

Abstract
Most retroviruses employ a frameshift mechanism during polyprotein synthesis to balance appropriate ratios of structural proteins and enzymes. To investigate the requirements for individual precursors in retrovirus assembly, we modified the polyprotein repertoire of Mason-Pfizer monkey virus (M-PMV) by mutating the frameshift sites to imitate the polyprotein organization of Rous sarcoma virus (Gag-Pro and Gag-Pro-Pol) or Human immunodeficiency virus (Gag and Gag-Pro-Pol). For the “Rous-like” virus, assembly was impaired with no incorporation of Gag-Pro-Pol into particles and for the “HIV-like” virus an altered morphogenesis was observed. A mutant expressing Gag and Gag-Pro polyproteins and lacking Gag-Pro-Pol assembled intracellular particles at a level similar to the wild-type. Gag-Pro-Pol polyprotein alone neither formed immature particles nor processed the precursor. All the mutants were non-infectious except the “HIV-like”, which retained fractional infectivity.
Keywords: Ribosomal frameshift, Retrovirus, Assembly, Mason-Pfizer monkey virus, Capsid
Introduction
The formation of a retrovirus particle is initiated by the expression of polyprotein precursors Gag, Pro and Pol, comprising all structural proteins and enzymes. The expression strategy controls the proper ratio of viral components in the immature particles originating either at the plasma membrane (type-C) or within the cytoplasm (type D). During or shortly after budding, retroviral Gag polyprotein precursor is specifically processed by the viral protease and the immature viral particle is rearranged to form mature, fully infectious virus.
The proteolytic processing of Mason-Pfizer monkey virus (M-PMV) Gag (Pr78) yields matrix protein (MA, p10), phosphoprotein (pp24/pp16–18), p12, capsid protein (CA, p27), nucleocapsid protein (NC, p14), and p4 (Bradac and Hunter, 1984). In addition to the Gag derived structural proteins the processing of the Gag-Pro polyprotein (Pr95) results in the release of an NC-dUTPase fusion protein and protease (PR). The Gag-Pro-Pol polyprotein (Pr180) yields reverse transcriptase (RT) and integrase (IN).
The differential synthesis of two (Gag, Gag-Pol) or three (Gag, Gag-Pro, Gag-Pro-Pol) polyprotein precursors per virus is regulated by ribosomal frameshifting from one reading frame to the (−1) one (Jacks and Varmus, 1985). The exception is the gammaretrovirus genus that utilizes suppression of a termination codon during translation of pro and pol (Yoshinaka et al., 1985). Frameshifting requires two cis-acting signals in the retroviral mRNA, a slippery sequence for the shift of the ribosome and a structural element several nucleotides downstream of the slippery sequence, which enhances the efficiency of the slip (Jacks et al., 1988). A fraction of the ribosomes translating gag of the M-PMV from unspliced genomic transcript undergoes the (−1) frameshift (with frequency ~10–15%) upstream of the end of the gag gene allowing translation through the pro stop codon. A second (−1) frameshift (2–3%) yields the pol region in fusion with both the gag and pro products i.e. Gag-Pro-Pol precursor. In contrast to M-PMV, most retroviruses employ only one frameshift event to synthesize only Gag and Gag-Pol precursors.
In addition to regulating the ratios of polyproteins, frameshifting provides a presence of common transport signal to all Gag related polyproteins for their targeting to the site of assembly (Farabaugh, 1996; Farabaugh, 2000). In contrast to betaretroviruses (B- and D-type retroviruses) that form immature particles within the cytoplasm, the C-type retroviruses concentrate their Gag and Gag-Pol polyproteins at the plasma membrane where assembly and budding occur simultaneously. M-PMV a prototype of D-type morphogenesis, transports its Gag-related polyproteins to the pericentriolar region, where they assemble into immature capsids that are transported via the vesicular transport system to the plasma membrane while acquiring env-gene product (Choi et al., 1999; Sfakianos et al., 2003; Sfakianos and Hunter, 2003; Vlach et al., 2008).
Retroviral protease activity levels vary inversely to their concentration in the virion, controlled by its gene position in the genome. For instance, in HIV the protease coding sequence is located at the 5′ end of the pol gene and it is thus expressed only in the pol reading frame. In avian retroviruses (ASLV, RSV), it is encoded at the 3′-end of the gag ORF, and it is thus present both in Gag and Gag-Pol molecules. The lower specific activity of the RSV protease compared to the HIV may be related to its higher concentration in RSV (Grinde et al., 1992). The active site of the RSV protease contains a serine in contrast to other retroviruses that contain threonine. Substitution of the threonine for the serine in the active site results in a 2-fold increase of catalytic activity of RSV protease (Ridky et al., 1996). In betaretroviruses (M-PMV and mouse mammary tumor virus) and deltaretroviruses (human T-cell lymphotropic virus), the protease is encoded in a separate reading frame between the gag and pol genes and intermediate protease concentrations are therefore incorporated into virions.
Alterations of a proper ratio of the polyprotein precursors dramatically impair the assembly, budding and processing (Hung et al., 1998; Karacostas et al., 1993; Park and Morrow, 1991;Shehu-Xhilaga et al., 2001). To further investigate the requirement for specific polyprotein precursors in assembly, maturation and virion infectivity, we mutated the frameshift sites in the M-PMV genome to produce only Gag and Gag-Pro-Pol or Gag-Pro and Gag-Pro-Pol polyproteins and to generate mutants mimicking HIV or RSV, respectively. Their properties were compared to the wild-type M-PMV and its D26N mutant with an inactive protease.
Results
Synthesis, processing, and release of M-PMV wild-type and mutant polyproteins
All wild-type M-PMV structural proteins and enzymes are synthesized as polyprotein precursors Gag (G), Gag-Pro (GP) and Gag-Pro-Pol (GPP). To study the role of polyprotein precursors ratios in assembly, maturation and virion infectivity, a series of vectors with mutated frameshift sites in the M-PMV genome was prepared (see Fig. 1). The insertion of a single A at the gag-pro frameshift site prevented the translation of sole Gag polyprotein. Resulting formation of Gag-Pro and Gag-Pro-Pol polyproteins (GP-GPP) mimics the organization of polyprotein precursors in Rous sarcoma virus (RSV) with protease at the 3′ end of Gag (Fig. 1). The insertion of T in pro-pol frameshift site yielded only the Gag and Gag-Pro-Pol in G-GPP mutant (Fig. 1). This imitates the strategy in HIV where protease is encoded in the 5′ terminus of pol. In addition two control constructs, GPP, expressing only the Gag-Pro-Pol precursor as a consequence of mutations in both frameshift sites, and G-GP, with termination codon at the end of gag-pro region were prepared (Fig. 1).
