

Abstract
Despite the progress in developing new therapeutic modalities, cancer remains one of the leading diseases causing human mortality. This is mainly attributed to the inability to diagnose tumors in their early stage. By the time the tumor is confirmed, the cancer may have already metastasized, thereby making therapies challenging or even impossible. It is therefore crucial to develop new or to improve existing diagnostic tools to enable diagnosis of cancer in its early or even pre-syndrome stage. The emergence of nanotechnology has provided such a possibility. Unique physical and physiochemical properties allow nanoparticles to be utilized as tags with excellent sensitivity. When coupled with the appropriate targeting molecules, nanoparticle-based probes can interact with a biological system and sense biological changes on the molecular level with unprecedented accuracy. In the past several years, much progress has been made in applying nanotechnology to clinical imaging and diagnostics, and interdisciplinary efforts have made an impact on clinical cancer management. This article aims to review the progress in this exciting area with emphases on the preparation and engineering techniques that have been developed to assemble “smart” nanoprobes.
Keywords: cancer diagnosis, biomarkers, nanotechnology, nanomedicine, bioconjugation, surface modification, imaging
1. Introduction
Cancer accounts for approximately one fourth of all deaths in the United States [1]. Prognosis of cancer patients depends largely on the time and accuracy of finding the primary and any dormant metastasis sites. Identification of these abnormal sites in an early enough stage can significantly reduce the mortality rate. For example, for breast cancer patients, the 5-year survival rate is 98% if the tumor can be diagnosed in stages 0 and I and drops to 85% for stage II and merely 20% for stage IV tumors [2]. Pancreatic cancer, on the other hand, is usually associated with a high mortality rate; about 75% of pancreatic cancer patients die within 1 year after diagnosis [3]. This high death rate stems from the recessive traits exhibited by pancreatic cancer and a lack of an effective early detection means. Needless to say, there is an urgent need for new tools that can detect cancer in the early or even pre-syndrome stage. Significant progress has been made recently thanks to advances in proteomics and genomics. These burgeoning techniques have identified various biomarkers whose abnormal regulations are closely associated with tumorigenesis and progression [4-6]. In addition, new screening technologies have been established allowing the identification of antibodies, peptide sequences, and nucleic acid aptamers with high affinity towards a specific biomarker [7, 8].
Nanotechnology, which is a multidisciplinary science involving chemistry, biochemistry, physics, and materials science, has found uses in a wide spectrum of medicine-related applications. These include nanoparticle-based imaging, drug delivery, biosensoring, and hyperthermia, and in the past decade, a number of these nanoparticle-based techniques have been translated into clinics [9]. Materials exhibit unique physical and biochemical properties when their dimensions are reduced to between several to hundreds of nanometers. By taking advantage of these unique characteristics, one can develop nanodevices that can sense and monitor biological events with unprecedented efficiency and sensitivity. When coupled to the aforementioned targeting ligands, one can produce “smart” nanoprobes that can interact with a biological system and sense changes on the molecular level. This can take place in vivo, where nanoprobes are systematically administrated, accumulated in tumors through ligand-biomarker interaction, and send out signals for sensitive diagnostic imaging. The nanoprobes are also useful to in vitro diagnosis and analysis of biological samples such as saliva, blood, and urine. Both applications are invaluable and hold great promise in revolutionizing cancer management [9]. The objective of this article is to review the recent progress in this exciting area. We will begin by discussing common tumor biomarkers and their corresponding targeting molecules, followed by a systematic description of the design and fabrication of magnetic nanoparticle-, quantum dot-, upconversion nanoparticle-, and gold nanoparticle-based diagnostic techniques, as well as their potential applications to cancer detection.
2. Tumor biomarkers
Tumor biomarkers are substances found in blood, urine, stool, or tissues of patients with cancer. They are most commonly proteins that are elevated in either cancer cells or other cells in the body in response to cancerous conditions. Measuring biomarker levels is one of the most common methods to detect, diagnose, and manage cancer.
Based on their location, protein biomarkers can be divided into three categories: intracellular, extracellular, and those that are on the cell membrane. For nanoparticle-enabled diagnosis, different biomarkers are targeted for different applications. For imaging, it is more common to target cell membrane protein biomarkers. For in vitro diagnosis, it is more convenient to target proteins that are soluble and shed into the circulation. Intracellular proteins, on the other hand, are less targeted given the relatively large size of nanoparticles and hence, their inability to pass through the cell membrane. Due to a long and ever growing list of biomarkers, it is not possible to cover all of them in this article. Instead, we will focus on introducing several biomarkers that have been well studied and are commonly targeted in validating nanoparticle-based imaging and diagnosis techniques.
2.1. EGFR and HER2
The epidermal growth factor receptor (EGFR) is a 170-kDa protein expressed on the membrane of many types of epithelial cancers [10, 11]. EGFR has long been regarded as a tumor biomarker because its mutation is often associated with autonomous cell growth, inhibition of apoptosis, and metastases [12, 13]. The epidermal growth factor (EGF)-EGFR interaction is among the first growth factor ligand-receptors studied [14]. EGFR has been found to be a member of a receptor tyrosine kinase (TK) family, the human epidermal growth factor receptor (HER) family. The other three members are HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) [15]. Hence, EGFR is also referred to as HER1 (ErbB1). Upon ligand binding, the EGFR pairs with either itself or another HER member such as HER2 form an active dimer. Dimerization stimulates protein-tyrosine kinase and activates the intracellular protein kinase cascade. Monoclonal antibody-based inhibitors that target EGFR have been developed that take advantage of EGFR’s important role in cancer development. The first example in this category is cetuximab, a chimeric monoclonal antibody now used clinically to treat patients with head and neck cancer as well as metastatic colorectal cancer. With high avidity, cetuximab binds with EGFR and sterically blocks its interaction with growth factors [16]. Other EGFR targeting antibody inhibitors including panitumumab, zalutumumab, nimotuzumab, and matuzumab, are currently in clinical trials. When coupled with nanoparticle-based probes, these antibodies can guide the probes to tumors to realize targeted imaging [17, 18].
HER2 has also been intensively studied as a tumor biomarker; in particular, its implications for breast cancer. Deregulation of HER2 is found in 20-25% of breast cancer patients, making it an important indicator of breast tumor malignancy [19, 20]. HER2 usually dimerizes with EGFR or HER3 [14], although it can also dimerize with itself at high expression levels [21]. Trastuzumab, an anti-HER2 monoclonal antibody, has been used clinically to treat breast cancers with HER2 overexpression [14]. It has been coupled with various kinds of nanoparticles and studied as a tumor imaging probe in a pre-clinical setting [22, 23]. Pertuzumab, another HER2 targeting monoclonal antibody, has recently received FDA approval for the treatment of HER2-positive metastatic breast cancer.
2.2. VEGFR
The vascular endothelial growth factor (VEGF) is another family of growth factors that play a key role in angiogenesis, the process of forming new blood vessels. Angiogenesis is critical to physiological development and also is frequently implicated in pathological processes, including tumor progression [24]. The VEGF family is composed of 7 members, VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, VEGF-F, and placenta growth factor [25]. The most important one is VEGF-A, which is a homodimeric, disulfide-bound glycoprotein existing in several isoforms with different numbers of amino acid residues [26]. The most studied isoforms are VEGF165, VEGF121, and VEGF110. VEGF165 is implicated mostly in angiogenesis and vascular permeability whereas VEGF121 and VEGF110 are permeability factors [27]. VEGF-A acts on the vascular endothelium to stimulate endothelial cell mitogenesis and cell migration [27]. The receptors to VEGF-A are mainly Flt-1 (VEGFR-1) and Flk-1/KDR (VEGFR-2). VEGFR-1 is involved in physiological and developmental angiogenesis [25]. VEGFR-2 is a more important player in tumor progression and is critical to mitogenic, angiogenic, and permeability enhancement [28].
Overexpression of VEGF/VEGFR is observed from many tumor types and is indicated as a poor prognostic marker. To inhibit the interaction, one strategy is to sequester VEGF with an antibody in order to prevent VEGF from accessing VEGFR. Bevacizumab is a human monoclonal antibody against all isoforms of VEGF-A and the first antigenic agent receiving FDA approval for cancer management. Similarly, ranibizumab, a monoclonal antibody fragment (Fab) of bevacizumab, is used clinically for mascular degeneration treatment. A second approach is to employ an antibody that targets VEGFR. Upon binding, VEGFR is sterically blocked from interacting with VEGF. Several anti-VEGFR-2 antibodies, including IMC-1C11, IMC-1121B, and CDP791, have entered clinical development [29]. VEGF, anti-VEGF antibodies, and anti-VEGFR antibodies are all possible targeting ligands for tumor imaging in vivo.
2.3. Integrins
Integrins constitute a family of cell adhesion molecules [30]. They are always present as heterodimers comprising of one α and one β subunit. In mammals, 18 α-subunits and 8 β-subunits have been characterized. These subunits assemble into at least 24 integrin receptors that mediate the interactions between the cell and surrounding tissues [27]. Integrins are found upregulated on most tumor vasculature and for certain cancer types, also on tumor cell membranes. They are important players in tumor angiogenesis and metastasis [31]. In particular, integrin αvβ3 has been widely studied as a tumor biomarker [27] for ovarian [32], breast [33], lung [34], and prostate [35] cancer detection. Integrin αvβ3 binds to arginine-glycine-aspartic acid (RGD), which is a three-amino-acid sequence [36]. RGD dimers, tetramers, and octamers have also been studied for integrin αvβ3 targeting [37, 38]. RGD and its derivatives have been coupled to the surface of many types of nanoparticles for tumor imaging and site-specific drug delivery [27]. It is worth mentioning that in many animal models, integrin αvβ3 is upregulated on both tumor vasculature and tumor cells, and targeting occurs at both places when a small-molecule-based imaging probe is used. With regard to nanoparticle-based probes, especially those with a relatively large size, extravasation is a limiting factor and targeting typically occurs on the surface of endothelial cells. In addition to αvβ3, other members in the integrin family have been studied as tumor biomarkers. Examples include αvβ6 in the detection and staging of ovarian carcinomas [32] and integrin α5β1 in early stage lung cancer diagnosis [34].
2.4. Folate receptor
Folic acid belongs to the vitamin B family. It is a required nutrient for all living cells and participates in the biosynthesis of nucleotide bases. Folic acid shows high affinity (KD < 1 nM) to folate receptors (FR) including FR-a and FR-b, both of which are 38 kDa membrane-bound proteins [39]. FRs are expressed at low levels in normal tissues and mostly restricted to epithelial cells [40]. On the other hand, FRs are found overexpressed in several tumor types, particularly in ovarian and endometrial cancers [40]. Studies have shown that folic acid binds to tumor cells 20 times more than normal epithelial cells or fibroblasts [41], and this tumor-associated disregulation makes FR an attractive tumor biomarker.
Folic acid can be easily coupled to a peptide or a macromolecule with insignificant effects to the affinity to FR [42]. Various kinds of nanoparticles have been coupled to folic acid and studied for tumor imaging [40]. FR is expressed at a relatively high level in the kidneys [40]. Imaging and therapy with folic acid-small molecule conjugates can be impeded by this feature due to high kidney uptake. However, this issue is of minor concern with nanoparticle-based probes due to their inaccessibility to these regions.
2.5. MMPs
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases. There are at least 20 members in this protease family and they are capable of degrading multiple components of the extracellular matrix (ECM) [43]. Based on their substrate specificity, MMPs can be classified as collagenases, gelatinases, stromelysins, and so on. Another subset is MMPs 14 to 17 which contain a transmembrane domain and are known as membrane-associated MMPs (MT-MMPs). In normal tissues, MMPs are expressed at a very low level [43], but when active tissue remolding occurs, MMPs are rapidly transcribed, secreted, and activated [44]. Such activation is usually associated with a pathological process such as cancer. Indeed, high MMP expression levels are found in many types of tumors and are expressed by the host stromal cells and sometimes by tumor cells [45]. For instance, MMP2 is highly expressed in breast cancers and an increased ratio of active MMP2 is usually indicative of a higher malignancy [46, 47]. MMPs 1, 3, 9, 14 are also key players in tumor cell growth, metastasis, and angiogenesis [43].
These implications make MMPs an important class of tumor biomarkers. In the previous decade, efforts were made to prepare MMP-targeted tumor imaging probes. Targeting can be achieved using a high affinity peptide. For instance, Zhu et al. found that a peptide sequence, HWKHLHNTKTFL, had high MT1-MMP binding affinity and when coupled with a dye molecule, could efficiently target tumors [48]. Koivunen et al. reported a short target peptide, CTTHWGFTLC, which could specifically bind MMP-2 and MMP-9 thus inhibiting the migration of human endothelium and cancer cells [49]. An alternative approach is to take advantage of the enzymatic function of MMPs and develop an MMP-activatable imaging probe that displays amplified signals in the tumor region. Gold nanoparticle-based MMP activatable imaging probes, for instance, have recently been reported by Lee and Xie et al. [50, 51].
2.6. PSMA
The prostate-specific membrane antigen (PSMA) is a 100-kDa type II transmembrane glycoprotein composed of a short and amino-terminal cytoplasmic tail (19 amino acids), hydrophobic transmembrane domain (24 amino acids), and large extracellular domain (707 amino acids) at the carboxy-terminus [52, 53]. PSMA is expressed at a low or moderate level in most hyperplastic and benign prostates, but is highly up-regulated in malignant tissues [54]. The increased PSMA expression is related to tumor aggressiveness, and the highest PSMA levels are usually found from high-grade tumors, metastatic lesions, and androgen-independent disease [55]. PSMA is also found abundantly expressed on neovasculature of non-prostatic solid tumors, including lung, colon, breast, renal, liver, and pancreatic carcinomas as well as sarcomas and melanoma [55]. Antibodies against PSMA such as 7E1, J591 and MDX-070 are now used clinically for either imaging or therapeutic purposes [55]. In addition to antibodies, other motifs have been discovered for PSMA targeting. Langer’s group, for instance, has been working on aptamer- [56, 57] and small molecule-based [54,55] ligands with high avidity towards PSMA. One of the technologies developed by the group, BIND-014, recently entered clinical trial for solid tumor therapy. BIND-014, a polylactide (PLA)-based nanoparticle bearing a cytotoxic drug docetaxel, uses S-2-[3-[5-amino-1-carboxypentyl]-ureido]-pentanedioic acid, a small molecule with high selectivity to PSMA for tumor targeting [54,55].
2.7. Phosphatidylserine
Cancer cells are essentially cells that grow out of control and lose the physiological balance of renewal and senescence. Most therapeutic approaches work by inducing apoptosis to cancerous cells. A widely acknowledged apoptosis biomarker is phosphatidylserine (PS), which in normal cells is fastened on the cytosolic side of the cell membrane, but is flipped to the exterior side when the cell undergoes apoptosis [58]. Measuring the PS level is a proven means to monitor the cell apoptotic level and evaluate the response of cancerous cells to a treatment [59]. High level of PS has also been found on tumor vascular endothelium [60]. Positive PS staining was observed on 4% to 40% tumor vasculature but not on normal tissues. Boasting high selectivity toward PS, Annexin-V is the most commonly used apoptosis probe. Flow cytometry assays with effusions from ovarian carcinoma patients showed good correlation between Annexin-V level and stage, overall survival (p = 0.005), as well as progression-free survival (p = 0.013) [61]. Annexin-V can be labeled with radio- or optical-tags for tumor detection or monitoring of therapeutic response [62].
In this section, we summarized some common biomarkers targeted for cancer detection. However, it is still debatable whether some of these proteins can be called early-detection biomarkers. Nonetheless, they are the most commonly targeted from the perspective of nanoparticle-based imaging/delivery technology. Active research is being conducted to identify other protein or even non-protein-based signatures that better represent early stage cancer [63, 64]. Therefore, it will not be surprising to see in the near future an increased number of reports pertaining to the development of nanoparticle-based probes/carriers that target these novel biomarkers.
3. Engineering of nanoparticles for tumor detection
Nanoparticles can be used as probes in in vivo imaging, biosensing, and immunostaining because nanoparticle-based probes offer many advantages. First of all, they deliver high sensitivity. Many nanoscale materials show unique magnetic, optical, or acoustic properties and they can be further imparted with other types of imaging functionalities to result in probes with multimodal abilities. Secondly, their size is appropriate. Most nanoparticles have a size that is above the threshold of renal clearance (<7 nm) allowing them to remain in circulation for a relatively long time before reaching the desired targets [65, 66]. On the other hand, nanoparticles are also small enough to penetrate many biological barriers such as endothelial barriers, cell membranes, or even nuclear envelopes to efficiently interact with biological systems on the molecular level [67]. Thirdly, they are multivalent. Multiple targeting ligands can be tethered onto one nanoparticle surface. This leads to a larger rate of receptor binding and smaller rate of dissociation, both contributing to higher tumor uptake and longer retention time [68, 69], the so-called multivalency effect. Moreover, it is possible to impart more than one type of targeting ligand [70, 71]. Recent studies have confirmed improved tumor cell selectivity from such a dually-targeting targeted approach [72-74]. Fourthly, they offer combined therapy and diagnosis. In addition to imaging functionalities, therapeutics can be loaded onto nanoplatforms. The resulting nanoparticles, so-called nano-theranostics, have both imaging and therapeutic capabilities [75]. Nano-theranostics are appealing for potentially allowing therapy response to be monitored in real-time by imaging methods, an emerging concept in modern personalized medicine [76].
In the following section, recent progress on nanoparticle-based probes/sensors for cancer diagnosis is reviewed. Owing to limited space, we will only describe magnetic nanoparticles, quantum dots, gold nanoparticles, and upconversion nanoparticles. Readers who are interested in other materials such as carbon nanotubes and graphene are referred to other excellent reviews [77-81].
3.1. Magnetic nanoparticles
Magnetic nanoparticles have been investigated extensively in the field of biomedicine and one of their important applications is contrast probes used in magnetic resonance imaging (MRI). Magnetic nanoparticles create microscopic field gradients in a strong magnetic field; causing shortening and diphase of longitudinal (T1) or transverse relaxation times (T2 and T2*) of nearby nuclei, most commonly protons. This induces hyper- (for T1) or hypo-intensities (for T2 and T2*) on MRI maps and by doing so, highlighting the areas that are concentrated with the particles. Magnetic nanoparticles, particularly iron oxide nanoparticles (IONPs), are used in liver imaging, lymph node imaging, and cell tracking. Recently, devices with extremely high sensitivity using magnetic nanoparticles as probes have been developed. In this section, we will describe the synthesis, surface modification, and biological applications of magnetic nanoparticles with a focus on IONPs which are by far the most common magnetic nanoparticles.
3.1.1. Synthesis of magnetic nanoparticles
3.1.1.1. Iron oxide nanoparticles made by co-precipitation
Bulk iron oxide, either magnetite or hematite, is a ferromagnetic material with a strong magnetic moment. In the ambient environment, magnetite and hematite have magnetic moments of 90 and 76 emu g-1, respectively. However, both Fe3O4 and Fe2O3 nanoparticles show zero remnant magnetism in the ambient environment. This is because on such a small scales, thermal energy is sufficient to overcome the anisotropy energy of each small magnet thereby causing random fluctuations in the magnetization and an overall zero magnetic moment [82]. When an external magnetic field is applied, the nanoparticle magnets are easily magnetized and re-aligned to reach a saturation magnetic moment. However, when the magnetic field is turned off, the magnets are again randomized and the magnetism drops to zero. This phenomenon, termed superparamagnetism, is also observed from other types magnetic nanoparticles [82]. One of the most important reasons IONPs are so commonly used is the biocompatibility of iron oxide. Iron oxide has low toxicity and can be degraded by a biological system to become part of the body’s iron stores. Another reason is well-established surface chemistry. In the past decade, many surface engineering techniques have been developed that can modify IONPs, making them stable in physiological environments and easy to be conjugated with various bio-molecule species.
Traditionally, IONPs are synthesized by co-precipitation (Table 1) [83]. In brief, Fe(II) and Fe(III) precursors are dissolved in an aqueous solution into which an alkaline medium is added to induce particle formation. The size, shape, and composition of the magnetic nanoparticles depends on the type of the salts used (e.g. chlorides, sulfates, nitrates), Fe2+/Fe3+ ratio, reaction temperature, pH value, and ionic strength [83-85]. This synthetic approach is straightforward and has a high throughput, but the resulting particles exhibit low magnetism, typically in the range of 30 to 50 emu g-1 [86]. Bare IONPs are not stable and tend to aggregate. To improve their stability, it is common to include stabilizing agents in the reaction. They are usually polymers such as polyvinylpyrrolidone (PVP), dendrimers, polyaniline, and dextrans [87-93] which lower the surface energy and increase electrostatic repulsion, resulting in improved colloidal stability. Most IONPs used clinically, including Feridex, Combidex, Resovist, and Ferumoxytol, are made by the co-precipitation method using dextran and its derivatives as the coating materials [94, 95].
Table 1.
Summary of magnetic nanoparticles of various types
| Particle | Synthesis | Surface coating | r1 (mM-1 s-1) | r2 (mM-1 s-1) |
|---|---|---|---|---|
| Fe3O4 | co-precipitation | cross-linked aminated dextran [100] | 22.4 [100] | 76.7 [100] |
| Zn0.4Mn0.6Fe2O4 | thermal decomposition of metal chloride (MCl2, M= Zn2+, Mn2+, and Fe2+) and Fe(acac)3 | DMSA | N/A | 860 [114] |
| Fe | thermal decomposition of Fe(CO)5 | PEGylated dopamine | N/A | 220 [119] |
| MnO | Thermal decomposition of Mn-oleate complex | silica | 0.99 [148] | N/A |
| Mn NMOFs [154] | reverse-phase microemulsion using MnCl2 and [NMeH3]2(BDC) | PVP/silica | 4.6 [154] | 141.2 [154] |
| Gd-apoferritin | loading GdHPDO3A into apoferritins | apoferritin | 80±5 [157] | N/A |
| Metallofullerene | arc-burning of Gd2O3 and graphite | carbon | 207 [161] | N/A |
A sugar coating does not favor direct bio-molecule coupling, and to make the particles conjugation-friendly, a post-synthesis surface engineering step is usually required. A common approach is to treat the dextran-coated particles with epichlorohydrin and ammonia. This treatment produces a more rigid, cross-linked coating [96] and more importantly, introduces multiple amine groups on the particle surface that can be used for bio-conjugation [97-99]. The treated nanoparticles, termed cross-linked iron oxide, can be coupled to antibodies, peptides, aptamers, as well as drug molecules for the purpose of tumor imaging and therapy [100].
3.1.1.2. Magnetic nanoparticles made by thermal decomposition
Thermal decomposition has become a popular synthetic approach in IONP preparation (Table 1) [101, 102]. Instead of using an aqueous solution as the reaction medium, thermal decomposition takes place in an organic solvent with a high boiling point. In a typical synthesis, organometallic compounds such as Fe(CO)5, Fe(acac)3, and Fe-oleate are heated to a high temperature to induce nucleation and particle growth, as illustrated in Fig. 1 [103-105]. Surfactants, such as oleic acid (OA) and oleylamine (OAm), are also essential to the process because they dynamically interact with the growing particle surface and determine the monodispersity and morphology of the products. In general, thermal decomposition provides better product quality control over co-precipitation methods. By changing/adjusting the surfactant, precursor, precursor concentration, surfactant/precursor ratio, heating rate, and reaction temperature [106-109], IONPs with single crystallinity, narrow size distribution, and controllable size can be produced. For instance, Woo et al. prepared monodispersed IONPs by thermal decomposition of Fe(CO)5 in a solution of octyl ether and oleic acid [110]. Sun et al. heated Fe(acac)3 with 1,2-hexadecanediol in the presence of OA and OAm to make monodispersed IONPs with size of 4 to 20 nm (Fig. 1a) [111]. An increase in size can be achieved using a seed-mediated process using small particles as the seeds. Park et al. reported that monodispersed magnetic IONPs with a continuous size between 6 and 13 nm could be synthesized by a procedure similar to seed-mediated growth (Fig. 2) [106]. In this process, the metal-surfactant complexes generated in situ are thermally decomposed to form nanoparticles. A prominent size effect is observed from these pyrolysis-yielded nanoparticles. Generally, particles with a larger diameter possess larger magnetic moments due to spin disorder and canting on the particle surface [112]. For instance, 4, 6, 9, and 12 nm IONPs have magnetic moments of 25, 43, 80, and 102 emu g-1, respectively (Fig. 3) [113]. The difference in magnetism dramatically affects their ability as T2 contrast agents in MRI.
Fig. 1.
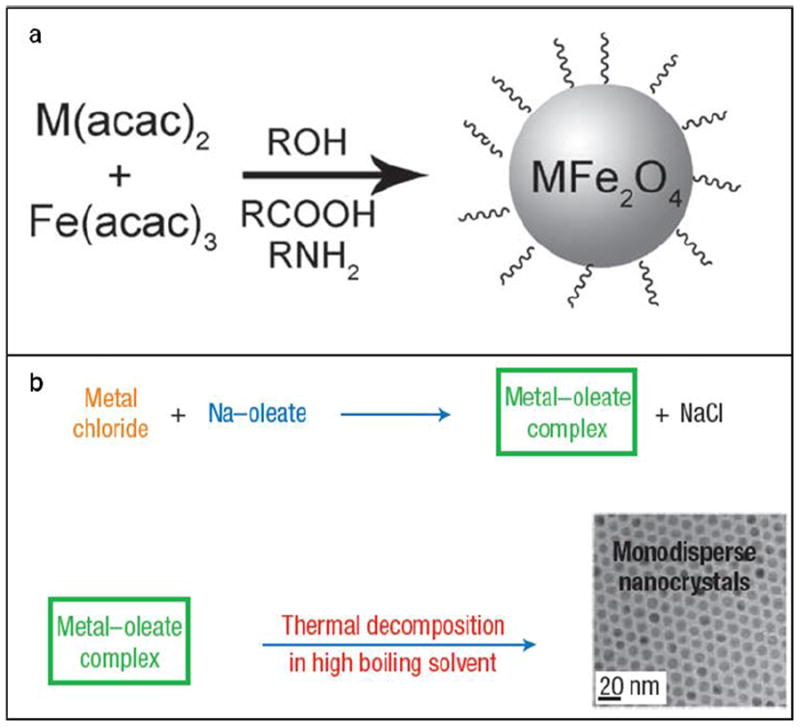
Schematic illustration of the synthesis of monodisperse magnetic nanoparticles using different precursors: (a) metal acetylacetonates, (b) metal–oleate. Reprinted with permission from refs. [111] and [149].
Fig. 2.

TEM images of IONPs synthesized by a thermal decomposition route with precise control of their diameters. (a) 6 nm, (b) 7 nm, (c) 8 nm, (d) 9 nm, (e) 10 nm, (f) 11 nm, (g) 12 nm, (h) 13 nm. Reproduced with permission from [106].
Fig. 3.

Nanoscale size effect of IONPs on MR signals. (a) TEM images of nanoparticles of 4, 6, 9, and 12 nm. (b) Size-dependent T2-weighted MR images of IONPs at 1.5 T. (c) Size-dependent changes in color-coded MR images based on T2 values. Reprinted with permission from [113].
The thermal decomposition method can be extended to prepare other magnetic nanoparticles with even larger magnetic moments. For example, MFe2O4 (M = Co, Mn, Ni) nanoparticles can be prepared by simply replacing one third of the Fe(acac)3 precursor with Co(acac)2/Mn(acac)2/Ni(acac)2 (Fig. 1). The nanoparticles produced possess different magnetic properties. MnFe2O4 nanoparticles with a size of 15 nm possess a magnetic moment of 74 emu g-1 which is higher than that of IONPs with the same size [111]. Other transition metals can be incorporated into the matrix as well. Cheon’s group reported the preparation of ZnxMn1-xFe2O4 nanoparticles. Zn2+ was introduced into the tetrahedral sites, and because Zn2+ resides mainly in the Td sites rather than Oh sites, magnetism is significantly increased in comparison with normal spinel metal ferrite nanoparticles [114]. In particular, Zn0.4Mn0.6Fe2O4 nanoparticles with a size of 15 nm have an extremely high magnetic moment of 175 emu g-1 and r2 value of 860 mM-1 s-1.
Synthesis of metallic nanoparticles such as Fe and Co have been reported [115]. Bulk Fe and Co have magnetic moments of 218 and 162 emu g-1, respectively, both of which are significantly larger than iron oxide’s. Metallic Fe nanoparticles have been synthesized by thermal decomposition of Fe(CO)5 in the presence of stabilizing surfactants such as polyisobutene and decalin [116, 117]. By adjusting the ratio of Fe(CO)5 to surfactants, Fe nanoparticles with a size of 2 to 10 nm and narrow size distribution (polydispersity of approximately 10%) have been fabricated. However, these Fe particles are not stable in the ambient environment. They are easily oxidized, leading to reduced magnetic susceptibility. Sun’s group reported a simple one-pot synthetic approach to make air-stable Fe nanoparticles [118, 119]. Briefly, Fe(CO)5 was decomposed in the presence of hexadecylammonium chloride, an oxidizing agent. This treatment led to the formation of a crystalline Fe3O4 shell that protected the Fe core from continued oxidation [120].
Synthesis of Co nanoparticles has been reported by a number of groups. Alivisatos and co-workers reported the synthesis of cobalt nanodisks by thermal decomposition of Co2(CO)8 [121, 122]. Co nanoparticles also suffer from the air-sensitivity problem. Bonnemann et al. described the synthesis of air stable monodisperse colloidal Co nanoparticles by thermolysis of Co2(CO)8 in the presence of aluminum alkyl compounds [123]. By varying the alkyl chain length of the organoaluminum compounds, the size of the Co particles can be tuned in the range of 3–11 nm.
Nanoscale particles of many other magnetic materials have been made. Hou’s group employed a facile wet-chemical route to synthesize Hägg iron carbide (Fe5C2) nanoparticles, in which bromide was found to be the key to inducing the conversion of Fe(CO)5 to Fe5C2 (Fig. 4) [124]. The Fe5C2 nanoparticles were ~20 nm in diameter (Fig. 4a) and had a core-shell structure (Fig. 4b). Dai’s group adopted a chemical vapor deposition method to synthesize FeCo/single-graphitic-shell nanocrystals [125] and Sun et al. reported the synthesis of FePt nanoparticles via thermal decomposition of Pt(acac)2 and Fe(CO)5 in benzyl ether [126].
Fig. 4.
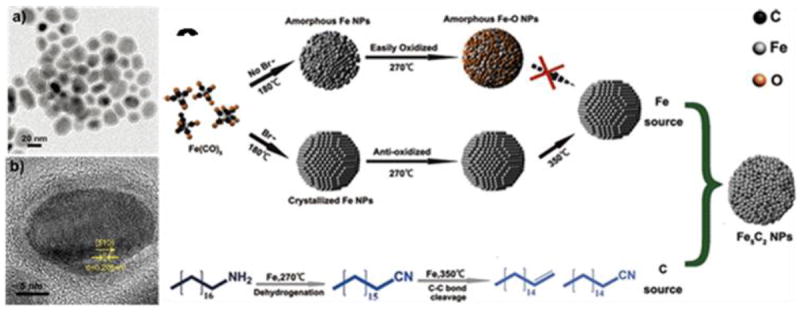
Fe5C2 nanopaticles synthesized by thermal decomposition. (a) TEM and (b) HRTEM images of 20 nm Fe5C2 nanopaticles. (c) schematic illustration of the formation mechanism of Fe5C2 nanopaticles. Reprinted with permission from [124].
