

Abstract
Alström syndrome (ALMS) is a rare autosomal recessive disorder caused by mutations in the ALMS1 gene. We report on two brothers, 2 and 3 years of age, diagnosed with Alström syndrome who initially presented in infancy with severe dilated cardiomyopathy during febrile respiratory infection. The disease course in the two siblings was marked by significant intra-familial variability. While cardiomyopathy in the older sibling has mainly resolved allowing for the discontinuation of medical therapy, heart function in the younger sibling continues to deteriorate despite maximal drug support with furosemide, carvedilol, captopril and aldospirone. Genetic analysis revealed homozygous mutations, c.8008C>T (R2670X), in ALMS1 resulting in premature protein truncation. This report further emphasizes the exceptional intra-familial variability of ALMS, mainly in the natural course of cardiac disease.
Keywords: Alström syndrome, dilated cardiomyopathy, autosomal recessive, ALMS1 gene
INTRODUCTION
Alström syndrome is a rare monogenic disease caused by recessively-inherited mutations in ALMS1 [2]. The disease is characterized by multi-systemic involvement including progressive cone-rod dystrophy eventually leading to complete blindness, sensorineural hearing loss, childhood obesity, multiple endocrine dysfunction, including type 2 diabetes mellitus, pulmonary disease, and variable involvement of other systems [10]. Dilated cardiomyopathy (DCM) is one of the major manifestations of the syndrome ranging from sudden-onset infantile congestive heart failure (CHF) and DCM, which often resolves with treatment, to adult onset cardiomyopathy, sometimes of the restrictive hypertrophic form, and CHF, with a poor prognosis. Of note, about one third of Alström patients will never develop cardiac involvement. CHF in infancy or early childhood, along with nystagmus and photophobia are strong evidence leading to the correct diagnosis of ALMS. Importantly, infantile CHF can recur in adolescence or adulthood with a poor prognosis for affected patients [10]. Herein, we present two brothers with ALMS with completely different courses of cardiac disease.
CASE REPORTS
The patients are two brothers born to healthy parents of Arab Muslim origin.
Case 1
This 4 year (y) old boy was born at term following an uncomplicated pregnancy and vaginal delivery. His birth weight was 2900g, He was first admitted at 4 weeks (w) of age with cyanotic spells and severe respiratory distress. Echocardiography showed dilated left atrium and left ventricle with 26% shortening fraction (normal range 30–40%) and mild tricuspid regurgitation resulting in a diagnosis of DCM. Medical treatment was started including furosemide, digoxin, and ACE inhibitors. Surprisingly, on follow-up, steady improvement of his cardiac function was observed, allowing gradual decrease of his medical treatment until complete resolution was achieved.
Photophobia, nystagmus and visual disturbance developed gradually during his second year of life with concomitant deterioration of visual acuity. At the age of 3y, electroretinogram (ERG) and visual evoked potential (VEP) studies revealed severe bilateral cone-rod dystrophy.
Beginning at the age of 2y, his weight increased gradually crossing weight centiles from the 50th centile to 4SD above the mean. This weight gain was not associated with appropriate height gain, as reflected by his BMI of 23.5 (3.5SD above mean) at 3y. His laboratory chemistries were unremarkable except for mild to moderate hypertriglyceridemia and hypercholesterolemia. Glucose levels, as well as liver and renal function were normal. In addition, he displayed mild to moderate motor delay, although no deficits were found on neurological examination. He attends a mainstream educational system but receives physical and occupational therapy. His last cardiologic and echocardiographic examination at the age of 4 y was normal with shortening fraction of 35% (figure 1B).
Figure 1.