Fig.1.

Organization of the M-PMV gag, pro, and pol genes (upper left corner) and M-PMV frameshift mutants. The open boxes represent open reading frames in the proviral DNA; the filled boxes indicate the corresponding polyprotein precursors. (−1) FS indicates the frameshift site.
In order to evaluate the expression and stability of the polyprotein precursors, transfected COS-1 cells were metabolically labeled and both the cell- and virus-associated proteins were immunoprecipitated after a pulse and 4 h chase with rabbit anti-M-PMV capsid protein serum (Fig. 2). All the anticipated precursors were found in the cells transfected with the mutant and the wild-type genomes (Fig. 2A). Despite the alteration of the frameshift sequences, a minor band corresponding approximately to the molecular weight of Gag-Pro (Pr95) was observed in the lysates of the cells transfected with mutants G-GPP or GPP (Fig. 2A). This suggests that an infrequent (−1) frameshift continued to occur resulting in a small amount of translation into the (−2) reading frame where there is a stop codon four amino acids downstream of the slippery sequence. The resulting polyprotein would therefore have a molecular mass of approximately 95 kDa.
Fig. 2.
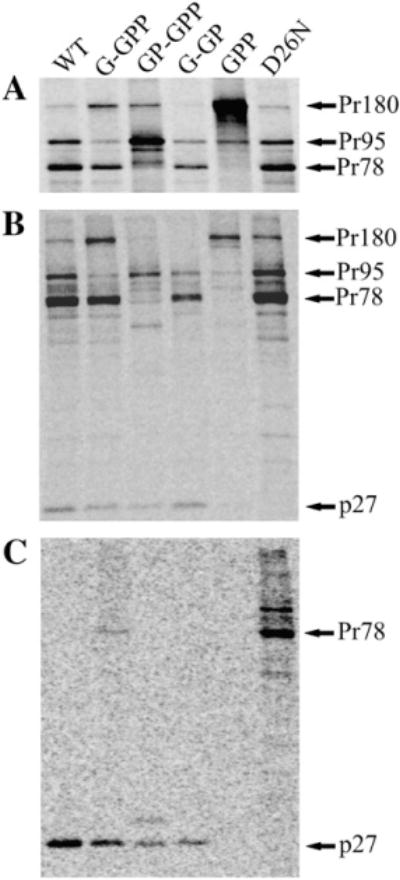
Synthesis and processing of the wild-type and mutant polyprotein precursors. COS-1 cells transfected with wild-type or mutant proviruses were metabolically labeled 24 h post-transfection with [35S]methionine-cysteine for 15 min and chased for 4 h. Intracellular proteins after the pulse (A) and chase (B) periods, as well as virus-associated proteins after the chase period (C) were immunoprecipitated using polyclonal anti-p27 (M-PMV CA) serum. The Gag-related polyprotein precursors and their major cleavage product the capsid protein are indicated: Pr180, Gag-Pro-Pol; Pr95, Gag-Pro; Pr78, Gag; p27, CA. D26N indicates a protease active site mutation in an otherwise the wild-type background.
Likewise, all the expected polyproteins for the wild-type and most of the mutants were found in the cells after the chase, indicating that there was no significant increase in activation of the viral protease or decrease in protein stability for most of the mutants (Fig. 2B). However, the Gag-related polyproteins of the GP-GPP and GPP mutants were significantly reduced in the cells during the chase period (Fig. 2B). This could be either due to nonspecific degradation within the cells or due to a higher level of protease produced by these mutants compared to the wild-type. To exclude this, a D26N protease active site mutant of GP-GPP vector was constructed. Analysis of this mutant showed a similar pattern of degradation products suggesting the role of cellular proteases for the observed degradation of polyproteins (Fig. 3). As expected, no mature CA protein was observed in the chased cells expressing this mutant. In contrast, the wild-type and all the other mutants except GPP displayed some intracellular capsid protein after the 4 h chase (Fig. 2B).
Fig. 3.

Assembly, release, and processing of the wild-type and mutant viral particles. COS-1 cells transfected with the wild-type and mutant genomes were metabolically labeled 24 h post-transfection with 35S methionine-cysteine for 15 min and chased for 30 min, 1 h and 2 h. The assembled polyprotein precursors were pelleted from the cell lysates by centrifugation through a 20% (w/v) cushion. The polyprotein precursors from the pellet (P), supernatant (S) and viral particles (V) were immunoprecipitated using polyclonal anti-p27 (M-PMV CA) serum, analyzed bySDS-PAGE, and quantified on a phosphorimager using ImageQuant software. An experiment with mock transfected cells was conducted as a negative control (Mock).
Released mature viral proteins in the culture medium of cells expressing the wild-type and all the mutants except GPP contained correctly processed CA protein (Fig. 2C). However, a small amount of virus-associated unprocessed Gag polyprotein in G-GPP and low level of CA-related protein of higher molecular weight in GP-GPP suggested less efficient maturation for these mutants (Fig. 2C). As expected, no cleavage occurred in the M-PMV D26N protease mutant. The GPP mutant producing only the Gag-Pro-Pol precursor was neither processed nor released into the medium even during an overnight chase (not shown).