In summary, nanoparticles of iron oxide and other types of magnetic materials have been produced mainly by co-precipitation and thermal decomposition. Co-precipitation is straightforward, repeatable, and has a high throughput. However, the particles produced have a relatively broad size distribution and suboptimal magnetism. Thermal decomposition allows better control of the particle size, shape, and composition, but requires stricter reaction conditions (high temperature, oxygen and water free, etc.). Another disadvantage of thermal decomposition is that the synthesized nanoparticles are coated with a thick alkyl coating and not water soluble. Subsequent surface modification needs to be performed before the particles can be used in biological applications.
3.1.2. Surface modification of magnetic nanoparticles
The two most commonly used surface modification approaches are ligand exchange and ligand addition. The former refers to the strategy of introducing a ligand with higher affinity to the particle surface. The latter involves the use of an amphiphilic substance that interacts with the existing hydrophobic alkyl layer via hydrophobic-hydrophobic interactions while leaving the hydrophilic section outward to suspend the particles, as shown in Fig. 5.
Fig. 5.
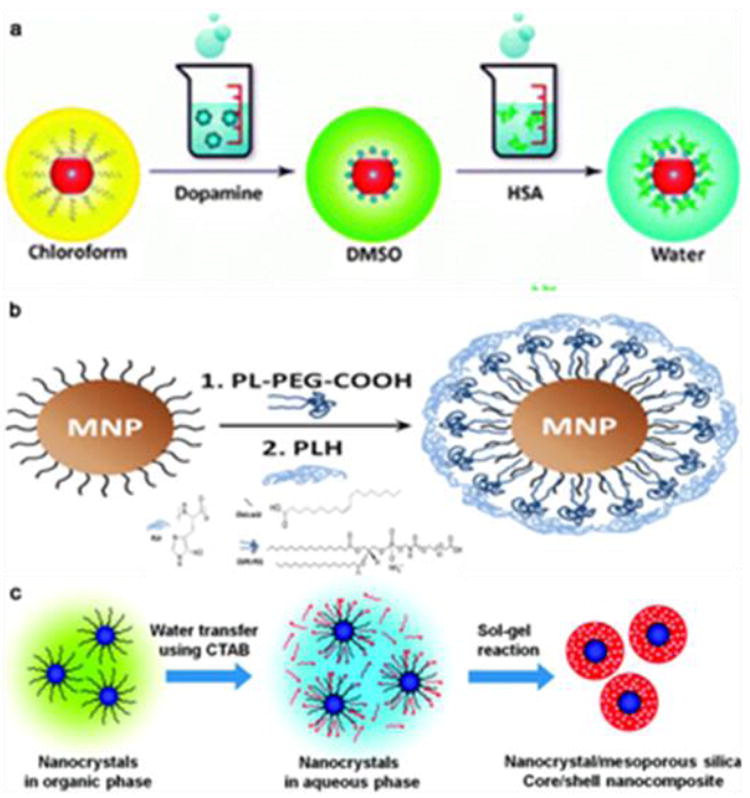
Representative IONPs surface modification methods. (a) Ligand exchange; (b) ligand addition; (c) silica coating. Reproduced by permission from ref. [134], [140], and [145].
3.1.2.1. Ligand exchange
Polymers such as poly(pyrrole), poly(aniline), poly(alkylcyanoacrylates), poly(methylidene malonate), poly(lactic acid), poly (glycolic acid), and poly(e-caprolactone) [127-131] have been studied as ligands to replace the original alkyl coating layer on the surface of magnetic nanoparticles. These polymers contain amine, carboxylate, phosphate, or hydroxyl end groups, and due to their multi-dentate nature, they can chemically adsorb onto the nanoparticle surface to form a protective coating. Alternatively, small molecule-based anchors that chelate with surface metals can be used as a replacing ligand. For IONPs, one good example is dopamine and its derivatives. This bidentate enediol compound can chelate with the under-coordinated surface Fe and convert it to a bulk-like lattice structure with an octahedral geometry [132]. This results in strong binding between the dopamine moiety and surface of the IONPs. This method was first reported by Xu et al. who successfully anchored nickel nitrilotriacetic acid (Ni-NTA) on the iron oxide shell of Co or SmCo5.2 nanoparticles using dopamine as the anchor [132]. Afterwards, Sun’s group reported surface-modifications of IONPs with PEGylated dopamine and evaluated the stability and potential applications of these PEGylated nanoconjugates [133]. Xie et al. reported a two-step surface modification approach for IONPs. In this process, the particles were first ligand-exchanged with dopamine and subsequently adsorbed with a layer of human serum albumin (HSA) as shown in Fig. 5a [134]. Another commonly used anchor compound is dimercaptosuccinic acid (DMSA), which is able to chelate with surface iron via the two carboxylic groups. These particles can be further stabilized by disulfide cross-linkages formed among the ligands [135]. Bio-molecules can be coupled onto the particle surface by utilizing the surface thiol groups. Herceptin, for instance, has been coupled onto DMSA modified particles [135].
It is common to observe particle aggregation during ligand exchange and phase transition. This is attributed to the dramatic polarity change on the particle surface which makes the particles more susceptible to clustering. Moreover, the interaction between the new ligand and particle surface is usually pH sensitive and can be labile in a harsh environment, such as that in endosomes/lysosomes. Nonetheless, ligand exchange remains a popular surface modification strategy in the field.
3.1.2.2. Ligand addition
Unlike ligand exchange, the native hydrophobic coatings are retained in ligand addition. The new ligands are usually amphiphilic materials such as poloxamine, poly(lactic-co-glycolic acid), poly(maleic anhydride alt-1-tetradecene) [136, 137], and phospholipids [138]. The hydrophobic segments, mostly hydrocarbons, intercalate/interact with the alkyl tails of the native ligands. The hydrophilic segments, usually containing PEG and/or multiple charged chemical groups, are pointed outward to improve water solubility. Ligand addition and phase transition are usually achieved by film hydration as shown in Fig. 5b [139, 140]. Using phospholipids as an example, the phospholipids and nanocrystals are first mixed in chloroform to form a homogeneous solution. The solvent is evaporated with a rotary evaporator to produce a film with uniformly distributed components. Afterwards, the film is hydrated and the components self-assemble into water-dispersible nanoparticles. This dispersing step usually requires energy input from heating and sonication [138].
The coating thickness impacts the relaxation times of the magnetic nanoparticles. LaConte et al. used PEGylated phospholipids of PEG chains with various lengths to modify IONPs. As the coating thickness increased, R2 decreased and R1 increased [141] due to physical exclusion of protons from the magnetic field and residence time of protons in the coating zone. It is worth mentioning that PEGylation is a common method used in nanoparticle surface modification [133, 138-141]. In the blood, nanoparticles can be easily adsorbed with a layer of opsonin proteins and as a result, recognized by the immune system and removed from circulation. PEGylation has proven to be able to stabilize nanoparticles, inhibit serum protein absorption, and extend particle circulation half-lives [138].
Small molecules, such as drug molecules, can be introduced along with IONPs into polymer/liposome matrices. For instance, Hu et al. fabricated core/shell particles containing an IONP core and a thermally responsive polymer shell into which hydrophobic drugs were encapsulated [142].
3.1.2.3. Silica coating
In addition to ligand addition and exchange with organic compounds, surface modification can also be performed using thick and dense inorganic materials. Compared to organic coatings, the rigid structure of an inorganic coating can better prevent the particle cores from contacting each other and the environment. The most commonly used inorganic coating is silica. For IONPs made by thermal decomposition, the coating usually occurs in a reverse microemulsion containing hydrophobic IONPs and tetraethylorthosilicate (TEOS) [143, 144]. TEOS hydrolyzes and condenses onto the particles to form a silica shell. Other silanes, such as N-(6-aminohexyl)-aminopropyltrimethoxysilane (AHAPS) and 3-(triethoxysilyl)propylsuccinic anhydride) (SSA), can be mixed with TEOS as the precursors. The resulting nanoparticles have surface amine or thiol groups suitable for further bio-conjugation. Alternatively, functional molecules can be pre-conjugated with silanes and encapsulated into the silica matrix. Dye molecules such as rhodamine B isothiocyanate (RITC), for instance, can first be coupled with 3-aminopropyltriethoxysilane (APTES) and co-condensed to form a fluorescent silica coating, as shown in Fig. 5c [145]. To further improve the physiological stability, PEGylation is usually added to the silica coating [146].
Functionality loading can also be achieved by using a mesoporous silica coating. This is formed by including cetrimonium bromide (CTAB) as a coating precursor [147]. CTAB works as a surfactant to transfer IONPs to the aqueous phase where TEOS condenses to form a silica coating on the IONPs. CTAB is then removed by refluxing gently in a hydrochloric acid/ethanol solution at 60 °C for 3 h. This produces 2-6 nm pores on the silica coating surface, into which small molecules can be easily encapsulated via physical interactions [148].
3.1.3. Magnetic nanoparticles-based T1 contrast probes
The aforementioned magnetic nanoparticles are made of ferromagnetic materials. These materials can significantly reduce the transverse relaxation time of protons and are primarily used as T2/T2* contrast agents. On the contrary, nanoparticles made of paramagnetic materials are used in T1 contrast probes. One example is MnO nanoparticles. MnO nanoparticles can be made by pyrolyzing Mn-oleate in a high boiling point solvent [22, 149]. By adjusting the heating time and temperature, monodispersed MnO nanoparticles with size variations of < 5% can be synthesized.
Like IONPs, the synthesized MnO nanoparticles are coated with a layer of OA/OAm which isolates the particle surface from the surroundings [150]. To become water soluble, both the ligand addition and ligand exchange routes have been investigated. A significant difference in MRI contrast ability has been found from particles surface-modified by different approaches. For example, an r1 relaxivity of 0.37 mM-1s-1 is observed from 20 nm MnO nanoparticles coated with PEGylated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) (DSPE-PEG) [151]. On the other hand, the same size MnO nanoparticles modified by a dopamine-plus-human serum albumin (HSA) approach showed a r1 of 1.97 mM-1s-1 [152]. The difference is due to the dependence of T1 reduction on the probe-water interaction. When a DSPE-PEG coating is used, a hydrophobic inner layer remains on MnO surface that isolates the cores from the surroundings. The dopamine-HSA coating, on the other hand, is more hydrophilic and compact, leading to more efficient water interactions on the MnO surface.
Mesoporous silica-coated hollow manganese oxide (HMnO@mSiO2) nanoparticles have also been studied as a T1 MRI contrast agent. The mesoporous shell enables optimal access of water molecules to the magnetic core and an r1 of 0.99 mM-1s-1 is observed at 11.7 T. These nanoparticles have been used to label adipose-derived mesenchymal stem cells (MSCs) with electroporation. Intracranial grafting of these HMnO@mSiO2-labeled MSCs enables serial MR monitoring of the cell transplants over a period of 14 days (Fig. 6) [148].
Fig. 6.
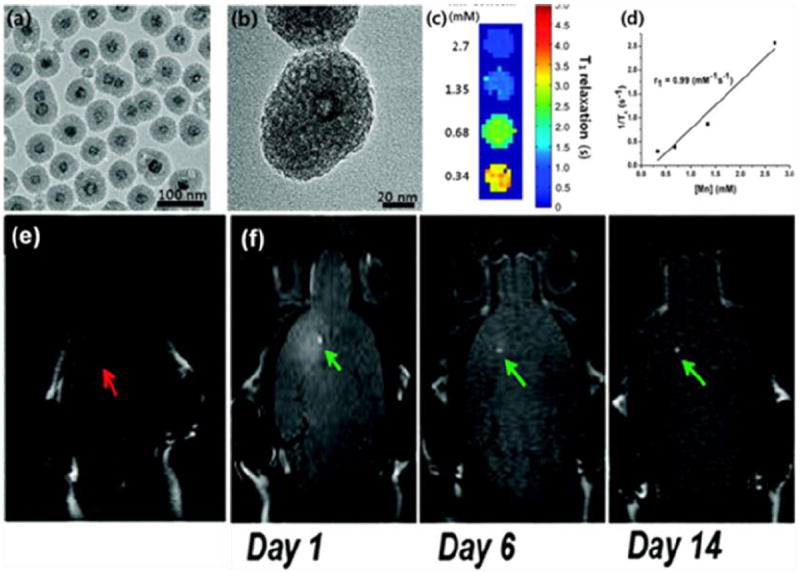
In vivo MRI tracking of transplanted adipose-derived MSCs bearing HMnO@mSiO2. (a) TEM and (b) HRTEM images of HMnO@mSiO2 nanoparticles. (c) T1 map of HMnO@mSiO2 nanoparticles of different concentrations at 11.7 T. (d) Plot of R1 versus Mn concentration. In vivo MRI results when (e) unlabeled MSCs and (f) HMnO@mSiO2-labeled MSCs were injected into small animal models. Reprinted with permission from [148].
Paramagnetic nanoparticles made from other materials and methods have also been studied. For instance, Lin’s group has reported the preparation of paramagnetic nanoscale metal-organic frameworks (NMOFs) [153, 154], a new class of isoreticular materials built from linking metal ions with well-defined coordination geometry using organic bridging ligands. Paramagnetic transition cations are built into these NMOFs and their potential as T1 contrast probes has been evaluated [153, 154]. For instance, Mn-containing NMOFs with controllable morphologies were synthesized using reverse-phase microemulsion techniques at room temperature and a surfactant-assisted procedure at 120 °C with microwave heating [154]. The resulting nanoparticles showed a modest r1 of 5-8 mM−1 s−1 on a per Mn basis. Similarly, Gd-containing NMOFs were prepared and the nanoparticles had an r1 relaxivity of 1.5 mM-1s-1 [153]. Luminescent lanthanide ions such as Eu and Tb can also be doped into the NMOF matrices to produce MR and optical dual functional imaging probes [155]. For both Mn- and Gd-containing NMOFs, the nanoparticles can be coated with silica and coupled with functional molecules [156]. In one study, a cyclic RGD peptide and a dye molecule were both covalently coupled onto silica-coated, Mn-containing NMOFs [154].
Protein-based nanoparticles can be encapsulated with Gd and used as T1 contrast agents. In particular, Gd has been encapsulated into apoferritin nanocages and the conjugates show a T1 relaxivity of about 80 ± 5 mM-1 s-1 at 20 MHz and 298 K. This value is almost 20 times higher than those of commercial Gd agents [157, 158].
Yin et al. described a metallofullerene nanoparticle-based MRI contrast agent [159, 160]. These nanoparticles are synthesized by arc-burning of composite rods consisting of Gd2O3 and graphite in a He atmosphere. The Gd cations are encapsulated inside the fullerene of the nanostructures. Using Gd3N@C80(OH)26(CH2CH2COOM)16 (M=Na or H) nanoparticles as an example, r1 is 207 mM-1s-1 and 50 times higher than that of commonly used Gd3+ complexes, including Omniscan and Magnevist [161].
3.1.4. Magnetic nanoparticles for cancer detection
3.1.4.1. Magnetic nanoparticle-based probes for tumor imaging
Magnetic nanoparticles made of ferromagnetic materials have been coupled with targeting motifs and studied for tumor homing and imaging. For instance, RGD has been coupled onto silica [37], triblock-copolymers [162], polyaspartic acid [163], and dextran [69, 164] coated IONPs and evaluated as MRI contrast probes. Coupling of multiple RGD on the particle surface results in a significant increase in the affinity to integrin αvβ3 due to the multivalent effect [165]. Taking polyaspartic acid (PASP)-coated IONPs as an example, the IC50 values of the RGD conjugates are 34 ± 5 nM, compared to 250 ± 60 nM of free RGD [68]. Chen et al. used a PEGylated triblock copolymer consisting of a polybutylacrylate segment, polyethylacrylate segment, polymethacrylic acid segment, and a hydrophobic hydrocarbon side chain to coat IONPs prepared by thermal decomposition (Fig. 7a). A cyclic RGD peptide, c(RGDyK), along with a NIRF dye IRDye800, were covalently coupled onto the particle surface. When the materials were tested on a U87MG subcutaneous model strong hypointensities were observed from the tumor areas and peaked at 4 hours post-injection (p.i.) (Fig. 7b). Postmortal immunohistological studies revealed nanoparticles in both the tumor endothelial cells and tumor cells (Fig. 7c) [162], both of which overexpressed integrin αvβ3 [165, 166]. Similar targeting profiles were observed from BT-20 (human breast cancer) tumor models with RGD-IONP conjugates [109,110].
Fig.7.
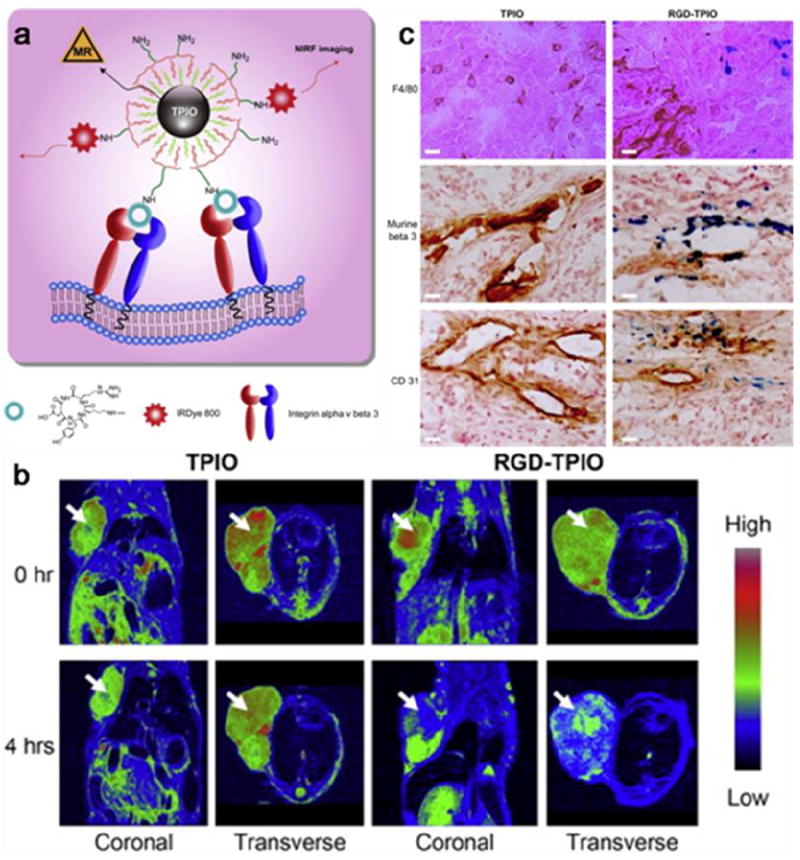
Triblock copolymer coated IONPs (TPIONPs) conjugated with RGD peptides and dye molecules for tumor targeting. (a) Schematic illustration of the optical/MR dual-modal imaging probes. (b) MR imaging of U87MG tumor-bearing mice injected with RGD–TPIONPs or TPIONPs. (c) Prussian blue and CD31/murine β3/F4/80 double staining withthe tumor sections. Reprinted with permission from ref. [162].
Other peptide-based targeting molecules have been studied. Chlorotoxin (CTX) was conjugated with IONPs and studied on 9L (rat gliosarcoma) xenograft models for tumor imaging [167]. The nanoparticles were found to induce a maximum R2 change of 16 s-1 in tumors after 12 h. It was subsequently shown in a transgenic mouse model that the CTX-IONPs could cross the blood-brain barrier (BBB) and specifically target brain tumors [168] without damaging the BBB. Although the exact mechanism is not clear, the BBB penetration is likely associated with CTX on the particle surface. Moreover, A54, CREKA, bombesin, F3, tumorstatin, and LHRH have been studied as targeting motifs for IONP-based imaging [169-172].
Magnetic nanoparticles have been coupled with antibodies [23]. Lee et al. coupled Herceptin with MnFe2O4 nanoparticles and evaluated the MR imaging capability on breast and ovarian tumor models [173]. Hadjipanayis et al. coupled IONPs with a purified antibody that selectively binds to the EGFR deletion mutant EGFRvIIIAb present in human glioblastoma multiforme cells [174]. In vivo MRI revealed hypointensities in or adjacent to intracranial human xenograft tumors and the IONPs continued producing contrast days later. In both studies, the antibodies had therapeutic abilities and the conjugates were therefore examples of nano-theranostics.
Magnetic nanoparticle-aptamer conjugates have also been reported. Hwang et al. conjugated the AS1411 aptamer (MF-AS1411) onto silica coated CoFe2O4 nanoparticles [175]. MF-AS1411 is a sequence that targets nucleolin, a cellular membrane protein highly expressed on the surface of many cancer types. The nanoparticle conjugates labeled with 67Ga were evaluated in vivo on a C6 glioma xenograft model. Accumulation in the tumor areas was observed by both MRI and single-photon emission computed tomography (SPECT) imaging. On the other hand, little tumor accumulation was found when the AS1411 mutant (where core nucleotides G in AS1411 aptamer were substituted with C) was coupled onto IONPs.
Paramagnetic nanoparticle-based T1 contrast probes have been studied in tumor imaging. Na et al. coupled MnO nanoparticles with Herceptin [22]. The conjugates were able to locate breast cancer metastasis in the brain, while those not conjugated with Herceptin could not. Crich et al. studied Gd-loaded apoferritin nanoparticles in tumor angiogenesis imaging using a two-step approach [157]. Firstly, a biotinylated derivative of C3d peptide was injected. With high affinity, C3d bound to the neural cell adhesion molecule (NCAM) that is overexpressed in tumor endothelial cell linings of human carcinomas. Secondly, biotinylated Gd-loaded apoferritin (Gd-Apo-Bio) and streptavidin were injected. With streptavidin as a cross-linker, Gd-Apo-Bio migrated to endothelial cells to induce hyperintensities in the T1-weighted MRI maps. However, in vivo imaging of tumors with nanoparticle-based T1 contrast agents is relatively scarce due to the relatively less prominent signal change and concerns with particle toxicity.
3.1.4.2. Magnetic nanoparticle-based biosensors
Detection and accurate measurement of biomarkers in biological samples are invaluable to healthcare. Devices employing magnetic nanostructures have shown marked progress toward this goal. The greatest advantage of magnetic nanoparticle-based biosensors over traditional diagnostic methods is the near absence of biological background interference. This enables the detection schemes to deliver high sensitivity on biological specimens.
Weissleder’s group has recently developed a technology called diagnostic magnetic resonance (DMR) which exploits magnetic nanoparticles as proximity sensors to modulate the spin-spin relaxation time of water molecules surrounding the molecularly-targeted nanoparticles [176]. When a few magnetic nanoparticles bind to the intended molecular target through affinity ligands, the particles form soluble nanoscale clusters causing a decrease in T2. Instead of performing analysis on traditional and expensive NMR equipment, the group has developed miniaturized easy-to-use systems that provide accurate and rapid readouts. With antibody-conjugated IONPs, they are able to profile mammalian cells using the expressions of a number of biomarkers such as Her2/neu and EGFR [177]. They have also demonstrated the potential of using DMR for multiplexed screening and 8 biomarkers can be simultaneously analyzed on a 2×4 microcoil array. In the early studies, dextran-coated IONPs with a suboptimal r2 (62 mM-1s-1) were used. They later employed MnFe2O4 nanoparticles with a much higher r2 (420 mM-1s-1) as the probes, and this significantly improved the sensitivity. As few as 2 cancer cells in 1-μL volumes of unprocessed fine-needle aspirates of tumors can be detected and analyzed for biomarker expression [178]. This sensitivity is comparable to that of clinical methods such as flow cytometry and Western blot, but is performed in a much shorter time (< 15 min) [178].
Conjugation chemistry also impacts the sensitivity of DMR. Instead of using the traditional bioconjugation techniques, a new conjugation technology between 1,2,4,5-tetrazine (Tz) and trans-cyclooctene (TCO) has been evaluated [179, 180]. Because of the small size of the coupling reagents, a higher multiplicity of antibodies per nanoparticle is observed [181]. This leads to a higher number of particles binding to cells and in turn, improves the detection sensitivity [181]. A clinical study has recently been performed using this method [178]. The DMR system was used to analyze cells obtained by fine-needle aspirates from suspected lesions in 50 patients. With a four-protein signature, 96% accuracy in cancer diagnosis was accomplished. This is a large improvement over clinical immunohistochemistry which boasted an accuracy rate of 84%. Further tests conducted on 20 patients yielded an accuracy rate of 100%. The time required to perform the analysis is reduced from three days, for the clinical method, to less than an hour for DMR. This technology holds great clinical promise pertaining to the analysis of a wide range of targets including whole cells, proteins, DNA/mRNA, metabolites, drugs, viruses, and bacteria [176].
Another magnetic nanoparticle-based biosensor technology has been developed by Wang et al. based on the magnetoresistive (GMR) sensor [182]. Using protein analysis as an example, in a typical assay, a captured antibody for the desired analyte is attached to a magnetoresistive sensor. The samples are added to the sensor but only the targeted analytes remain by interaction with the captured antibody. A second, biotinylated antibody is then introduced to bind to the previously captured analytes. Finally streptavidin-coated magnetic nanoparticles are applied and immobilized on the sensor via the biotin-streptavidin interaction. The attached nanoparticles alter the voltage in the magnetoresistive sensor yielding quantitative signals. Initial studies show that multiple protein-based biomarkers can be detected at fM concentration levels with a dynamic range of more than four orders of magnitude [182]. The detection limit can be further improved by adopting an amplification step to lower the detection limit down to 50 aM, which is 1,000 times lower than that of ELISA [183]. Gaster et al. used this system to monitor the dynamic changes of CEA, VEGF, and EpCAM in a human colorectal cancer xenograft mouse model [183]. A strong correlation between the CEA concentrations and tumor growth was observed. On the other hand, ELISA failed to detect the CEA level change as the concentrations were below the detection limit. The GMR-based biosensor technology has been improved and a recent publication shows that it can be scaled to over 100,000 sensors per cm² to measure the binding kinetics of various proteins with high spatial and temporal resolution, as shown in Fig. 8 [184]. Moreover, the detection limit has been improved to as low as 20 zeptomoles of the solute.
Fig. 8.
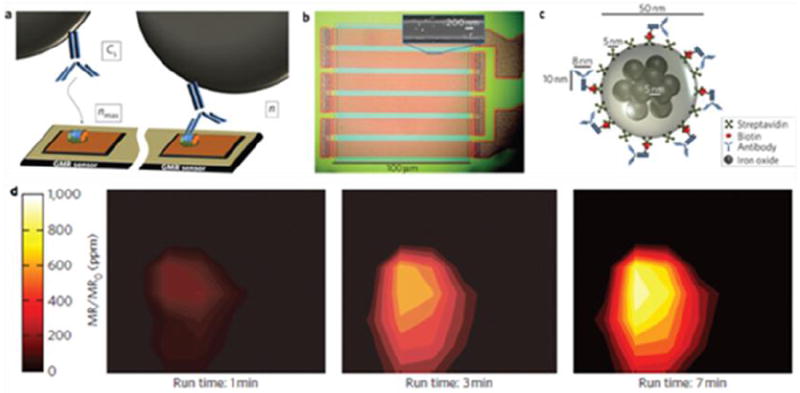
Magnetic nanoparticle-based biosensors. (a) Schematic representation of antibody–antigen binding on the GMR sensor surface. (b) Optical micrograph showing the GMR sensor architecture. Inset: SEM image of one stripe of the GMR sensor that is bound with magnetic nanoparticle tags. (c) Schematic representation of a magnetically labeled antibody, drawn to scale. (d) Visualization of CEA protein surface concentration at different times using a high-density GMR sensor array. Reprinted with permission from ref. [184].
3.2. Quantum dots
3.2.1. Optical properties of quantum dots
Optical fluorescence imaging is used in tumor diagnosis, especially in preclinical circumstances. Fluorescence microscopy has advanced the fields of immunology and cellular biology. The recent advent of small animal whole-body fluorescence imaging systems offers a sensitive and cost-effective tool in preclinical cancer studies and has found widespread use in biomarker discovery and drug development. An intrinsic drawback of fluorescence imaging is limited tissue penetration. On account of scattering and absorption, light transmission is severely attenuated when traveling both in and out of tissues. This leads to a shallow penetration depth of typically less than 1 cm [185]. Using fluorophores with absorption and emission in the near-infrared region can alleviate the energy loss to a certain extent, but in general, in vivo fluorescence imaging has been limited to small animal models with targets close to the skin. Notably, the limited penetration does not exclude the clinical use of fluorescence imaging. Fluorescence imaging has found extensive uses in immunostaining for analyzing patient samples. Moreover, extensive research is being conducted to develop fluorescence-based imaging technologies at endoscopic and intraoperative settings, where tissue penetration is less a concern.
Organic dye molecules were overwhelmingly used in fluorescence imaging in the past, but recently, inorganic nanoparticle-based fluorophores have been found to offer advantages over traditional organic dyes. One of the most studied fluorescent nanoparticles is quantum dots (QDs), which are semiconductor nanoparticles composed of groups II and VI elements (e.g. CdSe and CdTe) or groups III and V elements (e.g. InP and InAs). Bulk semiconductors have relatively small band gaps (less than 4 eV), thus behave like insulators at ambient conditions and exhibiting electrical conductivity only under external stimulation. QDs, on the other hand, have a physical size of 2 to 10 nanometers in diameter, which is smaller than the exciton Bohr radius. This results in a 3-dimensional quantum confinement of charge carriers, limiting the number of possible energy states that an electron can occupy. In most cases, relaxation of an excited electron in QDs results in the release of the band gap energy in the form of light in the visible or near-infrared (NIR) region [186, 187]. This fluorescence mechanism gives rise to unique absorption and emission profiles. Unlike organic fluorophores which yield an emission spectrum that is usually a mirror image of the absorption one, QDs exhibit a broad absorption spectrum but a narrow and usually symmetrical emission spectrum. This allows a group of QDs with distinctly different emissions to be excited by the same light source, making them ideal tags for multiplex imaging.
The emission from QDs is size dependent (Fig. 6a). A smaller particle size leads to an increased degree of confinement and in turn, increases the difference in energy between the discrete ground and excited states [188-191]. Taking CdSe nanoparticles as an example, a red shift is observed when the QD size increases from 1.5 to 8 nm [192, 193]. The emission also depends on the composition of the QDs. For example, in ZnxHg1–xSe QDs, the emission can be tuned by adjusting the composition to cover most of the visible and NIR region [194]. On top of the semiconductor core, a ZnS or CdS shell is usually deposited. The shell is several atomic layers thick and passivates the photoactive cores of the QDs [195, 196]. With a wider band gap, the shell efficiently confines the excitation to the core to reduce the nonradiative relaxation pathways and increase the quantum yield (QY) [197]. The shell thickness can have a significant impact on the photo-physical properties of the QDs. While thinner shells (1–2 monolayers) usually produce higher QY, thicker shells (4–6 monolayers) protect the cores more effectively from photooxidation and degradation [196, 198]. A thicker shell also reduces QD “blinking”. QD blinking is an intermittence in light emission associated with charge trapping and un-trapping at surface defects in a nanomaterial or due to charge ejection from the QD (Auger ionization) followed by recombination [199-201]. Reduced blinking is desired in diagnosis since blinking may cause signal fluctuations in ultrasensitive detection, loss of distance information when movement of a single molecule is observed, and spectral jumping (change in the emission peak position) [202].
In general, QDs have high absorption coefficients and QYs [203]. They can be 10-20 times brighter on an individual basis than organic dyes [204]. QDs are also much more photostable, as shown in Fig. 9b. Wu et al. compared the photostability of QD 608–streptavidin and Alexa 488–streptavidin with specimens mounted with glycerol or the antifade mounting medium Vectashield [205]. Without an antifade medium, the fluorescence intensity of Alexa 488 diminished to 50% of the initial intensity at 10 s, and to 10% at 60 s. With protection by the antifade reagents, Alexa 488 retained 80% of the initial intensity at 60 s and 55% at the end of 3 minutes of illumination. For QD 608–streptavidin, the intensity was 104% of the initial intensity from the glycerol-mounted specimens and 97% from the antifade medium–mounted specimens at the end of the illumination. Gao et al. showed that QDs were several thousand times more stable against photobleaching than organic dyes (e.g. Texas red) under the same excitation conditions (Fig. 9b) [206]. These observations suggest the advantages of QDs over organic dyes, especially for quantitative analysis and longitudinal tracking (Fig. 9c) [206].