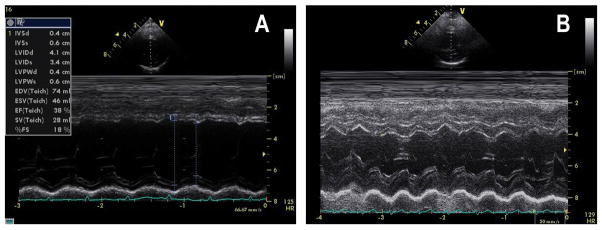
2D guided left ventricular M-mode echocardiography showing in case 1 at 4y normal left ventricular function and left ventricular fractional shortening of 35%(B), and case 2 at 3y with dilated left ventricle, reduced left ventricular function with fractional shortening of 18% (A). S=systole, D=diastole, LV=left ventricle, RV-right ventricle, LVPW=left ventricle posterior wall, IVS=inter-ventricular septum, FS=fractional shortening
Case 2
This 2.4 y old patient is the younger brother of case 1. He was born spontaneously at 35 w by vaginal delivery, after an unremarkable pregnancy, with birth weight of 2750g (10th centile). He first presented at 4 months (m) of age, with severe respiratory distress. His disease course was complicated by evolving CHF requiring admission to the pediatric intensive care unit (PICU) due to severe dilation of the left ventricle with systolic shortening fraction of 10%. The patient was given mechanical ventilation and aggressive treatment with furosemide, digoxin, aldacton and captopril. Subsequently, he has had several episodes of febrile illnesses and pneumonia, with severe reduction of his cardiac function, that required admission to the PICU and titrating of his heart medications. Similar to his older brother, photophobia and nystagmus evolved gradually towards the end of the first year of life. Initially, ophthalmologic evaluation disclosed hypermetropia but otherwise his examination, in particular the optic discs, was normal. ERG and VEP studies at 2.5 years of age were compatible with severe cone-rod dystrophy. Laboratory assessment including serum glucose, and lipid profile and liver and renal function was unremarkable. He also displayed generalized developmental delay, mainly fine and gross motor function, which was partly attributed to his cardiac dysfunction disrupting his well being and to the recurrent admissions during febrile illnesses.
At present, at age 3y, the patient’s weight is 10kg (3SD below mean). He is currently being treated with the combination of furosemide, carvedilol, aldospirone and captopril. His most recent cardiologic examination shows left apical deviation with an apical systolic murmur and echocardiography reveals dilated left ventricle with marked global reduction of systolic function and fractional shortening of 18% (Figure 1A).
Microarray and sequence analysis of ALMS1 identified both siblings to be homozygous for a nonsense mutation in exon 10 of ALMS1, c.8008C>T, that is predicted to result in early truncation of the ALMS1 protein (p.R2637X). [12] Both parents were carriers for the mutation. This mutation has been previously reported in other patients with Alström syndrome [1,11].
DISCUSSION
DCM is a myocardial disorder characterized by a dilated left ventricle and systolic dysfunction that commonly results in CHF. There are various causes of DCM, the most common of which is myocarditis. Other causes can include neuromuscular disorders, familial isolated DCM (inherited as either autosomal recessive, autosomal dominant, or x-linked), inborn errors of metabolism and multi-systemic genetic syndromes [13]. Therefore, the identification of the underlying cause of an isolated case of DCM is a major challenge both for the general pediatrician and the pediatric cardiologist. This task may be simplified by the co-occurrence of other affected systems that may indicate a specific genetic syndrome and/or the existence of similar affected family members which suggest a presumable mode of inheritance. The two siblings presented initially with CHF in infancy due to severe DCM. At that time, an autosomal recessive disorder was predicted, but the lack of other progressive manifestations failed to deliver a specific diagnosis. Only later, when retinal dystrophy was identified, the combination of infantile DCM and early onset cone-rod retinal degeneration strongly suggested Alström syndrome, which was further confirmed by the identification of homozygous ALMS1 mutations in both siblings.
The complete resolution of DCM in the older sibling is of major significance. This is even more striking when compared with the severe cardiac disease progression in the younger sibling. Spontaneous resolution of myocardial function has been reported in a subset of pediatric myocarditis patients [3]. Although the early descriptions of Alström syndrome failed to include cardiomyopathy as a frequent feature, the common prevalence of DCM in ALMS during infancy (almost 50%) has been increasingly recognized in the past few decades [9,10]. Surprisingly, of those who survive, most will make an apparently full or near complete recovery [1,9]. Therefore, resolving cardiomyopathy, especially when combined with ophthalmologic abnormalities such as photophobia, nystagmus or retinal changes should alert the physician to pursue ALMS1 genetic analysis.
The intra-familial variability of cardiac presentation between the two siblings is a remarkable finding. A similar variability was previously shown by Hoffman et al who reported four siblings with Alström syndrome. Three siblings presented with infantile DCM, two of them resolved completely, one displayed near full recovery but later experienced some deterioration in his cardiac function. The fourth sibling experienced the most severe course, culminating in end stage heart failure following an unsuccessful heart transplantation [6]. Our report further emphasizes this familial variability, both in the course and prognosis of cardiac disease. The clinical variability may be in part due to modifying alleles of genes that interact with ALMS to alter the onset and course of cardiac disease in Alström Syndrome.