The assembly of individual mutants was monitored by fractionation of cell-associated viral proteins and virus-associated proteins followed by immunoprecipitation using anti-CA antibody from samples chased in various times (Fig. 3). The percentage of Gag-related precursors in the pellet and in the cellular supernatant is compared in Fig. 4. Nearly 70% of the intracellular wild-type M-PMV Gag-related polyproteins were found in the pelleted fraction after the 15 min pulse (Fig. 4) and this relative amount remained constant during the first hour of the chase. Over 50% of the wild-type precursor molecules were specifically cleaved during the two-hour chase period (Figs. 3 and 4). In contrast, the initial assembly phase of the G-GPP mutant was slower as only 20% of polyproteins were pelleted suggesting that they were incorporated into the particles during 15 min pulse. However, the proportion of pelletable polyproteins reached the wild-type level after 30 min chase. Most of this material likely represented assembled particles since it was detected in the culture medium after the two-hour chase period. The GP-GPP and G-GP mutants displayed an intermediate phenotype between the wild-type and G-GPP as about half of the radiolabeled precursors was in the pelletable fraction after the pulse period. However, majority of the intracellular G-GPP, GP-GPP and G-GP precursors was converted to a pelletable form within the two-hour chase. Likewise for GPP, about 50% of the radiolabeled Gag-Pro-Pol precursors were found in the pelletable fraction after the 15 min pulse period and the amount of the pelletable material of this mutant exceeded that of the wild-type and other mutants during the 2 h chase period, but no released proteins were observed in the medium (Fig. 3).
Fig. 4.
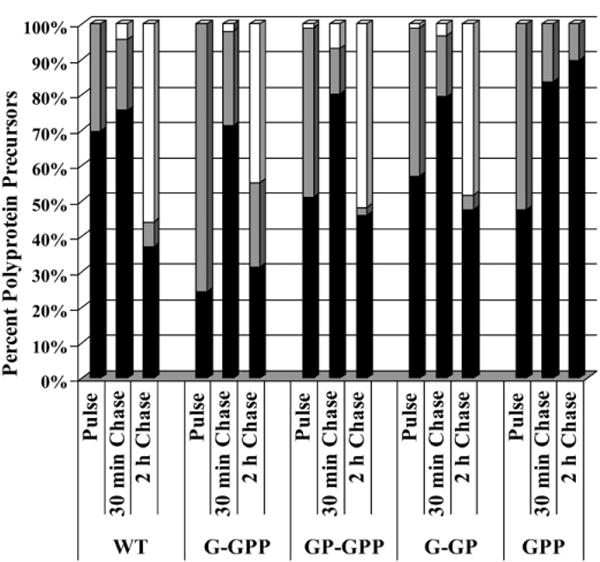
The assembly of intracytoplasmic particles. COS-1 cells transfected with the wildtype and mutant genomes were metabolically labeled 24 h post-transfection with [35S] methionine-cysteine for 15 min and chased for 30 min and 2 h. The assembled polyprotein precursors were pelleted from cell lysates by centrifugation through a 20% (w/v) cushion. Viral proteins from the pellet, supernatant and culture medium were immunoprecipitated and quantified using ImageQuant software. The sum of intensities of all Gag-related polyproteins plus that of the capsid protein at each time point was regarded as 100%. Black- and grey-portions of bars indicate the percentage of intracellular Gag-related precursors present in the pellet and supernatant form the cell lysates, respectively. The amounts of released (extracellular) viral proteins represent the white portions of the bars.
Since the pelletable cellular fractions might contain assembled particles but also aggregates, we performed an equilibrium centrifugation and EM analysis to assess their relative proportion. All immunoprecipitated material of the GPP mutant migrated to the bottom of the sucrose gradient (25–55% (w/w)), suggesting formation of aggregates or large clusters of particles. The presence of aggregates was supported by the fact that no particles were detectable by the electron microscopy (data not shown). The wild-type and the G-GPP proteins migrated to the fraction of sucrose density ~1.21 g/ml, indicative of the formation of assembled immature particles (Klikova et al., 1995; Sakalian et al., 1996). A surprisingly small portion of GP-GPP mutant polyproteins banded at this density whilst most of the polyproteins were detected at the gradient bottom. A pulse chase experiment with a GP-GPP/D26N double mutant (Fig. 3) showed that released particles of GP-GPP contained only the Gag-Pro polyprotein, suggesting that immature particles consisting of both polyproteins were release-defective or that Gag-Pro-Pol was not incorporated into Gag-Pro particles.
We further quantified and compared relative levels of total polyproteins present in the COS-1 cells and of those assembled into immature particles (Fig. 5). Multiple autoradiography analyses revealed an intracellular expression ratio of ~87:12:0.7 for Gag:Gag-Pro:Gag-Pro-Pol for the wild-type. However, the proportions incorporated into immature particles were different at ~89:8:2.6, indicating an increased preferential incorporation of the Gag-Pro-Pol precursor. Similar results were obtained for the mutant G-GPP where the Gag-Pro-Pol precursor expression represented ~8.6%, but its incorporation reached about 21.6%. No specific cleavage products were found by autoradiography of labeled proteins of the intracellular wild-type and G-GPP mutant particles.
Fig. 5.

Comparison of the wild-type and G-GPP mutant precursor ratios in pulse-labeled cells and in isolated immature capsids. COS-1 cells were metabolically labeled 24 h post-transfection with [35S]methionine-cysteine. Cell lysates and immature particles isolated from cell lysates of pulse-labeled cells were immunoprecipitated using polyclonal anti-p27 (M-PMV CA) serum and the viral proteins were analyzed by phosphorimager. Three independent experiments were performed and are shown in the gels below the table under the corresponding data column. Table data give the average with the standard deviation. The Pr78, Pr95 and Pr180 polyproteins contain 5, 13 and 27 methionines, respectively.
The half-time of processing of the wild-type polyprotein precursors was approximately 2 h, which is in accordance with previously published data (Sfakianos and Hunter, 2003; Stansell et al., 2004). The polyproteins formed from the GP-GPP and G-GP mutants were processed similarly (determined from the gels in Fig. 3). The polyproteins of the G-GPP mutant were cleaved less efficiently with the half-time of approximately 2.5 h, which is in agreement with the fact that released particles contained about 17% (molar) of unprocessed Gag after 4 h. This is in contrast to the wild-type and other mutants, capable of particle release, with fully processed released precursors (Fig. 2C).
Altered polyprotein ratios affect morphogenesis of mutant particles
To determine the morphogenesis of the frameshift mutants, the localization of the intracellular particles in COS-1 cells was analyzed by thin section transmission electron microscopy (Fig. 6). The wild-type immature capsids were found both within the cytoplasm and budding from the plasma membrane (Figs. 6A, B). Similarly, the G-GP mutant formed intracytoplasmic particles with the wild-type morphology that budded from the cell surface (Figs. 6C, D).
Fig. 6.
