

Abstract
D-penicillamine (3,3-Dimethyl-D-cysteine; DP) is an FDA-approved redox-active D-cysteine-derivative with antioxidant, disulfide-reducing, and metal chelating properties used therapeutically for the control of copper-related pathology in Wilson’s disease and reductive cystine-solubilization in cystinuria. Based on the established sensitivity of metastatic melanoma cells to pharmacological modulation of cellular oxidative stress, we tested feasibility of using DP for chemotherapeutic intervention targeting human A375 melanoma cells in vitro and in vivo. DP treatment induced caspase-dependent cell death in cultured human metastatic melanoma cells (A375, G361) without compromising viability of primary epidermal melanocytes, an effect not observed with the thiol-antioxidants N-acetyl-L-cysteine (NAC) and dithiothreitol. Focused gene expression array analysis followed by immunoblot detection revealed that DP rapidly activates the cytotoxic unfolded protein response (UPR; involving phospho-PERK, phospho-eIF2α, Grp78, CHOP, and Hsp70) and the mitochondrial pathway of apoptosis with p53 upregulation and modulation of Bcl-2 family members (involving Noxa, Mcl-1, and Bcl-2). DP (but not NAC) induced oxidative stress with early impairment of glutathione homeostasis and mitochondrial transmembrane potential. SiRNA-based antagonism of PMAIP1 expression blocked DP-induced upregulation of the proapoptotic BH3-only effector Noxa and prevented downregulation of the Noxa-antagonist Mcl-1, rescuing melanoma cells from DP-induced apoptosis. Intraperitoneal administration of DP displayed significant antimelanoma activity in a murine A375 xenograft model. It remains to be seen if melanoma cell-directed induction of UPR and apoptosis using DP or improved DP-derivatives can be harnessed for future chemotherapeutic intervention.
Keywords: Metastatic melanoma, D-penicillamine, Noxa (PMAIP1), Mcl-1, unfolded protein response (UPR), apoptosis
Introduction
Metastatic melanoma is a malignant tumor originating from neural crest-derived melanocytes that causes the majority of skin cancer-related deaths, creating an urgent need for the identification of improved molecular agents targeting metastatic melanoma cells [1]. Despite recent clinical advances based on the availability of targeted therapies, curative chemotherapeutic intervention remains an unmet challenge of ongoing melanoma drug discovery and development [2, 3]. Cancer cell–selective induction of apoptosis through pharmacological modulation of proteotoxic and oxidative stress has recently emerged as a promising strategy for anticancer intervention [4–11]. Based on earlier research aiming at the identification of experimental cancer chemotherapeutics that induce melanoma cell apoptosis through induction of cytotoxic oxidative stress [6, 9, 12–15], the potential antimelanoma activity of the redox drug D-penicillamine (3,3-dimethyl-D-cysteine; DP) was the focus of this study. DP is a FDA-approved D-cysteine-derivative with antioxidant, disulfide-reducing, and metal chelating properties used therapeutically for the control of copper-related pathology in Wilson’s disease, lead poisoning, cystinuria, and rheumatoid arthritis [16, 17]. Cancer cell-directed cytotoxicity of DP has been observed earlier, attributed to a variety of molecular mechanisms of this pleiotropic drug that displays activity as thiol-based antioxidant, disulfide-directed reducing agent, copper-depleting metal chelator, copper-dependent pro- and antioxidant, and nucleophilic scavenger [18–24]. Cancer cell-selective induction of apoptosis by DP has been attributed to thiol-based antioxidant activity [22], whereas prooxidant cytotoxic mechanisms involving production of reactive oxygen species (ROS) have been shown to be operative when DP is administered together with redox-active copper ions [21, 24].
Here, we have examined the molecular mechanisms underlying DP-induced melanoma cell apoptosis. We show for the first time that this FDA-approved drug (I) induces apoptotic cell death in cultured human metastatic melanoma cells without compromising viability of primary epidermal melanocytes, (II) causes early upregulation of the unfolded protein response (UPR), (III) activates the mitochondrial pathway of apoptosis with modulation of Bcl-2 family members and causative involvement of NOXA (PMAIP1) upregulation, and (IV) displays significant antimelanoma activity in a murine A375 xenograft model.
Materials and methods
Chemicals
All chemicals were purchased from Sigma Chemical Co (St. Louis, MO, USA). The cell-permeable pancaspase inhibitor Z-VAD-fmk was from Calbiochem-Novabiochem (San Diego, CA, USA).
Cell culture
A375 and LOX-IMVI human melanoma cells from ATCC (Manassas, VA, USA) were cultured in RPMI medium containing 10% BCS and 2 mM L-glutamine. G361 human metastatic melanoma cells (ATCC) were cultured in McCoy’s 5a medium containing 10% BCS. Primary human epidermal melanocytes (adult skin, lightly pigmented: HEMa-LP from Cascade Biologics, abbreviated HEMa) were cultured using Medium 154 medium supplemented with HMGS-2 growth supplement. HEMa cells were passaged using recombinant trypsin/EDTA and defined trypsin inhibitor. Cells were maintained at 37 °C in 5% CO2, 95% air in a humidified incubator.
Human stress and toxicity pathfinder™ RT2 profiler™ PCR expression array
After pharmacological exposure, total cellular RNA (3×106 A375 cells) was prepared according to a standard procedure using the RNeasy kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed using the RT2 First Strand kit (Superarray, Frederick, MD, USA) and 5 μg total RNA as described previously [13]. The RT2 Human Stress and Toxicity Pathfinder™ PCR Expression Array (SuperArray) profiling the expression of 84 stress-related genes was run using the following PCR conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 sec alternating with 60 °C for 1 min (Applied Biosystems 7000 SDS). Gene-specific product was normalized to GAPDH and quantified using the comparative (ΔΔCt) Ct method as described in the ABI Prism 7000 sequence detection system user guide as published earlier [13, 25]. Expression values were averaged across three independent array experiments, and standard deviation was calculated for graphing.
DDIT3, PMAIP1, and BCL2 expression analysis by real time RT-PCR
For expression analysis by real time RT-PCR, total cellular RNA (3×106 cells) was prepared using the RNeasy kit from Qiagen (Valencia, CA, USA). Reverse transcription was performed using TaqMan Reverse Transcription Reagents (Roche Molecular Systems, Branchburg, NJ, USA) and 200 ng of total RNA in a 50 μl reaction. Reverse transcription was primed with random hexamers and incubated at 25 °C for 10 min followed by 48 °C for 30 min, 95°C for 5 min, and a chill at 4 °C. Each PCR reaction consisted of 3.75 μl of cDNA added to 12.5 μl of TaqMan Universal PCR Master Mix (Roche Molecular Systems), 1.25 μl of gene-specific primer/probe mix (Assays-by-Design; Applied Biosystems, Foster City, CA) and 7.5 μl of PCR water. PCR conditions were: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, alternating with 60 °C for 1 min using an Applied Biosystems 7000 SDS and Applied Biosystems’ Assays On Demand primers specific to DDIT3 (assay ID Hs00358796_g1), PMAIP1 (assay ID Hs00560402_m1), BCL2 (assay ID Hs00608023_m1), and GAPDH (assay ID Hs99999905_m1). Gene-specific product was normalized to GAPDH and quantified using the comparative (ΔΔCt) Ct method as described in the ABI Prism 7000 sequence detection system user guide [14]. Expression values were averaged across three independent experiments, and standard deviation was calculated for graphing.
siRNA-Transfection targeting PMAIP1 expression
A375 cells were transiently transfected with a 100 nmol pool of four small interfering RNA (siRNA) oligonucleotides (oligos) targeting PMAIP1 or a 100 nmol pool of four nontargeting siRNA oligos using the DharmaFECT 1 transfection reagent (Dharmacon RNA Technologies, Lafayette, Colorado, USA) following a standard procedure [13]. The sequences of siGENOME PMAIP1 SMARTpool (PMAIP1 siRNA) (GenBank: NM 021127) were AAACUGAACUUCCGGCAGA, AUUCUGUAUCCAAACUCU, CUGGAAGUCGAGUGUGCUA, and GCAAGAACGCUCAACCGAG. The oligos were resuspended in the Dharmacon 1x siRNA buffer and incubated in serum free media for 5 min. The oligos were incubated with the transfection reagent for 20 min before cellular treatment. Complete media was added to the siRNA oligo mixture and the cells were incubated with the siRNAs in appropriate cell culture conditions for 48 h. Cells were then re-transfected with another 100 nmol pool of four siRNA oligonucleotides targeting PMAIP1 or a 100 nmol pool of four nontargeting siRNA oligonucleotides. After another 24 h, cells were either harvested for confirmation of PMAIP1 knockdown by Noxa-immunoblot analysis or exposed to DHA followed by viability assessment using flow cytometric analysis of AV-FITC/PI stained cells.