Fig. 9.
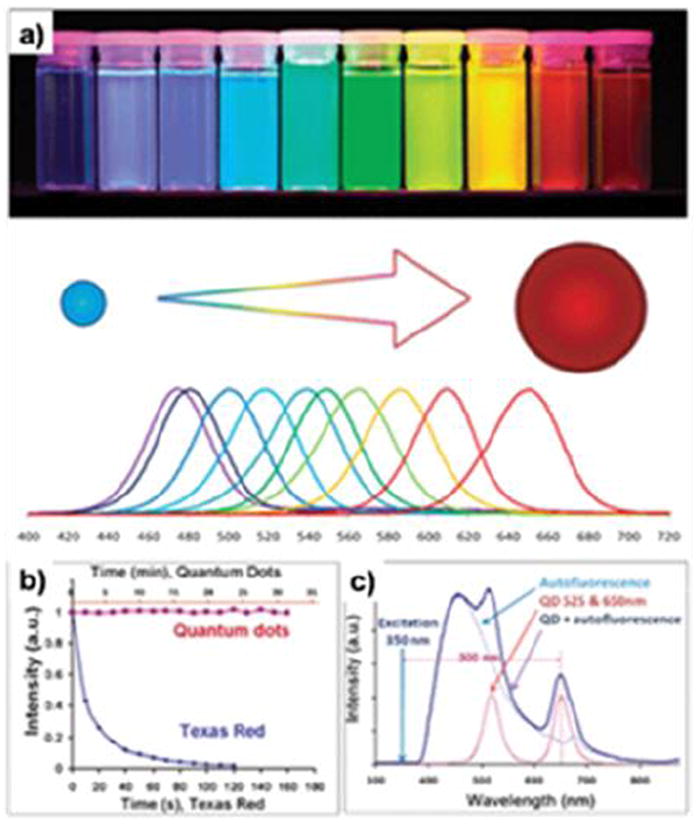
Photo-physical properties of QDs. (a) Size-dependent light emission. (b) Comparison of photostability between QDs and dye molecules. (c) Capability of absorbing high-energy (Uv-blue) light and emitting fluorescence with a large Stokes shift enables efficient separation of the QD signal over the fluorescent background. Reprinted with permission from ref. [202, 206].
3.2.2. Synthesis of quantum dots
Initially, QD synthesis was performed in aqueous solutions which yielded particles with relatively poor fluorescence efficiencies and large size variation [207, 208]. Advancements in synthetic procedures and surface chemistry have enabled production of water-soluble QDs with higher QYs (up to 40–50%) and narrower size distributions. For instance, the spectral emission widths are 50 nm from CdTe/CdSe particles and 19 nm from ZnSe QDs made by water-based approaches [207, 209, 210]. However, in general, aqueous synthesis is suboptimal in controlling the photo-physical and physiochemical properties of QDs.
In 1993, Bawendi and coworkers developed a high-temperature decomposition procedure to produce QDs, and the procedures are now widely used [211]. In this process, organometallic precursors are pyrolyzed at high temperature to induce nucleation and nanocrystal growth. The protecting ligands such as trioctyl phosphine/trioctyl phosphine oxide (TOP/TOPO) are involved in the reaction by interacting with the unsaturated surface metal atoms and preventing the formation of the bulk semiconductor. In a typical CdSe nanoparticle synthesis, the liquid precursors, e.g. a solution of Se and Cd(CH3)2, and capping ligands, e.g. tributylphosphine, are injected into an organic solvent comprised of TOPO and hexadecylamine at high temperature (~300 °C). To maximize the photo-physical properties, a layer of ZnS is usually grown on top of the CdSe cores. It is noted that Cd(CH3)2 is highly toxic and unstable, and special equipment and safety measures must be adopted. Alternatively, other Cd precursors (e.g. CdO) have been developed to allow QD preparation under milder conditions [212-214].
Various reaction parameters such as the precursor, precursor concentrations, solvent, temperature, and reaction time can be tuned to achieve controlled QD synthesis [215-219]. A selective precipitation step is commonly employed to produce nanoparticles with the desired size. Dabbousi et al. used selective precipitation to prepare CdSe/ZnS QDs with the size of cores ranging from 2.3 to 5.5 nm [195]. The emission from these QDs spans most of the visible spectrum from blue to red with QYs of 30-50%. Alternatively, size control can be achieved by gradually increasing the reaction temperature. Zlateva et al. reported large-scale synthesis of six colors of CdSe QDs in a single reaction by using a slow-increasing temperature gradient [220]. The QD fractions have a narrow size distribution and produce sharp photoluminescence spectra with good QY (45–70% in organic solvent) and an overall high productive yield (~95%) since every gradient is useful.
The photo-physical properties of QDs depend on the ratio of the metals. Zhong et al. prepared a series of alloyed ZnxCd1-xS (x = 0.10, 0.25, 0.36, 0.53) nanocrystals by pyrolyzing a mixture of CdO- and ZnO-oleic acid complexes with sulfur in octadecene [221]. As the Zn content was increased, the photoluminescence spectra blue-shifted systematically across the visible spectrum from 474 to 391 nm. Qu et al. found that for CdSe QDs, the initial Cd:Se ratio of the precursors was critical to the emission wavelength, QY, types of the bright points (sharp or flat), and sharpness of the luminescent peak [203]. In particular, a large excess of the selenium precursor was found necessary to achieve a high QY and narrow emission profile. Weiss et al. found that the ratio of Cd to Se (Cd/Se) in colloidal CdSe QDs is a function of the particle radius [133]. A Cd/Se ratio of 1.2:1 was observed from QDs with a radius larger than 3.3 nm. This number increased to 6.5 when the radius was reduced to 1.9 nm, associated with the blue-shifting of emission wavelength.
Much effort has been directed to the preparation of QDs with NIR emission. This is because tissue pigments, most importantly water and hemoglobin, exhibit minimum absorption in the NIR region. This makes the NIR region (650 nm to 900 nm) a relatively transparent spectrum window and ideal for in vivo fluorescence imaging [222]. Peng et al. reported a one-pot synthesis of InAs/CdSe QDs with bright, stable, and narrow NIR photoluminescence [194]. Kelley’s group prepared NIR PbS QDs with a QY of ~26% [223]. Alloyed QDs emitting in the NIR region such as CuIn5Se8 [224], CdxHg1-xTe [225, 226], and Cu-doped InP [227] have been synthesized [228-230]. However, Cd-free QDs in general have poorer stability and inferior photo-physical properties than Cd-containing ones [202]. Recently, Nie’s group found that lattice strain could be used to adjust the emission wavelength of QDs [231]. They prepared QDs by epitaxial deposition of a compressive shell (ZnSe or CdS) on a soft and small nanocrystalline core (CdTe). The lattice mismatch between the core and shell led to dramatic changes in both the conduction and valence band energies and in turn, a large spectral shift. These strain-tunable QDs exhibit narrow light emission with high QY (60%) across a broad range of visible and NIR wavelengths (500 nm to 1050 nm).
3.2.3. Surface modification of quantum dots
QDs made by thermal decomposition are highly hydrophobic. One common surface modification approach is to substitute the native TOPO/TOP coating with a bifunctional ligand comprised a surface-anchoring group (mostly thiols) and a hydrophilic functional group. Examples include negatively-charged carboxyl-terminated thiols such as mercaptoacetic (MAA) [232], mercaptopropionic (MPA) acids [233] and thiol-containing zwitterionic molecules such as cysteine [66, 234]. In addition to providing hydrophilicity, these terminal chemical groups can be utilized in conjugation with other biological species such as proteins, peptides, or nucleic acids. Despite the simplicity of the procedure, ligand exchange with monodentate surface ligands is often associated with compromised fluorescence efficiency, photochemical stability, and shelf life. Because of limited binding affinity, monodentate ligands tend to detach from the QD surface, leaving behind surface trap sites and causing nanoparticle aggregation [235, 236]. The stability can be dramatically improved by using multidentate ligands. Liu et al. used a di-thiol ligand, PEGylated dihydrolipoic acid (DHLA), to modify QDs. The surface modification process led to aqueous stable nanoparticles with a moderate drop in QY (from 65% to 43%) [237]. Sukhanova et al. reported the use of cysteine to modify QDs and further stabilization of the particles with poly(allylamine). They observed an increase in QY from 40% to 65% [238]. Jiang et al. improved the stability of a mercaptoundecanoic acid (MUA) shell by covalently cross-linking neighboring molecules with lysine [239]. However, crosslinking also resulted in a dramatic increase in the nanoparticle size (from 8.7 to 20.3 nm). Recently, Smith and Nie developed a new class of multidentate polymer coatings only 1.5–2 nm thick [240]. Consisting of a poly(acrylic acid) backbone grafted with multiple anchors (thiol and amine groups), this coating renders CdTe QDs biocompatible and colloidally stable, while keeping the final particle size between 5.6 and 9.7 nm.
Silane-based coatings have also been proposed [241, 242]. 3-(mercaptopropyl)trimethoxysilane (MPS) has been used as a ligand to displace the native TOPO molecules. Upon addition of base, the silanol groups are hydrolyzed and condense to form a stable and compact (1–5 nm thick) silica/siloxane shell rendering the particles hydrophilic. The QDs can also be encapsulated by silica using a reverse microemulsion coating method [243]. However, the resulting coating is usually thicker, typically larger than 25 nm. Co-polymers have also been used in QDs coatings. By using the ligand addition approach, the amphiphilic materials are coated onto the hydrophobic QD surface via hydrophobic-hydrophobic interactions, leaving the hydrophilic section free to interact with surrounding water molecules to suspend the particles. Gao et al. used an amphiphilic triblock copolymer to coat QDs [244]. The resulting QDs were very stable in water and able to be coupled with targeting agents such as antibodies in tumor imaging. This ligand addition approach usually produces QDs with a large hydrodynamic size.
3.2.4. Toxicity of quantum dots
In biomedical applications, the toxicity of the materials and their interaction with biological systems must be addressed. Extensive toxicity studies have been performed on QDs both in vitro and in vivo but there is no common consensus, partially due to the diversity of QDs. It is important that each type of QD is characterized individually as not only the constituent metals, but also the QD size, charge, and functional group should be considered as contributing factors to QD toxicity [245].
Protection layers and surface coatings can reduce the toxicity of QDs. This is because QDs deteriorate in the physiological environment and release toxic heavy metals into the surroundings. The organic coating or inorganic shell can inhibit or retard this process and consequently, reduce the toxicity. Derfus et al. studied the cytotoxicity of CdSe QDs and found that without a coating, the cytotoxicity of CdSe cores could be correlated with the liberation of free Cd2+ ions [246]. With the appropriate coating, CdSe QDs could be rendered nontoxic and used to track cell migration and reorganization. Lovric et al. found that CdTe QDs coated with MPA and cysteamine were much less toxic to rat pheochromocytoma cells (PC12) than uncoated ones [247]. However, the coating itself can be the source of toxicity. For instance, Hoshino et al. observed severe cytotoxicity from EL-4 (mouse lymphoma) cells when they were treated with a QD capping ligand, MUA at 100 μg/mL [248]. They believed that it was MUA, not the QD cores, that was the major cause of the cytotoxicity but their postulate is debatable [249]. A multidentate coating may help reduce the toxicity. Voura et al. treated B16F10 melanoma cells with DHLA-capped CdSe/ZnS QDs (5 μL/mL) and found no detectable reduction in cell growth [250]. Jaiswal et al. treated HeLa and Dictyostelium discoideum cells with DHLA-capped CdSe/ZnS QDs (400–600 nM) for one week and observed no adverse effects on the cell morphology and physiology [251]. This can be attributed to a more stable coating and reduced risk of the capping falling off.
The particle size also has an impact. Lovric et al. observed that smaller cationic QDs (2.2 ± 0.1 nm) induced more pronounced cytotoxicity than larger cationic QDs (5.2 ± 0.1 nm) [247]. Later studies revealed that the variation was associated with the different subcellular distribution pattern of the two particles. While larger QDs are mainly distributed in the cytosol, smaller QDs are localized to the nuclei which are more susceptible to damage. Toxicity can be caused by several factors including free Cd ions, free radicals, or QD interaction with intracellular components. To elucidate the mechanism, QDs were co-incubated with N-acetylcysteine (NAC; a known inhibitor of Cd toxicity) and Trolox (a water-soluble vitamin E). While NAC improves the viability, Trolox fails to do so, suggesting that the toxicity stems from Cd in lieu of the free radicals. Since it is generally believed that the released heavy metals are the main source of QD toxicity, researchers have developed non-Cd formulations such as InAs/ZnSe [252], InAs/InP/ZnSe [253], and Cu-In-Se/ZnS QDs [254]. Although there are still toxic elements such as indium and arsenic, the toxicity is significantly mitigated compared to those containing Cd, Hg, and Pb [255].
The size of QDs determines their distribution and circulation in a biological system and therefore affects the toxicity profiles. A reduced particle size may lead to a longer circulation half-life and lower liver uptake. However, further reducing the particle size to <10 nm will meet the threshold of renal filtration and urinary excretion. This may be desirable in imaging because the unbound probes will not stay in the body for a long time but instead be excreted via urine. An important study was conducted by Frangioni et al. who intravenously administered QDs into rodents and studied the size effect. They studied CdSe/ZnS nanoparticles coated with DHLA, cysteamine, cysteine, and PEGylated DHLA with hydrodynamic sizes ranging from 4.4 to 8.7 nm. A hydrodynamic size of 5.5 nm was found to be a critical point below which nanoparticles could be efficiently eliminated from the body through renal clearance [66]. Gao et al. prepared MPA coated InAs/InP/ZnSe QDs with a hydrodynamic size of less than 10 nm [256]. UV/Vis absorption and fluorescence emission analyses detected QDs in urine samples from the administrated animals, suggesting that QDs could be cleared through the urinary excretion.
The majority of in vivo studies with QDs have hitherto shown no obvious toxicity. Larson et al. observed no noticeable ill effects in mice injected with 20 nM and 1 μM solutions of CdSe/ZnS QDs [257]. Ballou et al. injected amphiphilic polymer coated QDs at a 20 pmol QD/g dose to mice [258]. The injected mice remained viable for 133 days and histology studies found no signs of necrosis at the sites of tissue deposition. More exciting news came recently from a pilot toxicity study on non-human primates which found no adverse response to intravenous injection of QDs (Fig. 11) [259]. The researchers injected rhesus macaques with phospholipid micelle encapsulated CdSe/CdS/ZnS QDs at a dose of 25 mg/kg. The blood and biochemical markers were found to be in the normal range and histology of major organs showed no abnormalities after 90 days. Two treated monkeys underwent continued evaluation and showed no ill effects for one year. However, it was observed that most of the initial cadmium dose remained in the liver, spleen, and kidneys after 90 days. The ultimate fate of the heavy metals and their long-term impact on the body require further analysis, but nonetheless, this encouraging pilot study suggests a potential role of QDs in clinical applications.
Fig. 11.
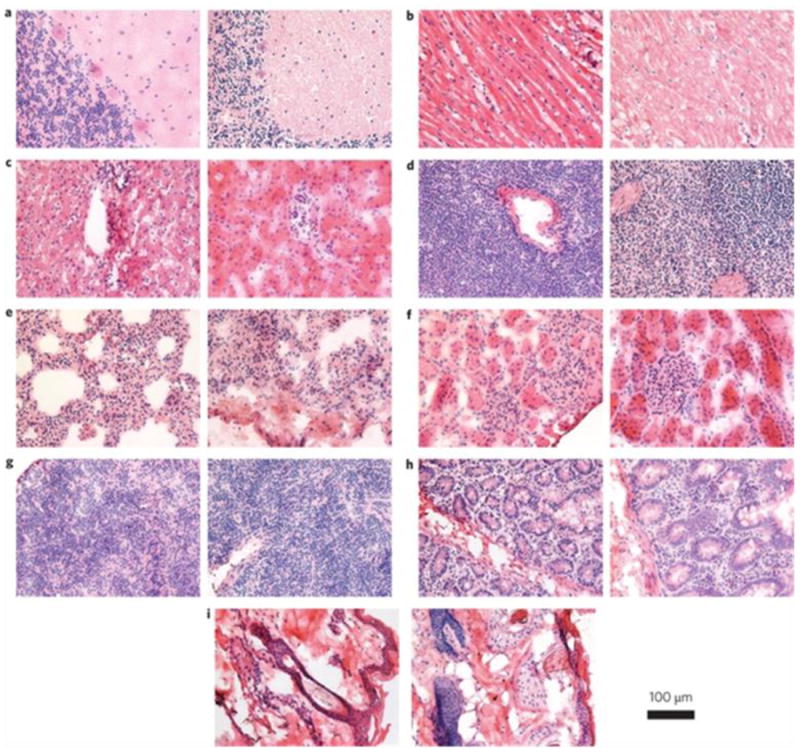
Histological images from the major organs of the rhesus macaques three months after intravenous injection of the QD formulation. In each pair, the left image is from the control animal and the right image is from a treated animal. Tissues were collected from brain (a), heart (b), liver (c), spleen (d), lung (e), kidney (f), lymph (g), intestine (h) and skin (i). Images were taken at ×40 magnification. Reprinted with permission from ref. [259].
3.2.5 Quantum dots for in vivo imaging
Despite controversies, tumor targeting is believed to benefit from a long circulation half-life, and for this purpose, a stable and thick coating offers many advantages [260]. A coating with high molecular weight PEG has been found to reduce rapid clearance of QDs by liver and bone marrow [258], and the use of branched PEG may further improve circulation [260, 261]. Amphiphilic triblock copolymer coated QDs show a circulation half-life of 5-8 h [260]. Nie et al. have coated CdSe/ZnS QDs with a triblock copolymer and conjugated onto the coating an anti-PSMA antibody [244]. In vivo imaging conducted on subcutaneous tumor models reveals good tumor accumulation attributable to the enhanced permeability and retention effect (EPR) as well as antibody-PSMA interaction (Fig. 12). Cai et al. conjugated c(RGDyK) onto polymer coated NIR QDs (705 nm) and studied tumor targeting on a subcutaneous U87MG human glioblastoma model [262]. The tumor fluorescence intensity reached a maximum at 6 h p.i. with good tumor-to-normal-tissue contrast, and the targeting was mainly caused by the RGD-integrin interaction. Recently, Gao et al. reported a dendron-coated InP/ZnS core/shell QD with 710 nm emission [263]. After coupling with a RGD dimer, the particles had a hydrodynamic size of ~12 nm, but unexpectedly, the fluorescent signals were found in urinary bladder and urine collected 90 min after administration. A pilot mouse toxicity study confirmed that the particles did not cause significant toxicity at their particular doses. The nanoparticles showed a long circulation half-life that allowed clear delineation of the tumor up to 28 hours p.i. (Fig. 13). Gao et al. also conjugated anti-HER2 antibody molecules onto phospholipid coated QDs [264] and the resulting conjugates were able to specifically target HER2 positive tumors with a peak tumor-to-background ratio of ~4 at the 4 h p.i. time point.
Fig. 12.
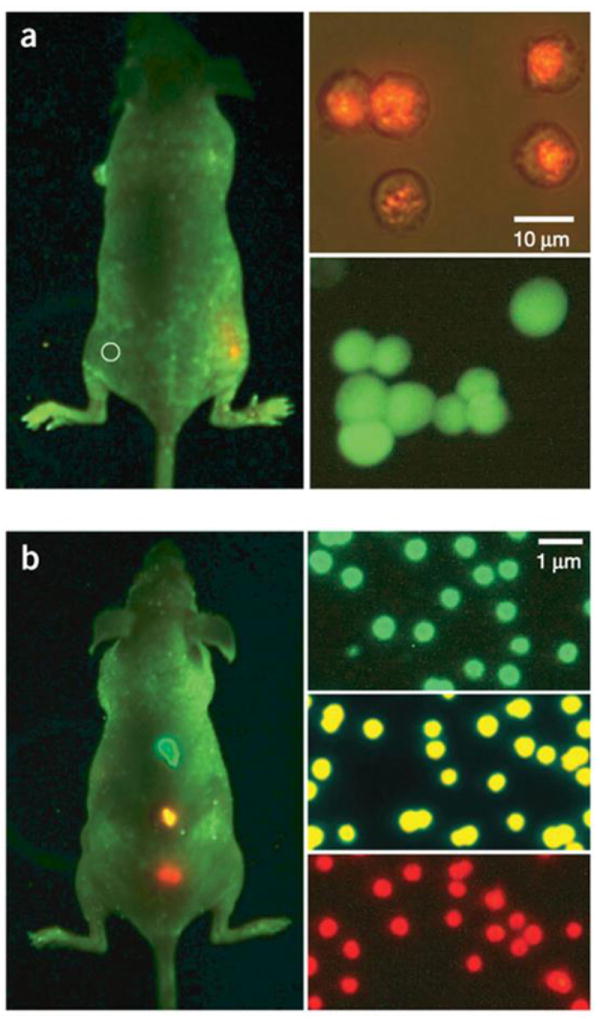
In vivo imaging of implanted QD-tagged tumor cells. (a) Bright QD tags enable visualization of tumor cells with a non-invasive whole-animal fluorescence imaging, whereas organic dye signal is indistinguishable from autofluorescence. (b) Imaging of subcutaneously implanted QD-loaded microbeads shows the potential for multiplexed in vivo cell detection and tracking. Reprinted with permission from ref. [244].
Fig. 13.
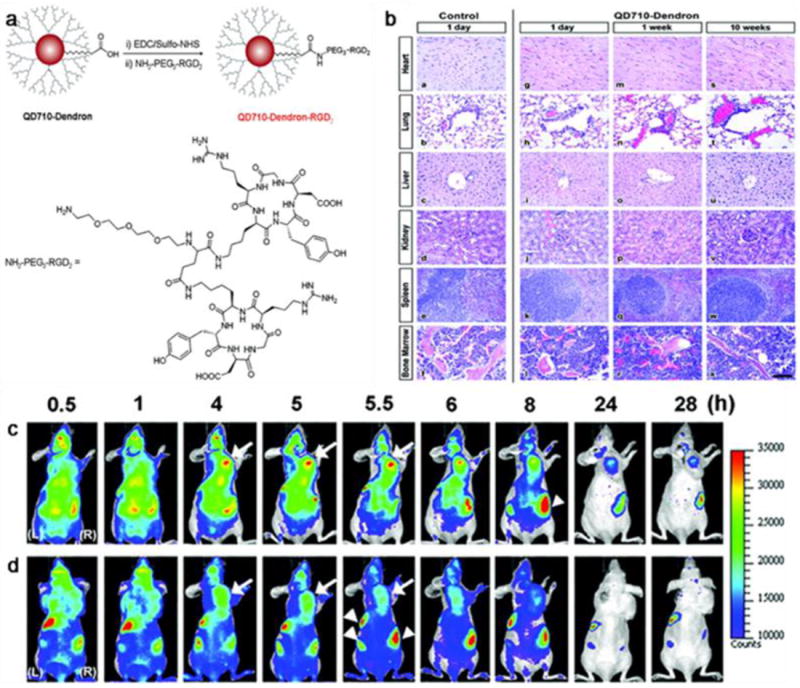
In vivo NIR fluorescence imaging results with QDs. (a) Structure and synthesis of QD710-Dendron-RGD2 conjugate. (b) Representative organ histology of PBS and QD710-Dendron treated animals. Scale bar, 100 μm. The dorsal images of SKOV3 tumor-bearing (arrows) mice (L, left side; R, right side) injected with (c) QD710-Dendorn-RGD2 (200 pmol) and (d) QD710-Dendron (200 pmol) at 0.5, 1, 4, 5, 5.5, 6, 8, 24, and 28 h p.i. Reprinted with permission from ref. [263].
Long circulating QDs can stay intact for months in living subjects and remain fluorescent, making repeated injection and imaging impossible. Reducing the particle size below the renal clearance threshold may be preferred from the perspective of reduced background and toxicity. Choi et al. used cysteine to modify QDs and couple them to the terminal carboxyl groups of either GPI (a small molecule targeting PSMA) or c(RDyK) [66]. A maximum of 5 to 10 GPI or cRGD could be conjugated to the QD surface while the hydrodynamic size of the particles was kept within 5.5 nm, the size threshold for efficient renal clearance. The QD-GPI conjugates were injected into bilateral subcutaneous tumor models implanted with both LNCap (PSMA positive prostate cancer cell line) and PC-3 (PSMA negative prostate cancer cell line). The QD-RGD conjugates were injected into animal models where M21 (human melanoma cells, integrin αvβ3 positive) and M21-L (integrin αvβ3 negative) were inoculated. In both scenarios, tumor accumulation occurred in positive tumors but not negative ones, and most of the unbound QDs were eliminated through the urine within 4 h, thereby providing a “clinically realistic” imaging window.
QDs have been studied with other imaging modalities to assess tumor targeting. Smith et al. used intravital microscopy to study in vivo extravasation and cellular binding with RGD-QD conjugates [265]. Unlike whole-body fluorescence imaging, intravital microscopy is used to examine a small area with high spatial (~0.5 μm) and temporal resolution. It is therefore a prefect technique to study the migration of nanoparticles in vessels and their interaction with the surroundings. RGD-QDs do not extravasate and are only bound as aggregates rather than individually to tumor vasculature, where integrin αvβ3 is overexpressed. Further studies disclose a similar binding feature in different tumor types [266]. Extravasation, on the other hand, is highly tumor-type dependent. The QDs used in the study were polymer-coated with a hydrodynamic size of ~20-25 nm. Whether or not the same conclusion is applicable to other types of QDs is unknown.
Cai et al. labeled RGD-QDs with 64Cu, a radioisotope for PET imaging, and studied the particles in subcutaneous U87MG models by both PET and fluorescence imaging [267]. Based on PET imaging results, RGD-QDs showed tumor accumulation rates of 2.2 ± 0.3 and 4.0 ± 1.0 %ID/g at 5 and 18 h p.i. respectively. Excellent linear correlation was achieved from the results measured by in vivo PET imaging and those measured by ex vivo NIRF imaging and tissue homogenate fluorescence (r2 = 0.93). Histologic examination revealed that the particles mainly targeted the tumor vasculature through RGD-integrin interaction with little extravasation.
3.2.6. Quantum dots for immunostaining
Histological techniques such as immunohistochemistry (IHC) are used in research and clinics to analyze cell or tissue specimens. Despite the advantages of in vivo imaging, histopathology continues to be essential to cancer diagnosis and staging. Although organic fluorophores have been widely used, their disadvantages such as quick photobleaching and inflexible excitation wavelengths are well documented [268]. QDs, with much better photostability and brightness, hold great potential in this field. With broad excitation wavelengths as well as narrow and tunable emission wavelengths, QDs allow multiplexed labeling of biomarkers in tissue samples. Ghazani et al. showed three-color staining of EGFR, E-cadherin, and cytokeratin with 655, 605, and 565 nm QDs on lung carcinoma xenografts [269]. Yzzhelyev et al. demonstrated multiplexed labeling of HER2, ER, and PR with QD655, 605, and 565 on fixed breast cancer cells [270]. By analyzing the fluorescence activities and taking into account the relative intensities of each type of QD, they were able to determine the relative abundance of the three markers. The results were in accordance with those from the conventional methods. The same group also labeled five biomarkers, HER2, ER, PR, EGFR and mTOR, simultaneously with 525 nm, 565 nm, 605 nm, 655 nm and 705 nm QDs in breast cancer cells [270]. The results correlated well with those obtained by traditional IHC, western blotting and FISH, thus corroborating the advantages and potential of QDs in histological studies.
Compared to in vivo applications, there are fewer restrictions on using QDs in a histology setting. The impeding issues in vivo, such as tissue autofluorescence and toxicity, are less of a concern when using QDs in immunostaining. However, with a relatively large size, QDs have limited access to intracellular compartments, making labeling cytoplasmic and nuclear markers challenging. Another issue is nonspecific interaction of particles. To achieve highly sensitive and quantitative analysis, it is critical that particles are immobilized on the specimens through ligand-biomarker interaction and the background signals are at a minimum. A majority of the non-specific binding arises from electrostatic interactions. To avoid this, a common strategy is to functionalize the QD surface with PEG. This however, leads to a larger particle size.
Nonetheless, quantitative molecular profiling with QDs has recently been studied clinically. Liu et al. used QD-based immunohistofluorescence (IHF) to analyze samples involving a confirmed case of Hodgkin’s lymphoma, two suspicious lymphomacases, and two with reactive lymph nodes (but not lymphoma). IHF was able to accurately detect Hodgkin’s lymphoma and to differentiate it from lymphoid hyperplasia [271]. Xu et al. compared QD-based IHF and IHC on quantifying the aldehyde dehydrogenase 1 (ALDH1A1) expression in primary tumor samples taken from 96 HNSCC patients including 50 with disease in the lymph nodes and 46 without (Fig. 14) [272]. The two methods were found to be comparable from the viewpoint of quantification but QD-IHF was more sensitive and objective than IHC.
Fig. 14.
Correlation between QD-based IHF and conventional IHC. (a) Aldehyde dehydrogenase 1 (ALDH1A1)-positive tumour; (b) ALDH1A1-negative tumour; (c) Correlation between ALDH1A1 expression levels measured by immunohistochemistry (IHC) and by QD-IHF. Calculated using Spearman’s rank correlation. (d) Box-Plot for ALDH1A1 in non-metastatic and metastatic groups. Reprinted with permission from ref. [272].
3.3. Upconversion nanoparticles
Upconversion nanoparticles (UCNPs) have recently emerged as a new type of optical nanoparticle. UCNPs absorb NIR light (commonly 980 nm) and emit with higher frequency. The anti-Stokes shift is caused by a nonlinear optical process in which two or more photons are almost simultaneously absorbed to provide sufficient energy for the upconversion. UCNPs show a much lower QY than QDs. Using bulk upconversion materials, the highest QY reported so far is 4.5% (260 mW cm2-) [177,178]. QYs for UCNPs are even smaller and the values are in the range of 0.005 to 0.3, with the highest measured value being 0.3% for 100 nm NaYF4:Yb3+, Er3+ nanoparticles [273]. UCNP-based imaging benifits from a clean background. For in vivo imaging, this may give rise to an increased signal-to-noise ratio and longer penetration depth.
3.3.1. Construction of upconversion nanoparticles
UCNPs are comprised of three components: a host matrix, sensitizer, and activator [274]. The three components play different roles but are all essential to the energy upconversion process. The host matrix is usually made of inorganic materials with low lattice photon energies to minimize non-radiative loss and maximize emission [275]. Many fluorides, oxides, phosphates, oxysulfides, and so on have been studied as host matrix materials to construct UCNPs [276]. Among them, fluorides are the most widely used due to the small photon energies and high chemical stability [275]. Sensitizers or activators are usually trivalent lanthanides incorporated into the inorganic crystalline host. Yb3+ is the most common sensitizer for its large absorption cross-section in the NIR region [277]. On the other hand, Er3+, Tm3+, and Ho3+ are the most popular activators because they possess multiple metastable states that are beneficial to energy conversion [277]. Both sensitizers and activators are at a low concentration in the overall nanostructure, usually ~10 mol% for the sensitizer and < 2 mol% for the activator [276]. Upon irradiation, the sensitizers are excited and transfer the energy to nearby activators to emit light.