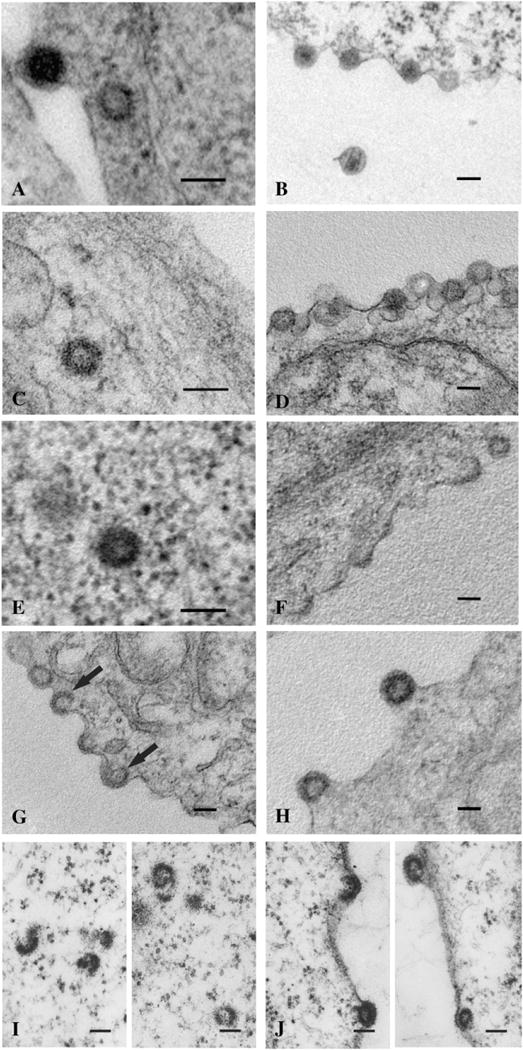
Frameshift mutants display altered assembly phenotypes. The wild-type (A, B) and the G-GP mutant (C, D) produce the normal intracytoplasmic immature particles (A, C) which can then be seen budding from the plasma membrane (B, D). Mutant G-GPP (E–H) also produced intracytoplasmic particles (E), but also an abundance of C-type-like assembling and budding structures at the plasma membrane (F–H). These structures were seen at various levels of completion with some appearing to continue assembling into the cytoplasm with visible plasma membrane support (G, black arrows). Mutant GP-GPP (I, J) produced only incomplete and aberrant intracytoplasmic structures (I); although the radius of curvature of these incomplete rings was similar to that of the wild-type immature particles. GP-GPP budding structures were also observed (J), some of which appeared C-type-like. The scale bars represent 100 nm.
Mutant G-GPP formed both intracytoplasmic particles (Fig. 6E) and C-type-like particles assembling at the plasma membrane of COS-1 cells (Figs. 6F–H). The latter type was present at various stages of the assembly, early stage (Fig. 6F) to late stage (Fig. 6H). Notably, we detected many incomplete particles at the plasma membrane consisting of segments extended into the cytoplasm without direct membrane contact (Fig. 6G, black arrows). This is in contrast to the assembly of common C-type particles (e.g. RSV) whose assembly requires the interaction of Gag related polyproteins with the plasma membrane. It is possible that these incomplete structures originated in the cytoplasm and were transported to the membrane rather than assembled at the plasma membrane itself. The GP-GPP mutant, lacking the Gag polyprotein, formed only a few incomplete intracytoplasmic particles composed of crescent-shaped segments with the same approximate diameter as the wild-type (Fig. 6I). Very few GP-GPP budding structures were observed (Fig. 6J), some with the C-type-like morphology.
To analyze the shape and size of immature G-GPP and GP-GPP particles, the cell lysates of COS-1 cells expressing the wild-type and both mutants were clarified and viral particles were purified through sucrose gradient and analyzed by electron microscopy. Isolated immature G-GPP particles were spherical with a surface appearance similar to those of the wild-type M-PMV (Figs. 7A, B). In comparison to the wild-type immature particles the G-GPP capsids differed in a diameter, which was slightly larger (90–130 nm) than that of the wild-type particles (80–100 nm). The gradient centrifugation analyses of the GP-GPP mutant revealed particles forming large aggregated clusters (Fig. 7C).
Fig. 7.
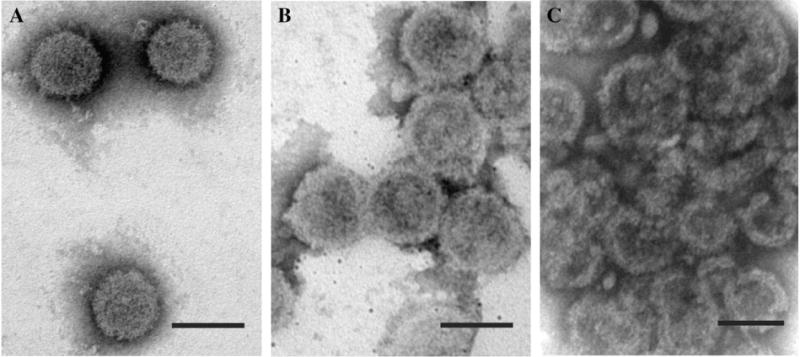
Isolated intracytoplasmic wild-type and mutant particles. Particles isolated by gradient sedimentation were negatively stained with 4% sodium silicotungstate. The wild-type (A) and G-GPP (B) particles were complete. GP-GPP (C) particles were largely incomplete segments similar to the structures seen in thin section. The scale bars represent 100 nm.
Influence of frameshift mutations on virus infectivity
Single round infectivity assay was performed to assess the impact of the altered proportions of polyproteins on viral replication. With the exception of G-GPP displaying about 20% of the wild-type infectivity, all the other mutants were non-infectious (data not shown). To verify that G-GPP was indeed replication competent, viral growth was monitored over 24 days post-infection by western blot. The RT assay was not used since the relative levels of reverse transcriptase in capsid of the mutants varied according to the altered frameshift sequences. The wild-type virus reached the maximum of the particle production on the ninth day post-infection with the level remaining constant thereafter (Fig. 8). The G-GPP mutant was indeed replication competent, but with a reduced efficiency. Over the course of the 24 day experiment the virus production slowly increased and did not reach a sustained maximum (Fig. 8).
Fig. 8.
