

Abstract
Idiopathic dilated cardiomyopathy (DCM) is a primary myocardial disorder characterized by ventricular chamber enlargement and systolic dysfunction. Twenty to fifty percent of idiopathic DCM cases are thought to have a genetic cause. Of more than 30 genes known to be associated with DCM, rare variants in the VCL and MYBPC3 genes have been reported in several cases of DCM. In this report, we describe a family with DCM and congenital abnormalities who carry a novel missense mutation in the VCL gene. More severely affected family members also possess a second missense variant in MYBPC3, raising the possibility that this variant may be a disease modifier. Interestingly, many of the affected individuals also have congenital defects, including two with bicuspid aortic valve with aortic regurgitation. We discuss the implications of the family history and genetic information on management of at-risk individuals with aortic regurgitation.
Keywords: Dilated cardiomyopathy, Vinculin, Myosin binding protein C, VCL, MYBPC
Introduction
Dilated cardiomyopathy (DCM) is a primary myocardial disorder characterized by ventricular chamber enlargement and systolic dysfunction. Idiopathic dilated cardiomyopathy is diagnosed when secondary causes have been excluded, and accounts for up to half of all patients presenting with a dilated phenotype.(1-4) DCM is relatively common, with an estimated prevalence of 1 in 2,500 persons. It is the third most common cause of heart failure and the most frequent indication for heart transplantation.(5) Reports thus far suggest that 20-50% of idiopathic DCM cases have a genetic cause, and more than 30 causal genes, encoding proteins with a wide range of functions, have been identified.(2, 5-8) The autosomal dominant mode of inheritance is estimated to account for 90% of familial DCM, with autosomal recessive and X-linked inheritance being considerably less common.(9) The vinculin (VCL) and myosin binding protein C (MYBPC3) genes, though not typically associated with familial DCM, have each been described in several cases of DCM.(10-13) Our report describes a family in which the proband and multiple related family members with dilated cardiomyopathy share a novel rare variant (c.2444A>G) in exon 17 of the VCL gene resulting in as substitution of arginine for lysine at position 815. The most severely affected family members also possess a previously reported the Arg177Cys variant in the MYPBC3 gene.
Case Presentation
The proband was a 69-year-old woman followed in heart transplant clinic with a complex cardiac history who had undergone heart transplantation 8 years earlier. Prior to that she had been followed at another facility where she had surgical repair of an ASD at the age of 53. At the time of surgery she underwent simultaneous coronary artery bypass grafting, reportedly due to diagnosed coronary disease. She later developed cardiomyopathy and had stable Class III heart failure symptoms for a number of years on medical therapy. Echocardiographic evaluation was technically difficult, but left ventricular (LV) function was reported as 45-50% (LV diastolic diameter 45 mm, LV systolic diameter 29 mm). Right ventricle was dilated with mildly reduced systolic function. There was no evidence of left ventricular non-compaction. Her symptoms progressed and she ultimately underwent orthotopic heart transplantation at the age of 61. The explanted heart was enlarged (11 × 10 × 7.5 cm with a total mass of 230 grams) and demonstrated a markedly dilated right ventricle and moderately dilated left ventricle. Pathologic examination of the coronary arteries showed only mild atherosclerotic disease (no vessel with >10% stenosis) and it appears that her prior bypass was unnecessary. There were widespread areas of fibrous subendocardial white discoloration of both ventricles, but no histopathologic examination was performed. At the time of transplantation, she was felt to have an advanced non-ischemic dilated (based on autopsy findings and poor quality of echocardiographic data) cardiomyopathy with associated widespread biventricular subendocardial fibrosis, a late-stage markedly remodeled and dilated RV that exhibited moderate systolic dysfunction at rest and a dilated LV that exhibited mild to moderate systolic dysfunction at rest.
There was a family history of dilated cardiomyopathy associated with minor congenital abnormalities, with the proband’s sister, father, and brother all affected (Figure 1). Her sister underwent surgical repair of a patent ductus arteriosus during childhood and died at age 53 from dilated cardiomyopathy. The father of the proband had died at the age of 58, also of dilated cardiomyopathy. The proband’s brother had a bicuspid aortic valve complicated by severe aortic insufficiency requiring aortic valve replacement at the age of 40. Despite valve replacement, he developed progressive systolic dysfunction and Class III heart failure, prompting evaluation in the advanced heart failure clinic for consideration of transplantation at the age of 57. Echocardiography at that time demonstrated a severely reduced left ventricular ejection fraction of 10-20% as well as ventricular dilation with a left ventricular end diastolic diameter of 57 mm. Fortunately, he responded well to cardiac resynchronization therapy with stable Class II symptoms and improvement in ejection fraction to 30%.