By adjusting the combination of the host, sensitizer and activator, the emission from UCNPs can be tuned to cover a broad spectrum range from violet to NIR [275]. For instance, a combination of NaYF4 nanocrystals with Yb3+ and Tm3+ results in UCNPs with blue emission. Yb3+/Er3+ co-doped UCNPs display green emission at 510-570 nm (centered at 525 and 550 nm) and red emission between 630 and 680 nm (centered at 660 nm) [278]. Yb3+/Ho3+ co-doped UCNPs exhibit three main bands centered at 541, 647 and 751 nm [279] and multiple dopants can result in UCNPs with a broader spectrum coverage. Wang et al. doped the NaYF4 matrix with Yb3+, Tm3+, and Er3+ and by adjusting the concentrations, obtained colloids with over a wide color range: bluish to whitish (NaYF4:Yb3+,Tm3+,Er3+) and greenish-yellowish to redish (NaYF4:Yb3+,Er3+), as shown in Fig. 15 [280].
Fig. 15.
UCNP multicolor fine-tuning with dopants: (a) NaYF4:Yb3+/Er3+ (18/2 mol%), (b) NaYF4:Yb3+/Tm3+ (20/0.2 mol%), (c) NaYF4:Yb3+/Er3+ (25–60/2 mol%), and (d) NaYF4:Yb3+/Tm3+/Er3+ (20/0.2/0.2–1.5 mol %) (10 mM). Luminescent photos of (e) NaYF4:Yb/Tm (20/0.2 mol%), (f–j) NaYF4:Yb3+/Tm3+/Er3+ (20/0.2/0.2–1.5 mol%), and (k–n) NaYF4:Yb3+/Er3+ (18–60/2 mol%). Reprinted with permission from ref. [280].
As mentioned above, UCNPs have relatively low QYs. Efforts have been made to improve the QYs by adjusting the particle composition and architecture. For UCNPs with the same composition, the QY is size-dependent and enhanced by an increased particle size. This is due to defects at the particle surface boundary that are detrimental to luminescence. Smaller particles have larger surface-to-volume ratios and are affected more by the effect. The surface effect was confirmed by Wang et al. who coated NaYF4:Yb3+,Tm3+ UCNPs with an inert thin-shell coating. The coating preserved the optical integrity of the nanoparticles and largely minimized surface quenching induced emission losses [281].
The size can also affect the relative emission intensities. For Y2O3:Yb3+,Er3+ UCNPs, the relative intensity of the blue and red emissions to the green emission increases gradually with decreasing particle size [282]. This is ascribed to the highly energetic vibrations of the surface-bound hydroxyl and carbonate groups that bridge the energetic gaps between different levels thereby enhancing the population of the red emitting levels [283]. Strong size-dependence of the spectral properties was also observed by Schietinger et al. with NaYF4:Yb3+,Er3+ UCNPs. In their study however, relatively more efficient green emission was observed when the particle size was decreased. The authors excluded surface effects in the mechanism and instead attributed this behavior to a change in the phonon-mediated processes in the emitting ions. They believed that both the confinement effect on the spatial distribution of phonon modes [284] and reduced spectral density of phonon states contributed to the size effect [285].
The QYs are also affected by the host matrix. In order to minimize non-radiative losses, fluorides, in particular NaYF4, are overwhelmingly used as the host matrix. However, at ambient pressure, there are two crystal phases of NaYF4, the cubic phase (α-phase) and hexagonal phase (β-phase). Although the hexagonal phase of NaYF4 is thermodynamically more stable, it is the cubic phase that is usually obtained during a typical UCNP synthesis. When converting α-NaYF4:Yb3+, Er3+ UCNPs to β-NaYF4:Yb3+, Er3+ UCNPs by post-synthesis annealing [286], an enhancement in fluorescence intensity by 40 fold was observed [287], suggesting β-NaYF4 as the more favorable phase. However, annealing usually leads to severe aggregation of particles. More practically, nanoparticles are made by thermal decomposition at a high temperature and for a long time in order to produce the hexagonal phase in one step.
3.3.2. Synthesis of upconversion nanoparticles
UCNPs can be synthesized in an aqueous solution by co-precipitation. Yi et al. utilized a co-precipitation method to prepare NaYF4:Yb3+,Er3+ UCNPs with the aid of ethylenediamine tetraacetic acid (EDTA) [287]. The metal-EDTA complex was quickly injected into a NaF solution under vigorous stirring to effect nucleation and formation of the cubic phase, α-NaYF4:Yb3+,Er3+ UCNPs. The particle size could be controlled from 37 to 166 nm by varying the ratio between EDTA and metals. However, as aforementioned, α-NaYF4 is suboptimal as a host matrix and the fluorescent intensity of the resulting UCNPs is weak. An annealing treatment is necessary to achieve the phase transition but this is usually accompanied by severe particle aggregation and coating carbonization, thus imposing limitations on the use of the products in a biological setting. Instead, β-NaYF4 UCNPs can be made through one-step thermal decomposition at a high temperature using a long reaction time. This is accomplished by a hydrothermal method in which the rare-earth and fluoride precursors are mixed in an aqueous solution and heated to high temperature in a sealed autoclave (often Teflon-lined). Li’s group developed a general hydrothermal protocol to synthesize nanoparticles [288-291], and a series of rare-earth fluoride nanocrystals with different hosts, crystal structures, sizes and shapes were prepared accordingly [292-315]. Liu et al. demonstrated that NaYF4:Yb3+,Er3+ UCNPs can be tuned in size, phase (cubic or hexagonal), and upconversion behavior by introducing trivalent lanthanide ions at precisely defined concentrations (Fig. 16) [316]. Zhang’s group prepared pure β-NaYF4:Yb3+,Er3+/Tm3+ nanocrystals with controllable shape and strong emission [317]. Schafer et al. reported the synthesis of β-NaYF4:Yb3+,Er3+ particles at ambient temperature by using Y2(CO3)3, Yb2(CO3)3, Er2(CO3)3, Na2CO3, and NH4F as precursors [318]. In addition to NaYF4, LaF3, NaLuF4, and NaYb1-xGdxF4-based UCNPs have also been prepared hydrothermally [319-323].
Fig. 16.

Simultaneous phase and size control of UCNPs through lanthanide doping. (a–c), TEM images of NaYF4:Yb3+/Er3+ (18/2 mol%) products in the absence of Gd3+ dopant ions. (d) HRTEM image of UCNPs. (e) Selected area electron diffraction pattern of UCNPs in (a). (f-h) TEM images of the NaYF4:Yb3+/Er3+ (18/2 mol%) products. (i) HRTEM image of UCNPs in (h). (j) DFT calculation. Scale bars are 500 nm in (a–c), 200 nm in (f–h) and 5 nm in (d) and (i). Reprinted with permission from ref. [316].
UCNPs are more commonly synthesized by thermal decomposition in organic solvents with a high boiling point. Zhang et al. made triangular LaF3 nanoplates by thermal decomposition of La(CF3COO)3 in octadecene with OA as the capping ligand [324]. This method was later extended to prepare different types of UCNPs. Boyer et al. synthesized α-NaYF4:Yb3+,Er3+/Tm3+ UCNPs using Na(CF3COO) and Re(CF3COO)3 as precursors [325, 326]. They showed that the rate of decomposition and particle formation could be controlled by the addition rate of precursors to the reaction solution. With a slow addition rate, the nucleation and growth stages of the NPs could be separated, and the resulting products showed a narrow size distribution. Mai et al. further studied the reaction mechanism and demonstrated that NaYF4:Yb3+,Er3+ UCNPs were formed via a unique delayed nucleation pathway [327]. They found that monodispersed β-NaYF4:Yb3+,Er3+ UCNPs with tunable sizes could be obtained from α-NaYF4:Yb3+,Er3+ monomers by restricting or enhancing the Ostwald ripening process in which the cubic-to-hexagonal phase transition process occurred at a delayed time. This synthetic protocol has been developed into a common synthesis technique and many other UCNPs have been synthesized using a similar approach. Ehlert et al. reported the synthesis of Er3+-, Tm3+- and Ho3+-doped NaYbF4 UCNPs via thermal decomposition [226] and Du et al. prepared NaMF3 (M = Mn, Co, Ni, Mg) and LiMAlF6 (M = Ca, Sr) nanocrystals [328].
In the above cases, the UCNPs are prepared in octadecene using OA and/or OAm as the capping ligand. Recently, Shan et al. synthesized β-NaYF4:Yb3+,Er3+/Tm3+/Ho3+ UCNPs using TOPO [329] which served as both the high-boiling solvent and capping reagent. The UCNPs showed good upconversion efficiency and narrow size distribution. More importantly, the energy barrier of the cubic-to-hexagonal phase transition was significantly reduced by this procedure which favored the β-phase NaYF4 formation. The same group later reported the synthesis of β-NaYF4:Yb3+,Er3+ UCNPs using octadecene as the solvent and both OA and TOP as capping reagents [330]. Based on a series of studies, a two-step growth mechanism for β-NaYF4 UCNPs is proposed. The first step involves a kinetically controlled precipitation stage in which α-NaYF4 UCNPs are formed. The second is a diffusion controlled growth stage in which α-NaYF4 are converted into β-NaYF4. Chen et al. prepared NaYF4:Yb3+,Er3+ UCNPs using Re-oleate complexes as the precursors and octadecene as the reaction solvent [319]. The phase transition could be tuned by controlling the reaction temperature. α-NaYF4:Yb3+,Er3+ UCNPs were obtained by heating at 210°C for 6 h whereas β-NaYF4:Yb3+,Er3+ UCNPs were prepared by heating at 260°C for 6 h.
Although thermal decomposition is an effective approach to fabricate monodispersed, single-crystalline, well-defined, and phase-pure UCNPs, some disadvantages remain, such as requiring rigorous (anhydrous and oxygen-free) and harsh (long reaction time and high reaction temperature) synthesis conditions. In addition, thermal decomposition of metal trifluoroacetates produces fluorinated and oxy-fluorinated carbon species that are considered toxic. Moreover, the capping ligands such as OA, OM, and TOPO have long hydrocarbon chains that make the synthesized UCNPs insoluble in water.
3.3.3. Surface modification of upconversion nanoparticles
In this section, we discuss UCNPs produced by thermal decomposition as this is the most popular method. Similar to IONPs and QDs, UCNPs made by thermal decomposition are coated with a hydrophobic layer and not readily applicable to biological applications. A post-synthesis surface modification step is needed to make them water soluble. This can be achieved by adding an amphiphilic coating layer. Speghini et al. deposited an oleate layer on OA-coated CaF2:Yb3+,Er3+/Ho3+/Tm3+ nanoparticles which then became water soluble [311]. Prud’homme et al. modified OA-coated NaYF4:Yb3+,Er3+ UCNPs with copolymers including poly(ethylene glycol)-block-poly(caprolactone), poly(ethylene glycol)-block-poly(lactic-coglycolic acid), and poly((ethylene glycol)-block-lactic acid). The copolymers could be assembled on the particle surface and transitioned the UCNPs to an aqueous phase [331]. Surface modification using a biomimetic phospholipid layer has also been reported [332].
UCNPs made from thermal decomposition are frequently coated with OAm or OA. The interaction between OAm and particle surface is relatively weak and can be replaced by ligands containing carboxyl or multiple amine groups. PEG-diacid(MW 600) [333], polyethylenimine (PEI) [334], and thioglycollic acid(TGA) [335, 336] have been employed to exchange the surface OAm of UCNPs. Similarly, NaYF4:Yb3+,Er3+ UCNPs with the coordinating solvent N-(2-hydroxyethyl)ethylenediamine (HEEDA) can be surface modified with ethane-1,1-diphosphonic acid (HEDP) [337]. On the other hand, OA has stronger interactions with lanthanide ions thereby requiring ligands with high affinity and usually multi-dentation to achieve ligand replacement. Examples include citrate [338, 339], polyacrylic acid (PAA) [331, 340-344], DMSA [345], 3-mercaptopropionic acid (3MA) [346, 347], poly(ethyleneglycol) (PEG)-phosphate [348], hexanedioic acid (HDA) [349], 1,10-decanedicarboxylic (DDA) and MUA [350]. OA-coated NaYF4:Yb3+,Er3+/Tm3+ UCNPs can be modified by PEG-phosphate to become water dispersible [348]. Murray et al. reported a generalized ligand exchange strategy using nitrosonium tetrafluoroborate (NOBF4) to replace the original ligands (OA or OAm) [351]. After the UCNPs were stabilized in polar and hydrophilic media, they were further modified by hydrophilic polymers like PVP to achieve water stability.
Other ligand modification strategies have also been explored. For example, NaYF4 UCNPs were coated with SiO2 by a common sol–gel process [287, 352]. 3-aminopropyltrimethoxysilane [353] and N-[3-(trimethoxysilyl)propyl]ethylenediamine (AEAPTMS) [354] are usually co-condensed with TEOS to provide the chemical functional groups for further conjugation. Chen et al. prepared carboxylic acid functionalized NaYF4 UCNPs by direct oxidation of the OA coating using the Lemieux–von Rudloff reagent [355]. This approach is unique because no new ligands are introduced to the particle surface.
3.3.4. Toxicity of upconversion nanoparticles
There have been relatively few studies on the toxicity of UCNPs, and based on archival reports, cells and animals appear to have good tolerance to UCNPs [356-361]. However, the compatibility of UCNPs depends on the particle composition as well as coating materials and these issues need to be studied separately. Zhang et al. studied the cytotoxicity of polyethyleneimine (PEI) [362] and silica-coated [359] UCNPs. On silica-coated UCNPs at 1 μg/mL for 24 h incubation, they observed cell viability of 93.4% for skeletal cells and 93.2% for bone marrow MSCs [359]. Hu et al. incubated KB cells with elevated concentrations of PEGylated UCNPs [357] and found that even at a high concentration of 250 μg/mL, the cell viability still remained above 80%. Zhou el al. synthesized citrate-coated UCNPs (cit-NPs) and evaluated the impact of cit-NPs on KB cells at various concentrations (100-500 mg/mL, Fig. 17) [339]. No significant difference in cell proliferation was found in the absence or presence of 100-500 mg/mL cit-NPs. Even when the materials were incubated at the highest dose (500 mg/mL) for 24 h, a cell viability of 81% was still observed. Recently, Li et al. intravenously (i.v.) injected UCNPs at 15 mg/kg to mice and studied the particle distribution and long-term toxicity [344]. They observed no apparent adverse effects for 115 days, suggesting that the particles were well-tolerated. Owing to the potential of in the field of imaging, there is a growing interest in understanding the toxicity and metabolism of UCNPs in living subjects. So far, it is generally believed that UCNPs are more tolerable than QDs as imaging probes in vivo.
Fig. 17.
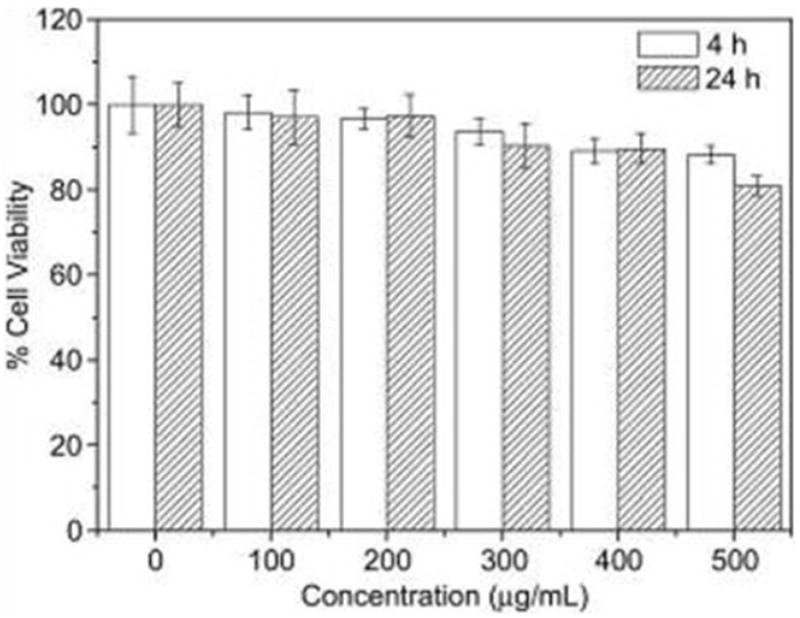
Cell viability reults from MTT assays with UCNPs. KB cells were cultured in the presence of 100-500 μg/mL UCNPs for 4 h and 24 h. Reprinted with permission from ref. [339].
3.3.5. Upconversion nanoparticles for tumor imaging
Boasting an ultralow background, UCNPs are potentially advantageous over QDs for in vivo imaging. Cheng et al. conducted a side-by-side comparison between UCNPs and QDs using small animals [363]. Despite a high QY, the detection limit of QDs was severely impeded by tissue autofluorescence and it was not possible to detect a concentration lower than 5 nM. On the other hand, UCNPs could be detected at a ~0.1 nM level.
UCNPs have been coupled with targeting peptides and antibodies. However, tumor selectivity by these ligand-UCNP conjugates in vivo has seldom been demonstrated. One of the first studies was performed by Xiong et al. who used folic acid as a targeting molecule and coupled it onto 6AA-coated NaYF4:Yb3+,Er3+ nanoparticles [364]. The particles were injected into a HeLa xenograft mouse model and imaging revealed significant difference from the control group to which amine-functionalized UCNPs were injected. The targeting selectivity was also confirmed by a blocking study in which free folic acid was injected along with UCNP probes. The same group also reported RGD conjugated UCNPs for tumor imaging [365]. In their investigation, NaYF4:Yb3+,Er3+,Tm3+ UCNPs were used and imaging was performed on a U87MG subcutaneous model. Good tumor to normal tissue contrast was observed by monitoring the emission at 800 nm at 24 h p.i. Li et al. reported CTX conjugated, NaYF4:Yb3+,Er3+/Ce2+ UCNPs for tumor imaging [366] and good tumor uptake was observed from both in vivo and ex vivo imaging.
There is an interest in developing UCNP-based probes for multimodality imaging. Zhou et al. evaluated NaGdF4:Yb3+,Er3+,Tm3+ [356] and Gd3+ labeled NaYF4:Yb3+,Er3+,Gd3+ UCNPs as MRI (T1) and upconversion luminescence (UCL) dual modal imaging probes [339]. These particles can even be labeled with 18F to achieve MRI/UCL/PET tri-modality imaging. Xia et al. developed NaYF4:Yb3+,Tm3+/FexOy core/shell nanoparticles and applied them to MR (T2) and UCL lymphatic imaging [367]. Xiao et al. coated UCNPs with a TaO shell and studied the particles in MRI/UCL/CT tri-modal imaging [368]. In the latter case, the water-soluble nanoparticles have an average size of about 30 nm and extraordinarily large longitudinal and transverse relaxivity values (r1=11.45mM-1s-1 and r2=147.3 mM-1s-1). A strong UCL signal can be measured by virtue of the high transparency of the TaOx shell (Fig. 18). Despite the promises, in vivo targeting with these multimodal probes has not been reported.
Fig. 18.
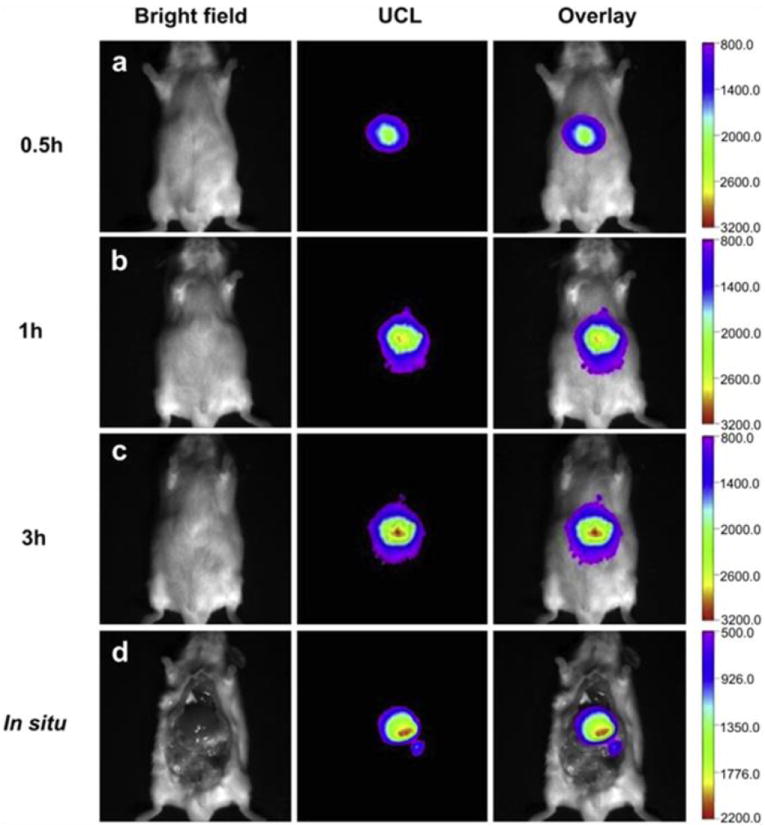
In vivo UCL images taken after injection of UCNP solutions into a mouse at (a) 0.5 h, (b) 1 h, and (c) 3 h. (d) In situ UCL imaging results taken 3 h after the particle injection. Reprinted with permission from ref. [368].
Overall, although UCNPs are an emerging class of materials with great potential in optical imaging, more development is needed before they can be applied to pre-clinical studies or clinical diagnosis. Ongoing studies focus on improving the QY and biocompatibility of UCNPs as well as the understanding of their pharmacokinetics and metabolism in vivo. It is also noted that almost all of the imaging data hitherto reported on UCNPs have been acquired on systems customer-adapted to accommodate the UCL feature. Efforts should be made to develop commercial microscopes and scanners dedicated to UCNP-based imaging.
3.4. Gold nanoparticles
3.4.1. Optical properties of gold nanoparticles
The synthesis of GNPs dates back to ancient Roman times but only recently have the biomedical applications of GNPs been extensively explored. Many studies have focused on surface plasmon resonance (SPR) which manifests as a strong absorption in the visible (and NIR) region [369]. This phenomenon is attributed to the collective oscillation of free electrons on the gold particles’ surfaces and only occurs when metallic particles have a diameter smaller than the light wavelength. With regard to 5 nm spherical GNPs, the SPR band is centered at ~520 nm [370]. The absorption is red-shifted to 517, 521, 533, and 575 nm when the GNP size increases to 9, 22, 48, and 99 nm, as shown in Fig. 19a [273,274]. The GNP size is found to impact the absorption cross-section. In addition to absorption, light scattering also occurs when GNPs are exposed to electromagnetic radiation. A larger particle size favors particle scattering and consequently diminishes absorption [371, 372]. The size effect is important when it comes to picking the right nanostructure for a particular application. In optical coherence tomography (OCT), large GNPs are preferred for stronger scattering and better signal-to-noise contrast. In GNP-mediated photothermal therapy, relatively small GNPs are used to take advantage of their higher absorption efficiency. The SPR of spherical GNPs is also highly dependent on the inter-particle distance. When GNPs aggregate, plasmon-plasmon interaction occurs, resulting in a substantial color change from red to blue [373]. This unique feature has been harnessed to develop colorimetric detection methods which will be described later in this review.
Fig. 19.
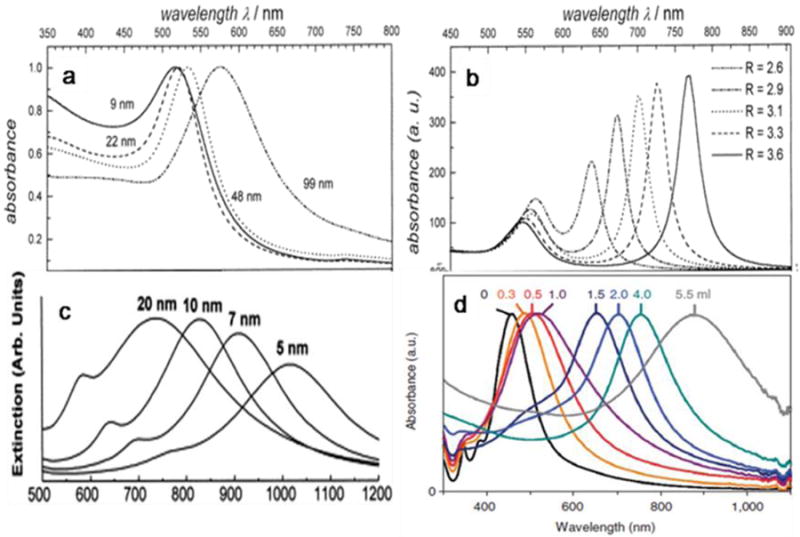
SPR absorption spectra of (a) spherical GNPs of different sizes, (b) GNRs of different length-to-diameter ratios, (c) GNSs of different thickness, and (d) GNCs. Reprinted with permission from ref. [369, 377, 410].
GNP geometry also has a large impact on the SPR. Using gold nanorods (GNRs) as an example, two plasmon bands are observed. The first one is the transverse plasmon band that corresponds to oscillations along the width of the nanorods usually found around ~520 nm. The second one is a longitudinal plasmon band caused by the oscillations along the length of the GNPs and its position is determined by the nanoparticle length-to-diameter ratio. For instance, GNRs with a length-to-diameter ratio of 5.2 (length of 48 nm and diameter of 9 nm) show a longitudinal band at 920 nm [374]. By tuning the length-to-diameter ratio of the GNRs, it is possible to achieve a longitudinal plasmon band that matches commercial lasers at 360 nm, 785 nm, and 1064 nm (Fig. 19b). Red-shifts have also been observed with gold nanoshells (GNSs) [375] and gold nanocages (GNCs), as shown in Figs. 19c and d [376]. The absorption is determined by the ratio of the thickness to diameter of the cavity (Fig. 19c). The smaller the ratio, the further the plasmon absorption shifts to the NIR region [377].
Non-spherical GNPs with NIR absorption are thought to have advantages over spherical GNPs in applications such as hyperthermia-based therapy because tissues are more transparent in the NIR spectrum window. However, recent studies show that spherical GNPs can be excited by radio frequency (RF) radiation, suggesting an alternative means to circumvent the tissue penetration problem [378]. For instance, Curley et al. showed that nonaggregated GNPs could serve as mediators to convert the RF energy to heat to cause damage [379].
3.4.2. Synthesis of gold nanoparticles
3.4.2.1. Synthesis of spherical gold nanoparticles
Spherical GNPs can be prepared in an aqueous solution by reducing HAuCl4. In the protocol introduced by Turkevitch et al. in 1951 [380], citrate was used as the reducing agent to convert Au3+ into Au0. This method is still widely used and spherical GNPs with diameters from 5 to 250 nm can be prepared [381, 382]. In addition to citrate, reducing agents such as sodium borohydride and hydrazine have been employed. Different capping ligands have also been exploited that passivate the particle surface and prevent particle aggregation [383-385].
Ultrasmall GNPs can be made by the Brust-Schiffrin method which takes places in a two-phase (water-toluene) solution in which AuCl4- is reduced by sodium borohydride with alkanethiols as the capping ligands [386]. The GNPs produced has diameters between 1 and 3 nm. This method has been replaced by a single-phase approach to prepare GNPs and other noble metal nanocrystals [294,295]. Briefly, the gold precursors are dissolved in toluene along with fatty acids or aliphatic amines. Tetrabutylammonium borohydride (TBAB) or a TBAB and hydrazine mixture is used as the reducing reagent. Monodispersed spherical GNPs with a diameter from 1 and 15 nm can be made, and since weak-binding ligands are employed, the nanocrystals can be readily surface-modified.
Concerning the toxicity of the reducing/capping reagents, efforts have been made to employ less toxic materials to prepare GNPs. Yin et al. used lactic acid as a reducing agent to produce GNPs [387]. The size and shape of the GNPs were modulated by varying the ratio of the gold precursors and lactic acid. Govindaraju et al. used β-glucosidase to prepare GNPs [292] and Dubey et al. produced GNPs using leaf extracts of Rosa rugosa in an aqueous media at room temperature [388]. Roy prepared 198Au nanoparticles by injecting H198AuCl4 into an aqueous biphasic system containing 2 M Na2SO4 and a 50% (w/w) solution of PEG4000 [389]. 198Au3+ was quantitatively extracted by the PEG-rich phase without the addition of an extracting agent. After extraction, Au3+ was reduced to Au0 to form spherical GNPs. Sun’s group synthesized GNPs in an organic phase by gently reducing HAuCl4·3H2O in OAm which was used as a mild reducing agent as well as surfactant [390, 391]. The prepared core/shell nanoparticles were coated with a layer of OAm and dispersed well in nonpolar solvents.
3.4.2.2. Synthesis of gold nanorods
GNRs were initially prepared by electrochemical and photochemical methods [392]. The use of wet chemical methods to prepare GNRs was pioneered by Murphy’s group [393, 394]. In the process, spherical GNPs are used as seeds and suspended in a solution of gold salt, ascorbic acid (AA, as a mild reducing agent), and CTAB (as a surfactant). CTAB is critical to the GNR synthesis by forming “soft” micellar templates for the nanorod growth. CTAB stabilizes the <110> faces to a greater extent than the <100> surfaces of the growing GNRs, resulting in a faster growth rate on the <100> surface and consequently one-dimensional growth [395]. The particle aspect ratio can be adjusted from 5 to 20 by changing the ratio of the GNP seed to the gold precursor. Jana et al. used silver ions during the seeded-growth procedure [396] and this method was later improved by Nikoobakht and El-Sayed to become an efficient GNR preparation protocol boasting 100% yield and excellent monodispersity [395]. The aspect ratios could be tailored by changing the quantity of Ag+ ions in the solution [397].
3.4.2.3. Synthesis of gold nanoshells
The synthesis of GNSs was first reported by the Halas group using silica nanoparticles as templates [377, 398]. A dielectric spherical silica core is grown by the Stöber method by reducing TEOS and APTES in ethanol under basic conditions [399] resulting in silica nanoparticles ranging from 50 to 500 nm. Small spherical GNPs (1–2 nm) are then adsorbed onto the silica surfaces and serve as nucleation sites. For shell formation, HAuCl4 is reduced to Au0 and deposited onto the particle surface, finally coalescing into a complete gold shell. The amount of HAuCl4 determines the final thickness of the GNSs, typically ranging from 5 to 30 nm. By changing the ratio of the core diameter to shell thickness, the plasmon resonance peak can be tuned from the visible to mid-infrared spectrum range [400-402].
Zhang’s group used cobalt nanoparticles as templates to grow GNSs [403]. The experiments were performed in an anaerobic environment to prevent Co oxidation. Independent of the cores, a critical issue is to form a complete and uniform shell otherwise a broad absorption spectrum is obtained. This approach is reproducible, as demonstrated by several independent studies.
Recently, Gao et al. developed a new technique to deposit ultrathin gold layers onto the surface of lipid vesicles and further, to virtually any discrete nanostructure or continuous surface [140, 404, 405]. Using magnetic nanoparticle coating as an example, IONPs with hydrophobic surface ligands are first solubilized into aqueous solutions using PEGylated phospholipids terminated with carboxylic groups. A layer of positively charged peptide is then electrostatically adsorbed onto the surface, which serves as a template for gold deposition. Afterwards, the gold salts are added and reduced to form the gold shell. The size of the shell can be tuned by adjusting the thickness of the lipid-PEG layer. This is achieved by stacking alternative layers of polyallylamine hydrochloride and sodium polystyrene sulfonate, as shown in Fig. 20.
Fig. 20.
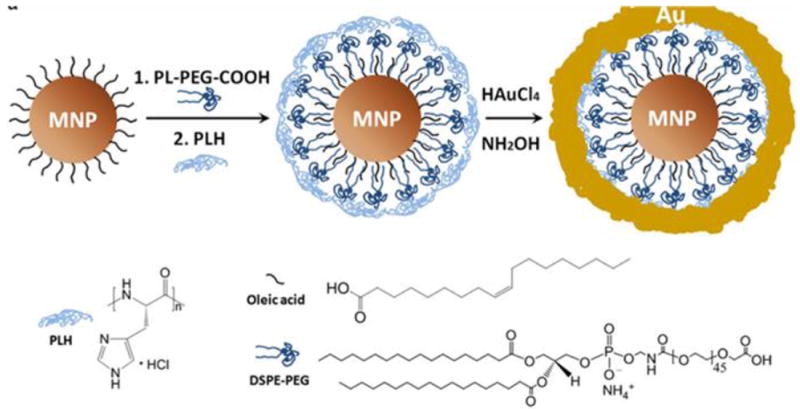
Schematic illustration of the formation of gold shells. Reprinted with permission from ref. [140].