Immunoblot analysis
Sample preparation, SDS-PAGE, transfer to nitrocellulose, and development occurred as described earlier [13, 25, 26]. Gel percentages were 15% (Noxa) and 12% (all other antigens). Antibodies were purchased from the following manufacturers: Cell Signaling Technology (Danvers, MA): anti-CHOP (mouse monoclonal); anti-PERK, anti-phospho-PERK (Thr980), anti-phospho-eIF2α, anti-eIF2α (total), anti-cytochrome C, anti-PARP, anti-Bcl-2 (rabbit monoclonal); anti-Bax, anti-PUMA, anti-Mcl-1 (rabbit polyclonal). Santa Cruz Biotechnology (Santa Cruz, CA): anti-p53 (mouse monoclonal); anti-GRP78, anti-ATF4, anti-BAK, anti-Bax, anti-PUMA, anti-Mcl-1 (rabbit polyclonal); anti-Bcl-2 (rabbit monoclonal). EMD Chemicals, Gibbstown, NJ: mouse anti-Noxa IgG (OP180); Enzo Life Sciences, Farmingdale, NY: anti-Hsp70 (mouse monoclonal). The following secondary antibodies were used: HRP-conjugated goat anti-rabbit antibody or HRP-conjugated goat anti-mouse antibody (Jackson Immunological Research, West Grove, PA). Equal protein loading was examined by β-actin-detection using a mouse anti-actin monoclonal antibody (Sigma).
Flow cytometric cell death analysis
Viability and induction of cell death (early and late apoptosis/necrosis) were examined by annexin-V-FITC (AV)/propidium iodide (PI) dual staining of cells followed by flow cytometric analysis as published previously [12]. Cells (100,000) were seeded on 35 mm dishes and received drug treatment 24 hours later. Cells were harvested at various time points after treatment and cell staining was performed using an apoptosis detection kit according to the manufacturer’s specifications (APO-AF, Sigma, St. Louis, MO, USA).
Flow cytometric detection of cleaved procaspase-3, phospho-p53 (Ser15), and phospho-H2A.X
Treatment-induced proteolytic caspase-3 activation and formation of phospho-p53 (Ser15) and phospho-H2A.X were examined in cultured A375 human melanoma cells using antibodies directed against cleaved/activated caspase-3 (Asp 175), phospho-p53 (Ser15), and phospho-histone H2A.X (Ser139) (Alexa Fluor 488 conjugates, Cell Signaling, Danvers, MA, USA) followed by flow cytometric analysis as published recently [12, 25].
Detection of intracellular oxidative stress by flow cytometric analysis
Induction of intracellular oxidative stress by DP was analyzed by flow cytometry using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) as a sensitive non-fluorescent precursor dye according to a published standard procedure [12].
Determination of reduced cellular glutathione content
Intracellular reduced glutathione was measured using the GSH-Glo Glutathione assay kit (Promega; San Luis Obispo, CA) as described recently [27]. Cells were seeded at 100,000 cells/dish on 35 mm dishes. After 24 h, cells were treated with test compound. At selected time points after addition of test compound, cells were harvested by trypsinization and then counted using a Coulter counter. Cells were washed in PBS, and 10,000 cells/well (50 μl) were transferred onto a 96-well plate. A standard curve was prepared using a serial dilution of reduced glutathione. GSH-Glo reagent (50 μl) containing luciferin-NT and glutathione-S-transferase was then added followed by 30 min incubation. After addition of luciferin detection reagent to each well (100 μl) and 15 min incubation luminescence reading was performed using a BioTek Synergy 2 Reader (BioTek, Winooski, VT, USA). Data are normalized to GSH content in untreated cells and expressed as means ± SD (n=3).
Mitochondrial Transmembrane Potential
Mitochondrial transmembrane potential (Δψm) was assessed using the potentiometric dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) following a published procedure [12]. In brief, cells were trypsinized, washed in PBS, resuspended in 300 μl PBS containing 5 μg/ml JC-1 for 15 min at 37°C and 5% CO2 in the dark, then washed twice in PBS and resuspended in 300 μl PBS. Bivariate analysis was performed by flow cytometry with excitation at 488 nm, and mitochondrial function was assessed as JC-1 green (depolarized mitochondria, detector FL-1) or red (polarized mitochondria, detector FL-2) fluorescence.
Mitochondrial and cytosolic protein extraction for cytochrome c detection
Mitochondrial and cytosolic fractions were isolated using a cytochrome c EIA Kit (Enzo Life Sciences, Farmingdale, NY), and cytochrome c was then detected by immunoblot analysis (anti-cytochrome c, rabbit polyclonal, Cell Signaling Technology).
Comet assay (alkaline single cell electrophoresis)
The alkaline comet assay was performed on A375 melanoma cells according to the manufacturer’s instructions (Trevigen, Gaithersburg, MD, USA) as published recently [26, 27]. After treatment, cells (100,000 per 100 mm dish) were harvested by gently scraping and suspended in 500 μl PBS. An aliquot (50 μl) was mixed with low-melting-point agarose (450 μl) and spread on pretreated microscope slides. To allow DNA unwinding and expression of alkali-labile sites, slides were exposed to alkaline buffer (1 mM EDTA and 300 mM NaOH, pH >13) protected from light at room temperature for 45 min. Electrophoresis was conducted in the same alkaline buffer for 20 min at 300 mA. After electrophoresis, slides were rinsed three times in distilled H2O, then fixed in 70 % ethanol for 5 min. Cells were stained with SYBR™ Green and then visualized and analyzed using a fluorescence microscope (fluorescein filter) and CASP software. At least 75 tail moments for each group were analyzed in order to calculate the mean ± S.D. for each group.
Human A375 Melanoma SCID mouse xenograft model
A375 human melanoma cells were grown in HyQ RPMI-1640 media (HyClone) supplemented with 10% fetal bovine serum (Omega Scientific), and maintained in 5% CO2-95% air humidified atmosphere at 37°C. Subconfluent cells were harvested by using 0.25% trypsin-EDTA (HyClone). Cells (>90% viability) were resuspended at the concentration of 10 × 106 cells/100μl of sterile saline. A SCID mouse colony was developed at the University of Arizona using original SCID (C.B-17/IcrACCSCID) obtained from Taconic (Germantown, New York). The mice were housed in microisolator cages (Allentown Caging Equipment Company, Allentown, N.J.) and maintained under specific pathogen-free conditions. Every month mice were screened by ELISA serology for mycoplasma, mouse hepatitis virus, pinworms, and Sendai virus and tested negative. SCID mice 6–8 weeks of age were bled (<200 μl) by retroorbital puncture in order to screen for the presence of mouse IgG using an ELISA. Only mice with IgG levels ≤ 20 ug/ml were used for the experiments. The mice ate NIH-31 irradiated pellets (Tekland Premier, Madison, WI) and received autoclaved water. Animal facilities are approved by the Association for the Assessment and Accreditation of Laboratory Animal Care International and in accordance with United States Department of Agriculture, Department of Health and Human Services, and NIH regulations. A375 cell injections (10 × 106 cells) were given subcutaneously on the mouse’s lower right flank on day 0, and after tumors became established (~65 mm3) mice were pair-matched into the treatment groups. The following day, treatment with DP or carrier only was initiated. DP (in PBS; 1,200 mg/kg/d, i.p., b.i.d.; n=12)) was given on days 1–10 post the day of pair-matching, whereas control animals received carrier only (n=12). Subcutaneous tumors were measured twice weekly for tumor volume estimation (mm3) in accordance with the formula (a2 × b)/2 where a is the smallest diameter and b is the largest diameter. The mice were sacrificed individually by CO2 when the tumors reached a volume of 2000 mm3. Tumor growth curves were obtained by determining average tumor volumes until day 30 after cell injection, and data points were analyzed using the two-sided Student’s t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001). All procedures were completed in accordance with the University of Arizona Institutional Animal Care and Use Committee (IACUC) protocol (# 07-029, approved May 24, 2007).