Figure 1.
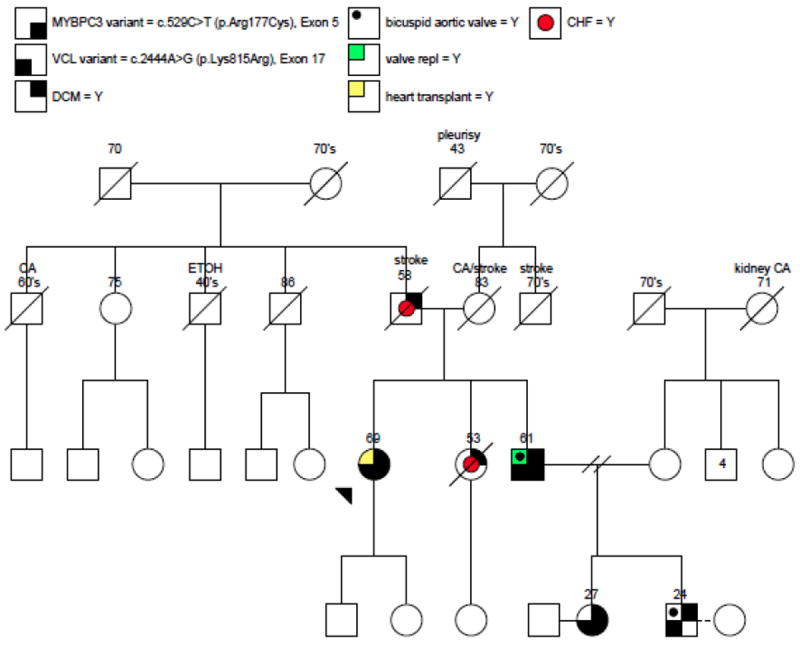
Pedigree of proband and affected family members
Proband is identified by arrow. Numbers above family members indicate age. Relevant clinical information provided as needed. CA=Cancer, ETOH = heavy alcohol use.
Owing to the family history of dilated cardiomyopathy, two asymptomatic siblings who were niece and nephew of the proband (age 27 and age 24 respectively; Figure 1) underwent screening echocardiography. The niece was found to have a mildly decreased ejection fraction of 45% with mild global hypokinesis (LVEDD 51 mm, LVESD 40 mm). There was no hypertrophy or ventricular non-compaction and valves were normal. Her brother, nephew of the proband, was found to have a bicuspid aortic valve with mild-moderate aortic insufficiency, mildly dilated left ventricle (LVEDD 60mm, LVESD 42mm), and preserved systolic function with an ejection fraction of 60%. On the basis of clinical data and family history, genetic testing was performed on the four available family members.
Genetic Testing
The DCM genetic screening of the proband was performed by the Laboratory for Molecular Medicine (Partners HealthCare Center for Personalized Genetic Medicine, Boston, MA, USA) using an array-based resequencing platform. This test screens the coding sequence and splice sites of 19 genes previously implicated in familial cardiomyopathy and has an estimated clinical sensitivity of 26-29% in individuals with clinical features of dilated cardiomyopathy (See Table 1).(14, 15) The proband was found to be heterozygous for two missense variants: a c.2444A>G (p.Lys815Arg) variant in exon 17 of the vinculin gene (VCL) and a c.529C>T (p.Arg177Cys) variant in exon 5 of the cardiac myosin binding protein 3 gene (MYBPC3).
Table 1.