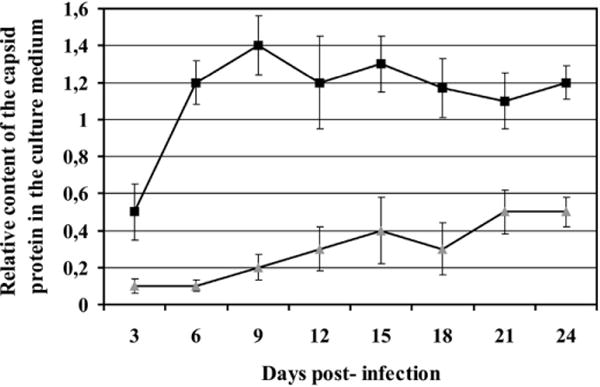
Determination of mutant infectivity. COS-1 cells were transfected with either the wild-type or G-GPP mutant. Culture medium was collected, the relative quantity of released particles was measured by western blot for CA, and normalized volume of medium was used to infect fresh COS-1 cells. Culture medium was collected at 3-day intervals post-infection. The relative content of capsid protein in the medium post-infection was similarly quantified by western blot analysis. Black squares and grey triangles indicate the wild-type and G-GPP mutant, respectively.
Discussion
We demonstrate here the effects of different M-PMV polyprotein ratios on the frameshift efficiency, assembly, release, and virus infectivity. The frameshift efficiencies have been determined for several retroviruses, including HIV-1, where its frequency represents about 5% in cells and 10% in vitro (Baril et al., 2003; Dinman et al., 2002; Dulude et al., 2002). Avian sarcoma leukemia virus (Arad et al., 1995) and Rous sarcoma virus (Jacks and Varmus, 1985) synthesize Gag-Pol at the level of 4.8% of Gag. Mouse mammary tumor virus which, similarly to M-PMV, contains separate pro reading frame between gag and pol, synthesizes up to 20% of Gag-Pro and about 2% of Gag-Pro-Pol polyprotein precursors compared to Gag, as determined in both in vitro and in vivo experiments (Chamorro et al., 1992; Jacks et al., 1987). About 10% efficiency of both frameshifts has been estimated for M-PMV (Sonigo et al., 1986). A more recent in vitro study suggested 16% efficiency for the M-PMV gag-pro frameshift (Sakalian et al., 1996). Our precise determination of the wild-type M-PMV Gag-related precursors in the cell lysates revealed 12% and 5.5% efficiency for the first and second frameshift, respectively, resulting in the synthesis of 87.1% of Gag, 12.1% of Gag-Pro and 0.7% of Gag-Pro-Pol precursors. Surprisingly, a detailed quantification of the Gag-related polyproteins in isolated M-PMV wild-type immature particles showed only 7.9% of Gag-Pro but 2.6% of Gag-Pro-Pol polyproteins. Similar results were obtained for the G-GPP mutant where the immature particles consisted of more than a twofold higher relative amount of Gag-Pro-Pol compared to its intracellular level. This enrichment of particles with Gag-Pro-Pol compared to its availability in the cells was surprising given its inability to independently form particles.
The GP-GPP and GPP mutants were considerably less stable than the wild-typeas observed during a four-hour chase (compare panels A and B in Fig. 2). One could speculate that this degradation might be caused by a higher level of viral protease. However, our experiments with the GP-GPP mutant containing an inactive protease showed similarly rapid degradation of its intracellular Gag-related polyproteins. Thus a possible explanation for the instability of the longer polyproteins could be that these mutants do not express the complete Gag but only C-terminally truncated polyprotein lacking p4 protein and 15 amino acids from NC. We have shown previously that this proline-rich protein is not essential for assembly of immature particles, since M-PMV Gag lacking the p4 protein efficiently assembles in vitro (Nermut et al., 2002; Rumlova-Klikova et al., 2000). However, the p4 domain in M-PMV Gag transiently interacts with a cytosolic chaperonin TRiC (Hong et al., 2001), which prevents aggregation and proteolytic degradation of newly translated proteins (Clarke, 2006; Kubota et al., 2006). The truncation of p4 triggered a degradation of the entire Gag and this mutant produced a lower level of self assembled capsids (Hong et al., 2001). In consonance with these interpretations a majority of GP-GPP intracytoplasmic particles was incomplete and formed aggregates. One can speculate that also the C-terminal sequence of nucleocapsid, which is not present in GP-GPP mutant, can contribute to the instability of polyprotein and to decrease the amount of cell-associated Gag polyproteins. However, the results from experiments in which the substantial part of the C-terminal sequence of M-PMV NC was removed and assembly of immature particles was not impaired (Michaela Rumlová, personal communication) show that the p4 protein assists very likely in the stability of newly synthesized Gag polyproteins and in correct assembly. Despite inefficient assembly, the presence of 27 kDa capsid protein in released particles indicated a partial maturation of GP-GPP mutant. Nevertheless, it was non-infectious and partially decelerated in the release. Released particles of GP-GPP mutant with inactive protease lacked Gag-Pro-Pol, likely to the impaired capability of large Gag-Pro-Pol polyprotein to co-assemble with Gag-Pro. One explanation could be that these Gag polyproteins are not correctly folded to an assembly competent form and proper intermolecular interactions between Gag-polyproteins do not arise before assembly. Another possible explanation for the instability of the longer polyproteins in the absence of the Gag polyprotein is their inefficient assembly due to steric problems associated with their accommodating into particles of ~90 nm diameter.
The GPP mutant, producing only the largest Gag-related polyprotein, did not form any detectable particles. Several studies have focused on expression of HIV Gag-Pol in the absence of Gag; however the results depended upon the expression system and the protease activity. In COS-1 cells (Park and Morrow, 1991; Smith et al., 1993), 293T cells (Chiu et al., 2002), and CV-1 cell (Karacostas et al., 1993) the expression of Gag-Pol alone resulted in intracellular protease activation, precursor processing, and failure of particle formation and budding. A low level of the reverse transcriptase activity was detected in the medium of the COS cells transfected with the HIV Gag-Pol, but particle formation was not confirmed (Park and Morrow, 1992). In contrast, RSV (Oertle et al., 1992) and M-MuLV (Felsenstein and Goff, 1988) Gag-Pol failed to form particles when expressed alone even though no precursor processing in the cells was observed. These reports demonstrate that intracytoplasmic Gag-Pol particle formation generally does not occur. Consistently with previously published data for other retroviruses (Gheysen et al., 1989; Haffar et al., 1990; Hoshikawa et al., 1991; Smith et al., 1993), our results confirmed that M-PMV Gag-Pro-Pol polyprotein is dispensable for the assembly.