Immunohistochemical characterization of tumor tissue from mice xenografts
At the time of analysis, tumors from SCID mice were harvested and tumor tissue sections were analyzed for cleaved (activated) procaspase 3 (Asp175) according to published procedures [14]. Briefly, tissues were harvested, fixed in 10 % neutral buffered formalin for 24 hours, processed and embedded in paraffin. Routine hematoxylin stain was performed on threemicron sections of tissue, and immunohistochemistry was performed using a rabbit polyclonal antibody to cleaved procaspase 3 (Asp175; large fragment) (#9661, Cell Signaling Technology; 1:100 dilution). Detection of primary antibody was performed on a Discovery XT Automated Immunostainer (Ventana Medical Systems,
Inc, Tucson, Arizona) using a biotinylated-streptavidin-HRP and DAB system. Hematoxylin counterstaining was also performed online. Following staining on the instrument, slides were dehydrated through graded alcohols to xylene and coverslipped with Pro-Texx mounting medium. Images were captured using an Olympus BX50 and Spot (Model 2.3.0) camera. Images were standardized for light intensity. No automated analysis of the data was performed.
Statistical analysis
Unless indicated differently, the results are presented as mean ± S.D. of at least three independent experiments. All data were analyzed employing one-way analysis of variance (ANOVA) with Tukey’s post hoc test using the Prism 4.0 software. Differences were considered significant at p ≤ 0.05. Means without a common letter differ.
Results
DP induces caspase-dependent cell death in human A375 metastatic melanoma cells but not in primary epidermal melanocytes
In an attempt to examine the potential antimelanoma activity of DP at the cellular level, differential cytotoxicity of DP directed against metastatic A375 melanoma cells and primary epidermal melanocytes (HEMa) was assessed by flow cytometric analysis using annexinV-FITC/propidium iodide-staining (Fig. 1). Exposure to DP (10 mM, 24 h) induced pronounced cell death in metastatic melanoma cells (Fig. 1A), but no significant impairment of viability was observed in DP-exposed primary epidermal melanocytes (Fig. 1B). Consistent with earlier observations, efficient induction of A375 cell death required exposure to millimolar concentrations DP and was not observed at concentrations below 1 mM (data not shown) [22]. Exposure to the thiol-antioxidant and DP-related cysteine-derivative N-acetyl-L-cysteine (NAC; 10 mM, 24 h) did not impair cell viability (Fig. 1C); similarly, the dithiol-based reducing agent dithiothreitol (DTT; 10 mM, 24 h) did not induce cell death (data not shown). DP-induction of cell death was efficiently blocked in the presence of the pan-caspase inhibitor zVADfmk (40 μM, 1 h preincubation) (Fig. 1C), and pronounced activation of caspase-3 (approximately two fold) was observed already within 6 h exposure time reaching a maximum after 24 h (approximately 15 fold) as assessed by flow cytometric detection of cleaved procaspase-3 (Fig. 1D), consistent with an apoptotic mode of DP-induced A375 melanoma cell death. Further analysis confirmed DP-induction of cell death in other metastatic melanoma cell lines (G361, LOX-IMVI) that was blocked by zVAD-fmk pretreament (Fig. 1E). Consistent with an apoptotic mode of DP-induced cell death, pronounced PARP cleavage detected by immunoblot analysis was detectable within 12 h exposure in A375 and G361 cells (Fig. 1F).
Fig. 1. DP but not NAC induces caspase-dependent cell death in human A375 metastatic melanoma cells.
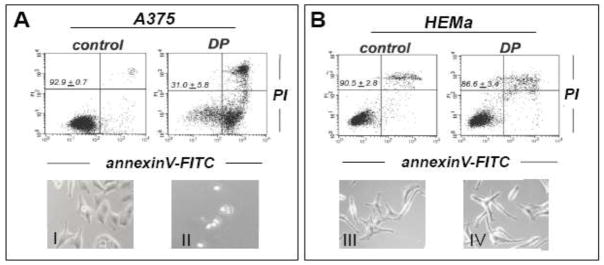
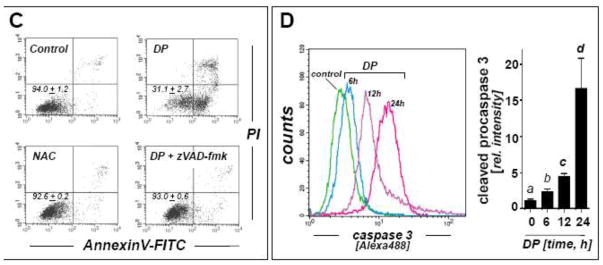
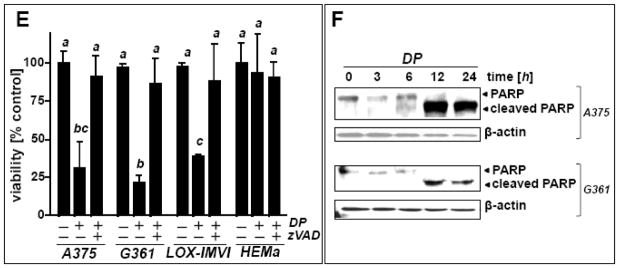
(A–B) Induction of A375 (panel A) and primary melanocyte (HEMa; panel B) cell death (DP; 10 mM, 24 h) was assessed by flow cytometric analysis after annexinV-FITC/propidium iodide-staining. The numbers indicate viable cells (AV−, PI−, lower left quadrant) in percent of total gated cells (mean ± SD, n=3). Representative light microscopy pictures taken after 24 h exposure to DP are shown in panels I–IV. (I and II: A375; III and IV: HEMa; I and III: control; II and IV: DP). (C) Induction of cell death in response to DP or NAC (10 mM, 24 h) in the presence or absence of zVADfmk (40 μM, 1 h preincubation). (D) DP-induced (10 mM, 6–24 h) caspase-3 activation as examined in A375 cells by flow cytometric detection using an Alexa 488-conjugated monoclonal antibody against cleaved procaspase-3. One representative experiment of three similar repeats (left panel) and quantitative analysis of three independent repeat experiments (right panel; mean ± SD) are displayed. (E) Metastatic melanoma cells (A375, G361, LOX-IMVI) and HEMa were exposed to DP (10 mM; 24 h) in the presence or absence of zVADfmk (40 μM, 1 h preincubation) and viability (mean ± SD, n=3) was determined as specified in (A). (F) DP-induced (10 mM, 3–24 h) PARP cleavage was examined by immunoblot analysis in A375 and G361 cells. Means without a common letter differ (p < 0.05)
Array analysis reveals DP-induced upregulation of cytotoxic stress response gene expression in A375 melanoma cells
To gain further mechanistic insight into the molecular events underlying melanoma cell-directed induction of cell death by DP, modulation of stress and toxicity response gene expression was examined in A375 cells exposed to DP (10 mM, 24 h exposure) using the RT2 Human Stress and Toxicity Profiler™ PCR Expression Array technology (SuperArray, Frederick, MD) (Fig. 2A).
Fig. 2. DP-induced transcriptional changes affecting stress response gene expression in A375 melanoma cells.


DP-induced (10 mM, 24 h) expression changes as identified by RT2 Human Stress and Toxicity Profiler™ PCR Expression Array analysis normalized to GAPDH. (A) The scatter blot depicts differential gene expression as detected by the RT2 Human Stress and Toxicity Profiler™ PCR Expression Array technology. Upper and lower lines: cut-off indicating fourfold up- or down-regulated expression, respectively. (B) Bar graph depiction of gene expression changes at the mRNA level. Analysis was performed in three independent repeats and analyzed using the two-sided Student’s t test (p < 0.001).
Out of 84 stress-related genes contained on the array DP-induced upregulation of expression affected 17 genes by at least three-fold over untreated control cells, indicative of genotoxic, proteotoxic, and oxidative stress as summarized in Fig. 2B. Induction of the p53-regulated gene growth arrest and DNA-damage-inducible, alpha [GADD45A (25.4 fold)], known to be responsive to genotoxic and other types of cytotoxic stress, was detected. Pronounced upregulation of gene expression indicative of the unfolded protein response (UPR) was observed including DNA damage inducible transcript 3 [DDIT3/GADD153 (53.8 fold)] and heat shock protein encoding genes [such as HSPA2 (15.1 fold), HSPA6 (9.4 fold), HSPA1L (3.4 fold), and HSPA1A (3.0 fold)]. Massive upregulation of the oxidative stress-responsive transcription factor and tumor suppressor early growth response protein 1 [EGR1 (46.7 fold)] was also detected. Expression changes also affected antioxidant defense enzyme encoding genes {upregulation of heme oxygenase 1 [HMOX1 (4.7 fold)] and glutathione S-transferase mu 3 [GSTM3 (4.3 fold)]} together with downregulation of catalase [CAT (6.2 fold)], consistent with the detection of DP-induced cellular oxidative stress (as detailed in Fig. 4A and B). In addition, DP-treatment caused altered expression of genes encoding enzymes involved in redox metabolism {flavin monooxygenases [FMO1 (34.0 fold), FMO5 (34.6 fold)]; cytochrome P450 monooxygenases [CYP1A1 (8.3 fold), CYP2E1 (5.3 fold)]} and inflammatory signaling {prostaglandin-endoperoxide synthase 1 [PTGS1 (22.7 fold)]; inducible nitric oxide synthase 2 [NOS2A (4.6 fold)]; chemokines [CCL3 (19.3 fold) and CCL4 (40.3 fold)]}.