Genes tested by DCM array based resequencing platform
| Gene | Full Name | Location/Function | Locus |
|---|---|---|---|
| MYBPC3 | Myosin-binding protein C | Sarcomere | 11p11.2 |
| MYH7 | Myosin, heavy chain 7 | Sarcomere | 14q12 |
| TNNT2 | Troponin t2, cardiac | Sarcomere | 1q32 |
| TNNI3 | Troponin i, cardiac | Sarcomere | 19q13.4 |
| TPM1 | Tropomyosin, alpha 1 | Sarcomere | 15q22.1 |
| ACTC | Alpha-Actinin | Sarcomere | 15q14 |
| LMNA | Lamin A/C | Nuclear Lamina | 1q21.2-3 |
| PLN | Phospholamban | Cardiac Ca++ | 6q22.1 |
| ATPase inhibitor | |||
| TAZ | Tafazzin | Mitochondrial cardiolipin transacylase | Xq28 |
| LDB3 | Cypher/ZASP | Z-disk | 10q22.2-q23.3 |
| ACTN2 | Actinin, alpha 2 | Z-disk | 1q42-q43 |
| CSRP3 | Cysteine and glycine-rich protein 3 | Z-disk | 11p15.1 |
| TCAP | Titin-cap | Z-disk | 17q12 |
| VCL | Vinculin | Z-disk | 10q22.1-q23 |
| ABCC9 | ATP-binding cassette, subfamily C, member 9 | ATP sensitive | 12p12.1 |
| K channel | |||
| CTF1 | Cardiotrophin 1 | Cytokine | 16p11.2-p11.1 |
| SGCD | Delta-Sarcoglycan | Dystrophin Assoc. Protein Complex | 5q33-q34 |
| DES | Desmin | Intermediate Filament | 2q35 |
| EMD | Emerin | Nuclear Lamina | Xq28 |
Targeted sequencing of the VCL and MYBPC3 genes of the proband’s brother, niece and nephew revealed that all four family members carried the p.Lys815Arg variant in the VCL gene, and three of the four (proband, brother and niece) also carried the p.Arg177Cys variant in the MYBPC3 gene.
Discussion
The VCL gene, which is highly conserved throughout vertebrate evolution, is located on chromosome 10q22-q23 and contains 22 exons.(16) VCL encodes two protein isoforms via alternate splicing of exon 19. Vinculin is ubiquitously expressed, while metavinculin, the larger isoform containing an additional 68 amino acids, is expressed only in cardiac and smooth muscle.(17, 18) Vinculin and metavinculin are cytoskeletal protein components of intercalated disks and are associated with cell-cell and cell-matrix junctions, where they are thought to participate in anchoring F-actin to the membrane and the transmission of contractile force between cardiac myocytes.(11) Animal data suggests that mutations in vinculin can result in dilated cardiomyopathy. Zebrafish morphants in which the vinculin gene has been knocked down display a DCM phenotype marked by progressive impairment in cardiac contractility, pericardial edema, and blood congestion.(19) A mouse model with cardiac myocyte specific inactivation of the vinculin gene displays dilated cardiomyopathy and death occur before 6 months of age, though sudden cardiac death was also described in this model at earlier ages and in the absence of contractile dysfunction.(20)
In humans, mutations in the VCL gene have been associated with both HCM and DCM.(11-13) Two VCL variants (Arg975Trp and Leu954del), both affecting the cardiac-specific isoform metavinculin, have previous been associated with DCM.(11) Both variants significantly altered metavinculin-mediated cross-linking of actin filaments in an in vitro assay, suggesting that disruption of force transmission at the thin filament-intercalated disc interface may be a mechanism by which mutations in metavinculin lead to DCM.(11) Later, the p.Arg975Trp variant was also identified in a patient with HCM, suggesting that different cardiomyopathy phenotypes may result from the same variant due to modifier genes and/or environmental stressors in cardiac remodeling.(13) However, the mechanisms underlying HCM and DCM are generally felt to be fundamentally different and it is unclear whether one variant can cause both phenotypes. A third VCL variant (p.Leu277Met), affecting the vinculin isoform, has also been described in a patient with HCM.(12) Alterations in the VCL gene affecting the expression of metavinculin have been identified in patients with DCM.(10, 11) Thus, though variants in the VCL gene have been found in individuals with HCM as well as DCM,(11-13) the number of studies is still small and the contribution of VCL to HCM and DCM as well as the types of variants that cause disease have not been well established.