Interestingly, the G-GPP mutant formed both intracytoplasmic and membrane bound particles slightly larger (90–130 nm) than the wild-type (Fig. 7B). However, the membrane associated particles differed from those typical for C-type retroviruses as their segments protruded into the cytoplasm suggesting that the association with the membrane was dispensable for the assembly or that they were transported to the membrane as partially assembled fragments (Fig. 6G, arrows). We favor the first hypothesis as incomplete particles were not found in the cytoplasm. The G-GPP processing was significantly delayed in comparison to the G-GP and GP-GPP mutants, which were correctly cleaved with a half-time of about 2 h. It is likely that the free C-terminus of the PR domain in the wild-type Gag-Pro facilitates its activation (Bauerova-Zabranska et al., 2005). In contrast, the restricted mobility of the protease C-terminus in the Gag-Pro-Pol polyprotein of G-GPP mutant might explain its delayed effect.
In conclusion, the results described here reveal that the stability of newly synthesized Gag polyproteins within the host cells significantly influences the efficient assembly of immature particles. The presence of complete Gag polyprotein seems to be crucial for the incorporation of longer Gag-related polyproteins into immature particles. The maintenance of a proper ratio of polyprotein precursors is important for infectivity. Interestingly we demonstrate that the ratio of incorporation of Gag, Gag-Pro, and Gag-Pro-Pol polyproteins into immature particles is not dictated solely by the proportion of their synthesis.
Materials and methods
Expression plasmids
To generate frameshift mutations in the M-PMV genome, a vector pBGUP, containing the gag, pro and pol reading frames (SacI–MscI fragment)in vector pSIT (Andreansky and Hunter, 1994), was used as a template for site-directed mutagenesis. Oligonucleotides M-PMV-FP: 5′-TACCACCCCATCAGGGAAAACGGGTGGAG-3′, and M-PMV FS: 5′-AAAGGTTTTGGAAATTTTTTAACTGCGGCCATTGAC-3′ were used to alter the first and second frameshift sites, respectively. These oligonucleotides were designed to create an insertion of an A and a T in positions 2317 and 3227 of pSARM4 (Song and Hunter, 2003), creating a read-through from gag to pro and from pro to pol, respectively. SacI–Eco72I fragments (2110 bp) of pBGUP, carrying the appropriate mutations were inserted into pSARM4. A dsDNA linker (5′-TTAACTAGTTAA-3), containing stop codons in all three reading frames and a SpeI recognition site, was inserted at the 3′-end of the pro open reading frame to create the G-GP mutant. All vectors were verified by DNA sequencing. The control D26N mutant has been previously described by Sommerfelt et al. (1992).
Cell lines and transfection
COS-1 (Sigma-Aldrich) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum in 5% CO2, at 37 °C. 40–50% confluent cells were transfected with the appropriate DNA using FuGENE 6 transfection reagent (Roche Molecular Biochemicals) according to the manufacturer’s instructions.
Metabolic labeling and immunoprecipitation
COS-1 cells were starved for 10 min in cysteine and methionine deficient DMEM 24 or 48 h post-transfection and then pulse-labeled for 15 min with 400 μCi/ml TRAN35S-LABEL labeling mix (ICN Biomedicals). After washing with DMEM and desired chase period in complete DMEM, the medium was collected, filtered and adjusted to 1% Triton X-100, 1% sodium deoxycholate and 0.1% concentration of sodium dodecyl sulfate (SDS). The chased cells, as well as the pulsed cells, were washed twice with cold Tris-buffered saline (TBS) and incubated with lysis buffer A (1% Triton X-100, 1% sodium deoxycholate, 50 mM sodium chloride, and 25 mM Tris–HCl, pH 8.0) at room temperature for 10 min, clarified by centrifugation at 14,000× g for 10 min and the resulting lysate was adjusted to 0.1% (w/v) SDS. The material was then subjected to centrifugation through a 20% (w/v) sucrose cushion at 540,000× g for 30 min at 4 °C. Samples were pre-cleared with Staphylococcus A, incubated overnight at 4 °C with polyclonal antibodies against M-PMV CA and immunoprecipitated for 30 min at room temperature with Staphylococcus A. Proteins were resolved by 12% SDS-PAGE and visualized by autoradiography. Protein concentrations were determined using the Typhoon Variable Mode Imager (Amersham Pharmacia Biotech) and ImageQuant software (Amersham Pharmacia Biotech). The band intensity was determined and adjusted to the number of methionines in the particular protein.
Electron microscopy
COS-1 cells scraped from the plate 24 h post-transfection were fixed for 30 min with 2.5% glutaraldehyde and 1% paraformaldehyde in 0.1 M sodium cacodylate buffer and post-fixed with 2% osmium tetroxide OsO4 for 1 h followed by staining with 4% saturated uranyl acetate. Material dehydrated in increasing acetone concentrations was sectioned. Negative staining of immature capsids was carried out with 4% sodium silicotungstate at pH 7.2. The samples were analyzed with a Jeol JEM-1010 at 80 kV.
Isolation of immature capsids
The method described previously (Parker and Hunter, 2000; Parker et al., 2001; Parker and Hunter, 2001) was used. Briefly; the labeled cells were washed with cold TBS and lysed for 30 min on ice in lysis buffer (10 mM Tris, 1 mM EDTA, 1% (v/v) Triton X-100). The cell lysate was clarified by centrifugation at 3000× g and loaded over a 35% to 75% (w/v) sucrose step gradient in 10 mM Tris buffer, pH 7.6 and centrifuged for 6 h, at 4 °C, at 25,000 rpm in a Sorvall TH641 rotor. The 35 to 75% sucrose interface was collected and dialyzed (cut off 14 kDa) against 10 mM Tris buffer (pH 7.6) overnight at 4 °C. The particles were pelleted at 22,000× g for 30 min at 4 °C and resuspended in the lysis buffer supplemented with 500 mM NaCl and further purified by centrifugation (25,000 rpm, SorvallTH641 rotor, 30 min, 4 °C) through a 5%–20% sucrose gradient. 1-ml fractions were analyzed by SDS-PAGE and the fractions most abundant in virus particles were dialyzed and centrifuged (22,000× g for 30 min at 4 °C). The purified particles were subjected to EM analysis. The material from the sucrose interface was further separated in a continuous 25–55% (w/w) sucrose gradient in lysis buffer supplemented with 500 mM NaCl by ultracentrifugation in a TLS-55 rotor (Beckman) for 2 h at 55,000 rpm and analyzed by SDS-PAGE.
