. 2012;134:134–137. doi: 10.1021/ja2097095. [DOI] [PubMed] [Google Scholar]
- 90.Zhang XG, Dai HX, Wasa M, Yu JQ. J Am Chem Soc. 2012;134:11948–11951. doi: 10.1021/ja305259n. [DOI] [PubMed] [Google Scholar]
- 91.Yu JQ, Giri R, Chen X. Org Biomol Chem. 2006;4:4041–4047. doi: 10.1039/b611094k. [DOI] [PubMed] [Google Scholar]
- 92.Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242–3272. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]
- 94.Wasa M, Engle KM, Yu JQ. Isr J Chem. 2010;50:605–616. doi: 10.1002/ijch.201000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mei TS, Kou L, Ma S, Engle KM, Yu JQ. Synthesis. 2012;44:1778–1791. doi: 10.1055/s-0031-1289766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Because Cu is known to readily participate in single-electron transfer events, it has historically been challenging to elucidate the mechanism for Cu-catalyzed cross-coupling reactions. This has also proven to be the case with Cu-catalyzed C–H functionalization reactions, and the mechanism of these transformations is the the topic of ongoing investigation. It is possible that these reactions involve a Cu(II)/Cu(III)/Cu(I) or Cu(III)/Cu(I) redox couple, the latter of which would be isoelectronic to the Pd(II)/Pd(0) redox couple. It is difficult, however, to generalize from individual mechanistic studies because there could be fundamental differences from one reaction to the next. For a detailed discussion of the mechanism of these transformations and related Cu-catalyzed cross-coupling reactions, see: King AE, Huffman LM, Casitas A, Costas M, Ribas X, Stahl SS. J Am Chem Soc. 2010;132:12068–12073. doi: 10.1021/ja1045378.For selected examples of C–H functionalization using Cu(II) precatalysts, see: Chen X, Hao XS, Goodhue CE, Yu JQ. J Am Chem Soc. 2006;128:6790–6791. doi: 10.1021/ja061715q.Brasche G, Buchwald SL. Angew Chem Int Ed. 2008;47:1932–1934. doi: 10.1002/anie.200705420.Ueda S, Nagasawa H. Angew Chem Int Ed. 2008;47:6411–6413. doi: 10.1002/anie.200801240.Do HQ, Daugulis O. J Am Chem Soc. 2009;131:17052–17053. doi: 10.1021/ja907479j.
- 97.For selected examples of Rh(III)-catalyzed C–H functionalization, see: Ueura K, Satoh T, Miura M. Org Lett. 2007;9:1407–1409. doi: 10.1021/ol070406h.Stuart DR, Bertrand-Laperle M, Burgess KMN, Fagnou K. J Am Chem Soc. 2008;130:16474–16475. doi: 10.1021/ja806955s.Patureau FW, Glorius F. J Am Chem Soc. 2010;132:9982–9983. doi: 10.1021/ja103834b.Hyster TK, Rovis T. J Am Chem Soc. 2010;132:10565–10569. doi: 10.1021/ja103776u.Presset M, Oehlrich D, Rombouts F, Molander GA. Org Lett. 2013 doi: 10.1021/ol400307d.
- 98.For selected examples of Ru(II)-catalyzed C–H functionalization, see: Weissman H, Song X, Milstein D. J Am Chem Soc. 2000;123:337–338. doi: 10.1021/ja003361n.Oi S, Fukita S, Hirata N, Watanuki N, Miyano S, Inoue Y. Org Lett. 2001;3:2579–2581. doi: 10.1021/ol016257z.Ackermann L. Org Lett. 2005;7:3123–3125. doi: 10.1021/ol051216e.Özdemir I, Demir S, Çetinkaya B, Gourlaouen C, Maseras F, Bruneau C, Dixneuf PH. J Am Chem Soc. 2008;130:1156–1157. doi: 10.1021/ja710276x.Padala K, Jeganmohan M. Org Lett. 2011;13:6144–6147. doi: 10.1021/ol202580e.
- 99.For early examples of an alternative approach to effect C–H functionalization using low-valent Ru(0) and Rh(I) catalysts, see: Murai S, Kakiuchi F, Sekine S, Tanaka Y, Kamatani A, Sonoda M, Chatani N. Nature. 1993;366:529–531.Trost BM, Imi K, Davies IW. J Am Chem Soc. 1995;117:5371–5372.Lenges CP, Brookhart M. J Am Chem Soc. 1999;121:6616–6623.Jun CH, Hong JB, Kim YH, Chung KY. Angew Chem Int Ed. 2000;39:3440–3442. doi: 10.1002/1521-3773(20001002)39:19<3440::aid-anie3440>3.0.co;2-1.Tan KL, Bergman RG, Ellman JA. J Am Chem Soc. 2001;123:2685–2686. doi: 10.1021/ja0058738.