While we cannot conclude definitively at this time that the c.2444A>G (p.Lys815Arg) VCL variant we have identified is responsible for DCM in our pedigree, this amino acid substitution may be poorly tolerated over the course of an individual’s lifetime. The Lysine-815 of the vinculin protein is highly conserved across distant vertebrate species, including mouse, chicken, and fish, suggesting that even a subtle amino acid substitution at this site might be consequential in terms of cardiac structure and function. Consistent with its pathogenic potential, the p.Lys815Arg variant described in the current pedigree has not been reported previously in the literature, nor was it identified in 328 chromosomes sequenced from healthy Caucasian controls from the Harvard-Partners Center for Genetics and Genomics. However, determining the clinical significance of missense variants can be challenging, as the impact to the protein can be difficult to discern. While data show that the majority of missense variants have an effect on the protein and that the overall population frequency correlates with the severity of the effect, it can be difficult to rule out that a variant is a benign change present in the population at low frequency.(21)
The MYBPC3 gene is located on chromosome 11p11 and encodes the cardiac myosin binding protein. While mutations in MYBPC3 are more commonly associated with hypertrophic cardiomyopathy, responsible for about 25% of HCM cases,(8) a few MYPBC3 mutations have been associated with DCM. For example, a p.Arg272Cys MYBPC3 variant has been described in two familial DCM cases and in a case of peripartum cardiomyopathy.(22-24) Another variant, p.Asn948Thr, has also been described in a patient with DCM patient that was absent in both 88 healthy controls and 137 HCM patients.(25)
Three of the four patients tested in our pedigree were found to carry a missense mutation in MYBPC3 (p.Arg177Cys) on exon 5. While the Arginine-177 is not absolutely conserved across difference species (chicken carries a Lysine), this is a very rare variant, not detected in >1500 individuals tested in the Harvard-Partners Center for Genetics and Genomics.
The p.Arg177Cys variant has been observed previously in one proband with HCM (26). However, it is unclear whether the same variant can cause both HCM and DCM. In addition, arginine (Arg) at position 177 is not 100% conserved across different species (chicken carries a lysine), reducing the likelihood that the change is pathogenic. This variant was also predicted to be benign using a novel computational tool, which was validated using a set of cardiomyopathy variants with well-established clinical significance. This tool’s benign interpretation is estimated to be correct 89% of the time, which suggests but does not prove that this variant is benign.(27) The pathogenic significance of the p.Arg177Cys MYBPC3 variant in our family cohort is not clear, especially since no family member tested carries this variant in isolation. However, those family members who carry both the VCL variant and the MYBPC3 variant appear to be more severely affected, suggesting that the MYBPC3 variant may be a disease modifier which exacerbates DCM progression in individuals made susceptible by the presence of the p.Lys815Arg VCL variant. Therefore, while the available evidence raises the possibility that the p.Arg177Cys variant may be insufficient to cause disease when present in isolation we cannot yet rule out that it may have a mild effect. To determine the pathological significance of the p.Arg177Cys MYBPC3 variant, in isolation or in association with the p.Lys815Arg VCL variant, it will be necessary to examine the genotypes and phenotypes of additional family members.
The finding of associated congenital abnormalities along with cardiomyopathy in our pedigree is very interesting. An atrial septal defect was present in the proband, who had evidence of advanced right ventricular remodeling - perhaps related to ASD, though the possibility of a genetic susceptibility to hemodynamic stress cannot be dismissed. Another relative had a patent ductus arteriosus, and bicuspid aortic valve was documented in two additional members. There is some evidence that bicuspid aortic valve, in particular, is heritable. Two reports have documented that among the first-degree relatives of probands with bicuspid aortic valve, the prevalence of bicuspid aortic valve is ~9%, compared to ~1% in the general population.(28, 29) Patients with bicuspid aortic valve commonly have other congenital abnormalities, most often coarctation of the aorta, though other anomalies including ventricular septal defects, atrial septal defects, patent ductus arteriosus, abnormalities of the mitral valve, aortic root dilation, and hypoplastic left heart syndrome have been described.(28) Bicuspid aortic valve has been linked to chromosome 15q25-26 in one analysis,(30) and variants in the NOTCH1 gene have been associated with a dominantly inherited phenotype that includes bicuspid aortic valve.(31) From the data available we cannot conclude whether the observed congenital abnormalities are related to variants in VCL or MYBPC3 or due to other, undetected, genetic or non-genetic cause. To our knowledge there is no known biologic mechanism that could link mutations in VCL or MYBPC3 to the defects in our pedigree. However, the observation of concomitant cardiomyopathy in the setting of VCL variants and congenital heart defects does raise titillating questions as to whether their presence could be a clinical clue to the etiology in patients with cardiomyopathy.