3.4.2.4. Synthesis of gold nanocages
The synthesis of GNCs was first reported by Xia et al. in 2002 [406] based on their previous success on making Ag nanocubes [407]. The GNCs were prepared by incubating Ag nanocubes with HAuCl4 in an aqueous solution [408-410]. Because of the favorable difference in the electrochemical potentials between Ag+/Ag (0.80 V) and AuCl4-/Au (1.00 V), a galvanic replacement occurred as follows:
| (1) |
A four-stage morphological change with Ag nanocubes was observed [408-410] and is explained by the following: (i) Initiation of Ag dissolution (this usually occurs at a site with poor protection such as a defect on the side face of a cube with sharp corners or the truncated corner(s) of a cube); (ii) Dissolution of bulk Ag from the interior of the particle through the initial site(s) and concurrent deposition of Au on the rest of the surface; (iii) Formation of a nanobox (for a sharp cube) or nanocage (for a truncated cube) with uniform wall thickness due to alloying between Au and Ag; (iv) Dealloying of Ag to generate pores on the walls of the nanobox eventually leading to the formation of GNCs (Fig. 21). This technique allows large-scale synthesis of GNCs with a wall thickness accuracy down to 0.5 nm. By adjusting the wall thickness, the localized surface plasmon resonance peaks can be fine-tuned in the range of 600-1200 nm [411]. The size of GNCs depends largely on the dimension of the Ag nanocage templates, which can be varied from 20 to 500 nm. As mentioned earlier, the size of the GNCs affects the relative magnitude of absorption or scattering and consequently the efficacy of the GNCs in imaging and therapy.
Fig. 21.
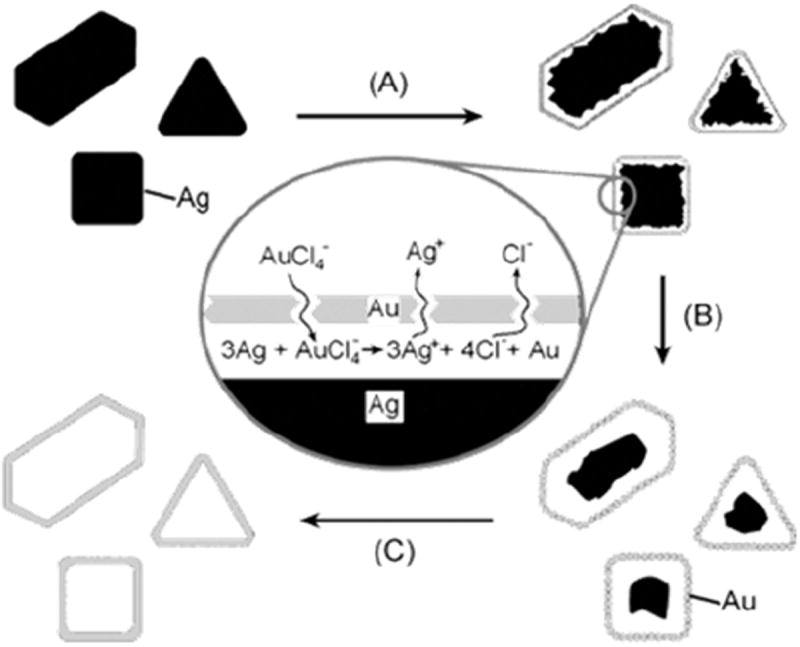
Schematic illustration of the formation of GNCs. Reprinted with permission from ref. [406].
The hollow GNCs can be used to load drug molecules of various kinds. The loading capacity can be tuned by adjusting the volume of the cavity and the pore size on the walls [412]. Xia et al. evaluated controlled drug release from the GNCs coated with a layer of thermally sensitive materials such as poly(N-isopropylacrylamide) (pNIPAAm) and its derivatives. These materials display dramatic conformational changes in response to small variations in temperature. Upon NIR laser irradiation or high-intensity focused ultrasound (HIFU), the local temperature is increased to trigger polymer collapse and release of pre-loaded therapeutics [413, 414]. Alternatively, controlled drug release can be achieved by filling the hollow interior with a phase-change material (PCM) such as 1-tetradecanol. Upon heating, PCM in the GNCs is melted to induce drug release into the surroundings [415].
3.4.3. Surface modification of gold nanoparticles
On account of the strong interactions between thiols and gold, surface modification and functionality anchoring have been performed mostly with thiolated compounds [416]. Many peptides [417], antibodies [418], and small molecule compounds [419] have been tethered onto GNP surfaces by taking advantage of the thiol-gold interaction. With regard to in vivo applications, PEGylation is usually necessary to reduce the non-specific interaction with the biological system and improve the particle circulation half-life. A long circulation time (t1/2= 17 h) was observed from GNRs coated with 5-kDa methyl-PEG-thiol [420] thus permitting efficient EPR-mediated tumor uptake. When these GNRs were injected at a dose of 20 mg/kg into MDA-MB-435 xenograft mouse models, tumor accumulation of ~ 7%ID/g at 72 h p.i. was observed. In general, GNPs coated with a long PEG chain (e.g. 5 kDa) are more stable than those coated with a short PEG chain (e.g. 2 kDa). Furthermore, PEG with a multidentate-thiol-anchor such as thioctic acid has better colloidal stability than monothiolated PEG [417-419, 421].
Due to a thick layer of CTAB, the longitudinal wall of GNRs is nearly inaccessible for thiolated species. In this case, the attack usually occurs at the terminal ends. MUA, 3-animo-5-mercapto-1,2,4-triazole (AMTAZ), and thiolated PEG have been used in GNR modification [422-425]. Hu et al. developed a multilayered coating that endows remarkable stability on GNRs [426]. They first coated CTAB capped GNRs with a silica shell. The resulting hydrophilic GNRs were subjected to a second silica growth process with octadecyltrimethoxysilane (OTMS) to be rendered hydrophobic. Finally, an amphiphilic polymer such as PEGylated phospholipid was assembled onto the particle surface to make the particles water soluble. This technique can be extended to stabilize other nanostructures that are not in the most thermodynamically or chemically stable states.
3.4.4. Toxicity of gold nanoparticles
One major advantage of GNPs in tumor imaging or therapy is their good biocompatibility. Previous studies have suggested that GNPs are either nontoxic or cause little toxicity to cells or small animals. Recent studies, however, disclosed size-dependent toxicity with GNPs [427]. Pan et al. systematically studied the cytotoxicity of triphenylphosphine coated GNPs with sizes from 0.8 to 15 nm on four cell lines: connective tissue fibroblasts, epithelial cells, macrophages, and melanoma cells. It was found that 1.4 nm GNPs were 60 time more toxic than 15 nm ones. This observation indicates variations in the cellular uptake kinetics and/or target specificities for GNPs of different sizes, and the difference may lead to a different toxicity profile. It is also possible that the toxicity may stem from interactions with cell membranes but not necessarily after internalization.
Although the gold cores are inert, the coatings may have an adverse impact on the biocompatibility [428]. This is particularly true for GNRs because the synthesis usually involves the use of CTAB which is cytotoxic and difficult to get rid of after the synthesis. Studies with CTAB-capped GNRs on human colon carcinoma cells (HT-29) show apparent cytotoxicity attributable to the free CTAB released from the particle surface to the solution [429]. Methods have been developed to mitigate the toxicity of GNRs [430]. For example, CTAB-capped GNRs were coated with a layer of polyelectrolyte to retard physical desorption of CTAB molecules [431, 432] and the resulting nanoconjugates showed substantially reduced cytotoxicity. Murphy’s group described an on-rod polymerization method [433]. They replaced the original CTAB coating with a CTAB analog, 11-(acryloyloxy) undecyltrimethyl ammonium bromide (p-CTAB) with a replacement rate of 77 ± 3%. The new ligand, p-CTAB, was polymerized in situ with a cationic free radical initiator. The GNRs produced were more stable than the CTAB-capped ones and less cytotoxic [433]. A similar approach was also applied to coat spherical GNPs with some success [434].
More biocompatible molecules such as PEG or phospholipids have been proposed to replace the CTAB coating [435]. Takahashi et al. extracted CTAB from an aqueous solution of GNRs with chloroform that contained phosphatidylcholine. This two-phase ligand replacement did not induce particle aggregation but yielded improved biocompatibility [426]. Achieving complete replacement of the CTAB coating, however, is challenging.
Owing to their relatively low toxicity, GNPs lead other nanostructures in the area of clinical translation. Clinical trials on GNS-based photothermal ablation of recurrent head and neck cancer are currently in progress [436]. Another formulation, recombinant human tumor necrosis factor-alpha (rhTNF) bearing GNPs, is in phase II clinical trial for advanced-stage cancer therapy [437].
3.4.5. Gold nanoparticles for cancer diagnosis
3.4.5.1. Gold nanoparticles for optical coherence tomography imaging
Optical coherence tomography (OCT) provides cross-sectional tomographic images of the internal microstructure of biological systems [334,335]. By measuring the backscattered or back-reflected light, OCT generates three dimensional images of a subject with micrometer resolution and in real time [438, 439]. With a large scattering cross-section and tunable optical resonance wavelengths, GNPs are good contrast agents for OCT [337,338], and in particular, GNRs, GNSs, and GNCs that have absorption and scattering in the NIR region are promising [440]. For instance, GNCs displaying 800 nm SPR have been studied as OCT contrast agents [441]. Phantom studies found a scattering cross-section of 8.10 × 10-16/m2 and absorption cross-section of 7.26×10-15/m2. Oldenburg et al. evaluated GNRs with maximum SPR at 775 nm from the viewpoint of OCT imaging [442]. The spectroscopic absorption profile of the GNRs was incorporated into a depth-resolved algorithm to map the relative GNR density in the OCT images. As a proof-of-concept demonstration, they successfully imaged GNR distributions in excised human breast carcinomas. Gobin et al. studied GNSs with a silica core of 119 nm diameter and a gold shell of 12 nm as OCT contrast agents [443]. Mie scattering theory predicts that the GNSs have approximately 67% of their extinction due to absorption and 33% due to scattering at 800 nm. In vivo imaging was performed after PEGylated GNSs had been injected into the subcutaneous tumor models. OCT imaging was performed 20 h later by directly applying the probe over the tumor skin. Substantial signal increase was observed from the tumor area, as shown in Fig. 22 [444]. Further laser irradiation led to complete tumor regression as a result of efficient hyperthermia [444, 445].
Fig. 22.
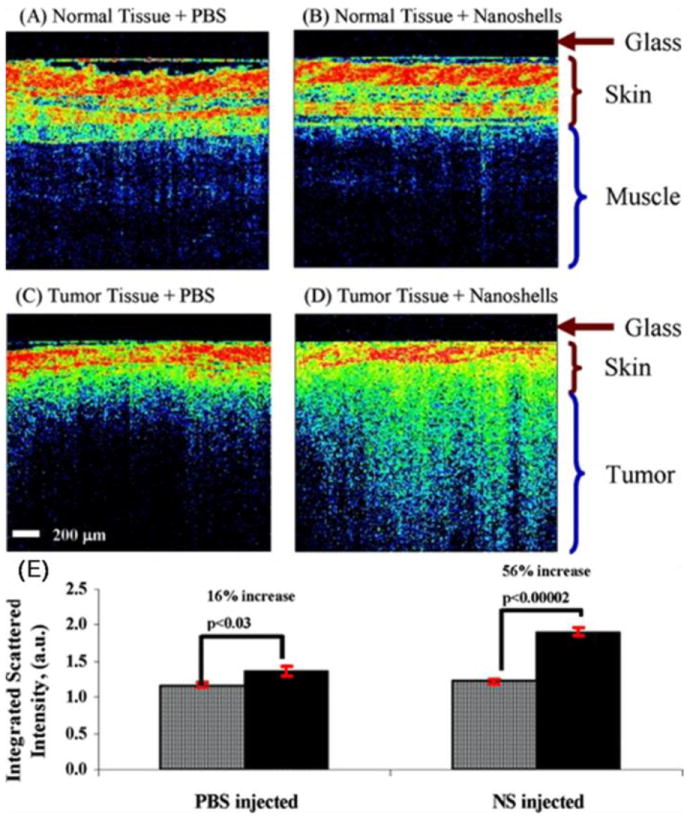
Representative OCT images from normal skin and muscle tissue areas of mice systemically injected with GNSs (A) or with PBS (B). Representative OCT images from tumors of mice systemically injected with GNSs (C) or with PBS (D). (E) Histogram graph of results from (A) to (D). Reprinted with permission from ref. [444].
3.4.5.2. Gold nanoparticles for photoacoustic imaging
Photoacoustic imaging (PA) is an emerging imaging modality combining high resolution and good optical contrast. In typical PA imaging, laser pulses are delivered to the areas-of-interest and by using the appropriate probes, some of the energy is absorbed and converted to heat, leading to thermoelastic expansion and ultrasonic emission. The signals are monitored to produce 2D or 3D images [446, 447]. PAT can detect objects embedded at depths as much as 7 cm, which is much deeper than possible by fluorescence imaging [448, 449]. Because of strong and tunable NIR absorption, GNCs, GNRs, and GNSs are promising contrast agents in this new imaging modality. For instance, Eghtedari et al. used GNRs in PA imaging [450]. They injected 25 μL GNRs at 1.25 pM into nude mice and detected the signals using a single-channel acoustic transducer. PA imaging could monitor the distribution of 100 μL of subcutaneously delivered GNRs at 125 pM. Notably, the irradiation can lead to a photon-induced shape transition of GNRs, mainly a rod-to-sphere conversion [451]. This transition results in a loss of contrast at the NIR wavelength of interest.
Yang et al. demonstrated PEGylated GNCs as intravascular contrast agents in PA imaging of the cerebral cortex in a rat model [452]. Gradual enhancement of the optical absorption in the cerebral cortex up to 81% was observed over the course of the experiment. The same group described the use of GNCs as probes in PA mapping of sentinel lymph nodes (SLN) [453], and the method was able to detect signals from as deep as 33 mm below the skin surface with good contrast. This depth is greater than the mean depth of SLNs in human beings, suggesting good clinical potential. Recently, Wilson et al. developed a novel PA agent composed of liquid perfluorocarbon (PFC) nanodroplets with encapsulated GNRs [454]. These droplets used optical absorption to achieve the following purposes: (1) Triggering the liquid-to-gas transition of a nanoscale PFC droplet; (2) Production of strong PA signals by vaporization; and (3) Prolonged thermal expansion signal from the GNRs. The gaseous phase of the PFC increases the acoustic impedance mismatch and ultrasound signal. Both in vitro and in vivo studies reveal that the nanodroplets are efficient contrast agents in both PA imaging and ultrasound imaging, as indicated in Fig. 23.
Fig. 23.
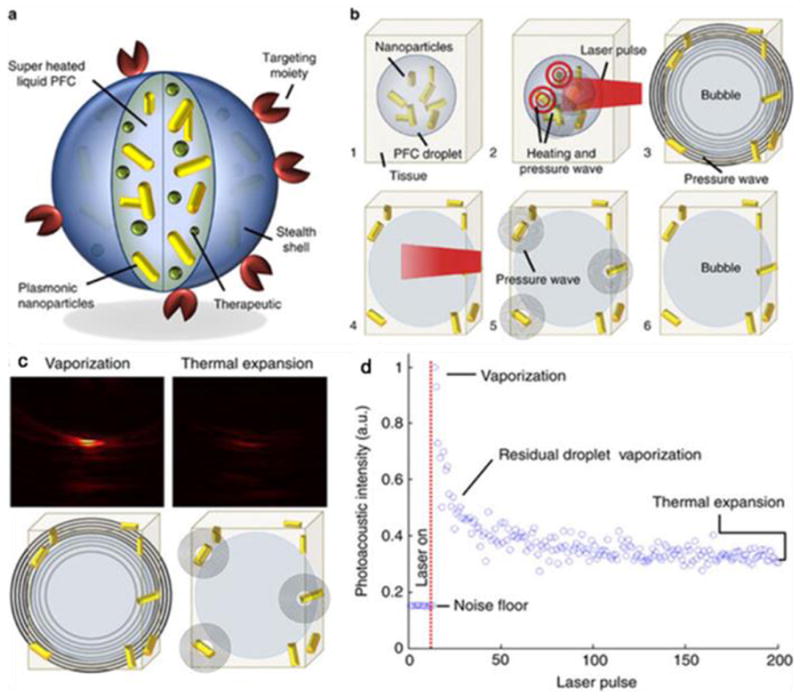
A novel particle-based contrast probe for PA imaging. (a) Diagram depicting the dual-contrast agent concept. (b) Step-by-step diagram of remote activation of the particle probes. (c) PA images reconstructed using vaporization-based and thermal expansion-based signals captured at one location. (d) Magnitude of pressure transients. Reprinted with permission from ref. [454].
3.4.5.3. Gold nanoparticles for Raman imaging
Surface-enhanced Raman scattering (SERS) refers to the enhancement of Raman-active vibrations of molecules when they are in close proximity to a metal surface [455]. The enhancement is attributed to both electromagnetic enhancement through excitation of localized surface plasmons [456] and chemical enhancement via charge-transfer [457]. It has been shown that the Raman signals can be enhanced by a factor of 1014-1015 when molecules adsorb on the GNP surface [458, 459], suggesting GNPs as attractive SERS probes. The degree of enhancement is influenced by the size, shape, and inter-particle distance. In addition to the high sensitivity, another reason that makes SERS an appealing method is the multiplexing capability. Because every molecule has its own spectral fingerprint, it is possible to image multiple biomarkers simultaneously, which could be challenging for other imaging modalities [460].
Lee et al. studied antibody conjugated Au/Ag core–shell nanoparticles for targeted tumor imaging with live HEK292 cells expressing PLGγ1 [461]. Zong et al. reported a dual mode cancer cell targeting probe based on CdTe QDs conjugated, silica coated Au/Ag core-shell nanorods. The nanorods could generate both SERS and fluorescence signals. The nanorods were conjugated with folic acid and used on both HeLa cells and MRC-5 cells, which were folate receptor positive and negative, respectively. Both SERS and fluorescence images confirmed the good targeting selectivity, as shown in Fig. 24 [462]. Jokerst et al. conjugated antibodies onto a gold-silica nanoparticle and evaluated the conjugates as SERS probes [463]. The nanoconjugates revealed EGFR-positive A431 tumors with a signal nearly 35 fold larger than that from EGFR-negative MDA-435S (human melanoma) tumors. The low-level EGFR expression in adjacent healthy tissue is 7 fold smaller than that observed from the positive tumors.
Fig. 24.
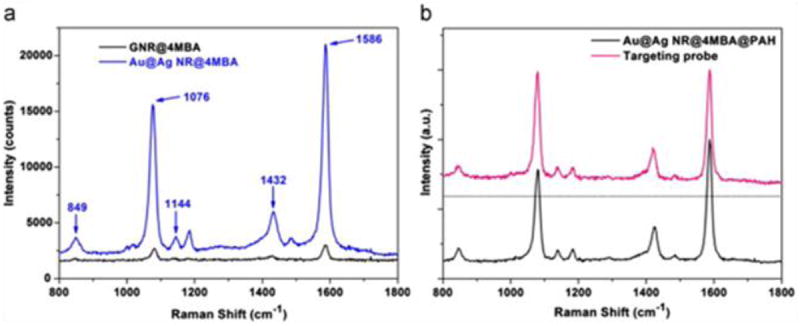
(a) SERS signals obtained from 4MBA-labeled GNRs and Au@Ag NRs; (b) SERS spectra of Au@Ag NR@4MBA@PAH and the targeting probe. Reprinted with permission from ref. [462].
Nie’s group reported SERS-based tumor imaging using PEGylated GNPs [436]. In the nanoconjugates, small-molecule Raman reporters were found to be stabilized, not displaced, by the PEG layer and the resulting SERS nanoparticles were considerably brighter than QDs in NIR spectrum window. When conjugated with single-chain variable fragment (ScFv) antibodies targeting EGFR, the particles accumulated in tumors and could be detected by a Raman detection system. In this particular study, small tumors (0.03 cm3) at a depth of 1-2 cm could be detected. Gambhir’s group developed MR-PA-SERS tri-modal imaging nanoparticles [464]; essentially a gold-silica-based SERS probe coated with Gd3+ ions. The probes showed picomolar sensitivity for all three modalities. When intravenously injected into glioblastoma-bearing mice, the probes accumulated in the tumor areas but not in the surrounding healthy tissues and were detected tri-modally. Raman imaging was used as an intraoperative imaging technique that allowed for guidance of tumor resection. The accuracy of Raman imaging was confirmed histologically, as shown in Fig. 25.
Fig. 25.
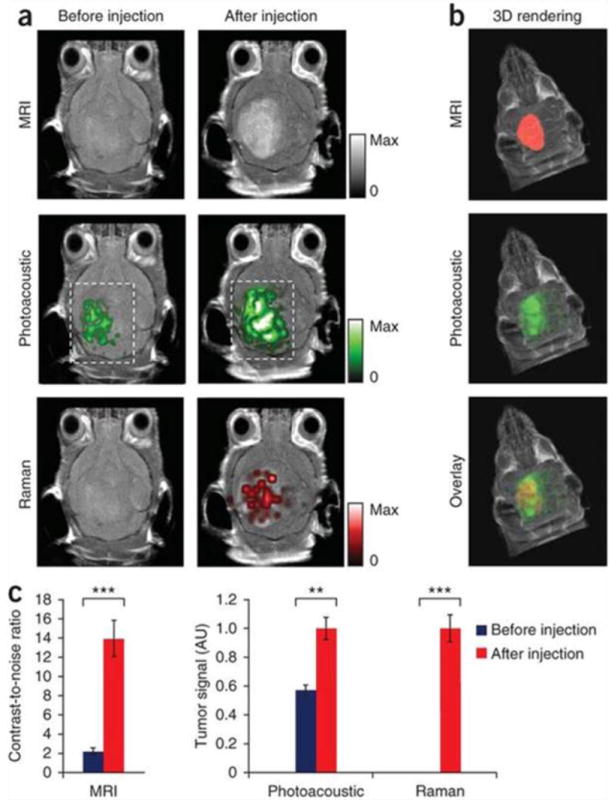
(a) Two-dimensional axial MRI, PA and Raman images. The post-injection images of all three modalities showed clear tumor visualization (dashed boxes outline the imaged area). (b) A three-dimensional (3D) rendering of MR images with the tumor segmented (red; top), an overlay of the three-dimensional PA images (green) over the MRI (middle) and an overlay of MRI, the segmented tumor and the PA images (bottom) showing good colocalization of the PA signal with the tumor. (c) Quantification of the signals in the tumor showing a significant increase in the MRI, PA, and Raman signals after as compared to before the injection. Reprinted with permission from ref. [464].
3.4.5.4. Gold nanoparticles for computed tomography imaging
GNPs also hold potential in CT imaging which relies on the differential levels of X-ray attenuation by tissues within the body to produce images [465]. Compared to light- or sound-based imaging techniques discussed above, CT imaging offers unlimited tissue penetration and higher spatial resolution. GNPs have attracted much attention in this area because their X-ray absorption coefficient is higher than that of iodine (e.g. Ultravist), which is commonly used to improve the contrast [466, 467]. Traditionally, CT is regarded as an anatomical imaging modality and not ideal for early stage tumor diagnosis. By conjugating with targeting molecules, GNPs may allow CT-based molecular imaging [468].
Kim et al. compared GNPs and Ultravist as CT contrast agents. When intravenously injected into rats, PEGylated GNPs showed a circulation half-life > 4 h, which was significantly longer than that of Ultravist (<10 min). On account of the improved circulation half-life and contrast, CT images acquired from rats using PEG-GNPs showed clearer delineation of the cardiac ventricles and great vessels. When they were tested on a hepatoma-bearing rat model, a high contrast (2-6 fold) was obtained between hepatoma and normal liver tissue in the CT images, suggesting that PEGylated GNPs are a potential CT contrast agent in blood pool imaging and hepatoma imaging [420, 469]. Recently, Reuveni et al. evaluated anti-EGFR conjugated GNPs (30 nm) in tumor CT imaging [470]. The GNRs were intravenously injected into nude mice implanted with human squamous cell carcinoma head and neck cancer. It was found that small tumors not detectable by anatomical CT were visible when using GNP as contrast agents (Fig. 26). However, a relatively large amount of GNPs needed to be injected to induce sufficient contrast on CT images, and some researchers believe that GNP-based CT imaging is not practical for real clinical applications [470].
Fig. 26.
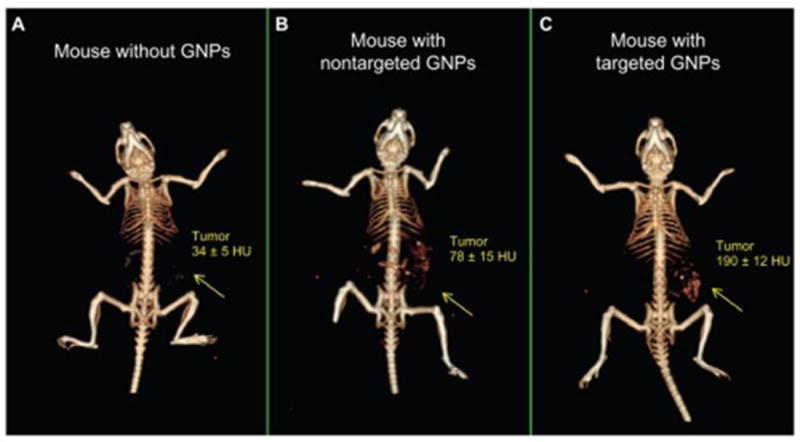
In vivo CT volume-rendered images of (A) mouse before injection of GNPs, (B) mouse 6 hours p.i. of nonspecific immunoglobulin G conjugated GNPs as a passive targeting experiment, and (C) mouse 6 hours p.i. Reprinted with permission from ref. [470].
3.4.5.5. Gold nanoparticles-based biosensors
Owing to their unique optical and electronic properties, GNPs have been intensively studied in the detection of biomarkers such as DNA and proteins in vitro. In particular, many efforts have been made to develop colorimetric biosensors by making use of the distance-dependent SPR of GNPs. Mirkin’s group first evaluated this idea by using two batches of 13-nm gold particles capped with thiolated, non-complementary DNA oligonucleotides [471, 472]. When targeting strands composed of sticky ends complementary to the two grafted sequences were introduced, cross-linking among particles occurred, resulting in a color change that could be monitored even by the naked eye. The sensitivity of the method was found to depend on the particle size and generally, large GNPs (e.g. 50 nm or 100 nm) delivered better sensitivity. They later reported a biochip by making use of the sharp melting transitions of GNP-labeled DNA assemblies. Using a sandwich-like assay involving an oligonucleotide-modified glass slide, a GNP-based probe, and targeted DNA, this method was able to detect single nucleotide mismatches with high selectivity [473, 474]. A limitation of the colorimetric method is the inherent inability of multiplexing. Mirkin’s group extended this technology by developing a Raman-based biosensor [475, 476]. The GNPs were labeled with both oligonucleotides and Raman-active dyes. After a silver treatment, the GNPs enabled SERS-based detection in a multiplex manner with high sensitivity.
GNP-based sensors have also been utilized to analyze protein targets. Mirkin and co-workers introduced a GNP-based bio-barcode assay for quantitative protein analysis [477]. Specifically, a magnetic microparticle conjugated with a detecting antibody and a GNP conjugated with both polyclonal antibodies for the target protein and oligonucleotides hybridized to barcode strands were used in the assay. The magnetic microparticles were applied to the analyte and addition of GNPs formed a sandwich-like complex if the target protein was present in the sample. These complexes were magnetically separated and the barcodes were released by heating. Subsequently, a silver-amplified sandwich assay similar to that described previously was performed to quantify the analytes. Multiple proteins at the mid-femtomolar concentrations could be determined [477].
Although spherical GNPs are used predominately, other gold nanostructures have been studied in in vitro biosensing. Hirsch et al. used GNSs to detect immunoglobulins in saline, serum, and whole blood [478]. Murphy’s group used biotin-avidin as a model system to show GNR-based particle aggregation and spectrum shift [478]. Lu et al. reported a highly sensitive two-photon scattering assay of cancer cells with an oval-shaped GNP conjugated with monoclonal anti-HER2/c-erb-2 antibody and S6 RNA aptamer [479]. When oval GNPs were mixed with the breast cancer SK-BR-3 cell line, a distinct color change occurred and the two-photon scattering intensity increased by about 13 times (Fig. 27).
Fig. 27.
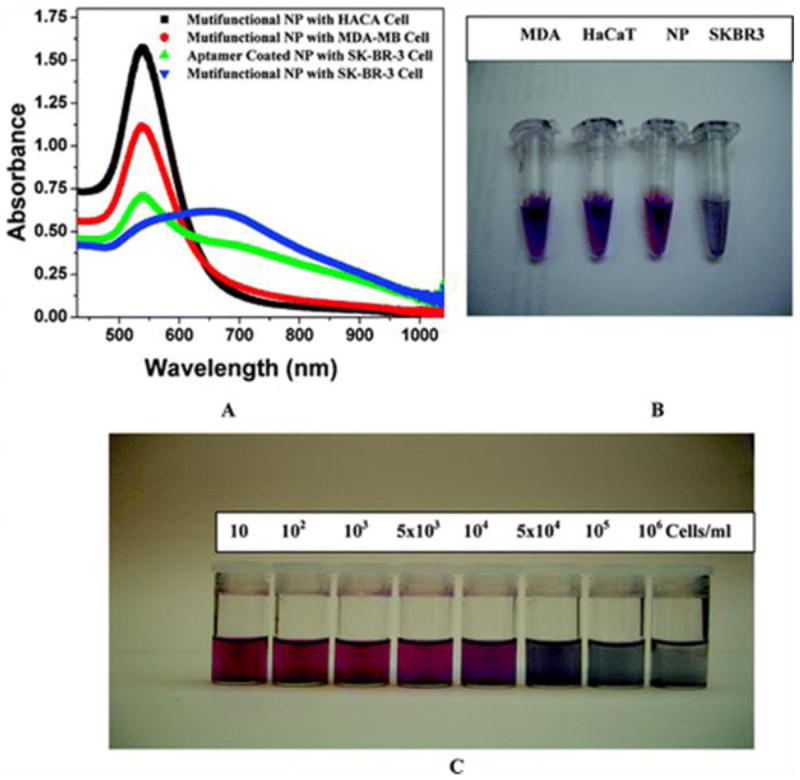
(A) Absorption profile variation of multifunctional oval-shaped GNPs due to the addition of different cancerous and noncancerous cells. (B) Photograph showing colorimetric change upon addition of different cancer cells (104 cells/mL). (C) Photograph demonstrating colorimetric change upon the addition of different numbers of SK-BR-3 cells. Reprinted with permission from ref. [479].
GNP-based Raman particles have also been widely used to detect biomarkers in cancer therapy [480]. Sha et al. reported a SERS-based cell detection assay that allowed rapid and direct enumeration of circulating tumor cells (CTCs) down to 50 tumor cells/mL in whole blood [481]. Recently, Wang et al. reported an EGF-conjugated, peptide-SERS-encoded GNPs for detecting CTCs in mouse and human blood samples with a high sensitivity of 5–50 cells/mL blood [482].