- 100.It is important to clarify that along a Pd(0)/Pd(II) catalytic cycle, the cross-coupling of C(aryl)–H bonds with aryl halides can be promoted with phosphine and NHC ligands. Analogous to traditional Pd(0)-catalyzed cross-coupling reactions, this type of reaction begins with oxidative addition of the aryl halide as the first elementary step in the catalytic cycle. Though Pd(II) salts are often used as precatalysts in these tranformations, in situ reduction generates a [Pd(0)Ln] species, which is the active catalyst. This is a distinct mechanistic manifold that is outside of the scope of this Review. For reviews discussing this mode of catalysis, see Ref. 92 and the following: Campeau LC, Stuart DR, Fagnou K. Aldrichimica Acta. 2007;40:35–41.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174–238. doi: 10.1021/cr0509760.For lead references using chiral [Pd(0) L*] catalysts in C–H functionalization, see: Albicker MR, Cramer N. Angew Chem Int Ed. 2009;48:9139–9142. doi: 10.1002/anie.200905060.Renaudat A, Jean-Gérard L, Jazzar R, Kefalidis CE, Clot E, Baudoin O. Angew Chem Int Ed. 2010;49:7261–7265. doi: 10.1002/anie.201003544.Nakanishi M, Katayev D, Besnard C, Kündig EP. Angew Chem Int Ed. 2011;50:7438–7441. doi: 10.1002/anie.201102639.Anas S, Cordi A, Kagan HB. Chem Commun. 2011;47:11483–11485. doi: 10.1039/c1cc14292e.Saget T, Lemouzy SJ, Cramer N. Angew Chem Int Ed. 2012;51:2238–2242. doi: 10.1002/anie.201108511.Martin N, Pierre C, Davi M, Jazzar R, Baudoin O. Chem Eur J. 2012;18:4480–4484. doi: 10.1002/chem.201200018.Saget T, Cramer N. Angew Chem Int Ed. 2012;51:12842–12845. doi: 10.1002/anie.201207959.
- 101.Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936–946. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074–1086. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ryabov AD, Sakodinskaya IK, Yatsimirsky AK. J Chem Soc Dalton Trans. 1985:2629–2638. [Google Scholar]
- 105.Gómez M, Granell J, Martinez M. Organometallics. 1997;16:2539–2546. [Google Scholar]
- 106.Davies DL, Donald SMA, Macgregor SA. J Am Chem Soc. 2005;127:13754–13755. doi: 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]
- 107.The mechanisms and associated nomenclature of metal-mediated C–H cleavage are topics of ongoing discussion in the literature. With Pd(II), oxidative addition, electrophilic palladation, and concerted metalation/deprotonation (CMD) have all been proposed, and based on our current knowledge the latter two seem to be opperative in the preponderance of cases. It is important to consider, however, that in actuality these broad categories likely represent extremes on a continuum of possible mechanistic scenarios. For example, one could reasonably argue that the “CMD” category actually encompasses nearly all base-assisted, redox-neutral C–H activation mechanisms. If proton removal at the transition state is not advanced compared to Pd–C bonding, the CMD pathway would possess electrophilic substitution character. If, on the other hand, proton removal is advanced compared to Pd–C bonding, the CMD pathway would have deprotonative character. As the community gains a deeper understanding of the true mechanism at play in each unique reaction, the adoption more precise nomenclature, such “CMD with deprotonative character” or “CMD with electrophilic substitution character” may be in order. To simplify discussion in the present review, we use “electrophilic palladation” and “CMD” in their traditional sense. The former we use to describe Pd(II)-mediated C(sp2)–H activation that likely involves a Wheland (arenium) intermediate. The latter we use to describe Pd(II)-mediated C(sp3)–H activation and C(sp2)–H activation with strongly deprotonative character (involving minimal charge build-up in the transition state).
- 108.We kindly acknowledge a referee for pointing out the different manfestations of CMD discussed in Ref. 107.