Fig. 4. DP-induced oxidative and mitochondriotoxic stress in A375 melanoma cells occurs in the absence of genotoxic stress.

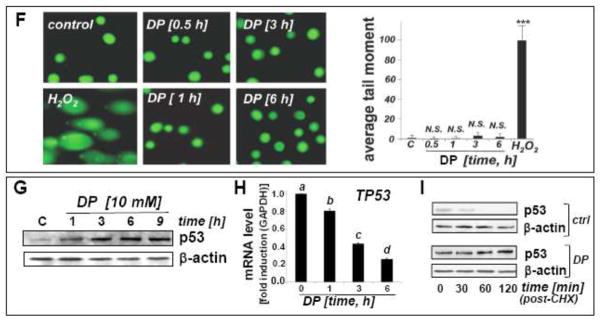
(A) Modulation of intracellular reduced glutathione content in A375 melanoma cells exposed to DP or NAC (10 mM, 0.5 – 6 h exposure). Glutathione content was normalized to untreated control (mean ± SD, n=3). Means without a common letter differ (p < 0.05). (B) DP-induction of intracellular oxidative stress as assessed by DCF flow cytometry. Cells were exposed to DP or NAC (10 mM, 6 h), and intracellular oxidative stress was assessed by 2′,7′-dichloro-dihydrofluorescein diacetate staining followed by flow cytometric analysis of DCF fluorescence. One representative experiment out of three similar repeats is shown. (C) Time course analysis of loss of mitochondrial transmembrane potential (Δψm) in response to DP (10 mM, up to 18 h) as assessed by flow cytometric analysis of JC-1 stained cells. Panels from one representative experiment of three similar repeats are shown, and numbers indicate cells with impaired Δψm (in percent of total gated cells) detected outside the circle (mean ± SD, n=3). (D) DP-induced apoptosis with or without pretreatment using cyclosporine A (CysA; 5 μM; 60 min) as examined by annexin V-PI flow-cytometric analysis. The numbers indicate viable cells (AV−, PI−, lower left quadrant) in percent of total gated cells (mean ± SD, n=3). (E) Immunodetection of cytochrome c in cytosolic (c) and mitochondrial (m) fractions prepared from DP-treated A375 cells (10 mM; 6–24 h). (F) A375 cells were exposed to DP (10 mM, 0.5–6 h), and DNA damage was detected using the comet assay as described in Materials and Methods. As a positive control, cells were exposed to H2O2 (100 μM, 30 min). Representative comet images (left panels) as visualized by fluorescence microscopy and quantitative analysis of average tail moments (bar graph) are shown. At least 75 tail moments for each group were analyzed in order to calculate the mean ± S.D. for each group. (G–H) DP-induced (10 mM; 1–9 h exposure) upregulation of p53 was assessed by immunoblot analysis (panel G) and real time RT-PCR [panel H; TP53; mean ± SD, n=3; means without a common letter differ (p < 0.05)]. (I) Time course analysis of p53 protein levels in cells first pretreated with DP (3h, 10 mM) or left untreated (ctrl). Both groups were then exposed to cycloheximide (CHX; 25 μM, 0 – 120 min) followed by immunoblot analysis.
The unfolded protein response (UPR) is induced at the mRNA and protein level in DP-exposed A375 melanoma cells
Led by upregulated expression of DDIT3, HSPA2, HSPA6, HSPA1L, and HSPA1A as observed by array analysis we further examined DP-treated A375 melanoma cells for the occurrence of ER stress and UPR at the mRNA and protein level (Fig. 3).
Fig. 3. DP-induced unfolded protein response in A375 melanoma cells.

(A) Cells were treated with DP (10 mM, up to 9 h). Upregulation of DDIT3 mRNA levels detected by real time RT-PCR analysis (upper panel). Modulation of UPR-marker proteins (CHOP, Hsp70, GRP78/Bip, phospho- and total PERK, phospho- and total eIF2α, ATF4) as detected by immunoblot analysis (lower panel). (B) DDIT3 mRNA levels detected by real time RT-PCR analysis in cells exposed to thapsigargin (T; 300 nM) or NAC (10 mM; up to 6 h). (C) Upregulation of selected UPR-marker proteins in cells exposed to T (300 nM, up to 6 h; upper panel), NAC (10 mM, up to 9 h; middle panel) or DTT (10 mM, up to 9 h; bottom panel) as detected by immunoblot analysis. Means without a common letter differ (p < 0.05)
Time course analysis by quantitative RT-PCR and immunoblot detection revealed early upregulation of CHOP (CCAAT/-enhancer-binding protein homologous protein), the transcription factor encoded by DDIT3 (Fig. 3A). DDIT3 upregulation occurred within 3 h continuous exposure, and mRNA levels exceeded control levels by almost 40-fold within 9 h exposure. As a positive control, CHOP protein upregulation was monitored in response to thapsigargin (T), an inducer of ER stress and UPR through inhibition of SERCA (sarco/endoplasmic reticulum)-Ca2+ ATPase (Fig. 3B and C) [15].
Consistent with UPR-induction by DP, increased cellular levels of heat shock proteins were detected. Rapid upregulation of GRP78 (78 kDa glucose-regulated protein, also known as BiP or heat shock 70kDa protein 5, a molecular chaperone and upstream UPR regulator located in the lumen of the ER) and inducible Hsp70 (encoded by HSPA1A) was observed (Fig. 3A). In contrast, no DP-induced quantitative changes at the protein level occurred with the UPR-associated transcription factor ATF4 [28]. Next, we examined DP-induced changes affecting eukaryotic translation initiation factor eIF2α, since cytotoxic ER stress is known to cause inhibitory phosphorylation of eIF2α through activation of PERK [double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase] [29]. Comparing total versus phospho-eIF2α protein levels by immunoblot analysis, DP-induced phosphorylation was detected within 1 h continuous exposure indicating rapid induction of ER stress-signaling (Fig. 3A). Consistent with the effects of DP treatment on eIF2α phosphorylation, PERK activation by autophosphorylation of its cytoplasmic kinase domain (Thr980) was detected at early time points (1–9 h) (Fig. 3A). In contrast, at equimolar concentrations, NAC was unable to induce upregulation of DDIT3 mRNA levels (Fig. 3B) or eIF2α phosphorylation (Fig. 3C, middle panel). Only a transient induction of eIF2α phosphorylation was observed using dithiothreitol (DTT), an established UPR-inducer known to cause ER stress through reductive protein unfolding (Fig. 3C, bottom panel) [30]. In contrast to DP-induced upregulation of Hsp70 protein levels (Fig. 3A), no upregulation of Hsp70 occurred in response to NAC or DTT treatment (Fig. 3C). Importantly, DP-induced modulation of Hsp70 and eIF2α phosphorylation was also observed in other metastatic melanoma cell lines including G361 (Fig. 5C)
Fig. 5. Modulation of Noxa (PMAIP1), Mcl-1, and Bcl-2 (BCL2) in DP-induced A375 melanoma cell apoptosis.


(A) DP-modulation (up to 10 mM, up to 9 h exposure) of the Bcl2-family of apoptosis regulators was examined in A375 melanoma cells by immunoblot analysis. For comparison, NAC (10 mM) was used (bottom panel). The middle panel depicts the dose response (0.1–10 mM) of DP-induced Noxa upregulation. (B) PMAIP1 mRNA detection in A375 cells using real time RT-PCR analysis [10 mM DP; 0–24 h; mean ± SD, n=3; means without a common letter differ (p < 0.05)]. (C) DP-modulation of Noxa, Mcl-1, Hsp70, phospho- and total eIF2α protein levels in G361 melanoma cells. (D) DP-modulation of Bcl-2 expression at the protein and mRNA [BCL2; mean ± SD, n=3; means without a common letter differ (p < 0.05)] level in A375 and G361 melanoma cells. (E) After DP exposure (10 mM, up to 9 h) with or without PMAIP1 knockdown (siPMAIP1), Noxa and Mcl-1 immunoblot detection was performed in A375 cells as specified in Materials and Methods. Bcl-2 downregulation by DP (6 h) was also examined in siPMAIP1- and siControl-treated A375 cells (bottom panels). (F) Genetic downregulation of PMAIP1 expression confers protection of A375 cells against DP-induced (10 mM, 36 h) cytotoxicity as detected by AV/PI flow cytometric analysis after siControl and siPMAIP1 treatment. Numbers indicate viable cells (AV−, PI−, lower left quadrant) in percent of total gated cells (mean ± SD, three determinations).