Our report of a novel variant VCL associated with familial DCM and congenital abnormalities provides further support for the idea that mutations in VCL can lead to DCM. Our study also suggests that rare variants in MYBPC3 may contribute to exacerbation of the disease phenotype in susceptible individuals. Interestingly, our family cohort also had significant family history concomitant congenital heart abnormalities, including patent ductus arteriosus, atrial septal defect and bicuspid aortic valve. Presently, it is not clear whether these congenital abnormalities are causally related to the VCL and MYBPC3 variants reported here or if they reflect the presence of an uncharacterized mutation in our family cohort. Finally, it is noted that proband was affected with an ASD and both the proband’s brother and nephew were affected by bicuspid aortic valves with aortic insufficiency, which may be been hemodynamic modifiers contributing to the disease progression. In terms of clinical management, our observation is particularly germane for the 24-year-old nephew, who is asymptomatic yet has already developed left ventricular chamber dilatation. Given the family history of progressive systolic heart failure and the presence of a potentially pathogenic VCL variant in the nephew, it may be prudent not only to closely monitor his left ventricular structure and function but also consider aortic valve repair or replacement earlier than otherwise indicated. Additionally, further study regarding the possible relationship of VCL related cardiomyopathy and congenital heart disease is warranted.
Acknowledgments
Genetic testing program at the Vanderbilt Center for Inherited Heart Disease was funded in part by the generous grant from the Edwards Lifesciences Fund. C.C.H. was funded by the Veterans Affairs Career Development Transition Award, VA Merit Award, National Institutes of Health grants 5U01HL100398 and 1R01HL104040.
Footnotes
Contribution of authors:
Quinn Wells: Acquisition, analysis and interpretation of data, manuscript drafting/revision
Natalie Ausborn: Acquisition, analysis and interpretation of data, manuscript drafting/revision
Birgit H Funke: Project design, acquisition, analysis and interpretation of data, manuscript revision
Jean P. Pfotenhauer: Acquisition, analysis and interpretation of data, manuscript revision
Joseph L. Fredi: Acquisition, analysis and interpretation of data, manuscript revision
Samantha Baxter: Acquisition, analysis and interpretation of data, manuscript revision
Thomas D. DiSalvo: Project conception and design, acquisition, analysis and interpretation of data, manuscript drafting/revision
Charles C. Hong: Project conception and design, acquisition, analysis and interpretation of data, manuscript revision
Conflicts of interest:
None of the authors report conflicts of interest.
References
- 1.Kasper EK, Agema WR, Hutchins GM, Deckers JW, Hare JM, Baughman KL. The causes of dilated cardiomyopathy: a clinicopathologic review of 673 consecutive patients. J Am Coll Cardiol. 1994 Mar 1;23(3):586–90. doi: 10.1016/0735-1097(94)90740-4. [DOI] [PubMed] [Google Scholar]
- 2.Fatkin D, Robyn O, Richmond Zara. Genetics of dilated cardiomyopathy. Heart Failure Clin. 2010:129–40. doi: 10.1016/j.hfc.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Manolio TA, Baughman KL, Rodeheffer R, Pearson TA, Bristow JD, Michels VV, et al. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop. Am J Cardiol. 1992 Jun 1;69(17):1458–66. doi: 10.1016/0002-9149(92)90901-a. [DOI] [PubMed] [Google Scholar]
- 4.Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000 Apr 13;342(15):1077–84. doi: 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006 Apr 11;113(14):1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 6.Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998 Jan;31(1):186–94. doi: 10.1016/s0735-1097(97)00434-8. [DOI] [PubMed] [Google Scholar]
- 7.Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992 Jan 9;326(2):77–82. doi: 10.1056/NEJM199201093260201. [DOI] [PubMed] [Google Scholar]
- 8.Marian AJ. The genetic basis of cardiomyopathy. Curr Cardiovasc Risk Reports. 2008;2:468–75. [Google Scholar]
- 9.Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet Med. 2010 Nov;12(11):655–67. doi: 10.1097/GIM.0b013e3181f2481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maeda M, Holder E, Lowes B, Valent S, Bies RD. Dilated cardiomyopathy associated with deficiency of the cytoskeletal protein metavinculin. Circulation. 1997 Jan 7;95(1):17–20. doi: 10.1161/01.cir.95.1.17. [DOI] [PubMed] [Google Scholar]
- 11.Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation. 2002 Jan 29;105(4):431–7. doi: 10.1161/hc0402.102930. [DOI] [PubMed] [Google Scholar]
- 12.Vasile VC, Ommen SR, Edwards WD, Ackerman MJ. A missense mutation in a ubiquitously expressed protein, vinculin, confers susceptibility to hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2006 Jul 7;345(3):998–1003. doi: 10.1016/j.bbrc.2006.04.151. [DOI] [PubMed] [Google Scholar]
- 13.Vasile VC, Will ML, Ommen SR, Edwards WD, Olson TM, Ackerman MJ. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab. 2006 Feb;87(2):169–74. doi: 10.1016/j.ymgme.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Medicine H-PCfPG. [2010 December 19];DCM CardioChip TEST. 2010 [Web Page]; Available from: http://pcpgm.partners.org/lmm/tests/cardiomyopathy/DCM.