- 109.Gant TG, Meyers AI. Tetrahedron. 1994;50:2297–2360. [Google Scholar]
- 110.Xia JB, You SL. Organometallics. 2007;26:4869–4871. [Google Scholar]
- 111.Takebayashi S, Shizuno T, Otani T, Shibata T. Beilstein J Org Chem. 2012;8:1844–1848. doi: 10.3762/bjoc.8.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giri R, Chen X, Yu JQ. Angew Chem Int Ed. 2005;44:2112–2115. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]
- 113.Giri R, Liang J, Lei JG, Li JJ, Wang DH, Chen X, Naggar IC, Guo C, Foxman BM, Yu JQ. Angew Chem Int Ed. 2005;44:7420–7424. doi: 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]
- 114.Giri R, Chen X, Hao XS, Li JJ, Liang J, Fan ZP, Yu JQ. Tetrahedron: Asymmetry. 2005;16:3502–3505. [Google Scholar]
- 115.Giri R, Lan Y, Liu P, Houk KN, Yu JQ. J Am Chem Soc. 2012;134:14118–14126. doi: 10.1021/ja304643e. [DOI] [PubMed] [Google Scholar]
- 116.Shi BF, Maugel N, Zhang YH, Yu JQ. Angew Chem Int Ed. 2008;47:4882–4886. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- 117.For original data corresponding to Entries 5 and 8, see: Maugel, N. L., Palladium-Catalyzed Alkylation, Arylation and Dehydrogenation of Unactivated C–H bonds. Syntheses of the Tetracyclic Aminoquinone Moiety of Marmycin A, (±)-Nosyberkol (Isotuberculosinol, Revised Structure of Edaxadiene), and (±)-Tuberculosinol. Ph.D. Thesis, Brandeis University, Waltham, MA, February 2011. For all other entries, see the Supporting Information of Ref. 116.
- 118.Navarro R, García J, Urriolabeitia EP, Cativiela C, Diaz-de-Villegas MD. J Organomet Chem. 1995;490:35–43. [Google Scholar]
- 119.Mono-N-protected amino acids have been used to control the stereoselectivity in stoichiometric cyclopalladation reactions. For examples, see: Sokolov VI, Troitskaya LL, Reutov OA. J Organomet Chem. 1977;133:C28–C30.Sokolov VI, Troitskaya LL, Reutov OA. J Organomet Chem. 1979;182:537–546.Sokolov VI, Troitskaya LL, Khrushchova NS. J Organomet Chem. 1983;250:439–446.Günay ME, Richards CJ. Organometallics. 2009;28:5833–5836.Dendele N, Bisaro F, Gaumony AC, Perrio S, Richards CJ. Chem Commun. 2012;48:1991–1993. doi: 10.1039/c2cc16864b.
- 120.Musaev DG, Kaledin A, Shi BF, Yu JQ. J Am Chem Soc. 2012;134:1690–1698. doi: 10.1021/ja208661v. [DOI] [PubMed] [Google Scholar]
- 121.Shi BF, Zhang YH, Lam JK, Wang DH, Yu JQ. J Am Chem Soc. 2010;132:460–461. doi: 10.1021/ja909571z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Prior to determining the absolute configuration of product (−)-127 as (R) (see Ref. 120), we put forward a working model for stereoinduction in Ref. 116. However, the model actually predicted preferential formation of the the opposite enantiomer. That model has since been abandoned, and a revised model was put forward in Refs. 120 and 121. The discussion herein is based on the revised model.