Taken together, these data demonstrate for the first time that DP treatment of metastatic melanoma cells induces ER stress/UPR characterized by upregulation of Hsp70 and the ER-chaperone GRP78, autophosphorylation of PERK, phosphorylation of eIF2α, and increased expression of CHOP detected at the mRNA and protein level.
Upregulation of cellular oxidative and mitochondriotoxic stress occurs in response to DP treatment
Led by modulation of stress response gene expression indicative of DP-induced oxidative stress (EGR1, HMOX1, NOS2A, CAT; Fig. 2), we further examined the occurrence of redox alterations in DP-treated A375 melanoma cells at early time points (Fig. 4A–B). Indeed, cellular reduced glutathione levels were depleted by more than 40 % within 6 h DP exposure (Fig. 4A). Moreover, using 2′,7′-dichloro-dihydrofluorescein diacetate detection of intracellular peroxide levels by flow cytometry, an approximately three-fold increase in DCF fluorescence intensity [(mean ± S.D.): control: 8.4 ± 1.0; DP: 28.5 ± 3.6; n=3; p<0.05] was observed after 6 h continuous exposure indicating the occurrence of cellular oxidizing species in response to DP (Fig. 4B), a finding consistent with prior research on DP-induced redox alterations targeting cancer cells [24]. In contrast, exposure to an equimolar concentration of the closely related cysteine-derivative and antioxidant NAC did not cause a reduction in the cellular glutathione pool (Fig. 4A), and a statistically significant reduction in DCF fluorescence by more than 50% versus untreated control cells [(mean ± S.D.): control: 8.4 ± 1.0; NAC: 3.8 ± 1.5; n=3; p<0.05] was observed after 6 h continuous exposure (Fig. 4B), an observation consistent with the antioxidant properties of NAC.
Next, flow cytometric analysis using the sensor dye JC-1 revealed an early impairment of mitochondrial integrity as indicated by loss of mitochondrial transmembrane potential (Δψm) that became already apparent at 6 h DP exposure (more than 50% of cells displaying loss of Δψm; Fig. 4C). Consistent with a causative involvement of mitochondriotoxicity in DP-induced apoptosis, immunodetection of cytochrome c (cytosolic versus mitochondrial) indicated massive mitochondrial release of this apoptotic key mediator, detectable within 6–12 h exposure time and completed within 24 h (Fig. 4E). Moreover, significant rescue of A375 cells from DP-induced apoptosis was achieved by cotreatment with cyclosporine A, a pharmacological inhibitor of mitochondrial permeability transition pore opening, providing further evidence in support of a causative involvement of mitochondrial events in DP-induced apoptosis (Fig. 4D) [31].
Next, led by expression changes affecting DNA-damage responsive genes (GADD45A, DDIT3, UNG; Fig. 2) we examined the occurrence of DP-induced impairment of genomic integrity (Fig. 4F). However, alkaline single cell electrophoresis (comet assay) of A375 melanoma cells did not indicate the induction of DNA damage in response to DP exposure at time points (up to 6 h) when induction of oxidative, proteotoxic, and mitochondriotoxic stress already had become apparent. Consistent with this finding, flow cytometric analysis of phospho-H2A.X status, an established marker of DNA strand breaks, did not reveal DP-induced changes affecting genomic integrity (data not shown) [26].
Earlier work has suggested the involvement of p53 expression in DP-induction of cancer cell death [22]. Indeed, in A375 melanoma cells, pronounced upregulation of cellular p53 protein levels was observed by immunoblot detection as early as within 1 h reaching a maximum at 9 h exposure time (Fig. 4G). However, phospho-p53 (Ser15), a common posttranslational modification in response to genotoxic stress, was not detectable in DP-treated cells by flow cytometric analysis using an Alexa488-conjugated monoclonal antibody (data not shown) [27]. Interestingly, DP-upregulation of cellular p53 protein levels occurred with pronounced downregulation of TP53 mRNA levels (detectable within 1 h exposure and down to 20% of untreated controls within 6 h) observed over the same time course (Fig. 4H). Further time course analysis examined p53 protein levels in cells first pretreated with DP (3h, 10 mM) or left untreated, followed by exposure to the translation inhibitor cycloheximide (CHX; 25 μM, 0–120 min; Fig 4I). In control cells (unexposed to DP) p53 protein was undetectable within 60 min after CHX exposure, a finding consistent with the rapid turnover of this protein documented before. In contrast, after DP-pretreatment, p53 levels were maintained throughout 120 min after CHX addition. These data suggest that a DP-induced increase in p53 protein levels (Fig. 4G) observed together with TP53 transcriptional downregulation (Fig. 4H) originates from blockade of protein turnover (Fig. 4I). However, more detailed mechanistic studies are necessary to further elucidate the mechanistic basis of DP-induced p53 protein upregulation.
Taken together, these data demonstrate that DP exposure causes early induction of oxidative and mitochondriotoxic (but not genotoxic) stress with upregulation of p53 levels detectable in metastatic melanoma cells.
DP modulates the expression of Bcl-2 family members Noxa, Mcl-1, and Bcl-2 involved in mitochondrial apoptosis of A375 melanoma cells
Following detection of impaired mitochondrial transmembrane potential, cytochrome c release, rescue from DP-induced cell death by cyclosporine A cotreatment, and upregulation of p53 protein levels at early time points, we further examined mitochondrial involvement in DP-induced apoptosis. First, p53-regulated Bcl2-family members potentially involved in mitochondria-dependent melanoma cell death were examined by quantitative RT-PCR and immunoblot analysis (Fig. 5A–C). Importantly, DP treatment (at concentrations ≥ 5 mM) caused massive upregulation of the proapoptotic BH3-domain only protein Noxa (encoded by PMAIP1) observed at the transcriptional (Fig. 5B) and protein level (Fig. 5A), an effect that was already apparent at 1 h exposure time. DP-induced Noxa upregulation was also observed in other human metastatic melanoma cell lines including G361 (Fig. 5C). In contrast, Noxa-modulation was not observed at equimolar concentrations of the cysteine-derivative NAC (Fig. 5A, bottom panel), consistent with the lack of apoptogenicity of NAC as observed above (Fig. 1C).
In contrast to DP-modulation of Noxa, the p53-regulated proapototic Bcl-2 family members Bax and Bak did not display any expression changes (Fig. 5A). Interestingly, the pro-apoptotic BH3-domain only proteins PUMAα (p53-upregulated modulator of apoptosis) and PUMAβ displayed downregulation at the protein level, a paradoxical effect known to occur in the context of apoptotic cell death, attributable to caspase-dependent degradation after commitment to mitochondrial apoptosis has occurred [32].
Consistent with Noxa-involvement in DP-induced mitochondrial apoptosis, we observed that the anti-apoptotic Bcl-2-homologue Mcl-1, known to antagonize Noxa proapototic activity, was downregulated at the protein level, an effect observed in A375 and G361 cells (Fig 5A, C, and E) [33–37]. We also detected DP-induced downregulation of the anti-apoptotic factor Bcl-2 (encoded by BCL2), observed at the transcriptional and protein level in A375 and G361 cells (Fig. 5D), an important determinant of cell viability known to be transcriptionally attenuated by CHOP (DDIT3) in response to ER stress (Fig. 3A–B) [38].
Genetic antagonism of PMAIP1 (Noxa) expression provides partial protection against DP-induced apoptosis in A375 metastatic melanoma cells
Based on the established key role of Noxa in the mitochondrial pathway of apoptosis [33–37], we then tested the hypothesis that upregulation of PMAIP1 expression may be causatively involved in DP-induced A375 melanoma cell death. To this end, genetic target modulation by siRNA (small interfering RNA) targeting PMAIP1 expression was employed (Fig. 5E–F). First, efficacy of PMAIP1 knockdown (siPMAIP1) was confirmed at the protein level (Fig. 5E). DP-induced upregulation of Noxa protein levels, as observed earlier in wildtype A375 control cells (Fig. 5A), could be blocked by prior transfection using siPMAIP1 oligonucleotides (Fig. 5E, top right panel), but was not suppressed when transfection occurred using non-targeting siRNA control reagent (siControl; Fig. 5E, top left panel). In parallel, DP-induced Mcl-1-downregulation occurred in siControl-treated cells (Fig. 5E, middle left panel) but not in siPMAIP1-treated cells (Fig. 5E, middle right panel), consistent with the established role of Noxa in causing Mcl-1 degradation.