The localization of ALMS1 to the centrosomes and basal bodies, the structure from which the primary cilium arises [5], implicates ALMS1 in ciliogenesis and/or normal ciliary function, and in intra-cellular trafficking and protein transport, and includes Alström syndrome among the growing list of ciliopathies [4]. Despite the putative role of ALMS1 in ciliary function, the pathophysiology of cardiomyopathy in Alström syndrome is not understood. While echocardiographic studies performed in a cohort of Alström patients demonstrated dilated cardiomyopathy [8], recent autopsy and cardiac MRI studies confirmed that the pathogenic process is mainly the result of myocardial fibrosis [7,9]. A recent study performed on ALMS1-deficient fibroblasts support these findings by showing an increased tendency for tissue fibrosis [14]. Most cases of myocardial fibrosis are gradual and non-reversible, thus the infantile onset of DCM and the high rate of resolution are difficult to explain. We thereby speculate that another pathogenic process may underly the infantile onset of cardiac Alström disease. At early stages of infancy, the DCM may be reversible, but as the patient becomes older and significant myocardial fibrosis occurs, it becomes irreversible and the prognosis is poor. Future studies are needed to clarify the evolution of myocardial disease and its heterogeneous course in Alström patients.
Acknowledgments
We thank the patients and their families for their participation in this study.
JDM, GBC, and JKN are supported by a grant from the National Institutes of Health HD036878. We are grateful to Alström Syndrome International and Alström Syndrome Canada for support for the Asper Ophthalmics microarray evaluation. The Jackson Laboratory institutional allele typing and sequencing shared services were supported by US. Public Health Service (PHS), National Institutes of Health (CA034196).
References
- 1.Bond J, Flintoff K, Higgins J, et al. The importance of seeking ALMS1 mutations in infants with dilated cardiomyopathy. J Med Genet. 2005;42:e10. doi: 10.1136/jmg.2004.026617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collin GB, Marshall JD, Ikeda A, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31:74–8. doi: 10.1038/ng867. [DOI] [PubMed] [Google Scholar]
- 3.Foerster SR, Canter CE, Cinar A, et al. Ventricular survival and survival are more favorable for myocarditis than for idiopathic dilated remodeling and cardiomyopathy in childhood: an outcomes study from the pediatric cardiomyopathy Registry. Circ Heart Fail. 2010;3:689–97. doi: 10.1161/CIRCHEARTFAILURE.109.902833. [DOI] [PubMed] [Google Scholar]
- 4.Girard D, Petrovsky N. Alström syndrome: insights into the pathogenesis of metabolic disorders. Nat Rev Endocrinol. 2011;7:77–88. doi: 10.1038/nrendo.2010.210. [DOI] [PubMed] [Google Scholar]
- 5.Hearn T, Spalluto C, Phillips VJ, et al. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes. 2005;54:1581–7. doi: 10.2337/diabetes.54.5.1581. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman JD, Jacobson Z, Young TL, et al. Familial variable expression of dilated cardiomyopathy in Alström syndrome: a report of four sibs. Am J Med Genet A. 2005;135:96–8. doi: 10.1002/ajmg.a.30688. [DOI] [PubMed] [Google Scholar]
- 7.Loudon MA, Bellenger NG, Carey CM, et al. Cardiac magnetic resonance imaging in Alström syndrome. Orphanet J Rare Dis. 2009;4:14. doi: 10.1186/1750-1172-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makaryus AN, Zubrow ME, Marshall JD, et al. Cardiac manifestations of Alström syndrome: echocardiographic findings. J Am Soc Echocardiogr. 2007;20:1359–63. doi: 10.1016/j.echo.2007.04.033. [DOI] [PubMed] [Google Scholar]
- 9.Marshall JD, Bronson RT, Collin GB, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675–83. doi: 10.1001/archinte.165.6.675. [DOI] [PubMed] [Google Scholar]
- 10.Marshall JD, Beck S, Maffei P, et al. Alström syndrome. Eur J Hum Genet. 2007;15:1193–202. doi: 10.1038/sj.ejhg.5201933. [DOI] [PubMed] [Google Scholar]
- 11.Minton JA, Owen KR, Ricketts CJ, et al. Syndromic obesity and diabetes: changes in body composition with age and mutation analysis of ALMS1 in 12 United Kingdom kindreds with Alstrom syndrome. J Clin Endocrinol Metab. 2006;91:3110–6. doi: 10.1210/jc.2005-2633. [DOI] [PubMed] [Google Scholar]
- 12.Pereiro I, Hoskins BE, Marshall JD, et al. arrayed Primer Extension (APEX) technology simplifies mutation detection in Bardet Biedl and Alström Syndrome. Eur J Hum Genet. 2011;19:485–8. doi: 10.1038/ejhg.2010.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–76. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 14.Zulato E, Favaretto F, Veronese C, et al. ALMS1-deficient fibroblasts over-express extra-cellular matrix components, display cell cycle delay and are resistant to apoptosis. PLoS One. 2011;6:e19081. doi: 10.1371/journal.pone.0019081. [DOI] [PMC free article] [PubMed] [Google Scholar]