4. Conclusion and perspectives
This article reviews recent developments in the engineering of nanoparticles used in cancer imaging and diagnosis. With unique physical properties, these nanostructures can revolutionize a number of imaging modalities. For instance, magnetic nanoparticles can serve as contrast probes in MRI, and QDs and UCNPs can function as probes in fluorescence imaging. Depending on the nanostructure, GNPs can find applications in OCT imaging, PA imaging, or CT imaging. Extensive work conducted on small animal models confirms the advantages of the nanoparticle-based probe. With regard to multimodality imaging, it is possible to integrate more than one imaging functionality into a nanoparticle formulation and the resulting conjugates can work as reporters for multiple imaging methods. Multimodality imaging is appealing because existing imaging modalities have their own sets of advantages and disadvantages. The combined use of more than one imaging system can potentially circumvent the drawbacks of each modality and reduce the chance of misdiagnosis [483]. Other than imaging functionalities, therapeutics can be loaded onto nanoplatforms to achieve nano-theranostics and readers are referred to several reviews on this topic [75, 484-487].
There has been significant progress in our ability to prepare nanoparticles with controlled size, composition, morphology, and physical properties. Their behavior in the biological environment depends on many parameters such as the presence of a surface coating, targeting molecules, overall particle size, delivery route, and so on. These issues are sometimes intertwined and the complication underscores the multi-disciplinary nature of nanobiotechnology. To achieve bench-to-bed-side translation, it requires combined expertise and efforts from researchers in chemistry, physics, biology, pharmacology, and engineering. In particular, attention should be paid to the safety of the nanomaterials. As novel materials, there are concerns about possible side effects. So far, only IONPs and GNPs have entered clinics/clinical trials in the attempt to validate their biosafety. With regard to other materials such as QDs and UCNPs, more investigations are needed to understand their metabolism in vivo as well as long-term impact on a biological system.
Most in vivo imaging work described in this review serves as proof-of-concept and is performed mostly on subcutaneous animal models with a large tumor volume and known overexpression of certain biomarkers. Future investigations should focus on more clinically-relevant models. Indeed, we expect nanoparticle-based imaging can improve cancer management by providing the following features: first, as afore-mentioned, providing good detection sensitivity to permit finding tumors in their early stage of development; second, accurate and dynamic monitoring of patients’ response to treatment; third, simultaneous measurement of the expression levels of multiple biomarkers in vivo and non-invasively. So far, however, we are still far from achieving these goals.
Compared to in vivo imaging, nano-based biosensors are more likely to gain immediate regulation approval. Unlike imaging, in which nanoparticles are systematically administrated, nano-biosensors are applied in an in vitro setting and do not invoke complications arising from toxicity and biodistribution. Many studies have demonstrated the higher sensitivity rendered by nano-biosensors compared to conventional clinical tools such as ELISA. However, whether or not the high sensitivity can be translated into capability in practice to identify cancer in an early or pre-syndrome stage requires further clinical development and corroboration.
Fig. 10.
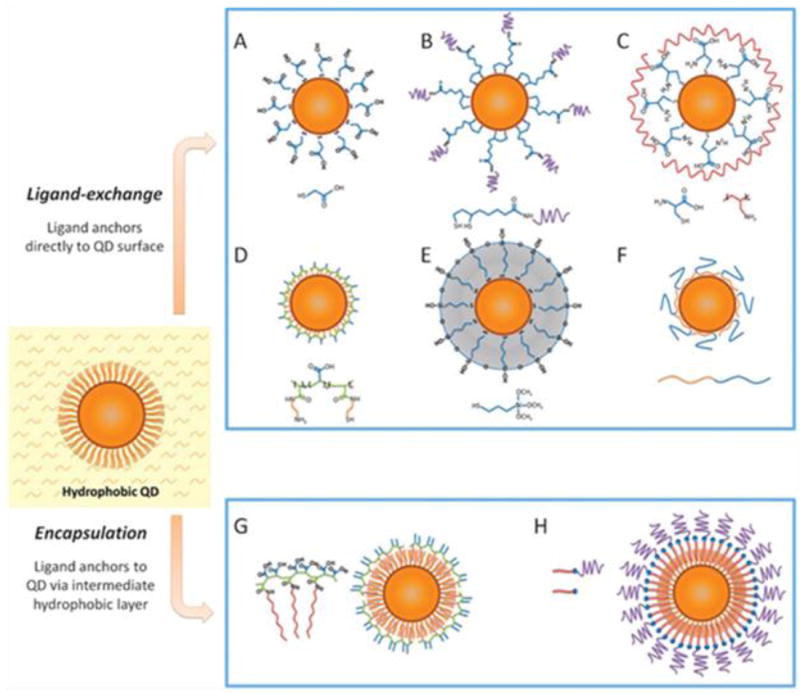
Routes for water-solubilization of hydrophobic QDs. (a–f) Ligand-exchange; (g–h) encapsulation. Reprinted with permission from ref. [202].
Acknowledgments
This work was jointly supported by an NCI/NIH R00 grant (5R00CA153772), a UGA startup grant, Hong Kong Research Grants Council (RGC) General Research Funds (GRF) No. CityU 112212 an City University of Hong Kong Applied Research Grant (ARG) No. 9667066. T. Todd was partially supported by a Philbrook scholarship.
References
- 1.Kochanek KD, Kirmeyer SE, Martin JA, Strobino DM, Guyer B. Pediatrics. 2012;129:338–348. doi: 10.1542/peds.2011-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 4.Jiang Y, Wang X. J Hematol Oncol. 2012;5:11. doi: 10.1186/1756-8722-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang S, Liu CC, Li W, Shen H, Laird PW, Zhou XJ. Nucleic Acids Res. 2012;40:9379–9391. doi: 10.1093/nar/gks725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatt AN, Mathur R, Farooque A, Verma A, Dwarakanath BS. Indian J Med Res. 2010;132:129–149. [PubMed] [Google Scholar]
- 7.James W. Curr Opin Pharmacol. 2001;1:540–546. doi: 10.1016/s1471-4892(01)00093-5. [DOI] [PubMed] [Google Scholar]
- 8.Weng CH, Huang CJ, Lee GB. Sensors (Basel) 2012;12:9514–9529. doi: 10.3390/s120709514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrari M. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 10.Sebastian S, Settleman J, Reshkin SJ, Azzariti A, Bellizzi A, Paradiso A. Biochim Biophys Acta. 2006;1766:120–139. doi: 10.1016/j.bbcan.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Arteaga C. Semin Oncol. 2003;30:3–14. [PubMed] [Google Scholar]
- 12.Schlessinger J. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 13.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 14.Cai W, Niu G, Chen X. Eur J Nucl Med Mol Imaging. 2008;35:186–208. doi: 10.1007/s00259-007-0560-9. [DOI] [PubMed] [Google Scholar]
- 15.Casalini P, Iorio MV, Galmozzi E, Menard S. J Cell Physiol. 2004;200:343–350. doi: 10.1002/jcp.20007. [DOI] [PubMed] [Google Scholar]
- 16.Dy GK, Adjei AA. Cancer. 2008;113:1857–1887. doi: 10.1002/cncr.23651. [DOI] [PubMed] [Google Scholar]
- 17.Li PC, Wang CR, Shieh DB, Wei CW, Liao CK, Poe C, Jhan S, Ding AA, Wu YN. Opt Express. 2008;16:18605–18615. doi: 10.1364/oe.16.018605. [DOI] [PubMed] [Google Scholar]
- 18.Kah JC, Kho KW, Lee CG, James C, Sheppard R, Shen ZX, Soo KC, Olivo MC. Int J Nanomedicine. 2007;2:785–798. [PMC free article] [PubMed] [Google Scholar]
- 19.Coussens L, Yang-Feng TL, Liao YC, Chen E, Gray A, McGrath J, Seeburg PH, Libermann TA, Schlessinger J, Francke U, et al. Science. 1985;230:1132–1139. doi: 10.1126/science.2999974. [DOI] [PubMed] [Google Scholar]
- 20.Olayioye MA. Breast Cancer Res. 2001;3:385–389. doi: 10.1186/bcr327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarden Y, Sliwkowski MX. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 22.Na HB, Lee JH, An K, Park YI, Park M, Lee IS, Nam DH, Kim ST, Kim SH, Kim SW, Lim KH, Kim KS, Kim SO, Hyeon T. Angew Chem Int Ed. 2007;46:5397–5401. doi: 10.1002/anie.200604775. [DOI] [PubMed] [Google Scholar]
- 23.Huh YM, Jun YW, Song HT, Kim S, Choi JS, Lee JH, Yoon S, Kim KS, Shin JS, Suh JS, Cheon J. J Am Chem Soc. 2005;127:12387–12391. doi: 10.1021/ja052337c. [DOI] [PubMed] [Google Scholar]
- 24.Ferrara N. Nat Rev Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 25.Ferrara N. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 26.Cai W, Chen X. Front Biosci. 2007;12:4267–4279. doi: 10.2741/2386. [DOI] [PubMed] [Google Scholar]
- 27.Cai W, Chen X. J Nucl Med. 2008;49(Suppl. 2):113S–128S. doi: 10.2967/jnumed.107.045922. [DOI] [PubMed] [Google Scholar]
- 28.Ferrara N. EXS. 2005:209–231. doi: 10.1007/3-7643-7311-3_15. [DOI] [PubMed] [Google Scholar]
- 29.Youssoufian H, Hicklin DJ, Rowinsky EK. Clin Cancer Res. 2007;13:5544s–5548s. doi: 10.1158/1078-0432.CCR-07-1107. [DOI] [PubMed] [Google Scholar]
- 30.Hynes RO. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 31.Hood JD, Cheresh DA. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed N, Riley C, Rice GE, Quinn MA, Baker MS. J Histochem Cytochem. 2002;50:1371–1380. doi: 10.1177/002215540205001010. [DOI] [PubMed] [Google Scholar]
- 33.Allen MD, Vaziri R, Green M, Chelala C, Brentnall AR, Dreger S, Vallath S, Nitch-Smith H, Hayward J, Carpenter R, Holliday DL, Walker RA, Hart IR, Jones JL. J Pathol. 2011;223:646–658. doi: 10.1002/path.2833. [DOI] [PubMed] [Google Scholar]
- 34.Dingemans AM, van den Boogaart V, Vosse BA, van Suylen RJ, Griffioen AW, Thijssen VL. Mol Cancer. 2010;9:152. doi: 10.1186/1476-4598-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCabe NP, De S, Vasanji A, Brainard J, Byzova TV. Oncogene. 2007;26:6238–6243. doi: 10.1038/sj.onc.1210429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 37.Zhang CF, Jugold M, Woenne EC, Lammers T, Morgenstern B, Mueller MM, Zentgraf H, Bock M, Eisenhut M, Semmler W, Kiessling F. Cancer Res. 2007;67:1555–1562. doi: 10.1158/0008-5472.CAN-06-1668. [DOI] [PubMed] [Google Scholar]
- 38.Wu Z, Li ZB, Chen K, Cai W, He L, Chin FT, Li F, Chen X. J Nucl Med. 2007;48:1536–1544. doi: 10.2967/jnumed.107.040816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elnakat H, Ratnam M. Adv Drug Deliv Rev. 2004;56:1067–1084. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Bioorg Med Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 41.Weitman SD, Lark RH, Coney LR, Fort DW, Frasca V, Zurawski VR, Jr, Kamen BA. Cancer Res. 1992;52:3396–3401. [PubMed] [Google Scholar]
- 42.Low PS, Henne WA, Doorneweerd DD. Acc Chem Res. 2008;41:120–129. doi: 10.1021/ar7000815. [DOI] [PubMed] [Google Scholar]
- 43.Scherer RL, McIntyre JO, Matrisian LM. Cancer Metastasis Rev. 2008;27:679–690. doi: 10.1007/s10555-008-9152-9. [DOI] [PubMed] [Google Scholar]
- 44.Sternlicht MD, Werb Z. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKerrow JH, Bhargava V, Hansell E, Huling S, Kuwahara T, Matley M, Coussens L, Warren R. Mol Med. 2000;6:450–460. [PMC free article] [PubMed] [Google Scholar]
- 46.Davies B, Waxman J, Wasan H, Abel P, Williams G, Krausz T, Neal D, Thomas D, Hanby A, Balkwill F. Cancer Res. 1993;53:5365–5369. [PubMed] [Google Scholar]
- 47.Vaisanen A, Tuominen H, Kallioinen M, Turpeenniemi-Hujanen T. J Pathol. 1996;180:283–289. doi: 10.1002/(SICI)1096-9896(199611)180:3<283::AID-PATH662>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 48.Zhu L, Wang H, Wang L, Wang Y, Jiang K, Li C, Ma Q, Gao S, Li W, Cai M, Niu G, Lee S, Yang W, Fang X, Chen X. J Control Release. 2011;150:248–255. doi: 10.1016/j.jconrel.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koivunen E, Arap W, Valtanen H, Rainisalo A, Medina OP, Heikkila P, Kantor C, Gahmberg CG, Salo T, Konttinen YT, Sorsa T, Ruoslahti E, Pasqualini R. Nat Biotechnol. 1999;17:768–774. doi: 10.1038/11703. [DOI] [PubMed] [Google Scholar]
- 50.Lee S, Cha EJ, Park K, Lee SY, Hong JK, Sun IC, Kim SY, Choi K, Kwon IC, Kim K, Ahn CH. Angew Chem Int Ed. 2008;47:2804–2807. doi: 10.1002/anie.200705240. [DOI] [PubMed] [Google Scholar]
- 51.Xie J, Zhang F, Aronova M, Zhu L, Lin X, Quan Q, Liu G, Zhang G, Choi KY, Kim K, Sun X, Lee S, Sun S, Leapman R, Chen X. ACS Nano. 2011;5:3043–3051. doi: 10.1021/nn200161v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Israeli RS, Powell CT, Fair WR, Heston WD. Cancer Res. 1993;53:227–230. [PubMed] [Google Scholar]
- 53.O’Keefe DS, Su SL, Bacich DJ, Horiguchi Y, Luo Y, Powell CT, Zandvliet D, Russell PJ, Molloy PL, Nowak NJ, Shows TB, Mullins C, Vonder Haar RA, Fair WR, Heston WD. Biochim Biophys Acta. 1998;1443:113–127. doi: 10.1016/s0167-4781(98)00200-0. [DOI] [PubMed] [Google Scholar]
- 54.Horoszewicz JS, Kawinski E, Murphy GP. Anticancer Res. 1987;7:927–935. [PubMed] [Google Scholar]
- 55.Olson WC, Heston WD, Rajasekaran AK. Rev Recent Clin Trials. 2007;2:182–190. doi: 10.2174/157488707781662724. [DOI] [PubMed] [Google Scholar]
- 56.Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R. Cancer Res. 2004;64:7668–7672. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- 57.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Proc Natl Acad Sci U S A. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verhoven B, Schlegel RA, Williamson P. J Exp Med. 1995;182:1597–1601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schilling D, Gehrmann M, Steinem C, De Maio A, Pockley AG, Abend M, Molls M, Multhoff G. FASEB J. 2009;23:2467–2477. doi: 10.1096/fj.08-125229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ran S, Thorpe PE. Int J Radiat Oncol Biol Phys. 2002;54:1479–1484. doi: 10.1016/s0360-3016(02)03928-7. [DOI] [PubMed] [Google Scholar]
- 61.Dong HP, Holth A, Kleinberg L, Ruud MG, Elstrand MB, Trope CG, Davidson B, Risberg B. American J Clin Pathol. 2009;132:756–762. doi: 10.1309/AJCPAVFA8J3KHPRS. [DOI] [PubMed] [Google Scholar]
- 62.Kenis H, Reutelingsperger C. Curr Pharm Des. 2009;15:2719–2723. doi: 10.2174/138161209788923903. [DOI] [PubMed] [Google Scholar]
- 63.Malik G, Ward MD, Gupta SK, Trosset MW, Grizzle WE, Adam BL, Diaz JI, Semmes OJ. Clin Cancer Res. 2005;11:1073–1085. [PubMed] [Google Scholar]
- 64.Ueda K, Fukase Y, Katagiri T, Ishikawa N, Irie S, Sato TA, Ito H, Nakayama H, Miyagi Y, Tsuchiya E, Kohno N, Shiwa M, Nakamura Y, Daigo Y. Proteomics. 2009;9:2182–2192. doi: 10.1002/pmic.200800374. [DOI] [PubMed] [Google Scholar]
- 65.Zhou C, Long M, Qin Y, Sun X, Zheng J. Angew Chem Int Ed. 2011;50:3168–3172. doi: 10.1002/anie.201007321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng MM, Cuda G, Bunimovich YL, Gaspari M, Heath JR, Hill HD, Mirkin CA, Nijdam AJ, Terracciano R, Thundat T, Ferrari M. Curr Opin Chem Biol. 2006;10:11–19. doi: 10.1016/j.cbpa.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 68.Lee HY, Li Z, Chen K, Hsu AR, Xu C, Xie J, Sun S, Chen X. J Nucl Med. 2008;49:1371–1379. doi: 10.2967/jnumed.108.051243. [DOI] [PubMed] [Google Scholar]
- 69.Montet X, Montet-Abou K, Reynolds F, Weissleder R, Josephson L. Neoplasia. 2006;8:214–222. doi: 10.1593/neo.05769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee JH, Lee K, Moon SH, Lee Y, Park TG, Cheon J. Angew Chem Int Ed. 2009;48:4174–4179. doi: 10.1002/anie.200805998. [DOI] [PubMed] [Google Scholar]
- 71.Cheon J, Lee JH. Acc Chem Res. 2008;41:1630–1640. doi: 10.1021/ar800045c. [DOI] [PubMed] [Google Scholar]
- 72.Du J, Lu WL, Ying X, Liu Y, Du P, Tian W, Men Y, Guo J, Zhang Y, Li RJ, Zhou J, Lou JN, Wang JC, Zhang X, Zhang Q. Mol Pharm. 2009;6:905–917. doi: 10.1021/mp800218q. [DOI] [PubMed] [Google Scholar]
- 73.Ying X, Wen H, Lu WL, Du J, Guo J, Tian W, Men Y, Zhang Y, Li RJ, Yang TY, Shang DW, Lou JN, Zhang LR, Zhang Q. J Control Release. 2010;141:183–192. doi: 10.1016/j.jconrel.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 74.Li Y, He H, Jia X, Lu WL, Lou J, Wei Y. Biomaterials. 2012;33:3899–3908. doi: 10.1016/j.biomaterials.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 75.Mura S, Couvreur P. Adv Drug Deliv Rev. 2012;64:1394–1416. doi: 10.1016/j.addr.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 76.Sawant RR, Jhaveri AM, Torchilin VP. Adv Drug Deliv Rev. 2012;64:1436–1446. doi: 10.1016/j.addr.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 77.Liu Z, Tabakman S, Welsher K, Dai H. Nano res. 2009;2:85–120. doi: 10.1007/s12274-009-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kostarelos K, Bianco A, Prato M. Nat Nanotechnol. 2009;4:627–633. doi: 10.1038/nnano.2009.241. [DOI] [PubMed] [Google Scholar]
- 79.Huang Y, He S, Cao W, Cai K, Liang XJ. Nanoscale. 2012;4:6135–6149. doi: 10.1039/c2nr31715j. [DOI] [PubMed] [Google Scholar]
- 80.Santos CM, Mangadlao J, Ahmed F, Leon A, Advincula RC, Rodrigues DF. Nanotechnology. 2012;23:395101. doi: 10.1088/0957-4484/23/39/395101. [DOI] [PubMed] [Google Scholar]
- 81.Shen H, Zhang L, Liu M, Zhang Z. Theranostics. 2012;2:283–294. doi: 10.7150/thno.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xie J, Lee S, Chen X. Adv Drug Deliv Rev. 2010;62:1064–1079. doi: 10.1016/j.addr.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Massart R. I EEE Trans Magn. 1981;17:1247–1248. [Google Scholar]
- 84.Bee A, Massart R, Neveu S. J Magn Magn Mater. 1995;149:6–9. [Google Scholar]
- 85.Babes L, Denizot B, Tanguy G, Le Jeune JJ, Jallet P. J Colloid Interface Sci. 1999;212:474–482. doi: 10.1006/jcis.1998.6053. [DOI] [PubMed] [Google Scholar]
- 86.Lu AH, Salabas EL, Schuth F. Angew Chem Int Ed. 2007;46:1222–1244. doi: 10.1002/anie.200602866. [DOI] [PubMed] [Google Scholar]
- 87.Mahtab F, Yu Y, Lam JWY, Liu JZ, Zhang B, Lu P, Zhang XX, Tang BZ. Adv Funct Mater. 2011;21:1733–1740. [Google Scholar]
- 88.Sadjadi MS, Fathi F, Farhadyar N, Zare K. J Nano Res. 2011;16:43–48. [Google Scholar]
- 89.Pan BF, Cui DX, Sheng Y, Ozkan CG, Gao F, He R, Li Q, Xu P, Huang T. Cancer Res. 2007;67:8156–8163. doi: 10.1158/0008-5472.CAN-06-4762. [DOI] [PubMed] [Google Scholar]
- 90.Tahmasebi E, Yamini Y, Mehdinia A, Rouhi F. J Sep Sci. 2012;35:2256–2265. doi: 10.1002/jssc.201200345. [DOI] [PubMed] [Google Scholar]
- 91.Corot C, Robert P, Idee JM, Port M. Adv Drug Deliv Rev. 2006;58:1471–1504. doi: 10.1016/j.addr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 92.Lee HY, Lee SH, Xu CJ, Xie J, Lee JH, Wu B, Koh AL, Wang XY, Sinclair R, Xwang S, Nishimura DG, Biswal S, Sun SH, Cho SH, Chen XY. Nanotechnology. 2008;19:165101. doi: 10.1088/0957-4484/19/16/165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li Z, Sun Q, Gao M. Angew Chem Int Ed. 2004;44:123–126. doi: 10.1002/anie.200460715. [DOI] [PubMed] [Google Scholar]
- 94.Harisinghani MG, Jhaveri KS, Weissleder R, Schima W, Saini S, Hahn PF, Mueller PR. Clin Radiol. 2001;56:714–725. doi: 10.1053/crad.2001.0764. [DOI] [PubMed] [Google Scholar]
- 95.Deddens LH, Van Tilborg GA, Mulder WJ, De Vries HE, Dijkhuizen RM. Cerebrovasc Dis. 2012;33:392–402. doi: 10.1159/000336116. [DOI] [PubMed] [Google Scholar]
- 96.Kang HW, Josephson L, Petrovsky A, Weissleder R, Bogdanov A., Jr Bioconjug Chem. 2002;13:122–127. doi: 10.1021/bc0155521. [DOI] [PubMed] [Google Scholar]
- 97.Banerjee SR. Abs Pap Am Chem Soc. 2004;228:U31–U31. [Google Scholar]
- 98.Glazer AN. Nature. 1996;381:290–290. [Google Scholar]
- 99.Gupta MN. Abs Pap Am Chem Soc. 1991;202:220-BIOT. [Google Scholar]
- 100.Tassa C, Shaw SY, Weissleder R. Acc Chem Res. 2011;44:842–852. doi: 10.1021/ar200084x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hao R, Xing R, Xu Z, Hou Y, Gao S, Sun S. Adv Mater. 2010;22:2729–2742. doi: 10.1002/adma.201000260. [DOI] [PubMed] [Google Scholar]
- 102.Hyeon T. Chem Commun. 2003:927–934. doi: 10.1039/b207789b. [DOI] [PubMed] [Google Scholar]
- 103.Green M. Chem Commun. 2005:3002–3011. doi: 10.1039/b501835h. [DOI] [PubMed] [Google Scholar]
- 104.Jeong U, Teng XW, Wang Y, Yang H, Xia YN. Adv Mater. 2007;19:33–60. [Google Scholar]
- 105.Hyeon T. Chem Commun. 2003:927–934. doi: 10.1039/b207789b. [DOI] [PubMed] [Google Scholar]
- 106.Park J, Lee E, Hwang NM, Kang M, Kim SC, Hwang Y, Park JG, Noh HJ, Kim JY, Park JH, Hyeon T. Angew Chem Int Ed. 2005;44:2872–2877. doi: 10.1002/anie.200461665. [DOI] [PubMed] [Google Scholar]
- 107.Hyeon T, Lee SS, Park J, Chung Y, Bin Na H. J Am Chem Soc. 2001;123:12798–12801. doi: 10.1021/ja016812s. [DOI] [PubMed] [Google Scholar]
- 108.Park J, An KJ, Hwang YS, Park JG, Noh HJ, Kim JY, Park JH, Hwang NM, Hyeon T. Nat Mater. 2004;3:891–895. doi: 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- 109.Jana NR, Chen YF, Peng XG. Chem Mater. 2004;16:3931–3935. [Google Scholar]
- 110.Woo K, Hong J, Ahn JP. J Magn Magn Mater. 2005;293:177–181. [Google Scholar]
- 111.Sun S, Zeng H, Robinson DB, Raoux S, Rice PM, Wang SX, Li G. J Am Chem Soc. 2004;126:273–279. doi: 10.1021/ja0380852. [DOI] [PubMed] [Google Scholar]
- 112.Khurshid H, Li WF, Phan MH, Mukherjee P, Hadjipanayis GC, Srikanth H. Appl Phys Lett. 2012;101:232405. [Google Scholar]
- 113.Jun YW, Huh YM, Choi JS, Lee JH, Song HT, Kim S, Yoon S, Kim KS, Shin JS, Suh JS, Cheon J. J Am Chem Soc. 2005;127:5732–5733. doi: 10.1021/ja0422155. [DOI] [PubMed] [Google Scholar]
- 114.Jang JT, Nah H, Lee JH, Moon SH, Kim MG. J Cheon, Angew Chem Int Ed. 2009;48:1234–1238. doi: 10.1002/anie.200805149. [DOI] [PubMed] [Google Scholar]
- 115.Murray CB, Sun SH, Doyle H, Betley T. MRS Bulletin. 2001;26:985–991. [Google Scholar]
- 116.Butter K, Kassapidou K, Vroege GJ, Philipse AP. J Colloid Interface Sci. 2005;287:485–495. doi: 10.1016/j.jcis.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 117.Butter K, Philipse AP, Vroege GJ. J Magn Magn Mater. 2002;252:1–3. [Google Scholar]
- 118.Peng S, Wang C, Xie J, Sun S. J Am Chem Soc. 2006;128:10676–10677. doi: 10.1021/ja063969h. [DOI] [PubMed] [Google Scholar]
- 119.Lacroix LM, Huls NF, Ho D, Sun X, Cheng K, Sun S. Nano Lett. 2011;11:1641–1645. doi: 10.1021/nl200110t. [DOI] [PubMed] [Google Scholar]
- 120.Song Q, Zhang ZJ. J Am Chem Soc. 2004;126:6164–6168. doi: 10.1021/ja049931r. [DOI] [PubMed] [Google Scholar]
- 121.Puntes VF, Zanchet D, Erdonmez CK, Alivisatos AP. J Am Chem Soc. 2002;124:12874–12880. doi: 10.1021/ja027262g. [DOI] [PubMed] [Google Scholar]
- 122.Puntes VF, Krishnan KM, Alivisatos AP. Science. 2001;291:2115–2117. doi: 10.1126/science.1057553. [DOI] [PubMed] [Google Scholar]
- 123.Bonnemann H, Brijoux W, Brinkmann R, Matoussevitch N, Waldofner N, Palina N, Modrow H. Inorg Chim Acta. 2003;350:617–624. [Google Scholar]
- 124.Yang C, Zhao H, Hou Y, Ma D. J Am Chem Soc. 2012;134:15814–15821. doi: 10.1021/ja305048p. [DOI] [PubMed] [Google Scholar]
- 125.Seo WS, Lee JH, Sun XM, Suzuki Y, Mann D, Liu Z, Terashima M, Yang PC, McConnell MV, Nishimura DG, Dai HJ. Nat Mater. 2006;5:971–976. doi: 10.1038/nmat1775. [DOI] [PubMed] [Google Scholar]
- 126.Kim J, Lee Y, Sun SH. J Am Chem Soc. 2010;132:4996–4997. doi: 10.1021/ja1009629. [DOI] [PubMed] [Google Scholar]
- 127.Wan MX, Li JC. J Poly Sci a-Poly Chem. 1998;36:2799–2805. [Google Scholar]
- 128.Butterworth MD, Bell SA, Armes SP, Simpson AW. J ColloidInterface Sci. 1996;183:91–99. [Google Scholar]
- 129.Tartaj P, Morales MP, Gonzalez-Carreno T, Veintemillas-Verdaguer S, Serna CJ. J Magn Magn Mater. 2005;290:28–34. [Google Scholar]
- 130.Zhang TR, Ge JP, Hu YP, Yin YD. Nano Lett. 2007;7:3203–3207. doi: 10.1021/nl071928t. [DOI] [PubMed] [Google Scholar]
- 131.Huang G, Zhang CF, Li SZ, Khemtong C, Yang SG, Tian RH, Minna JD, Brown KC, Gao JM. J Mater Chem. 2009;19:6367–6372. doi: 10.1039/b902358e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xu CJ, Xu KM, Gu HW, Zheng RK, Liu H, Zhang XX, Guo ZH, Xu B. J Am Chem Soc. 2004;126:9938–9939. doi: 10.1021/ja0464802. [DOI] [PubMed] [Google Scholar]
- 133.Xie J, Chen K, Lee HY, Xu CJ, Hsu AR, Peng S, Chen XY, Sun SH. J Am Chem Soc. 2008;130:7542–7543. doi: 10.1021/ja802003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xie J, Wang JH, Niu G, Huang J, Chen K, Li XG, Chen XY. Chem Commun. 2010;46:433–435. doi: 10.1039/b917195a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huh YM, Jun YW, Song HT, Kim S, Choi JS, Lee JH, Yoon S, Kim KS, Shin JS, Suh JS, Cheon J. J Am Chem Soc. 2005;127:12387–12391. doi: 10.1021/ja052337c. [DOI] [PubMed] [Google Scholar]
- 136.Torchilin VP, Trubetskoy VS. Adv Drug Deliv Rev. 1995;16:141–155. [Google Scholar]
- 137.Pellegrino T, Manna L, Kudera S, Liedl T, Koktysh D, Rogach AL, Keller S, Radler J, Natile G, Parak WJ. Nano Lett. 2004;4:703–707. [Google Scholar]
- 138.Hultman KL, Raffo AJ, Grzenda AL, Harris PE, Brown TR, O’Brien S. ACS Nano. 2008;2:477–484. doi: 10.1021/nn700400h. [DOI] [PubMed] [Google Scholar]
- 139.Tong S, Hou SJ, Ren BB, Zheng ZL, Bao G. Nano Lett. 2011;11:3720–3726. doi: 10.1021/nl201978c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jin YD, Jia CX, Huang SW, O’Donnell M, Gao XH. Nat Commun. 2010;1:41. doi: 10.1038/ncomms1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.LaConte LE, Nitin N, Zurkiya O, Caruntu D, O’Connor CJ, Hu X, Bao G. J Magn Reson Imaging. 2007;26:1634–1641. doi: 10.1002/jmri.21194. [DOI] [PubMed] [Google Scholar]
- 142.Hu SH, Chen YY, Liu TC, Tung TH, Liu DM, Chen SY. Chem Commun. 2011;47:1776–1778. doi: 10.1039/c0cc03998e. [DOI] [PubMed] [Google Scholar]