- 123.Wang DH, Engle KM, Shi BF, Yu JQ. Science. 2010;327:315–319. doi: 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kao LC, Sen A. J Chem Soc, Chem Commun. 1991:1242–1243. [Google Scholar]
- 125.Miura M, Tsuda T, Satoh T, Pivsa-Art S, Nomura M. J Org Chem. 1998;63:5211–5215. [Google Scholar]
- 126.Dangel BD, Johnson JA, Sames D. J Am Chem Soc. 2001;123:8149–8150. doi: 10.1021/ja016280f. [DOI] [PubMed] [Google Scholar]
- 127.Lee JM, Chang S. Tetrahedron Lett. 2006;47:1375–1379. [Google Scholar]
- 128.Wasa M, Engle KM, Yu JQ. J Am Chem Soc. 2009;131:9886–9887. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wasa M, Engle KM, Yu JQ. J Am Chem Soc. 2010;132:3680–3681. doi: 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wasa M, Engle KM, Lin DW, Yoo EJ, Yu JQ. J Am Chem Soc. 2011;133:19598–19601. doi: 10.1021/ja207607s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kutter E, Hansch C. J Med Chem. 1969;12:647–652. doi: 10.1021/jm00304a020. [DOI] [PubMed] [Google Scholar]
- 132.Sotomatsu T, Fujita T. J Org Chem. 1989;54:4443–4448. [Google Scholar]
- 133.Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2010;49:6169–6173. doi: 10.1002/anie.201002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shue RS. J Chem Soc D. 1971:1510–1511. [Google Scholar]
- 135.Dams M, DeVos DE, Celen S, Jacobs PA. Angew Chem Int Ed. 2003;42:3512–3515. doi: 10.1002/anie.200351524. [DOI] [PubMed] [Google Scholar]
- 136.Baxter RD, Sale D, Engle KM, Yu JQ, Blackmond DG. J Am Chem Soc. 2012;134:4600–4606. doi: 10.1021/ja207634t. [DOI] [PubMed] [Google Scholar]
- 137.Lu Y, Wang DH, Engle KM, Yu JQ. J Am Chem Soc. 2010;132:5916–5921. doi: 10.1021/ja101909t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang X, Lu Y, Dai HX, Yu JQ. J Am Chem Soc. 2010;132:12203–12205. doi: 10.1021/ja105366u. [DOI] [PubMed] [Google Scholar]
- 139.Lu Y, Leow D, Wang X, Engle KM, Yu JQ. Chem Sci. 2011;2:967–971. [Google Scholar]
- 140.Simmons EM, Hartwig JF. J Am Chem Soc. 2010;132:17092–17095. doi: 10.1021/ja1086547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Simmons EM, Hartwig JF. Nature. 2012;483:70–73. doi: 10.1038/nature10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xiao B, Gong TJ, Liu ZJ, Liu JH, Luo DF, Xu J, Liu L. J Am Chem Soc. 2011;133:9250–9253. doi: 10.1021/ja203335u. [DOI] [PubMed] [Google Scholar]
- 143.Wei Y, Yoshikai N. Org Lett. 2011;13:5504–5507. doi: 10.1021/ol202229w. [DOI] [PubMed] [Google Scholar]
- 144.Miura M, Tsuda T, Satoh T, Nomura M. Chem Lett. 1997;26:1103–1104. [Google Scholar]
- 145.Hennings DD, Iwasa S, Rawal VH. J Org Chem. 1997;62:2–3. doi: 10.1021/jo961876k. [DOI] [PubMed] [Google Scholar]
- 146.Pham MV, Ye B, Cramer N. Angew Chem Int Ed. 2012;51:10610–10614. doi: 10.1002/anie.201206191. [DOI] [PubMed] [Google Scholar]
- 147.Dai HX, Stepan AF, Plummer MS, Zhang YH, Yu JQ. J Am Chem Soc. 2011;133:7222–7228. doi: 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]
- 148.Iglesias Á, Álvarez R, de Lera ÁR, Muñiz K. Angew Chem Int Ed. 2012;51:2225–2228. doi: 10.1002/anie.201108351. [DOI] [PubMed] [Google Scholar]
- 149.Li G, Leow D, Wan L, Yu JQ. Angew Chem Int Ed. 2013;52:1245–1247. doi: 10.1002/anie.201207770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Leow D, Li G, Mei TS, Yu JQ. Nature. 2012;486:518–522. doi: 10.1038/nature11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schwarz H. Acc Chem Res. 1989;22:282–287. [Google Scholar]
- 152.Dietl N, Schlangen M, Schwarz H. Chem Eur J. 2011;17:1783–1788. doi: 10.1002/chem.201003041. [DOI] [PubMed] [Google Scholar]
- 153.Huang C, Chattopadhyay B, Gevorgyan V. J Am Chem Soc. 2011;133:12406–12409. doi: 10.1021/ja204924j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Novák P, Correa A, Gallardo-Donaire J, Martin R. Angew Chem Int Ed. 2011;50:12236–12239. doi: 10.1002/anie.201105894. [DOI] [PubMed] [Google Scholar]
- 155.Wang YN, Guo XQ, Zhu XH, Zhong R, Cai LH, Hou XF. Chem Commun. 2012;48:10437–10439. doi: 10.1039/c2cc34949c. [DOI] [PubMed] [Google Scholar]
- 156.Gao DW, Shi YC, Gu Q, Zhao ZL, You SL. J Am Chem Soc. 2013;135:86–89. doi: 10.1021/ja311082u. [DOI] [PubMed] [Google Scholar]
- 157.Cheng XF, Li Y, Su YM, Yin F, Wang J, Sheng J, Vora HU, Wang X, Yu JQ. J Am Chem Soc. 2013;135:1236–1239. doi: 10.1021/ja311259x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cong X, You J, Gao G, Lan J. Chem Commun. 2013;49:662–664. doi: 10.1039/c2cc37291f. [DOI] [PubMed] [Google Scholar]
- 159.For a recent example in which mono-N-protected amino acid ligands were used in Rh(III)-catalyzed C–H functionalization, see Ref. 97e.