Next, the effect of siPMAIP1 intervention on DP-induced apoptosis was examined in A375 cells (Fig. 5F). Flow cytometric analysis indicated that significant cytoprotection against prolonged exposure to DP (10 mM, 36 h) was achieved in siPMAIP1-transfected (viability: 44.8 ± 6.4 %) versus siControl-transfected (viability: 18.8 ± 4.9 %; p< 0.05) cells. Moreover, in siPMAIP1-transfected cells progression of DP-induced cell death was significantly delayed as obvious from an increased proportion of compromised cells in early (lower right quadrant: AV+/PI−) versus late apoptosis (upper right quadrant: AV+/PI+), whereas most siControl-treated cells displaying DP-induced loss of viability were already in late apoptosis (AV+/PI+) at the time of flow cytometric analysis. Pronounced cytoprotection and delay of apoptotic progression achieved by genetic antagonism targeting PMAIP1 expression support the conclusion that Noxa/Mcl-1 modulation is an important causative factor in DP-induced mitochondrial apoptosis of melanoma cells. However, further analysis revealed that DP-induced downregulation of cellular Bcl-2 protein levels (Fig. 5D) was not suppressed by siPMAIP1 treatment (Fig. 5E, right bottom panel), consistent with the finding that siPMAIP1-based intervention only partially blocked DP-induced melanoma cell death. Taken together, these findings demonstrate the causative involvement of Noxa-/Mcl1-modulation in DP-induced melanoma cell apoptosis that also involves Noxa-independent pathways of caspase-mediated cell death including Bcl-2.
DP inhibits tumor growth in a human A375 melanoma murine xenograft model
Earlier research suggests anticancer activity of DP observed in vitro and in vivo. We therefore tested DP as a potential inhibitor of melanoma tumor growth in a murine xenograft model of the disease (Fig. 6). Indeed, daily intraperitoneal administration at high doses (1,200 mg/kg/day) induced a pronounced suppression of tumor growth in human A375 melanoma xenograft-bearing SCID mice, which reached the level of statistical significance (p < 0.05 versus PBS-treated control) between days 18 and 27 after A375 cell injection. At the end of the treatment period (day 27) average tumor weights of DP-treated animals were approximately 70% lower than those of PBS-treated controls (Fig. 6A). Immunohistochemical analysis of xenograft tumor tissue harvested at the end of the treatment regimen indicated pronounced cleavage of procaspase 3 (Asp175) in response to DP exposure [percentage of activated caspase 3-positive cells per total number of nuclei: 8.21 ± 6.81 (control tumors) versus 23.46 ± 9.70 (DP-treated tumors); mean ± S.D.; n=3; p < 0.05; Fig. 6C], consistent with a pro-apoptotic effect of systemic DP intervention. Importantly, during DP treatment, no compound-related adverse reactions or average weight loss that would be indicative of toxic systemic effects were observed as compared to carrier-treated control mice (Fig. 6B).
Fig. 6. DP inhibits tumor growth in a human melanoma SCID-mouse xenograft model.

Human A375 melanoma cells (10 × 106) were implanted s.c. into the right flank of SCID mice. 14 days after cell injection animals were pair-matched (65 mm3 average tumor size) and daily treatment (DP, in PBS; 1,200 mg/kg/d, i.p., b.i.d.; n=12) was initiated. Control animals (n=12) received PBS only. (A) Tumor growth curves were obtained by determining average tumor volumes until day 27 after cell injection. Data points are depicted as means ± SEM; statistical comparison between individual data points (DP-treated versus control; days 16–27) was performed using the two-sided Student’s t test (p < 0.05). (B) Mean body weight was monitored during the duration of the experiment and expressed as % change from the average weight obtained on the day of pair matching. (C) Immunohistochemical staining for cleaved procaspase 3 (Asp175) using primary tumor tissue of DP-treated and PBS-treated mice harvested on the last day of drug treatment (n=3, each group). Paraformaldehyde-fixed, paraffin-embedded 5 μm sections were analyzed using a rabbit polyclonal antibody to activated caspase 3 followed by biotinylated-streptavidin-HRP/DAB visualization. Hematoxylin counterstaining was also performed. A photograph representative of three high power fields taken per tissue section is shown. Bar graph displays quantitative analysis of IHC (percentage of activated caspase 3-positive cells per total number of nuclei; mean ± S.D.; n=3; p < 0.05).
Discussion
Based on prior research documenting the ability of DP to impair redox homeostasis and viability of cancer cells, we tested the antimelanoma activity of DP in vitro and in vivo. DP (but not equimolar concentrations of the thiol-antioxidant NAC) induced caspase-dependent cell death in cultured human metastatic A375 melanoma cells without compromising viability of primary epidermal melanocytes (Fig. 1). Early responses to DP exposure involved upregulation of cellular levels of p53 and its downstream target Noxa, a proapototic BH3-only protein member of the Bcl-2 family that facilitates mitochondrial outer membrane permeabilization in melanoma cells, observed at the protein and mRNA (PMAIP1) level (Figs. 4G and 5A–C)[15, 33–37]. In parallel, downregulation of Mcl-1, the antiapoptotic Bcl-2 family member known to antagonize Noxa-facilitation of membrane permeabilization, was observed (Fig. 5A, C, and E)[39]. Consistent with a mitochondrial mode of DP-induced apoptosis, loss of mitochondrial transmembrane potential became apparent within 6 h exposure accompanied by mitochondrial release of cytochrome c (4C and E), and partial rescue from DP-induced cell death was achieved by cotreatment with the inhibitor of mitochondrial membrane permeabilization cyclosporin A (Fig. 4D),
Noxa upregulation/Mcl-1 downregulation upstream of subsequent mitochondrial outer membrane permeabilization and cytochrome C release has recently emerged as a promising key mechanism underlying potent apoptogenicity of various experimental drugs targeting metastatic melanoma [15, 33–37]. We therefore decided to further explore the causative involvement of Noxa in DP-induced melanoma cell death (Fig. 5E and F). Significant yet incomplete protection against DP-induced apoptosis was observed when Noxa (PMAIP1) expression was suppressed using a genetic siRNA-based approach. However, DP-induced downregulation of Bcl-2, another major antiapoptotic factor modulated by DP exposure, was not antagonized (Fig. 5D and E, bottom panels), an observation consistent with the limited protective effect achieved by siPMAIP1-based intervention (Fig. 5F).
After excluding rapid induction of genotoxic stress as a causative factor that might underlie early upregulation of p53 (Fig. 4F), stress response gene expression array analysis revealed that DP exposure elicits the unfolded protein response (UPR), a finding further confirmed by immunoblot detection of proteins involved in mediating the response to cytotoxic ER stress (Hsp70, GRP78/Bip, autophosphorylated PERK, phosphorylated eIF2α, CHOP) (Figs. 3 and 5C) [29]. The UPR serves as a cytoprotective survival pathway, maintaining ER homeostasis in response to cytotoxic stimuli that cause accumulation of unfolded and/or misfolded proteins within the ER including heat shock, oxidative stress, energy crisis, calcium dysregulation, and proteasome inhibition [4, 5, 10, 11, 29, 40]. However, under conditions of prolonged and extensive ER stress the UPR contributes to initiation of apoptosis that involves early upregulation of p53 and CHOP, a proapoptotic transcription factor (encoded by DDIT3) that modulates the mitochondrial pathway of apoptosis by downregulating expression of Bcl-2 (BCL2) [5, 38, 41]. Indeed, within one hour of continuous DP exposure, UPR signaling in A375 melanoma cells was rapidly initiated as evidenced by autophosphorylation of PERK (Thr980) and phosphorylation of the PERK substrate eIF2α (Fig. 3A). eIF2α-Phosphorylation downstream of ER stress-induced PERK activation is a key UPR signaling event, thought to underlie UPR associated inhibition of general cap-dependent translation, while allowing mRNAs with an internal ribosomal entry sites (IRES) (such as that one encoding CHOP) to be translated [5]. In our experiments, pronounced upregulation of DDIT3 mRNA and CHOP protein levels occurred in response to DP detected within 3h exposure (Fig. 3A), a finding consistent with earlier research demonstrating DP-induced ER stress with CHOP upregulation in human epithelial HeLa carcinoma cells [42]. In addition, the upstream ER stress sensor and UPR initiator GRP78, an ER chaperone that regulates PERK kinase activity and is itself subject to UPR-associated upregulation, was increased at the protein level (Fig. 3A) [29]. Importantly, DP-induced ER stress was sustained over a 9 h observation period as indicated by persistent upregulation of GRP78 and CHOP protein levels and by phosphorylation of PERK and eIF2α Fig. 3A). In contrast, ER stress caused by the reducing agent DTT, known to act via reductive protein unfolding, was only transient as evidenced by induction of eIF2α-phosphorylation, detected at 1 h exposure time yet reduced to constitutive levels at later time points (Fig. 3C, bottom panel), and NAC treatment was not associated with eIF2α-phosphorylation (Fig. 3C, middle panel) [30].