- 143.Yi DK, Lee SS, Papaefthymiou GC, Ying JY. Chem Mater. 2006;18:614–619. [Google Scholar]
- 144.Yi DK, Selvan ST, Lee SS, Papaefthymiou GC, Kundaliya D, Ying JY. J Am Chem Soc. 2005;127:4990–4991. doi: 10.1021/ja0428863. [DOI] [PubMed] [Google Scholar]
- 145.Kim J, Kim HS, Lee N, Kim T, Kim H, Yu T, Song IC, Moon WK, Hyeon T. Angew Chem Inte Ed. 2008;47:8438–8441. doi: 10.1002/anie.200802469. [DOI] [PubMed] [Google Scholar]
- 146.De Palma R, Peeters S, Van Bael MJ, Van den Rul H, Bonroy K, Laureyn W, Mullens J, Borghs G, Maes G. Chem Mater. 2007;19:1821–1831. [Google Scholar]
- 147.Lee JE, Lee N, Kim T, Kim J, Hyeon T. Acc Chem Res. 2011;44:893–902. doi: 10.1021/ar2000259. [DOI] [PubMed] [Google Scholar]
- 148.Kim T, Momin E, Choi J, Yuan K, Zaidi H, Kim J, Park M, Lee N, McMahon MT, Quinones-Hinojosa A, Bulte JWM, Hyeon T, Gilad AA. J Am Chem Soc. 2011;133:2955–2961. doi: 10.1021/ja1084095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Park J, An K, Hwang Y, Park JG, Noh HJ, Kim JY, Park JH, Hwang NM, Hyeon T. Nat Mater. 2004;3:891–895. doi: 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- 150.Zhen ZP, Xie J. Theranostics. 2012;2:45–54. doi: 10.7150/thno.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shin JM, Anisur RM, Ko MK, Im GH, Lee JH, Lee IS. Angew Chem Inte Ed. 2009;48:321–324. doi: 10.1002/anie.200802323. [DOI] [PubMed] [Google Scholar]
- 152.Huang J, Xie J, Chen K, Bu L, Lee S, Cheng Z, Li X, Chen X. Chem Commun. 2010;46:6684–6686. doi: 10.1039/c0cc01041c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Taylor KML, Jin A, Lin WB. Angew Chem Inte Ed. 2008;47:7722–7725. doi: 10.1002/anie.200802911. [DOI] [PubMed] [Google Scholar]
- 154.Taylor KML, Rieter WJ, Lin WB. J Am Chem Soc. 2008;130:14358–14359. doi: 10.1021/ja803777x. [DOI] [PubMed] [Google Scholar]
- 155.Rieter WJ, Taylor KM, Lin W. J Am Chem Soc. 2007;129:9852–9853. doi: 10.1021/ja073506r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Horcajada P, Chalati T, Serre C, Gillet B, Sebrie C, Baati T, Eubank JF, Heurtaux D, Clayette P, Kreuz C, Chang JS, Hwang YK, Marsaud V, Bories PN, Cynober L, Gil S, Ferey G, Couvreur P, Gref R. Nat Mater. 2010;9:172–178. doi: 10.1038/nmat2608. [DOI] [PubMed] [Google Scholar]
- 157.Geninatti Crich S, Bussolati B, Tei L, Grange C, Esposito G, Lanzardo S, Camussi G, Aime S. Cancer Res. 2006;66:9196–9201. doi: 10.1158/0008-5472.CAN-06-1728. [DOI] [PubMed] [Google Scholar]
- 158.Aime S, Frullano L, Geninatti Crich S. Angew Chem Int Ed. 2002;41:1017–1019. doi: 10.1002/1521-3773(20020315)41:6<1017::aid-anie1017>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 159.Yin JJ, Lao F, Fu PP, Wamer WG, Zhao Y, Wang PC, Qiu Y, Sun B, Xing G, Dong J, Liang XJ, Chen C. Biomaterials. 2009;30:611–621. doi: 10.1016/j.biomaterials.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yin JJ, Lao F, Meng J, Fu PP, Zhao Y, Xing G, Gao X, Sun B, Wang PC, Chen C, Liang XJ. Mol Pharmacol. 2008;74:1132–1140. doi: 10.1124/mol.108.048348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shu CY, Corwin FD, Zhang JF, Chen ZJ, Reid JE, Sun MH, Xu W, Sim JH, Wang CR, Fatouros PP, Esker AR, Gibson HW, Dorn HC. Bioconjug Chem. 2009;20:1186–1193. doi: 10.1021/bc900051d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen K, Xie J, Xu HY, Behera D, Michalski MH, Biswal S, Wang A, Chen XY. Biomaterials. 2009;30:6912–6919. doi: 10.1016/j.biomaterials.2009.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen K, Li ZB, Wang H, Cai WB, Chen XY. Eur J Nucl Med Mol Imaging. 2008;35:2235–2244. doi: 10.1007/s00259-008-0860-8. [DOI] [PubMed] [Google Scholar]
- 164.Montet XM, Weissleder XR, Josephson L. Bioconjug Chem. 2006;17:905–911. doi: 10.1021/bc060035+. [DOI] [PubMed] [Google Scholar]
- 165.Niu G, Chen XY. Theranostics. 2011;1:30–47. doi: 10.7150/thno/v01p0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chen XY. Theranostics. 2011;1:28–29. doi: 10.7150/thno/v01p0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sun C, Veiseh O, Gunn J, Fang C, Hansen S, Lee D, Sze R, Ellenbogen RG, Olson J, Zhang M. Small. 2008;4:372–379. doi: 10.1002/smll.200700784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Veiseh O, Sun C, Fang C, Bhattarai N, Gunn J, Kievit F, Du K, Pullar B, Lee D, Ellenbogen RG, Olson J, Zhang M. Cancer Res. 2009;69:6200–6207. doi: 10.1158/0008-5472.CAN-09-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Veiseh O, Sun C, Gunn J, Kohler N, Gabikian P, Lee D, Bhattarai N, Ellenbogen R, Sze R, Hallahan A, Olson J, Zhang MQ. Nano Lett. 2005;5:1003–1008. doi: 10.1021/nl0502569. [DOI] [PubMed] [Google Scholar]
- 170.Leuschner C, Kumar CS, Hansel W, Soboyejo W, Zhou J, Hormes J. Breast Cancer Res Treat. 2006;99:163–176. doi: 10.1007/s10549-006-9199-7. [DOI] [PubMed] [Google Scholar]
- 171.Reddy GR, Bhojani MS, McConville P, Moody J, Moffat BA, Hall DE, Kim G, Koo YE, Woolliscroft MJ, Sugai JV, Johnson TD, Philbert MA, Kopelman R, Rehemtulla A, Ross BD. Clin Cancer Res. 2006;12:6677–6686. doi: 10.1158/1078-0432.CCR-06-0946. [DOI] [PubMed] [Google Scholar]
- 172.Lee S, Xie J, Chen X. Chem Rev. 2010;110:3087–3111. doi: 10.1021/cr900361p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lee JH, Huh YM, Jun YW, Seo JW, Jang JT, Song HT, Kim S, Cho EJ, Yoon HG, Suh JS, Cheon J. Nat Med. 2007;13:95–99. doi: 10.1038/nm1467. [DOI] [PubMed] [Google Scholar]
- 174.Hadjipanayis CG, Machaidze R, Kaluzova M, Wang L, Schuette AJ, Chen H, Wu X, Mao H. Cancer Res. 2010;70:6303–6312. doi: 10.1158/0008-5472.CAN-10-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hwang do W, Ko HY, Lee JH, Kang H, Ryu SH, Song IC, Lee DS, Kim S. J Nucl Med. 2010;51:98–105. doi: 10.2967/jnumed.109.069880. [DOI] [PubMed] [Google Scholar]
- 176.Shao H, Min C, Issadore D, Liong M, Yoon TJ, Weissleder R, Lee H. Theranostics. 2012;2:55–65. doi: 10.7150/thno.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lee H, Sun E, Ham D, Weissleder R. Nat Med. 2008;14:869–874. doi: 10.1038/nm.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Haun JB, Castro CM, Wang R, Peterson VM, Marinelli BS, Lee H, Weissleder R. Sci Transl Med. 2011;3:71ra16. doi: 10.1126/scitranslmed.3002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tassa C, Liong M, Hilderbrand S, Sandler JE, Reiner T, Keliher EJ, Weissleder R, Shaw SY. Lab Chip. 2012;12:3103–3110. doi: 10.1039/c2lc40337d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Devaraj NK, Hilderbrand S, Upadhyay R, Mazitschek R, Weissleder R. Angew Chem Int Ed. 2010;49:2869–2872. doi: 10.1002/anie.200906120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Nat Nanotechnol. 2010;5:660–665. doi: 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Osterfeld SJ, Yu H, Gaster RS, Caramuta S, Xu L, Han SJ, Hall DA, Wilson RJ, Sun S, White RL, Davis RW, Pourmand N, Wang SX. Proc Natl Acad Sci U S A. 2008;105:20637–20640. doi: 10.1073/pnas.0810822105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gaster RS, Hall DA, Nielsen CH, Osterfeld SJ, Yu H, Mach KE, Wilson RJ, Murmann B, Liao JC, Gambhir SS, Wang SX. Nat Med. 2009;15:1327–1332. doi: 10.1038/nm.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gaster RS, Xu L, Han SJ, Wilson RJ, Hall DA, Osterfeld SJ, Yu H, Wang SX. Nat Nanotechnol. 2011;6:314–320. doi: 10.1038/nnano.2011.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kovar JL, Simpson MA, Schutz-Geschwender A, Olive DM. Anal Biochem. 2007;367:1–12. doi: 10.1016/j.ab.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 186.Alivisatos AP. J Phys Chem. 1996;100:13226–13239. [Google Scholar]
- 187.Rossetti R, Nakahara S, Brus LE. J Chem Phys. 1983;79:1086–1088. [Google Scholar]
- 188.Baskoutas S, Terzis AF. J Appl Phys. 2006;99:013708. [Google Scholar]
- 189.Neeleshwar S, Chen CL, Tsai CB, Chen YY, Chen CC, Shyu SG, Seehra MS. Phys Rev B. 2005;71:201307. [Google Scholar]
- 190.Sharma M, Sharma AB, Mishra N, Pandey RK. Mater Res Bulletin. 2011;46:453–459. [Google Scholar]
- 191.Mathew S, Saran AD, Bhardwaj BS, Joseph SA, Radhakrishnan P, Nampoori VPN, Vallabhan CPG, Bellare JR. J Appl Phys. 2012;111:013708. [Google Scholar]
- 192.Norris DJ, Bawendi MG. Phys Rev B Condens Matter. 1996;53:16338–16346. doi: 10.1103/physrevb.53.16338. [DOI] [PubMed] [Google Scholar]
- 193.Jasieniak J, Smith L, van Embden J, Mulvaney P, Califano M. J Phys Chem. 2009;C113:19468–19474. [Google Scholar]
- 194.Liu FC, Chen YM, Lin JH, Tseng WL. J Colloid Interface Sci. 2009;337:414–419. doi: 10.1016/j.jcis.2009.05.044. [DOI] [PubMed] [Google Scholar]
- 195.Dabbousi BO, RodriguezViejo J, Mikulec FV, Heine JR, Mattoussi H, Ober R, Jensen KF, Bawendi MG. J Phys Chem B. 1997;101:9463–9475. [Google Scholar]
- 196.Peng XG, Schlamp MC, Kadavanich AV, Alivisatos AP. J Am Chem Soc. 1997;119:7019–7029. [Google Scholar]
- 197.Chan WC, Maxwell DJ, Gao X, Bailey RE, Han M, Nie S. Curr Opin Biotechnol. 2002;13:40–46. doi: 10.1016/s0958-1669(02)00282-3. [DOI] [PubMed] [Google Scholar]
- 198.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nat Mater. 2005;4:435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 199.Franceschetti A, Zunger A. Phys Rev Lett. 1997;78:915–918. [Google Scholar]
- 200.Nirmal M, Dabbousi BO, Bawendi MG, Macklin JJ, Trautman JK, Harris TD, Brus LE. Nature. 1996;383:802–804. [Google Scholar]
- 201.Pinaud F, Michalet X, Bentolila LA, Tsay JM, Doose S, Li JJ, Iyer G, Weiss S. Biomaterials. 2006;27:1679–1687. doi: 10.1016/j.biomaterials.2005.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zrazhevskiy P, Sena M, Gao X. Chem Soc Rev. 2010;39:4326–4354. doi: 10.1039/b915139g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Qu L, Peng X. J Am Chem Soc. 2002;124:2049–2055. doi: 10.1021/ja017002j. [DOI] [PubMed] [Google Scholar]
- 204.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Methods. 2008;5:763–775. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 205.Wu X, Liu H, Liu J, Haley KN, Treadway JA, Larson JP, Ge N, Peale F, Bruchez MP. Nat Biotechnol. 2003;21:41–46. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]
- 206.Gao XH, Yang LL, Petros JA, Marshal FF, Simons JW, Nie SM. Curr Opin Biotech. 2005;16:63–72. doi: 10.1016/j.copbio.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 207.Zeng R, Zhang T, Liu J, Hu S, Wan Q, Liu X, Peng Z, Zou B. Nanotechnology. 2009;20:095102. doi: 10.1088/0957-4484/20/9/095102. [DOI] [PubMed] [Google Scholar]
- 208.Lesnyak V, Dubavik A, Plotnikov A, Gaponik N, Eychmuller A. Chem Commun. 2010;46:886–888. doi: 10.1039/b919986a. [DOI] [PubMed] [Google Scholar]
- 209.Zheng YG, Yang ZC, Ying JY. Adv Mater. 2007;19:1475–1479. [Google Scholar]
- 210.Law WC, Yong KT, Roy I, Ding H, Hu R, Zhao WW, Prasad PN. Small. 2009;5:1302–1310. doi: 10.1002/smll.200801555. [DOI] [PubMed] [Google Scholar]
- 211.Murray CB, Norris DJ, Bawendi MG. J Am Chem Soc. 1993;115:8706–8715. [Google Scholar]
- 212.Peng ZA, Peng X. J Am Chem Soc. 2001;123:183–184. doi: 10.1021/ja003633m. [DOI] [PubMed] [Google Scholar]
- 213.Peng XG. Adv Mater. 2003;15:459–463. [Google Scholar]
- 214.Peng ZA, Peng XG. J Am Chem Soc. 2001;123:1389–1395. doi: 10.1021/ja003633m. [DOI] [PubMed] [Google Scholar]
- 215.Smith AM, Nie SM. Acc Chem Res. 2010;43:190–200. doi: 10.1021/ar9001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Alivisatos AP. Science. 1996;271:933–937. [Google Scholar]
- 217.Kim S, Fisher B, Eisler HJ, Bawendi M. J Am Chem Soc. 2003;125:11466–11467. doi: 10.1021/ja0361749. [DOI] [PubMed] [Google Scholar]
- 218.Pietryga JM, Schaller RD, Werder D, Stewart MH, Klimov VI, Hollingsworth JA. J Am Chem Soc. 2004;126:11752–11753. doi: 10.1021/ja047659f. [DOI] [PubMed] [Google Scholar]
- 219.Smith AM, Gao X, Nie S. Photochem Photobiol. 2004;80:377–385. doi: 10.1562/0031-8655(2004)080<0377:QDNFIV>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 220.Zlateva G, Zhelev Z, Bakalova R, Kanno I. Inorg Chem. 2007;46:6212–6214. doi: 10.1021/ic062045s. [DOI] [PubMed] [Google Scholar]
- 221.Zhong X, Feng Y, Knoll W, Han M. J Am Chem Soc. 2003;125:13559–13563. doi: 10.1021/ja036683a. [DOI] [PubMed] [Google Scholar]
- 222.Ma Q, Su X. Analyst. 2010;135:1867–1877. doi: 10.1039/c0an00233j. [DOI] [PubMed] [Google Scholar]
- 223.Hinds S, Myrskog S, Levina L, Koleilat G, Yang J, Kelley SO, Sargent EH. J Am Chem Soc. 2007;129:7218–7219. doi: 10.1021/ja070525s. [DOI] [PubMed] [Google Scholar]
- 224.Allen PM, Bawendi MG. J Am Chem Soc. 2008;130:9240–9241. doi: 10.1021/ja8036349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Wang H, Lou S, Tang Z, Xu W, Shang H, Shen H, Li LS. Dalton Trans. 2012;41:12726–12732. doi: 10.1039/c2dt31602a. [DOI] [PubMed] [Google Scholar]
- 226.Wang M, Mi CC, Zhang YX, Liu JL, Li F, Mao CB, Xu SK. J Phys Chem C. 2009;113:19021–19027. doi: 10.1021/jp906394z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Xie R, Peng X. J Am Chem Soc. 2009;131:10645–10651. doi: 10.1021/ja903558r. [DOI] [PubMed] [Google Scholar]
- 228.Zhong XH, Zhang ZH, Liu SH, Han MY, Knoll W. J Phys Chem B. 2004;108:15552–15559. [Google Scholar]
- 229.Protiere M, Reiss P. Small. 2007;3:399–403. doi: 10.1002/smll.200600581. [DOI] [PubMed] [Google Scholar]
- 230.Zhong XH, Feng YY, Knoll W, Han MY. J Am Chem Soc. 2003;125:13559–13563. doi: 10.1021/ja036683a. [DOI] [PubMed] [Google Scholar]
- 231.Smith AM, Mohs AM, Nie S. Nat Nanotechnol. 2009;4:56–63. doi: 10.1038/nnano.2008.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Chan WC, Nie S. Science. 1998;281:2016–2018. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 233.Yu X, Chen L, Deng Y, Li K, Wang Q, Li Y, Xiao S, Zhou L, Luo X, Liu J, Pang D. J Fluoresc. 2007;17:243–247. doi: 10.1007/s10895-007-0163-7. [DOI] [PubMed] [Google Scholar]
- 234.Law WC, Yong KT, Roy I, Ding H, Hu R, Zhao W, Prasad PN. Small. 2009;5:1302–1310. doi: 10.1002/smll.200801555. [DOI] [PubMed] [Google Scholar]
- 235.Aldana J, Wang YA, Peng X. J Am Chem Soc. 2001;123:8844–8850. doi: 10.1021/ja016424q. [DOI] [PubMed] [Google Scholar]
- 236.Algar WR, Krull UJ. Chemphyschem. 2007;8:561–568. doi: 10.1002/cphc.200600686. [DOI] [PubMed] [Google Scholar]
- 237.Liu W, Howarth M, Greytak AB, Zheng Y, Nocera DG, Ting AY, Bawendi MG. J Am Chem Soc. 2008;130:1274–1284. doi: 10.1021/ja076069p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sukhanova A, Devy J, Venteo L, Kaplan H, Artemyev M, Oleinikov V, Klinov D, Pluot M, Cohen JH, Nabiev I. Anal Biochem. 2004;324:60–67. doi: 10.1016/j.ab.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 239.Jiang W, Mardyani S, Fischer H, Chan WCW. Chem Mater. 2006;18:872–878. [Google Scholar]
- 240.Smith AM, Nie S. J Am Chem Soc. 2008;130:11278–11279. doi: 10.1021/ja804306c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bruchez M, Jr, Moronne M, Gin P, Weiss S, Alivisatos AP. Science. 1998;281:2013–2016. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- 242.Gerion D, Pinaud F, Williams SC, Parak WJ, Zanchet D, Weiss S, Alivisatos AP. J Phys Chem B. 2001;105:8861–8871. [Google Scholar]
- 243.Selvan ST, Tan TT, Ying JY. Adv Mater. 2005;17:1620–1625. [Google Scholar]
- 244.Gao X, Cui Y, Levenson RM, Chung LW, Nie S. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 245.Hardman R. Environ Health Perspect. 2006;114:165–172. doi: 10.1289/ehp.8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Derfus AM, Chan WCW, Bhatia SN. Nano Lett. 2004;4:11–18. doi: 10.1021/nl0347334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lovric J, Bazzi HS, Cuie Y, Fortin GR, Winnik FM, Maysinger D. J Mol Med. 2005;83:377–385. doi: 10.1007/s00109-004-0629-x. [DOI] [PubMed] [Google Scholar]
- 248.Lovric J, Bazzi HS, Cuie Y, Fortin GRA, Winnik FM, Maysinger D. J Mol Med. 2005;83:377–385. doi: 10.1007/s00109-004-0629-x. [DOI] [PubMed] [Google Scholar]
- 249.Hoshino A, Hanaki K, Suzuki K, Yamamoto K. Biochem Biophys Res Commun. 2004;314:46–53. doi: 10.1016/j.bbrc.2003.11.185. [DOI] [PubMed] [Google Scholar]
- 250.Voura EB, Jaiswal JK, Mattoussi H, Simon SM. Nat Med. 2004;10:993–998. doi: 10.1038/nm1096. [DOI] [PubMed] [Google Scholar]
- 251.Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Nat Biotechnol. 2003;21:47–51. doi: 10.1038/nbt767. [DOI] [PubMed] [Google Scholar]
- 252.Zimmer JP, Kim SW, Ohnishi S, Tanaka E, Frangioni JV, Bawendi MG. J Am Chem Soc. 2006;128:2526–2527. doi: 10.1021/ja0579816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Xie R, Chen K, Chen X, Peng X. Nano Res. 2008;1:457–464. doi: 10.1007/s12274-008-8048-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Li L, Daou TJ, Texier I, Tran TKC, Nguyen QL, Reiss P. Chem Mater. 2009;21:2422–2429. [Google Scholar]
- 255.Aswathy RG, Yoshida Y, Maekawa T, Kumar DS. Anal Bioanal Chem. 2010;397:1417–1435. doi: 10.1007/s00216-010-3643-6. [DOI] [PubMed] [Google Scholar]
- 256.Gao J, Chen K, Xie R, Xie J, Lee S, Cheng Z, Peng X, Chen X. Small. 2010;6:256–261. doi: 10.1002/smll.200901672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Science. 2003;300:1434–1436. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 258.Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Bioconjug Chem. 2004;15:79–86. doi: 10.1021/bc034153y. [DOI] [PubMed] [Google Scholar]
- 259.Ye L, Yong KT, Liu L, Roy I, Hu R, Zhu J, Cai H, Law WC, Liu J, Wang K, Liu Y, Hu Y, Zhang X, Swihart MT, Prasad PN. Nat Nanotechnol. 2012;7:453–458. doi: 10.1038/nnano.2012.74. [DOI] [PubMed] [Google Scholar]
- 260.Chen K, Xie J, Xu H, Behera D, Michalski MH, Biswal S, Wang A, Chen X. Biomaterials. 2009;30:6912–6919. doi: 10.1016/j.biomaterials.2009.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Prencipe G, Tabakman SM, Welsher K, Liu Z, Goodwin AP, Zhang L, Henry J, Dai H. J Am Chem Soc. 2009;131:4783–4787. doi: 10.1021/ja809086q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Cai W, Shin DW, Chen K, Gheysens O, Cao Q, Wang SX, Gambhir SS, Chen X. Nano Lett. 2006;6:669–676. doi: 10.1021/nl052405t. [DOI] [PubMed] [Google Scholar]
- 263.Gao J, Chen K, Luong R, Bouley DM, Mao H, Qiao T, Gambhir SS, Cheng Z. Nano Lett. 2012;12:281–286. doi: 10.1021/nl203526f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Gao J, Chen K, Miao Z, Ren G, Chen X, Gambhir SS, Cheng Z. Biomaterials. 2011;32:2141–2148. doi: 10.1016/j.biomaterials.2010.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Smith BR, Cheng Z, De A, Koh AL, Sinclair R, Gambhir SS. Nano Lett. 2008;8:2599–2606. doi: 10.1021/nl080141f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Smith BR, Cheng Z, De A, Rosenberg J, Gambhir SS. Small. 2010;6:2222–2229. doi: 10.1002/smll.201001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Cai W, Chen K, Li ZB, Gambhir SS, Chen X. J Nucl Med. 2007;48:1862–1870. doi: 10.2967/jnumed.107.043216. [DOI] [PubMed] [Google Scholar]
- 268.Probst J, Dembski S, Milde M, Rupp S. Expert Rev Mol Diagn. 2012;12:49–64. doi: 10.1586/erm.11.86. [DOI] [PubMed] [Google Scholar]
- 269.Ghazani AA, Lee JA, Klostranec J, Xiang Q, Dacosta RS, Wilson BC, Tsao MS, Chan WC. Nano Lett. 2006;6:2881–2886. doi: 10.1021/nl062111n. [DOI] [PubMed] [Google Scholar]
- 270.Yezhelyev MV, Al-Hajj A, Morris C, Marcus AI, Liu T, Lewis M, Cohen C, Zrazhevskiy P, Simons JW, Rogatko A, Nie S, Gao X, O’Regan RM. Adv Mater. 2007;19:3146–3151. [Google Scholar]
- 271.Liu J, Lau SK, Varma VA, Kairdolf BA, Nie S. Anal Chem. 2010;82:6237–6243. doi: 10.1021/ac101065b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Xu J, Muller S, Nannapaneni S, Pan L, Wang Y, Peng X, Wang D, Tighiouart M, Chen Z, Saba NF, Beitler JJ, Shin DM, Chen ZG. Eur J Cancer. 2012;48:1682–1691. doi: 10.1016/j.ejca.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Boyer JC, van Veggel FC. Nanoscale. 2010;2:1417–1419. doi: 10.1039/c0nr00253d. [DOI] [PubMed] [Google Scholar]
- 274.Wang F, Liu X. Chem Soc Rev. 2009;38:976–989. doi: 10.1039/b809132n. [DOI] [PubMed] [Google Scholar]
- 275.Zhou J, Liu Z, Li F. Chem Soc Rev. 2012;41:1323–1349. doi: 10.1039/c1cs15187h. [DOI] [PubMed] [Google Scholar]
- 276.Wang M, Abbineni G, Clevenger A, Mao C, Xu S. Nanomedicine. 2011;7:710–729. doi: 10.1016/j.nano.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Ang LY, Lim ME, Ong LC, Zhang Y. Nanomedicine. 2011;6:1273–1288. doi: 10.2217/nnm.11.108. [DOI] [PubMed] [Google Scholar]
- 278.Ehlert O, Thomann R, Darbandi M, Nann T. ACS Nano. 2008;2:120–124. doi: 10.1021/nn7002458. [DOI] [PubMed] [Google Scholar]
- 279.Ye XC, Collins JE, Kang YJ, Chen J, Chen DTN, Yodh AG, Murray CB. Proc Nat Acad Sci USA. 2010;107:22430–22435. doi: 10.1073/pnas.1008958107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wang F, Liu X. J Am Chem Soc. 2008;130:5642–5643. doi: 10.1021/ja800868a. [DOI] [PubMed] [Google Scholar]
- 281.Wang F, Wang JA, Liu XG. Angew Chem Int Ed. 2010;49:7456–7460. doi: 10.1002/anie.201003959. [DOI] [PubMed] [Google Scholar]
- 282.Bai X, Song HW, Pan GH, Lei YQ, Wang T, Ren XG, Lu SZ, Dong B, Dai QL, Fan L. J Phys Chem C. 2007;111:13611–13617. [Google Scholar]
- 283.Xiao SG, Yang XL. Mod Phys Lett B. 2011;25:265–272. [Google Scholar]
- 284.Liu GK, Zhuang HZ, Chen XY. Nano Lett. 2002;2:535–539. [Google Scholar]
- 285.Schietinger S, Menezes Lde S, Lauritzen B, Benson O. Nano Lett. 2009;9:2477–2481. doi: 10.1021/nl901253t. [DOI] [PubMed] [Google Scholar]
- 286.Wang M, Abbineni G, Clevenger A, Mao CB, Xu SK. Nanomedicine-Nanotechn Biol Med. 2011;7:710–729. doi: 10.1016/j.nano.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Yi GS, Lu HC, Zhao SY, Yue G, Yang WJ, Chen DP, Guo LH. Nano Lett. 2004;4:2191–2196. [Google Scholar]
- 288.Wang X, Zhuang J, Peng Q, Li Y. Nature. 2005;437:121–124. doi: 10.1038/nature03968. [DOI] [PubMed] [Google Scholar]
- 289.Wang X, Zhuang J, Peng Q, Li Y. Inorg Chem. 2006;45:6661–6665. doi: 10.1021/ic051683s. [DOI] [PubMed] [Google Scholar]
- 290.Wang L, Li Y. Nano Lett. 2006;6:1645–1649. doi: 10.1021/nl060684u. [DOI] [PubMed] [Google Scholar]
- 291.Wang LY, Li YD. Chem Mater. 2007;19:727–734. [Google Scholar]
- 292.Zhang F, Wan Y, Yu T, Zhang F, Shi Y, Xie S, Li Y, Xu L, Tu B, Zhao D. Angew Chem Int Ed. 2007;46:7976–7979. doi: 10.1002/anie.200702519. [DOI] [PubMed] [Google Scholar]
- 293.Zhang F, Li J, Shan J, Xu L, Zhao D. Chemistry. 2009;15:11010–11019. doi: 10.1002/chem.200900861. [DOI] [PubMed] [Google Scholar]
- 294.Zhang F, Zhao D. ACS Nano. 2009;3:159–164. doi: 10.1021/nn800533v. [DOI] [PubMed] [Google Scholar]
- 295.Xu ZH, Li CX, Yang PP, Zhang CM, Huang SS, Lin J. Crys Grow Des. 2009;9:4752–4758. [Google Scholar]
- 296.Yang LW, Han HL, Zhang YY, Zhong JX. J Phys Chem C. 2009;113:18995–18999. [Google Scholar]
- 297.Li JJ, Yang LW, Zhang YY, Zhong JX, Sun CQ, Chu PK. Opt Mater. 2011;33:882–887. [Google Scholar]
- 298.Chen D, Huang P, Yu Y, Huang F, Yang A, Wang Y. Chem Commun. 2011;47:5801–5803. doi: 10.1039/c0cc05722c. [DOI] [PubMed] [Google Scholar]
- 299.Wang YH, Cai RX, Liu ZH. Crystengcomm. 2011;13:1772–1774. [Google Scholar]
- 300.Wang LY, Li P, Li YD. Adv Mater. 2007;19:3304–3307. [Google Scholar]
- 301.Hu H, Chen Z, Cao T, Zhang Q, Yu M, Li F, Yi T, Huang C. Nanotechnology. 2008;19:375702. doi: 10.1088/0957-4484/19/37/375702. [DOI] [PubMed] [Google Scholar]
- 302.Zhang CM, Ma PA, Li CX, Li GG, Huang SS, Yang DM, Shang MM, Kang XJ, Lin J. J Mater Chem. 2011;21:717–723. [Google Scholar]
- 303.Sun JY, Xian JB, Du HY. J Phys Chem Solids. 2011;72:207–213. [Google Scholar]
- 304.Sun JY, Xian JB, Xia ZG, Du HY. J Rare Earths. 2010;28:219–221. [Google Scholar]
- 305.Wang GF, Peng Q, Li YD. Chem Commun. 2010;46:7528–7529. doi: 10.1039/c0cc02109a. [DOI] [PubMed] [Google Scholar]
- 306.Chuai XH, Zhang DS, Zhao D, Zheng KZ, He CF, Shi F, Wang LL, Chen H, Qin WP. Mater Lett. 2011;65:2368–2370. [Google Scholar]
- 307.Li D, Ding CR, Song G, Lu SZ, Zhang Z, Shi YU, Shen H, Zhang YL, Ouyang HQ, Wang H. J Phys Chem C. 2010;114:21378–21384. [Google Scholar]
- 308.Zeng JH, Su J, Li ZH, Yan RX, Li YD. Adv Mater. 2005;17:2119–2123. [Google Scholar]