- 160.Moritani I, Fujiwara Y. Tetrahedron Lett. 1967;8:1119–1122. [Google Scholar]
- 161.Fujiwara Y, Moritani I, Matsuda M, Teranishi S. Tetrahedron Lett. 1968;9:3863–3865. [Google Scholar]
- 162.Fujiwara Y, Moritani I, Danno S, Asano R, Teranishi S. J Am Chem Soc. 1969;91:7166–7169. doi: 10.1021/ja01053a047. [DOI] [PubMed] [Google Scholar]
- 163.Asano R, Moritani I, Fujiwara Y, Teranishi S. J Chem Soc D. 1970:1293. doi: 10.1021/ja01053a047. [DOI] [PubMed] [Google Scholar]
- 164.Tsuji J, Nagashima H. Tetrahedron. 1984;40:2699–2702. [Google Scholar]
- 165.Jia C, Lu W, Kitamura T, Fujiwara Y. Org Lett. 1999;1:2097–2100. doi: 10.1021/ol006156l. [DOI] [PubMed] [Google Scholar]
- 166.Jia C, Piao D, Oyamada J, Lu W, Kitamura T, Fujiwara Y. Science. 2000;287:1992–1995. doi: 10.1126/science.287.5460.1992. [DOI] [PubMed] [Google Scholar]
- 167.Yokota T, Tani M, Sakaguchi S, Ishii Y. J Am Chem Soc. 2003;125:1476–1477. doi: 10.1021/ja028903a. [DOI] [PubMed] [Google Scholar]
- 168.Kubota A, Emmert MH, Sanford MS. Org Lett. 2012;14:1760–1763. doi: 10.1021/ol300281p. [DOI] [PubMed] [Google Scholar]
- 169.Fujiwara Y, Moritani I, Asano R, Tanaka H, Teranishi S. Tetrahedron. 1969;25:4815–4818. [Google Scholar]
- 170.Fujiwara Y, Asano R, Moritani I, Teranishi S. J Org Chem. 1976;41:1681–1683. [Google Scholar]
- 171.Fujiwara Y, Maruyama O, Yoshidomi M, Taniguchi H. J Org Chem. 1981;46:851–855. [Google Scholar]
- 172.Itahara T, Ikeda M, Sakakibara T. J Chem Soc, Perkin Trans 1. 1983:1361–1363. [Google Scholar]
- 173.Ferreira EM, Stoltz BM. J Am Chem Soc. 2003;125:9578–9579. doi: 10.1021/ja035054y. [DOI] [PubMed] [Google Scholar]
- 174.Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J Am Chem Soc. 2002;124:1586–1587. doi: 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]
- 175.Zhang YH, Shi BF, Yu JQ. J Am Chem Soc. 2009;131:5072–5074. doi: 10.1021/ja900327e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nishimura T, Onoue T, Ohe K, Uemura S. Tetrahedron Lett. 1998;39:6011–6014. [Google Scholar]
- 177.Nishimura T, Onoue T, Ohe K, Uemura S. J Org Chem. 1999;64:6750–6755. doi: 10.1021/jo9906734. [DOI] [PubMed] [Google Scholar]
- 178.Nishimura T, Ohe K, Uemura S. J Am Chem Soc. 1999;121:2645–2646. [Google Scholar]
- 179.Fix SR, Brice JL, Stahl SS. Angew Chem Int Ed. 2002;41:164–166. doi: 10.1002/1521-3773(20020104)41:1<164::aid-anie164>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 180.Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew Chem Int Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196. [DOI] [PubMed] [Google Scholar]
- 181.Trend RM, Ramtohul YK, Stoltz BM. J Am Chem Soc. 2005;127:17778–17788. doi: 10.1021/ja055534k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schiffner JA, Oestreich M. Eur J Org Chem. 2011:1148–1154. [Google Scholar]
- 183.Kandukuri SR, Oestreich M. J Org Chem. 2012;77:8750–8755. doi: 10.1021/jo301088f. [DOI] [PubMed] [Google Scholar]
- 184.Stuart DR, Fagnou K. Science. 2007;316:1172–1175. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]
- 185.Wang C, Rakshit S, Glorius F. J Am Chem Soc. 2010;132:14006–14008. doi: 10.1021/ja106130r. [DOI] [PubMed] [Google Scholar]