Prior studies have suggested the predominant role of redox modulation in cancer cell-directed cytotoxicity of DP [21, 22, 24, 43]. In our own experiments, cellular levels of reduced glutathione, a sensitive marker of cellular oxidative stress, dropped only after prolonged exposure to DP (6 h) (Fig. 4A), a time point at which pronounced modulation of p53 (Fig. 4G), phospho-eIF2α (Fig. 3A) and Noxa (Fig. 5A) had already occurred (all observable at 1 h exposure). Similarly, monitoring cellular oxidative stress levels using DCF fluorescence indicated that elevation of peroxide levels occurred only after 6 h exposure time (Fig. 4B). Moreover, cell membrane-permeable antioxidants and metal chelators [including NAC, (10 mM), pegylated catalase (400 u/ml), pegylated SOD (1000 u/ml), deferoxamine (20 μM); added 1 h before DP exposure] did not protect A375 melanoma cells from DP-induced cell death (10 mM, 24 h; data not shown), further disqualifying the early induction of oxidative stress as an upstream event in DP-induced apoptosis.
It is well established that pharmacological activity of DP in cystinuria, a hereditary disorder characterized by the formation of urogenital L-cystine stones, depends on reductive formation of mixed disulfides between DP and L-cysteine resulting in solubilization of precipitated L-cystine [44]. Similarly, serum immunoglobulin fragmentation by reductive disulfide cleavage has been involved in the immunosuppressive activity of DP [45]. Therefore, it is tempting to speculate that DP-induced early cellular changes (ER stress/UPR) may occur as a result of the reducing activity of DP, causing cellular stress through the reductive unfolding of proteins. Indeed, reactivity of DP, both as an antioxidant and as a disulfide cleaving agent, has been shown to depend on formation of a nucleophilic DP-thiolate species, and thiolate formation by thiol compounds has been shown to depend on the pKa value associated with the respective thiol-group [46]. Among the thiol compounds that we tested for UPR-induction (as evidenced by eIF2α phosphorylation), DP (pKa: 7.9) displayed the highest activity, whereas DTT (pKa: 9.1) only induced a transient response and NAC (pKa: 9.5) was completely inactive (Fig. 3C).
After initial reductive protein unfolding by DP, cellular oxidative stress observed by us at later time points could then originate from multiple downstream sources such as ROS leakage from loss of mitochondrial transmembrane potential (Fig. 4C) and ROS formation by ER-associated redox enzymes. Indeed, expression of cytochrome P450 monooxygenases (encoded by CYP1A1, CYP2E1) and flavin monooxygenases (encoded by FMO1 and FMO5), known to be involved in oxidative metabolism of sulfur-containing endogenous substrates and xenobiotics that occurs with production of ROS, was upregulated at the mRNA level by DP (Fig. 2) [47–49]. Interestingly, a role of flavin monooxygenases in oxidative protein refolding in response to cellular reductive stress has been described earlier [50, 51], and a similar molecular pathway may be initiated by DP in melanoma cells, where initial reductive protein unfolding could cause FMO1 and FMO5 upregulation followed by refolding and ROS formation. However, the relative causative role of DP-induced ER stress, mitochondrial impairment, and redox dysregulation and their sequential involvement in melanoma cell apoptosis remain to be elucidated by future experiments.
It has been demonstrated before that intratumoral injection of a long-acting benzyl ester of D-penicillamine inhibits the growth of melanoma tumors in mice [52]. In our murine SCID A375 melanoma model, significant inhibition of tumor growth was observed upon systemic administration of DP at very high daily doses (i.p.; 1,200 mg/kg/d) (Fig. 6A), and significant proteolytic activation of caspase 3 occurred in tumor tissue in response to drug treatment (Fig. 6C). In this context it should be mentioned that no statistically significant antimelanoma effects were observed in an independent murine xenograft study administering DP in combination with tPA (tissue plasminogen activator), a result that may be related to the lower dose regimen selected in that study [53]. Moreover, in a previous phase II clinical trial, DP administered at FDA-approved doses did not improve survival in patients with glioblastoma multiforme [54]. However, more recent drug development efforts have aimed at improving DP chemotherapeutic efficacy by optimization of intracellular delivery, and systemic administration of DP poly-L-glutamic acid conjugates displayed significant activity in a murine leukemia model [55]. Therefore, feasibility of harnessing DP-induced UPR-induction and apoptogenicity for future chemotherapeutic intervention targeting metastatic melanoma remains to be established.
Acknowledgments
Supported in part by grants from the National Institutes of Health [R01CA122484, R03CA167580, ES007091, ES006694, Arizona Cancer Center Support Grant CA023074].
Footnotes
Declaration of interest: The authors report no conflicts of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
References
- 1.Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist. 2011;16(1):5–24. doi: 10.1634/theoncologist.2010-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aplin AE, Kaplan FM, Shao Y. Mechanisms of resistance to RAF inhibitors in melanoma. J Invest Dermatol. 2011;131(9):1817–1820. doi: 10.1038/jid.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol. 2009;625(1–3):234–246. doi: 10.1016/j.ejphar.2009.06.064. [DOI] [PubMed] [Google Scholar]
- 6.Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal. 2009;11(12):3013–3069. doi: 10.1089/ars.2009.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tew KD, Townsend DM. Redox platforms in cancer drug discovery and development. Curr Opin Chem Biol. 2011;15(1):156–161. doi: 10.1016/j.cbpa.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 9.Qiao S, Lamore SD, Cabello CM, Lesson JL, Munoz-Rodriguez JL, Wondrak GT. Thiostrepton is an inducer of oxidative and proteotoxic stress that impairs viability of human melanoma cells but not primary melanocytes. Biochem Pharmacol. 2012;83(9):1229–40. doi: 10.1016/j.bcp.2012.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20(3):400–413. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wondrak GT. NQO1-activated phenothiazinium redox cyclers for the targeted bioreductive induction of cancer cell apoptosis. Free Radic Biol Med. 2007;43(2):178–190. doi: 10.1016/j.freeradbiomed.2007.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabello CM, Bair WB, 3rd, Bause AS, Wondrak GT. Antimelanoma activity of the redox dye DCPIP (2,6-dichlorophenolindophenol) is antagonized by NQO1. Biochem Pharmacol. 2009;78(4):344–354. doi: 10.1016/j.bcp.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabello CM, Bair WB, 3rd, Lamore SD, Ley S, Bause AS, Azimian S, et al. The cinnamon-derived Michael acceptor cinnamic aldehyde impairs melanoma cell proliferation, invasiveness, and tumor growth. Free Radic Biol Med. 2009;46(2):220–231. doi: 10.1016/j.freeradbiomed.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cabello CM, Lamore SD, Bair WB, 3rd, Qiao S, Azimian S, Lesson JL, et al. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Invest New Drugs. 2011 doi: 10.1007/s10637-011-9676-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy RS, Fisher M, Alter JN. Penicillamine: Review and cutaneous manifestations. J Am Acad Dermatol. 1983;8:548–558. doi: 10.1016/s0190-9622(83)70062-9. [DOI] [PubMed] [Google Scholar]
- 17.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369(9559):397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 18.Wondrak GT, Cervantes-Laurean D, Roberts MJ, Qasem JG, Kim M, Jacobson EL, et al. Identification of alpha-dicarbonyl scavengers for cellular protection against carbonyl stress. Biochem Pharmacol. 2002;63(3):361–373. doi: 10.1016/s0006-2952(01)00915-7. [DOI] [PubMed] [Google Scholar]