Single-round infectivity assay
The single-round infectivity of M-PMV molecular clones was determined as described by Song and Hunter (2003). Briefly, COS-1 cells were cotransfected with pMTΔE containing either the wild-type or the mutant provirus modified to contain the HIV-1 tat gene in place of the env gene, and with the glycoprotein expression vector pTMO. Culture medium was filtered through a 0.45 μm filter 48 h post-transfection and analyzed for relative capsid protein content by western blot. Thirty to 40% confluent GHOST cells (HOS-CD4/LTR-hGFP) were infected overnight with harvested media, normalized for CA. GHOST cells were washed with and resuspended in PBS, pH 7.4 supplemented with 5 mM EDTA 48 h post-infection and analyzed for the expression of GFP by flow cytometry.
Virus infectivity
The infectivity was determined as the level of CA protein released from infected cells. COS-1 cells were washed twice thoroughly with complete DMEM 6 h post-transfection and incubated with fresh medium for 48 h. The medium was harvested, filtered and analyzed by western blot. 30%–40% confluent COS-1 cells were infected with virus-containing culture medium normalized for CA. The cells were washed 12 h post-infection with complete DMEM and cultivated for another 24 days. The culture medium was harvested at 3 day intervals and virus particles pelleted through a 20% (w/w) sucrose cushion and analyzed by western blot using a rabbit polyclonal antibody against M-PMV CA, anti-rabbit HRP-conjugated antibody and Super Signal West Pico Chemiluminescent Substrate (Pierce). The signals recorded on a CCD camera were analyzed using AIDA Image Analyzer 2D Densitometry software (BioTech a.s.).
Acknowledgments
This work was supported by research project grants of Czech Ministry of Education 1M6837805002, M6138896301, MSM 6046137305, Z 40550506, ME 904, Czech Science Foundation Grants SCO/06/E001, Grant Agency of AS CR KAN200100801, KAN208240651 and grant # CA 27834 from the National Institutes of Health.
References
- Andreansky M, Hunter E. Phagemid pSIT permits efficient in-vitro mutagenesis and tightly controlled expression in Escherichia coli. BioTechniques. 1994;16:626–630. [PubMed] [Google Scholar]
- Arad G, Barmeir R, Kotler M. Ribosomal frameshifting at the Gag-Pol junction in avian leukemia sarcoma-virus forms a novel cleavage site. FEBS Lett. 1995;364:1–4. doi: 10.1016/0014-5793(95)00302-p. [DOI] [PubMed] [Google Scholar]
- Baril M, Dulude D, Steinberg SV, Brakier-Gingras L. The frameshift stimulatory signal of human immunodeficiency virus type 1 group O is a pseudoknot. J Mol Biol. 2003;331:571–583. doi: 10.1016/S0022-2836(03)00784-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerova-Zabranska H, Stokrova J, Strisovsky K, Hunter E, Ruml T, Pichova I. The RNA binding G-patch domain in retroviral protease is important for infectivity and D-type morphogenesis of Mason-Pfizer monkey virus. J Biol Chem. 2005;280:42106–42112. doi: 10.1074/jbc.M508031200. [DOI] [PubMed] [Google Scholar]
- Bradac J, Hunter E. Polypeptides of Mason-Pfizer monkey virus. 1. Synthesis and processing of the gag-gene products. Virology. 1984;138:260–275. doi: 10.1016/0042-6822(84)90350-7. [DOI] [PubMed] [Google Scholar]
- Chamorro M, Parkin N, Varmus HE. An RNA pseudoknot and an optimal heptameric shift site are required for highly efficient ribosomal frameshifting on a retroviral messenger-RNA. Proc Natl Acad Sci USA. 1992;89:713–717. doi: 10.1073/pnas.89.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu HC, Yao SY, Wang CT. Coding sequences upstream of the human immunodeficiency virus type 1 reverse transcriptase domain in gag-pol are not essential for incorporation of the Pr160(gag-pol) into virus particles. J Virol. 2002;76:3221–3231. doi: 10.1128/JVI.76.7.3221-3231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi G, Park S, Choi B, Hong ST, Lee JY, Hunter E, Rhee SS. Identification of a cytoplasmic targeting retention signal in a retroviral Gag polyprotein. J Virol. 1999;73:5431–5437. doi: 10.1128/jvi.73.7.5431-5437.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR. Cytosolic chaperonins: a question of promiscuity. Mol Cell. 2006;24:165–167. doi: 10.1016/j.molcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Dinman JD, Richter S, Plant EP, Taylor RC, Hammell AB, Rana TM. The frameshift signal of HIV-1 involves a potential intramolecular triplex RNA structure. Proc Natl Acad Sci USA. 2002;99:5331–5336. doi: 10.1073/pnas.082102199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulude D, Baril M, Brakier-Gingras L. Characterization of the frameshift stimulatory signal controlling a programmed-1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res. 2002;30:5094–5102. doi: 10.1093/nar/gkf657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farabaugh PJ. Programmed translational frameshifting. Microbiol Rev. 1996;60:103–134. doi: 10.1128/mr.60.1.103-134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farabaugh PJ. Translational frameshifting: implications for the mechanism of translational frame maintenance. Prog Nucleic Acid Res Mol Biol. 2000;64:131–170. doi: 10.1016/s0079-6603(00)64004-7. [DOI] [PubMed] [Google Scholar]
- Felsenstein KM, Goff SP. Expression of the gag-pol fusion protein of Moloney murine leukemia-virus without gag protein does not induce virion formation or proteolytic processing. J Virol. 1988;62:2179–2182. doi: 10.1128/jvi.62.6.2179-2182.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheysen D, Jacobs E, Deforesta F, Thiriart C, Francotte M, Thines D, Dewilde M. Assembly and release of Hiv-1 precursor Pr55Gag virus-like particles from recombinant baculovirus infected insect cells. Cell. 1989;59:103–112. doi: 10.1016/0092-8674(89)90873-8. [DOI] [PubMed] [Google Scholar]