- 186.Izawa Y, Stahl SS. Adv Synth Catal. 2010;352:3223–3229. doi: 10.1002/adsc.201000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Izawa Y, Pun D, Stahl SS. Science. 2011;333:209–213. doi: 10.1126/science.1204183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rentner J, Breinbauer R. Chem Commun. 2012;48:10343–10345. doi: 10.1039/c2cc35758e. [DOI] [PubMed] [Google Scholar]
- 189.Kravtsova SV, Romm IP, Stash AI, Belsky VK. Acta Crystallogr, Sect C: Cryst Struct Commun. 1996;52:2201–2204. [Google Scholar]
- 190.Zhang S, Shi L, Ding Y. J Am Chem Soc. 2011;133:20218–20229. doi: 10.1021/ja205294y. [DOI] [PubMed] [Google Scholar]
- 191.Wasa M, Chan KSL, Zhang XG, He J, Miura M, Yu JQ. J Am Chem Soc. 2012;134:18570–18572. doi: 10.1021/ja309325e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zaitsev VG, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 193.Giri R, Maugel N, Foxman BM, Yu JQ. Organometallics. 2008;27:1667–1670. [Google Scholar]
- 194.Nakao Y. Synthesis. 2011:3209–3219. [Google Scholar]
- 195.Moore EJ, Pretzer WR, O’Connell TJ, Harris J, LaBounty L, Chou L, Grimmer SS. J Am Chem Soc. 1992;114:5888–5890. [Google Scholar]
- 196.Godula K, Sezen B, Sames D. J Am Chem Soc. 2005;127:3648–3649. doi: 10.1021/ja042510p. [DOI] [PubMed] [Google Scholar]
- 197.Kawashima T, Takao T, Suzuki H. J Am Chem Soc. 2007;129:11006–11007. doi: 10.1021/ja074224u. [DOI] [PubMed] [Google Scholar]
- 198.Lewis JC, Bergman RG, Ellman JA. J Am Chem Soc. 2007;129:5332–5333. doi: 10.1021/ja070388z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Berman AM, Lewis JC, Bergman RG, Ellman JA. J Am Chem Soc. 2008;130:14926–14927. doi: 10.1021/ja8059396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N. Tetrahedron Lett. 2002;43:5649–5651. [Google Scholar]
- 201.Mkhalid IAI, Coventry DN, Albesa-Jove D, Batsanov AS, Howard JAK, Perutz RN, Marder TB. Angew Chem Int Ed. 2006;45:489–491. doi: 10.1002/anie.200503047. [DOI] [PubMed] [Google Scholar]
- 202.Murphy JM, Liao X, Hartwig JF. J Am Chem Soc. 2007;129:15434–15435. doi: 10.1021/ja076498n. [DOI] [PubMed] [Google Scholar]
- 203.Fischer DF, Sarpong R. J Am Chem Soc. 2010;132:5926–5927. doi: 10.1021/ja101893b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hurst TE, Macklin TK, Becker M, Hartmann E, Kügel W, Parisienne-La Salle J-C, Batsanov AS, Marder TB, Snieckus V. Chem Eur J. 2010;16:8155–8161. doi: 10.1002/chem.201000401. [DOI] [PubMed] [Google Scholar]
- 205.Li BJ, Shi ZJ. Chem Sci. 2011;2:488–493. [Google Scholar]
- 206.Cho JY, Iverson CN, Smith MR., III J Am Chem Soc. 2000;122:12868–12869. [Google Scholar]
- 207.Nakao Y, Kanyiva KS, Hiyama T. J Am Chem Soc. 2008;130:2448–2449. doi: 10.1021/ja710766j. [DOI] [PubMed] [Google Scholar]
- 208.Tsai CC, Shih WC, Fang CH, Li CY, Ong TG, Yap GPA. J Am Chem Soc. 2010;132:11887–11889. doi: 10.1021/ja1061246. [DOI] [PubMed] [Google Scholar]
- 209.Nakao Y, Yamada Y, Kashihara N, Hiyama T. J Am Chem Soc. 2010;132:13666–13668. doi: 10.1021/ja106514b. [DOI] [PubMed] [Google Scholar]
- 210.Grigg R, Savic V. Tetrahedron Lett. 1997;38:5737–5740. [Google Scholar]
- 211.Gürbüz N, Özdemir I, Çetinkaya B. Tetrahedron Lett. 2005;46:2273–2277. [Google Scholar]