- 19.Lengfelder E, Elstner EF. Determination of the superoxide dismutating activity of D-penicillamine copper. Hoppe Seylers Z Physiol Chem. 1978;359(6):751–757. doi: 10.1515/bchm.1978.359.1.751. [DOI] [PubMed] [Google Scholar]
- 20.Cohen JF, Elberling JA, DeMaster EG, Lin RC, Nagasawa HT. N-Terminal Dipeptides of D(−)-Penicillamine as Sequestration Agents for Acetaldehyde. J Med Chem. 2000;43:1029–1033. doi: 10.1021/jm9902741. [DOI] [PubMed] [Google Scholar]
- 21.Gupte A, Mumper RJ. An investigation into copper catalyzed D-penicillamine oxidation and subsequent hydrogen peroxide generation. J Inorg Biochem. 2007;101(4):594–602. doi: 10.1016/j.jinorgbio.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 22.Havre PA, O’Reilly S, McCormick JJ, Brash DE. Transformed and tumor-derived human cells exhibit preferential sensitivity to the thiol antioxidants, N-acetyl cysteine and penicillamine. Cancer Res. 2002;62(5):1443–1449. [PubMed] [Google Scholar]
- 23.Wondrak GT, Jacobson MK, Jacobson EL. Antimelanoma activity of apoptogenic carbonyl scavengers. J Pharmacol Exp Ther. 2006;316(2):805–814. doi: 10.1124/jpet.105.094953. [DOI] [PubMed] [Google Scholar]
- 24.Gupte A, Mumper RJ. Copper chelation by D-penicillamine generates reactive oxygen species that are cytotoxic to human leukemia and breast cancer cells. Free Radic Biol Med. 2007;43(9):1271–1278. doi: 10.1016/j.freeradbiomed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Cabello CM, Bair WB, 3rd, Ley S, Lamore SD, Azimian S, Wondrak GT. The experimental chemotherapeutic N(6)-furfuryladenosine (kinetin-riboside) induces rapid ATP depletion, genotoxic stress, and CDKN1A (p21) upregulation in human cancer cell lines. Biochem Pharmacol. 2009;77(7):1125–1138. doi: 10.1016/j.bcp.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamore SD, Cabello CM, Wondrak GT. The topical antimicrobial zinc pyrithione is a heat shock response inducer that causes DNA damage and PARP-dependent energy crisis in human skin cells. Cell Stress Chaperones. 2010;15(3):309–322. doi: 10.1007/s12192-009-0145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabello CM, Lamore SD, Bair WB, Davis AL, Azimian SM, Wondrak GT. DCPIP (2,6-dichlorophenolindophenol) as a genotype-directed redox chemotherapeutic targeting NQO1*2 breast carcinoma. Free Radic Res. 2011;45(3):276–292. doi: 10.3109/10715762.2010.526766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem. 2010;285(43):33165–33174. doi: 10.1074/jbc.M110.167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 30.Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 31.Eriksson O, Fontaine E, Petronilli V, Bernardi P. Inhibition of the mitochondrial cyclosporin A-sensitive permeability transition pore by the arginine reagent phenylglyoxal. FEBS Lett. 1997;409(3):361–364. doi: 10.1016/s0014-5793(97)00549-8. [DOI] [PubMed] [Google Scholar]
- 32.Keuling AM, Andrew SE, Tron VA. Inhibition of p38 MAPK enhances ABT-737-induced cell death in melanoma cell lines: novel regulation of PUMA. Pigment Cell Melanoma Res. 2010;23(3):430–440. doi: 10.1111/j.1755-148X.2010.00698.x. [DOI] [PubMed] [Google Scholar]
- 33.Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65(14):6282–6293. doi: 10.1158/0008-5472.CAN-05-0676. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez Y, Verhaegen M, Miller TP, Rush JL, Steiner P, Opipari AW, Jr, et al. Differential regulation of Noxa in normal melanocytes and melanoma cells by proteasome inhibition: Therapeutic implications. Cancer Res. 2005;65(14):6294–6304. doi: 10.1158/0008-5472.CAN-05-0686. [DOI] [PubMed] [Google Scholar]
- 35.Qin JZ, Xin H, Sitailo LA, Denning MF, Nickoloff BJ. Enhanced killing of melanoma cells by simultaneously targeting Mcl-1 and NOXA. Cancer Res. 2006;66(19):9636–9645. doi: 10.1158/0008-5472.CAN-06-0747. [DOI] [PubMed] [Google Scholar]
- 36.Weber A, Kirejczyk Z, Potthoff S, Ploner C, Hacker G. Endogenous Noxa Determines the Strong Proapoptotic Synergism of the BH3-Mimetic ABT-737 with Chemotherapeutic Agents in Human Melanoma Cells. Transl Oncol. 2009;2(2):73–83. doi: 10.1593/tlo.08223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu KS, Lee Y, Kim CM, Park EC, Choi J, Lim DS, et al. The protease inhibitor, elafin, induces p53-dependent apoptosis in human melanoma cells. Int J Cancer. 2010;127(6):1308–1320. doi: 10.1002/ijc.25125. [DOI] [PubMed] [Google Scholar]
- 38.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang CC, Lucas K, Avery-Kiejda KA, Wade M, deBock CE, Thorne RF, et al. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res. 2008;68(16):6708–6717. doi: 10.1158/0008-5472.CAN-08-0349. [DOI] [PubMed] [Google Scholar]
- 40.Xu W, Trepel J, Neckers L. Ras, ROS and proteotoxic stress: a delicate balance. Cancer Cell. 2011;20(3):281–282. doi: 10.1016/j.ccr.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281(11):7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 42.Guan D, Xu Y, Yang M, Wang H, Wang X, Shen Z. N-acetyl cysteine and penicillamine induce apoptosis via the ER stress response-signaling pathway. Mol Carcinog. 2010;49(1):68–74. doi: 10.1002/mc.20578. [DOI] [PubMed] [Google Scholar]
- 43.Jeitner TM, Lawrence DA. Mechanisms for the cytotoxicity of cysteamine. Toxicol Sci. 2001;63(1):57–64. doi: 10.1093/toxsci/63.1.57. [DOI] [PubMed] [Google Scholar]
- 44.DeBerardinis RJ, Coughlin CR, 2nd, Kaplan P. Penicillamine therapy for pediatric cystinuria: experience from a cohort of American children. J Urol. 2008;180(6):2620–2623. doi: 10.1016/j.juro.2008.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perrett D. The metabolism and pharmacology of D-penicillamine in man. J Rheumatol Suppl. 1981;7:41–50. [PubMed] [Google Scholar]
- 46.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27(3–4):322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 47.Kim HR, Lee GH, Cho EY, Chae SW, Ahn T, Chae HJ. Bax inhibitor 1 regulates ER-stress-induced ROS accumulation through the regulation of cytochrome P450 2E1. J Cell Sci. 2009;122(Pt 8):1126–1133. doi: 10.1242/jcs.038430. [DOI] [PubMed] [Google Scholar]
- 48.Suh JK, Poulsen LL, Ziegler DM, Robertus JD. Yeast flavin-containing monooxygenase generates oxidizing equivalents that control protein folding in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 1999;96(6):2687–2691. doi: 10.1073/pnas.96.6.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther. 2005;106(3):357–387. doi: 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poulsen LL, Ziegler DM. Microsomal mixed-function oxidase-dependent renaturation of reduced ribonuclease. Arch Biochem Biophys. 1977;183(2):563–570. doi: 10.1016/0003-9861(77)90391-5. [DOI] [PubMed] [Google Scholar]
- 51.Suh JK, Robertus JD. Yeast flavin-containing monooxygenase is induced by the unfolded protein response. Proc Natl Acad Sci U S A. 2000;97(1):121–126. doi: 10.1073/pnas.97.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chvapil M, Dorr R. Single intratumoral injection of long-acting benzyl ester of D-penicillamine inhibits the growth of melanoma tumor in mice. Anticancer Drugs. 2005;16(7):757–762. doi: 10.1097/01.cad.0000171767.59187.c2. [DOI] [PubMed] [Google Scholar]
- 53.de Groot-Besseling RR, Ruers TJ, Lamers-Elemans IL, Maass CN, de Waal RM, Westphal JR. Angiostatin generating capacity and anti-tumour effects of D-penicillamine and plasminogen activators. BMC Cancer. 2006;6:149. doi: 10.1186/1471-2407-6-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brem S, Grossman SA, Carson KA, New P, Phuphanich S, Alavi JB, et al. Phase 2 trial of copper depletion and penicillamine as antiangiogenesis therapy of glioblastoma. Neuro Oncol. 2005;7(3):246–253. doi: 10.1215/S1152851704000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wadhwa S, Mumper RJ. Intracellular delivery of the reactive oxygen species generating agent D-penicillamine upon conjugation to poly-L-glutamic acid. Mol Pharm. 2010;7(3):854–862. doi: 10.1021/mp1000058. [DOI] [PubMed] [Google Scholar]