- 309.Fan XP, Pi DB, Wang F, Qiu JR, Wang MQ. I EEE Trans Nanotech. 2006;5:123–128. [Google Scholar]
- 310.Wang GF, Peng Q, Li YD. J Am Chem Soc. 2009;131:14200–14201. doi: 10.1021/ja906732y. [DOI] [PubMed] [Google Scholar]
- 311.Pedroni M, Piccinelli F, Passuello T, Giarola M, Mariotto G, Polizzi S, Bettinelli M, Speghini A. Nanoscale. 2011;3:1456–1460. doi: 10.1039/c0nr00860e. [DOI] [PubMed] [Google Scholar]
- 312.Sun JY, Xian JB, Du HY. Appl Surf Sci. 2011;257:3592–3595. [Google Scholar]
- 313.Sun JY, Xian JB, Zhang XY, Du HY. J Rare Earths. 2011;29:32–38. [Google Scholar]
- 314.Pang T, Cao WH, Xing MM, Luo XX, Yang XF. Opt Mater. 2011;33:485–489. [Google Scholar]
- 315.Zeng JH, Xie T, Li ZH, Li YD. Cryst Grow Des. 2007;7:2774–2777. [Google Scholar]
- 316.Wang F, Han Y, Lim CS, Lu Y, Wang J, Xu J, Chen H, Zhang C, Hong M, Liu X. Nature. 2010;463:1061–1065. doi: 10.1038/nature08777. [DOI] [PubMed] [Google Scholar]
- 317.Li Z, Zhang Y. Nanotechnology. 2008;19:345606. doi: 10.1088/0957-4484/19/34/345606. [DOI] [PubMed] [Google Scholar]
- 318.Schafer H, Ptacek P, Eickmeier H, Haase M. Adv Funct Mater. 2009;19:3091–3097. [Google Scholar]
- 319.Wei Y, Lu FQ, Zhang XR, Chen DP. Chem Mater. 2006;18:5733–5737. [Google Scholar]
- 320.Wang M, Liu JL, Zhang YX, Hou W, Wu XL, Xu SK. Mater Lett. 2009;63:325–327. [Google Scholar]
- 321.Liu CH, Sun J, Wang H, Chen D. Scripta Mater. 2008;58:89–92. [Google Scholar]
- 322.Shi F, Wang JS, Zhai XS, Zhao D, Qin WP. Crystengcomm. 2011;13:3782–3787. [Google Scholar]
- 323.Chen DQ, Yu YL, Huang F, Yang AP, Wang YS. J Mater Chem. 2011;21:6186–6192. [Google Scholar]
- 324.Zhang YW, Sun X, Si R, You LP, Yan CH. J Am Chem Soc. 2005;127:3260–3261. doi: 10.1021/ja042801y. [DOI] [PubMed] [Google Scholar]
- 325.Boyer JC, Vetrone F, Cuccia LA, Capobianco JA. J Am Chem Soc. 2006;128:7444–7445. doi: 10.1021/ja061848b. [DOI] [PubMed] [Google Scholar]
- 326.Boyer JC, Cuccia LA, Capobianco JA. Nano Lett. 2007;7:847–852. doi: 10.1021/nl070235+. [DOI] [PubMed] [Google Scholar]
- 327.Mai HX, Zhang YW, Sun LD, Yan CR. J Phys Chem C. 2007;111:13730–13739. [Google Scholar]
- 328.Du YP, Zhang YW, Yan ZG, Sun LD, Gao S, Yan CH. Chem Asian J. 2007;2:965–974. doi: 10.1002/asia.200700054. [DOI] [PubMed] [Google Scholar]
- 329.Shan J, Qin X, Yao N, Ju Y. Nanotechnology. 2007;18:445607. [Google Scholar]
- 330.Shan JN, Ju YG. Nanotechnology. 2009;20:155101. [Google Scholar]
- 331.Budijono SJ, Shan JN, Yao N, Miura Y, Hoye T, Austin RH, Ju YG, Prud’homme RK. Chem Mater. 2010;22:311–318. [Google Scholar]
- 332.Li LL, Zhang RB, Yin LL, Zheng KZ, Qin WP, Selvin PR, Lu Y. Angew, Chem Int Ed. 2012;51:6121–6125. doi: 10.1002/anie.201109156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Yi GS, Chow GM. Adv Funct Mater. 2006;16:2324–2329. [Google Scholar]
- 334.Yi GS, Peng YF, Gao ZQ. Chem Mater. 2011;23:2729–2734. [Google Scholar]
- 335.Li D, Dong BA, Bai X, Wang Y, Song HW. J Phys Chem C. 2010;114:8219–8226. [Google Scholar]
- 336.Dong BA, Xu S, Sun JA, Bi S, Li D, Bai X, Wang Y, Wang LP, Song HW. J Mater Chem. 2011;21:6193–6200. [Google Scholar]
- 337.Schafer H, Ptacek P, Kompe K, Haase M. Chem Mater. 2007;19:1396–1400. [Google Scholar]
- 338.Cao TY, Yang TS, Gao Y, Yang Y, Hu H, Li FY. Inorg Chem Commun. 2010;13:392–394. [Google Scholar]
- 339.Zhou J, Yu M, Sun Y, Zhang X, Zhu X, Wu Z, Wu D, Li F. Biomaterials. 2011;32:1148–1156. doi: 10.1016/j.biomaterials.2010.09.071. [DOI] [PubMed] [Google Scholar]
- 340.Chen G, Ohulchanskyy TY, Law WC, Agren H, Prasad PN. Nanoscale. 2011;3:2003–2008. doi: 10.1039/c0nr01018a. [DOI] [PubMed] [Google Scholar]
- 341.Liu C, Wang Z, Jia H, Li Z. Chem Commun. 2011;47:4661–4663. doi: 10.1039/c1cc10597c. [DOI] [PubMed] [Google Scholar]
- 342.Zhang CL, Yuan YX, Zhang SM, Wang YH, Liu ZH. Angew Chem Int Ed. 2011;50:6851–6854. doi: 10.1002/anie.201100769. [DOI] [PubMed] [Google Scholar]
- 343.Naccache R, Vetrone F, Mahalingam V, Cuccia LA, Capobianco JA. Chem Mater. 2009;21:717–723. [Google Scholar]
- 344.Xiong L, Yang T, Yang Y, Xu C, Li F. Biomaterials. 2010;31:7078–7085. doi: 10.1016/j.biomaterials.2010.05.065. [DOI] [PubMed] [Google Scholar]
- 345.Chen QT, Wang X, Chen FH, Zhang QB, Dong B, Yang H, Liu GX, Zhu YM. J Mater Chem. 2011;21:7661–7667. [Google Scholar]
- 346.Kumar R, Nyk M, Ohulchanskyy TY, Flask CA, Prasad PN. Adv Funct Mater. 2009;19:853–859. [Google Scholar]
- 347.Nyk M, Kumar R, Ohulchanskyy TY, Bergey EJ, Prasad PN. Nano Lett. 2008;8:3834–3838. doi: 10.1021/nl802223f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Boyer JC, Manseau MP, Murray JI, van Veggel FC. Langmuir. 2010;26:1157–1164. doi: 10.1021/la902260j. [DOI] [PubMed] [Google Scholar]
- 349.Zhang QB, Song K, Zhao JW, Kong XG, Sun YJ, Liu XM, Zhang YL, Zeng QH, Zhang H. J ColloidInterface Sci. 2009;336:171–175. doi: 10.1016/j.jcis.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 350.Shen J, Sun LD, Zhang YW, Yan CH. Chem Commun. 2010;46:5731–5733. doi: 10.1039/c0cc00814a. [DOI] [PubMed] [Google Scholar]
- 351.Dong A, Ye X, Chen J, Kang Y, Gordon T, Kikkawa JM, Murray CB. J Am Chem Soc. 2011;133:998–1006. doi: 10.1021/ja108948z. [DOI] [PubMed] [Google Scholar]
- 352.Abdul Jalil R, Zhang Y. Biomaterials. 2008;29:4122–4128. doi: 10.1016/j.biomaterials.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 353.Shan JN, Ju YG. Appl Phys Lett. 2007;91:123103. [Google Scholar]
- 354.Jiang S, Zhang Y, Lim KM, Sim EK, Ye L. Nanotechnology. 2009;20:155101. doi: 10.1088/0957-4484/20/15/155101. [DOI] [PubMed] [Google Scholar]
- 355.Chen Z, Chen H, Hu H, Yu M, Li F, Zhang Q, Zhou Z, Yi T, Huang C. J Am Chem Soc. 2008;130:3023–3029. doi: 10.1021/ja076151k. [DOI] [PubMed] [Google Scholar]
- 356.Zhou J, Sun Y, Du XX, Xiong LQ, Hu H, Li FY. Biomaterials. 2010;31:3287–3295. doi: 10.1016/j.biomaterials.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 357.Hu H, Yu MX, Li FY, Chen ZG, Gao X, Xiong LQ, Huang CH. Chem Mater. 2008;20:7003–7009. [Google Scholar]
- 358.Jiang S, Zhang Y, Lim KM, Sim EKW, Ye L. Nanotechnology. 2009;20:155101–155109. doi: 10.1088/0957-4484/20/15/155101. [DOI] [PubMed] [Google Scholar]
- 359.Jalil RA, Zhang Y. Biomaterials. 2008;29:4122–4128. doi: 10.1016/j.biomaterials.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 360.Lim SF, Riehn R, Tung CK, Ryu WS, Zhuo R, Dalland J, Austin RH. Nanotechnology. 2009;20:405701. doi: 10.1088/0957-4484/20/40/405701. [DOI] [PubMed] [Google Scholar]
- 361.Hilderbrand SA, Shao F, Salthouse C, Mahmood U, Weissleder R. Chem Commun. 2009:4188–4190. doi: 10.1039/b905927j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Chatteriee DK, Rufalhah AJ, Zhang Y. Biomaterials. 2008;29:937–943. doi: 10.1016/j.biomaterials.2007.10.051. [DOI] [PubMed] [Google Scholar]
- 363.Cheng LA, Yang K, Zhang SA, Shao MW, Lee ST, Liu ZA. Nano Res. 2010;3:722–732. [Google Scholar]
- 364.Xiong LQ, Chen ZG, Yu MX, Li FY, Liu C, Huang CH. Biomaterials. 2009;30:5592–5600. doi: 10.1016/j.biomaterials.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 365.Xiong L, Chen Z, Tian Q, Cao T, Xu C, Li F. Anal Chem. 2009;81:8687–8694. doi: 10.1021/ac901960d. [DOI] [PubMed] [Google Scholar]
- 366.Yu XF, Sun Z, Li M, Xiang Y, Wang QQ, Tang F, Wu Y, Cao Z, Li W. Biomaterials. 2010;31:8724–8731. doi: 10.1016/j.biomaterials.2010.07.099. [DOI] [PubMed] [Google Scholar]
- 367.Xia A, Gao Y, Zhou J, Li C, Yang T, Wu D, Wu L, Li F. Biomaterials. 2011;32:7200–7208. doi: 10.1016/j.biomaterials.2011.05.094. [DOI] [PubMed] [Google Scholar]
- 368.Xiao Q, Bu W, Ren Q, Zhang S, Xing H, Chen F, Li M, Zheng X, Hua Y, Zhou L, Peng W, Qu H, Wang Z, Zhao K, Shi J. Biomaterials. 2012;33:7530–7539. doi: 10.1016/j.biomaterials.2012.06.028. [DOI] [PubMed] [Google Scholar]
- 369.Link S, El-Sayed MA. J Phys Chem B. 1999;103:8410–8426. [Google Scholar]
- 370.Daniel MC, Astruc D. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 371.Yguerabide J, Yguerabide EE. Anal Biochem. 1998;262:157–176. doi: 10.1006/abio.1998.2760. [DOI] [PubMed] [Google Scholar]
- 372.Horvath H. J Quant Spectro Radia Transfer. 2009;110:787–799. [Google Scholar]
- 373.Boisselier E, Astruc D. Chem Soc Rev. 2009;38:1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 374.Wu HY, Chu HC, Kuo TJ, Kuo CL, Huang MH. Chem Mater. 2005;17:6447–6451. [Google Scholar]
- 375.Lal S, Clare SE, Halas NJ. Acc Chem Res. 2008;41:1842–1851. doi: 10.1021/ar800150g. [DOI] [PubMed] [Google Scholar]
- 376.Xia YN, Li WY, Cobley CM, Chen JY, Xia XH, Zhang Q, Yang MX, Cho EC, Brown PK. Acc Chem Res. 2011;44:914–924. doi: 10.1021/ar200061q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Oldenburg SJ, Averitt RD, Westcott SL, Halas NJ. Chem Phys Lett. 1998;288:243–247. [Google Scholar]
- 378.Glazer ES, Zhu C, Massey KL, Thompson CS, Kaluarachchi WD, Hamir AN, Curley SA. Clin Cancer Res. 2010;16:5712–5721. doi: 10.1158/1078-0432.CCR-10-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Raoof M, Corr SJ, Kaluarachchi WD, Massey KL, Briggs K, Zhu C, Cheney MA, Wilson LJ, Curley SA. Nanomedicine. 2012;8:1096. doi: 10.1016/j.nano.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Turkevich J, Stevenson PC, Hillier J. Discuss Faraday Soc. 1951:55–75. [Google Scholar]
- 381.Frens G. Nature-Phys Sci. 1973;241:20–22. [Google Scholar]
- 382.Kimling J, Maier M, Okenve B, Kotaidis V, Ballot H, Plech A. J Phys Chem B. 2006;110:15700–15707. doi: 10.1021/jp061667w. [DOI] [PubMed] [Google Scholar]
- 383.Murphy CJ, San TK, Gole AM, Orendorff CJ, Gao JX, Gou L, Hunyadi SE, Li T. J Phys Chem. 2005;B109:13857–13870. doi: 10.1021/jp0516846. [DOI] [PubMed] [Google Scholar]
- 384.Hussain ST, Iqbal M, Mazhar M. J Nano Res. 2009;11:1383–1391. [Google Scholar]
- 385.Esumi K, Kameo A, Suzuki A, Torigoe K. Colloids Surfaces a-Physicochem Eng Aspects. 2001;189:155–161. [Google Scholar]
- 386.Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. J Chem Soc -Chem Commun. 1994:801–802. [Google Scholar]
- 387.Yin XJ, Chen SG, Wu AG. Micro & Nano Lett. 2010;5:270–273. [Google Scholar]
- 388.Dubey SP, Lahtinen M, Sillanpaa M. Colloids Surfaces a-Physicochem Eng Aspects. 2010;364:34–41. [Google Scholar]
- 389.Roy K, Lahiri S. Green Chem. 2006;8:1063–1066. [Google Scholar]
- 390.Xu C, Xie J, Ho D, Wang C, Kohler N, Walsh EG, Morgan JR, Chin YE, Sun S. Angew Chem Int Ed. 2008;47:173–176. doi: 10.1002/anie.200704392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Yu H, Chen M, Rice PM, Wang SX, White RL, Sun S. Nano Lett. 2005;5:379–382. doi: 10.1021/nl047955q. [DOI] [PubMed] [Google Scholar]
- 392.Yu YY, Chang SS, Lee CL, Wang CRC. J Phys Chem B. 1997;101:6661–6664. [Google Scholar]
- 393.Jana NR, Gearheart L, Murphy CJ. J Phys Chem B. 2001;105:4065–4067. [Google Scholar]
- 394.Gole A, Murphy CJ. Chem Mater. 2004;16:3633–3640. [Google Scholar]
- 395.Nikoobakht B, El-Sayed MA. Chem Mater. 2003;15:1957–1962. [Google Scholar]
- 396.Jana NR, Gearheart L, Murphy CJ. Adv Mater. 2001;13:1389–1393. [Google Scholar]
- 397.Xia YN, Xiong YJ, Lim B, Skrabalak SE. Angew Chem Int Ed. 2009;48:60–103. doi: 10.1002/anie.200802248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Hirsch LR, Gobin AM, Lowery AR, Tam F, Drezek RA, Halas NJ, West JL. Ann Biomed Engineer. 2006;34:15–22. doi: 10.1007/s10439-005-9001-8. [DOI] [PubMed] [Google Scholar]
- 399.Stober W, Fink A, Bohn E. J Colloid Interface Sci. 1968;26:62–69. [Google Scholar]
- 400.Halas NJ. MRS Bulletin. 2005;30:362–367. [Google Scholar]
- 401.Radloff C, Halas NJ. Nano Lett. 2004;4:1323–1327. [Google Scholar]
- 402.Bardhan R, Mukherjee S, Mirin NA, Levit SD, Nordlander P, Halas NJ. J Phys Chem C. 2010;114:7378–7383. [Google Scholar]
- 403.Zhang JZ. J Phys Chem Lett. 2010;1:686–695. [Google Scholar]
- 404.Jin YD, Gao XH. Nat Nanotech. 2009;4:571–576. doi: 10.1038/nnano.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Jin YD, Gao XH. J Am Chem Soc. 2009;131:17774–17776. doi: 10.1021/ja9076765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Sun YG, Mayers BT, Xia YN. Nano Lett. 2002;2:481–485. [Google Scholar]
- 407.Sun YG, Xia YN. Science. 2002;298:2176–2179. doi: 10.1126/science.1077229. [DOI] [PubMed] [Google Scholar]
- 408.Sun YG, Xia YN. J Am Chem Soc. 2004;126:3892–3901. doi: 10.1021/ja039734c. [DOI] [PubMed] [Google Scholar]
- 409.Skrabalak SE, Chen JY, Sun YG, Lu XM, Au L, Cobley CM, Xia YN. Acc Chem Res. 2008;41:1587–1595. doi: 10.1021/ar800018v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Skrabalak SE, Au L, Li XD, Xia YN. Nat Protocols. 2007;2:2182–2190. doi: 10.1038/nprot.2007.326. [DOI] [PubMed] [Google Scholar]
- 411.Chen JY, Yang MX, Zhang QA, Cho EC, Cobley CM, Kim C, Glaus C, Wang LHV, Welch MJ, Xia YN. Adv Funct Mater. 2010;20:3684–3694. doi: 10.1002/adfm.201001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Cobley CM, Au L, Chen J, Xia Y. Expert Opin Drug Deliv. 2010;7:577–587. doi: 10.1517/17425240903571614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Yavuz MS, Cheng Y, Chen J, Cobley CM, Zhang Q, Rycenga M, Xie J, Kim C, Song KH, Schwartz AG, Wang LV, Xia Y. Nat Mater. 2009;8:935–939. doi: 10.1038/nmat2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Li W, Cai X, Kim C, Sun G, Zhang Y, Deng R, Yang M, Chen J, Achilefu S, Wang LV, Xia Y. Nanoscale. 2011;3:1724–1730. doi: 10.1039/c0nr00932f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Moon GD, Choi SW, Cai X, Li W, Cho EC, Jeong U, Wang LV, Xia Y. J Am Chem Soc. 2011;133:4762–4765. doi: 10.1021/ja200894u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chem Rev. 2005;105:1103–1169. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 417.Lu W, Xiong C, Zhang G, Huang Q, Zhang R, Zhang JZ, Li C. Clin Cancer Res. 2009;15:876–886. doi: 10.1158/1078-0432.CCR-08-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Melancon MP, Lu W, Yang Z, Zhang R, Cheng Z, Elliot AM, Stafford J, Olson T, Zhang JZ, Li C. Mol Cancer Therapeutics. 2008;7:1730–1739. doi: 10.1158/1535-7163.MCT-08-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Lu W, Zhang G, Zhang R, Flores LG, 2nd, Huang Q, Gelovani JG, Li C. Cancer Res. 2010;70:3177–3188. doi: 10.1158/0008-5472.CAN-09-3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.von Maltzahn G, Park JH, Agrawal A, Bandaru NK, Das SK, Sailor MJ, Bhatia SN. Cancer Res. 2009;69:3892–3900. doi: 10.1158/0008-5472.CAN-08-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Zhang G, Yang Z, Lu W, Zhang R, Huang Q, Tian M, Li L, Liang D, Li C. Biomaterials. 2009;30:1928–1936. doi: 10.1016/j.biomaterials.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Cao J, Galbraith EK, Sun T, Grattan KTV. SensorsActuat B-Chem. 2012;169:360–367. [Google Scholar]
- 423.Sahu D, Chu HC, Yang PJ, Lin HC. Macromol Chem Phys. 2012;213:1550–1558. [Google Scholar]
- 424.Jung Y, Reif R, Zeng Y, Wang RK. Nano Lett. 2011;11:2938–2943. doi: 10.1021/nl2014394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Dai Q, Coutts J, Zou JH, Huo Q. Chem Commun. 2008:2858–2860. doi: 10.1039/b804797a. [DOI] [PubMed] [Google Scholar]
- 426.Hu XG, Gao XH. Phys Chem Chem Phys. 2011;13:10028–10035. doi: 10.1039/c0cp02434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Pan Y, Neuss S, Leifert A, Fischler M, Wen F, Simon U, Schmid G, Brandau W, Jahnen-Dechent W. Small. 2007;3:1941–1949. doi: 10.1002/smll.200700378. [DOI] [PubMed] [Google Scholar]
- 428.Ghandehari H, Malugin A, Herd H, Gormley A, Yu T. Abs Pap Am Chem Soc. 2011;241:21. [Google Scholar]
- 429.Alkilany AM, Nagaria PK, Hexel CR, Shaw TJ, Murphy CJ, Wyatt MD. Small. 2009;5:701–708. doi: 10.1002/smll.200801546. [DOI] [PubMed] [Google Scholar]
- 430.Alkilany AM, Murphy CJ. J Nano Res. 2010;12:2313–2333. doi: 10.1007/s11051-010-9911-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Hauck TS, Ghazani AA, Chan WC. Small. 2008;4:153–159. doi: 10.1002/smll.200700217. [DOI] [PubMed] [Google Scholar]
- 432.Leonov AP, Zheng J, Clogston JD, Stern ST, Patri AK, Wei A. ACS Nano. 2008;2:2481–2488. doi: 10.1021/nn800466c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Alkilany AM, Nagaria PK, Wyatt MD, Murphy CJ. Langmuir. 2010;26:9328–9333. doi: 10.1021/la100253k. [DOI] [PubMed] [Google Scholar]
- 434.Alkilany AM, Murphy CJ. Langmuir. 2009;25:13874–13879. doi: 10.1021/la901270x. [DOI] [PubMed] [Google Scholar]
- 435.Takahashi H, Niidome Y, Niidome T, Kaneko K, Kawasaki H, Yamada S. Langmuir. 2006;22:2–5. doi: 10.1021/la0520029. [DOI] [PubMed] [Google Scholar]
- 436.Qian X, Peng XH, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, Yang L, Young AN, Wang MD, Nie S. Nat Biotechnol. 2008;26:83–90. doi: 10.1038/nbt1377. [DOI] [PubMed] [Google Scholar]
- 437.Libutti SK, Paciotti GF, Byrnes AA, Alexander HR, Jr, Gannon WE, Walker M, Seidel GD, Yuldasheva N, Tamarkin L. Clin Cancer Res. 2010;16:6139–6149. doi: 10.1158/1078-0432.CCR-10-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Fujimoto JG, Pitris C, Boppart SA, Brezinski ME. Neoplasia. 2000;2:9–25. doi: 10.1038/sj.neo.7900071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Tearney GJ, Brezinski ME, Bouma BE, Boppart SA, Pitris C, Southern JF, Fujimoto JG. Science. 1997;276:2037–2039. doi: 10.1126/science.276.5321.2037. [DOI] [PubMed] [Google Scholar]
- 440.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Proc Nat Acad Sci USA. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Chen J, Saeki F, Wiley BJ, Cang H, Cobb MJ, Li ZY, Au L, Zhang H, Kimmey MB, Li XD, Xia YN. Nano Lett. 2005;5:473–477. doi: 10.1021/nl047950t. [DOI] [PubMed] [Google Scholar]
- 442.Oldenburg AL, Hansen MN, Ralston TS, Wei A, Boppart SA. J Mater Chem. 2009;19:6407. doi: 10.1039/b823389f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Lal S, Clare SE, Halas NJ. Acc Chem Res. 2008;41:1842–1851. doi: 10.1021/ar800150g. [DOI] [PubMed] [Google Scholar]
- 444.Gobin AM, Lee MH, Halas NJ, James WD, Drezek RA, West JL. Nano Lett. 2007;7:1929–1934. doi: 10.1021/nl070610y. [DOI] [PubMed] [Google Scholar]
- 445.Loo C, Lin A, Hirsch L, Lee MH, Barton J, Halas NJ, West J, Drezek R. Tech Cancer Res Treatment. 2004;3:33–40. doi: 10.1177/153303460400300104. [DOI] [PubMed] [Google Scholar]
- 446.Wang LHV, Hu S. Science. 2012;335:1458–1462. doi: 10.1126/science.1216210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Wang LV. Med Phys. 2008;35:5758–5767. doi: 10.1118/1.3013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Ku G, Wang LHV. Opt Lett. 2005;30:507–509. doi: 10.1364/ol.30.000507. [DOI] [PubMed] [Google Scholar]
- 449.Wang XD, Pang YJ, Ku G, Xie XY, Stoica G, Wang LHV. Nat Biotech. 2003;21:803–806. doi: 10.1038/nbt839. [DOI] [PubMed] [Google Scholar]
- 450.Eghtedari M, Oraevsky A, Copland JA, Kotov NA, Conjusteau A, Motamedi M. Nano Lett. 2007;7:1914–1918. doi: 10.1021/nl070557d. [DOI] [PubMed] [Google Scholar]
- 451.Wei CW, Huang SW, Wang CR, Li PC. IEEE Trans Ultrason Ferroelectr Freq Control. 2007;54:1131–1141. doi: 10.1109/tuffc.2007.367. [DOI] [PubMed] [Google Scholar]
- 452.Yang XM, Skrabalak SE, Li ZY, Xia YN, Wang LHV. Nano Lett. 2007;7:3798–3802. doi: 10.1021/nl072349r. [DOI] [PubMed] [Google Scholar]
- 453.Song KH, Kim CH, Cobley CM, Xia YN, Wang LV. Nano Lett. 2009;9:183–188. doi: 10.1021/nl802746w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Wilson K, Homan K, Emelianov S. Nat Commun. 2012;3:618. doi: 10.1038/ncomms1627. [DOI] [PubMed] [Google Scholar]
- 455.Fleischm M, Hendra PJ, Mcquilla AJ. Chem Phys Lett. 1974;26:163–166. [Google Scholar]
- 456.Jeanmaire DL, Vanduyne RP. J Electroanal Chem. 1977;84:1–20. [Google Scholar]
- 457.Albrecht MG, Creighton JA. J Am Chem Soc. 1977;99:5215–5217. [Google Scholar]
- 458.Lu Y, Liu GL, Kim J, Mejia YX, Lee LP. Nano Lett. 2005;5:119–124. doi: 10.1021/nl048232+. [DOI] [PubMed] [Google Scholar]
- 459.Liu GL, Lu Y, Kim J, Doll JC, Lee LP. Adv Mater. 2005;17:2683–2688. [Google Scholar]
- 460.Keren S, Zavaleta C, Cheng Z, de la Zerda A, Gheysens O, Gambhir SS. Proc National Acad Sci USA. 2008;105:5844–5849. doi: 10.1073/pnas.0710575105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Lee S, Kim S, Choo J, Shin SY, Lee YH, Choi HY, Ha S, Kang K, Oh CH. Anal Chem. 2007;79:916–922. doi: 10.1021/ac061246a. [DOI] [PubMed] [Google Scholar]
- 462.Zong S, Wang Z, Yang J, Wang C, Xu S, Cui Y. Talanta. 2012;97:368–375. doi: 10.1016/j.talanta.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 463.Jokerst JV, Miao Z, Zavaleta C, Cheng Z, Gambhir SS. Small. 2011;7:625–633. doi: 10.1002/smll.201002291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Kircher MF, de la Zerda A, Jokerst JV, Zavaleta CL, Kempen PJ, Mittra E, Pitter K, Huang R, Campos C, Habte F, Sinclair R, Brennan CW, Mellinghoff IK, Holland EC, Gambhir SS. Nat Med. 2012;18:829–834. doi: 10.1038/nm.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.James ML, Gambhir SS. Physiol Rev. 2012;92:897–965. doi: 10.1152/physrev.00049.2010. [DOI] [PubMed] [Google Scholar]
- 466.Cai QY, Kim SH, Choi KS, Kim SY, Byun SJ, Kim KW, Park SH, Juhng SK, Yoon KH. Invest Radio. 2007;42:797–806. doi: 10.1097/RLI.0b013e31811ecdcd. [DOI] [PubMed] [Google Scholar]
- 467.Kim D, Park S, Lee JH, Jeong YY, Jon S. Nanomedicine-Nanotech Bio Med. 2007;3:352–352. [Google Scholar]
- 468.Popovtzer R, Agrawal A, Kotov NA, Popovtzer A, Balter J, Carey TE, Kopelman R. Nano Lett. 2008;8:4593–4596. doi: 10.1021/nl8029114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Kim D, Park S, Lee JH, Jeong YY, Jon S. J Am Chem Soc. 2007;129:7661–7665. doi: 10.1021/ja071471p. [DOI] [PubMed] [Google Scholar]
- 470.Reuveni T, Motiei M, Romman Z, Popovtzer A, Popovtzer R. Int J Nanomedicine. 2011;6:2859–2864. doi: 10.2147/IJN.S25446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature. 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 472.Filippova M, Ross LS, Gill SS. Insect Mol Biol. 1998;7:223–232. doi: 10.1111/j.1365-2583.1998.00069.x. [DOI] [PubMed] [Google Scholar]
- 473.Elghanian R, Storhoff JJ, Mucic RC, Letsinger RL, Mirkin CA. Science. 1997;277:1078–1081. doi: 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- 474.Jin RC, Wu GS, Li Z, Mirkin CA, Schatz GC. J Am Chem Soc. 2003;125:1643–1654. doi: 10.1021/ja021096v. [DOI] [PubMed] [Google Scholar]
- 475.Cao YWC, Jin RC, Mirkin CA. Science. 2002;297:1536–1540. doi: 10.1126/science.297.5586.1536. [DOI] [PubMed] [Google Scholar]
- 476.Mirkin C. Abs Pap Am Chem Soc. 2003;225:U963–U964. [Google Scholar]
- 477.Stoeva SI, Lee JS, Thaxton CS, Mirkin CA. Angew Chem Int Ed. 2006;45:3303–3306. doi: 10.1002/anie.200600124. [DOI] [PubMed] [Google Scholar]
- 478.Hirsch LR, Jackson JB, Lee A, Halas NJ, West J. Anal Chem. 2003;75:2377–2381. doi: 10.1021/ac0262210. [DOI] [PubMed] [Google Scholar]
- 479.Lu WT, Arumugam R, Senapati D, Singh AK, Arbneshi T, Khan SA, Yu HT, Ray PC. ACS Nano. 2010;4:1739–1749. doi: 10.1021/nn901742q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Qian X, Zhou X, Nie S. J Am Chem Soc. 2008;130:14934–14935. doi: 10.1021/ja8062502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Sha MY, Xu H, Natan MJ, Cromer R. J Am Chem Soc. 2008;130:17214–17215. doi: 10.1021/ja804494m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Wang X, Qian XM, Beitler JJ, Chen ZG, Khuri FR, Lewis MM, Shin HJC, Nie SM, Shin DM. Cancer Res. 2011;71:1526–1532. doi: 10.1158/0008-5472.CAN-10-3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Louie AY. Chem Rev. 2010;110:3146–3195. doi: 10.1021/cr9003538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Veld RHI, Storm G, Hennink WE, Kiessling F, Lammers T. Nanoscale. 2011;3:4022–4034. doi: 10.1039/c1nr10733j. [DOI] [PubMed] [Google Scholar]
- 485.Lammers T, Kiessling F, Hennink WE, Storm G. Mol Pharmaceutics. 2010;7:1899–1912. doi: 10.1021/mp100228v. [DOI] [PubMed] [Google Scholar]
- 486.Kievit FM, Zhang MQ. Adv Mater. 2011;23:217–247. doi: 10.1002/adma.201102313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Wang LS, Chuang MC, Ho JA. Int J Nanomedicine. 2012;7:4679–4695. doi: 10.2147/IJN.S33065. [DOI] [PMC free article] [PubMed] [Google Scholar]