- 212.Wasa M, Worrell BT, Yu JQ. Angew Chem Int Ed. 2010;49:1275–1277. doi: 10.1002/anie.200906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Campeau LC, Rousseaux S, Fagnou K. J Am Chem Soc. 2005;127:18020–18021. doi: 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]
- 214.Cho SH, Hwang SJ, Chang S. J Am Chem Soc. 2008;130:9254–9256. doi: 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]
- 215.Wu J, Cui X, Chen L, Jiang G, Wu Y. J Am Chem Soc. 2009;131:13888–13889. doi: 10.1021/ja902762a. [DOI] [PubMed] [Google Scholar]
- 216.Xi P, Yang F, Qin S, Zhao D, Lan J, Gao G, Hu C, You J. J Am Chem Soc. 2010;132:1822–1824. doi: 10.1021/ja909807f. [DOI] [PubMed] [Google Scholar]
- 217.Tan Y, Barrios-Landeros F, Hartwig JF. J Am Chem Soc. 2012;134:3683–3686. doi: 10.1021/ja2122156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Larivée A, Mousseau JJ, Charette AB. J Am Chem Soc. 2007;130:52–54. doi: 10.1021/ja710073n. [DOI] [PubMed] [Google Scholar]
- 219.Mousseau JJ, Bull JA, Charette AB. Angew Chem Int Ed. 2010;49:1115–1118. doi: 10.1002/anie.200906020. [DOI] [PubMed] [Google Scholar]
- 220.Wei Y, Kan J, Wang M, Su W, Hong M. Org Lett. 2009;11:3346–3349. doi: 10.1021/ol901200g. [DOI] [PubMed] [Google Scholar]
- 221.Wei Y, Su W. J Am Chem Soc. 2010;132:16377–16379. doi: 10.1021/ja109383e. [DOI] [PubMed] [Google Scholar]
- 222.Lin BL, Labinger JA, Bercaw JE. Can J Chem. 2009;87:264–271. [Google Scholar]
- 223.McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org Lett. 2011;13:2830–2833. doi: 10.1021/ol200784y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Perch NS, Widenhoefer RA. J Am Chem Soc. 1999;121:6960–6961. [Google Scholar]
- 225.Tsujihara T, Takenaka K, Onitsuka K, Hatanaka M, Sasai H. J Am Chem Soc. 2009;131:3452–3453. doi: 10.1021/ja809965e. [DOI] [PubMed] [Google Scholar]
- 226.Takenaka K, Akita M, Tanigaki Y, Takizawa S, Sasai H. Org Lett. 2011;13:3506–3509. doi: 10.1021/ol201314m. [DOI] [PubMed] [Google Scholar]
- 227.He W, Yip KT, Zhu NY, Yang D. Org Lett. 2009;11:5626–5628. doi: 10.1021/ol902348t. [DOI] [PubMed] [Google Scholar]
- 228.Jiang F, Wu Z, Zhang W. Tetrahedron Lett. 2010;51:5124–5126. [Google Scholar]
- 229.Yang G, Shen C, Zhang W. Angew Chem Int Ed. 2012;51:9141–9145. doi: 10.1002/anie.201203693. [DOI] [PubMed] [Google Scholar]
- 230.Eberson L, Jonsson E. Acta Chem Scand Ser B. 1974;28:771–776. [Google Scholar]
- 231.Eberson L, Jönsson L. J Chem Soc, Chem Commun. 1974:885–886. [Google Scholar]
- 232.Eberson L, Jönsson L. Acta Chem Scand Ser B. 1976;30:361–364. [Google Scholar]
- 233.Eberson L, Jönsson L. Liebigs Ann Chem. 1977:233–241. [Google Scholar]
- 234.Jintoku T, Taniguchi H, Fujiwara Y. Chem Lett. 1987;16:1159–1162. [Google Scholar]
- 235.Campbell AN, Meyer EB, Stahl SS. Chem Commun. 2011;47:10257–10259. doi: 10.1039/c1cc13632a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Diao T, Wadzinski TJ, Stahl SS. Chem Sci. 2012;3:887–891. doi: 10.1039/C1SC00724F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hickman AJ, Sanford MS. ACS Catal. 2011;1:170–174. [Google Scholar]