- 15.Zimmerman RS, Cox S, Lakdawala NK, Cirino A, Mancini-DiNardo D, Clark E, et al. A novel custom resequencing array for dilated cardiomyopathy. Genet Med. 2010 May;12(5):268–78. doi: 10.1097/GIM.0b013e3181d6f7c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moiseyeva EP, Weller PA, Zhidkova NI, Corben EB, Patel B, Jasinska I, et al. Organization of the human gene encoding the cytoskeletal protein vinculin and the sequence of the vinculin promoter. J Biol Chem. 1993 Feb 25;268(6):4318–25. [PubMed] [Google Scholar]
- 17.Belkin AM, Ornatsky OI, Kabakov AE, Glukhova MA, Koteliansky VE. Diversity of vinculin/meta-vinculin in human tissues and cultivated cells. Expression of muscle specific variants of vinculin in human aorta smooth muscle cells. J Biol Chem. 1988 May 15;263(14):6631–5. [PubMed] [Google Scholar]
- 18.Koteliansky VE, Ogryzko EP, Zhidkova NI, Weller PA, Critchley DR, Vancompernolle K, et al. An additional exon in the human vinculin gene specifically encodes meta-vinculin-specific difference peptide. Cross-species comparison reveals variable and conserved motifs in the meta-vinculin insert. Eur J Biochem. 1992 Mar 1;204(2):767–72. doi: 10.1111/j.1432-1033.1992.tb16692.x. [DOI] [PubMed] [Google Scholar]
- 19.Vogel B, Meder B, Just S, Laufer C, Berger I, Weber S, et al. In-vivo characterization of human dilated cardiomyopathy genes in zebrafish. Biochem Biophys Res Commun. 2009 Dec 18;390(3):516–22. doi: 10.1016/j.bbrc.2009.09.129. [DOI] [PubMed] [Google Scholar]
- 20.Zemljic-Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, et al. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol. 2007 Nov;27(21):7522–37. doi: 10.1128/MCB.00728-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007 Apr;80(4):727–39. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morales A, Painter T, Li R, Siegfried JD, Li D, Norton N, et al. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation. 2010 May 25;121(20):2176–82. doi: 10.1161/CIRCULATIONAHA.109.931220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehlermann P, Weichenhan D, Zehelein J, Steen H, Pribe R, Zeller R, et al. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3 gene. BMC Med Genet. 2008;9:95. doi: 10.1186/1471-2350-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010 Apr;3(2):155–61. doi: 10.1161/CIRCGENETICS.109.912345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M, et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun. 2002 Oct 18;298(1):116–20. doi: 10.1016/s0006-291x(02)02374-4. [DOI] [PubMed] [Google Scholar]
- 26.Genomics of Cardiovascular Development, Adaptation, and Remodeling. [2011 May];NHLBI Program for Genomic Applications, Harvard Medical School. Available from: http://www.cardiogenomics.org.
- 27.Jordan DM, Kiezun A, Baxter SM, Agarwala V, Green RC, Murray MF, et al. Development and validation of a computational method for assessment of missense variants in hypertrophic cardiomyopathy. Am J Hum Genet. 2011 Feb 11;88(2):183–92. doi: 10.1016/j.ajhg.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004 Jul 7;44(1):138–43. doi: 10.1016/j.jacc.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 29.Huntington K, Hunter AG, Chan KL. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol. 1997 Dec;30(7):1809–12. doi: 10.1016/s0735-1097(97)00372-0. [DOI] [PubMed] [Google Scholar]
- 30.Goh D, Han L, Judge D. Linkage of familial bicuspid aortic valve with aortic aneurysm to chromosome 15q. Am J Hum Genet. 2002;(Suppl 71):11. Abstr. [Google Scholar]
- 31.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005 Sep 8;437(7056):270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]