- 238.Zhu C, Falck JR. Org Lett. 2011;13:1214–1217. doi: 10.1021/ol200093f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kirchberg S, Tani S, Ueda K, Yamaguchi J, Studer A, Itami K. Angew Chem Int Ed. 2011;50:2387–2391. doi: 10.1002/anie.201007060. [DOI] [PubMed] [Google Scholar]
- 240.Steinmetz M, Ueda K, Grimme S, Yamaguchi J, Kirchberg S, Itami K, Studer A. Chem Asian J. 2012;7:1256–1260. doi: 10.1002/asia.201101011. [DOI] [PubMed] [Google Scholar]
- 241.Yamaguchi K, Yamaguchi J, Studer A, Itami K. Chem Sci. 2012;3:2165–2169. [Google Scholar]
- 242.Shibahara F, Yamauchi T, Yamaguchi E, Murai T. J Org Chem. 2012;77:8815–8820. doi: 10.1021/jo301621t. [DOI] [PubMed] [Google Scholar]
- 243.Ma Y, You J, Song F. Chem Eur J. 2013;19:1189–1193. doi: 10.1002/chem.201203354. [DOI] [PubMed] [Google Scholar]
- 244.Jiang TS, Wang GW. Org Lett. 2013;15:788–791. doi: 10.1021/ol303440y. [DOI] [PubMed] [Google Scholar]
- 245.Izawa Y, Zheng C, Stahl SS. Angew Chem Int Ed. 2013;52:3672–3675. doi: 10.1002/anie.201209457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ye M, Gao GL, Yu JQ. J Am Chem Soc. 2011;133:6964–6967. doi: 10.1021/ja2021075. [DOI] [PubMed] [Google Scholar]
- 247.Subsequent to our work, an example of Pd(II)-catalyzed C2-selective pyridine C–H olefination was reported: Wen P, Li Y, Zhou K, Ma C, Lan X, Ma C, Huang G. Adv Synth Catal. 2012;354:2135–2140. In the absence of an ancillary ligand, N-coordination appears to facilitate C–H cleavage at the proximal C2 position.
- 248.Ye M, Gao GL, Edmunds AJF, Worthington PA, Morris JA, Yu JQ. J Am Chem Soc. 2011;133:19090–19093. doi: 10.1021/ja209510q. [DOI] [PubMed] [Google Scholar]
- 249.In a recent report, aryl tosylates were also found to be effective coupling partners in this transformation: Dai F, Gui Q, Liu J, Yang Z, Chen X, Guo R, Tan Z. Chem Commun. 2013 doi: 10.1039/C3CC41066H.
- 250.De Leon A, Pons J, Solans X, Font-Bardia M. Acta Crystallogr, Sect E: Struct Rep Online. 2007;63:m2164. doi: 10.1107/S1600536808008337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Milani B, Scarel A, Zangrando E, Mestroni G, Carfagna C, Binotti B. Inorg Chim Acta. 2003;350:592–602. [Google Scholar]
- 252.Rotondo A, Bruno G, Cusumano M, Rotondo E. Inorg Chim Acta. 2009;362:4767–4773. [Google Scholar]
- 253.Holzbock J, Sawodny W, Thewalt U. Z Anorg Allg Chem. 2000;626:2563–2568. [Google Scholar]
- 254.Tebbe KF, Gräfe-Kavoosian A, Freckmann B. Z Naturforsch, B: J Chem Sci. 1996;51:999–1006. [Google Scholar]
- 255.Ma L, Smith RC, Protasiewicz JD. Inorg Chim Acta. 2005;358:3478–3482. [Google Scholar]
- 256.Quagliano JV, Schubert L. Chem Rev. 1952;50:201–260. [Google Scholar]
- 257.Coe BJ, Glenwright SJ. Coord Chem Rev. 2000;203:5–80. [Google Scholar]
- 258.Gorelsky SI. Organometallics. 2012;31:794–797. [Google Scholar]
- 259.Petit A, Flygare J, Miller AT, Winkel G, Ess DH. Org Lett. 2012;14:3680–3683. doi: 10.1021/ol301521n. [DOI] [PubMed] [Google Scholar]
- 260.Ye M, Edmunds AJF, Morris JA, Sale D, Zhang Y, Yu J-Q. Chem Sci. 2013 doi: 10.1039/C3SC50184A.For closely related reactions reported by other groups, see: Hattori K, Yamaguchi K, Yamaguchi J, Itami K. Tetrahedron. 2012;68:7605–7612.Ben-Yahia A, Naas M, El Kazzouli S, Essassi EM, Guillaumet G. Eur J Org Chem. 2012:7075–7081.