- Grinde B, Cameron CE, Leis J, Weber IT, Wlodawer A, Burstein H, Bizub D, Skalka AM. Mutations that alter the activity of the Rous-sarcoma virus protease. J Biol Chem. 1992;267:9481–9490. [PubMed] [Google Scholar]
- Haffar O, Garrigues J, Travis B, Moran P, Zarling J, Hu SL. Human immunodeficiency virus-like, nonreplicating, gag-env particles assemble in a recombinant vaccinia virus expression system. J Virol. 1990;64:2653–2659. doi: 10.1128/jvi.64.6.2653-2659.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Choi G, Park S, Chung AS, Hunter E, Rhee SS. Type D retrovirus gag polyprotein interacts with the cytosolic chaperonin TRiC. J Virol. 2001;75:2526–2534. doi: 10.1128/JVI.75.6.2526-2534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshikawa N, Kojima A, Yasuda A, Takayashiki E, Masuko S, Chiba J, Sata T, Kurata T. Role of the gag and pol genes of human-immunodeficiency-virus in the morphogenesis and maturation of retrovirus-like particles expressed by recombinant vaccinia virus — an ultrastructural-study. J Gen Virol. 1991;72:2509–2517. doi: 10.1099/0022-1317-72-10-2509. [DOI] [PubMed] [Google Scholar]
- Hung M, Patel P, Davis S, Green SR. Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J Virol. 1998;72:4819–4824. doi: 10.1128/jvi.72.6.4819-4824.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Varmus HE. Expression of the Rous-sarcoma virus pol gene by ribosomal frameshifting. Science. 1985;230:1237–1242. doi: 10.1126/science.2416054. [DOI] [PubMed] [Google Scholar]
- Jacks T, Townsley K, Varmus HE, Majors J. 2 efficient ribosomal frameshifting events are required for synthesis of mouse mammary-tumor virus gag-related polyproteins. Proc Natl Acad Sci USA. 1987;84:4298–4302. doi: 10.1073/pnas.84.12.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Madhani HD, Masiarz FR, Varmus HE. Signals for ribosomal frameshifting in the Rous-sarcoma virus gag-pol region. Cell. 1988;55:447–458. doi: 10.1016/0092-8674(88)90031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karacostas V, Wolffe EJ, Nagashima K, Gonda MA, Moss B. Overexpression of the Hiv-1 gag-pol polyprotein results in intracellular activation of Hiv-1 protease and inhibition of assembly and budding of virus-like particles. Virology. 1993;193:661–671. doi: 10.1006/viro.1993.1174. [DOI] [PubMed] [Google Scholar]
- Klikova M, Rhee SS, Hunter E, Ruml T. Efficient in-vivo and in-vitro assembly of retroviral capsids from gag precursor proteins expressed in bacteria. J Virol. 1995;69:1093–1098. doi: 10.1128/jvi.69.2.1093-1098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S, Kubota H, Nagata K. Cytosolic chaperonin protects folding intermediates of G ss from aggregation by recognizing hydrophobic ss-strands. Proc Natl Acad Sci USA. 2006;103:8360–8365. doi: 10.1073/pnas.0600195103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nermut MV, Bron P, Thomas D, Rumlova M, Ruml T, Hunter E. Molecular organization of Mason-Pfizer monkey virus capsids assembled from gag polyprotein in Escherichia coli. J Virol. 2002;76:4321–4330. doi: 10.1128/JVI.76.9.4321-4330.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertle S, Bowles N, Spahr PF. Complementation studies with Rous-sarcoma virus gag and gag-pol polyprotein mutants. J Virol. 1992;66:3873–3878. doi: 10.1128/jvi.66.6.3873-3878.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Morrow CD. Overexpression of the gag-pol precursor from human-immunodeficiency-virus type-1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J Virol. 1991;65:5111–5117. doi: 10.1128/jvi.65.9.5111-5117.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Morrow CD. The nonmyristylated Pr160Gag-Pol polyprotein of human-immunodeficiency-virus type-1 interacts with Pr55Gag and is incorporated into virus-like particles. J Virol. 1992;66:6304–6313. doi: 10.1128/jvi.66.11.6304-6313.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SD, Hunter E. A cell-line-specific defect in the intracellular transport and release of assembled retroviral capsids. J Virol. 2000;74:784–795. doi: 10.1128/jvi.74.2.784-795.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SD, Hunter E. Activation of the Mason-Pfizer monkey virus protease within immature capsids in vitro. Proc Natl Acad Sci. 2001;98:14631–14636. doi: 10.1073/pnas.251460998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SD, Wall JS, Hunter E. Analysis of Mason-Pfizer monkey virus gag particles by scanning transmission electron microscopy. J Virol. 2001;75:9543–9548. doi: 10.1128/JVI.75.19.9543-9548.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridky TW, BizubBender D, Cameron CE, Weber IT, Wlodawer A, Copeland T, Skalka AM, Leis J. Programming the Rous sarcoma virus protease to cleave new substrate sequences. J Biol Chem. 1996;271:10538–10544. doi: 10.1074/jbc.271.18.10538. [DOI] [PubMed] [Google Scholar]
- Rumlova-Klikova M, Hunter E, Nermut MV, Pichova I, Ruml T. Analysis of Mason-Pfizer monkey virus gag domains required for capsid assembly in bacteria: role of the N-terminal proline residue of CA in directing particle shape. J Virol. 2000;74:8452–8459. doi: 10.1128/jvi.74.18.8452-8459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakalian M, Parker SD, Weldon RA, Hunter E. Synthesis and assembly of retrovirus Gag precursors into immature capsids in vitro. J Virol. 1996;70:3706–3715. doi: 10.1128/jvi.70.6.3706-3715.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfakianos JN, Hunter E. M-PMV capsid transport is mediated by Env/Gag interactions at the pericentriolar recycling endosome. Traffic. 2003;4:671–680. doi: 10.1034/j.1600-0854.2003.00126.x. [DOI] [PubMed] [Google Scholar]
- Sfakianos JN, LaCasse RA, Hunter E. The M-PMV cytoplasmic targeting-retention signal directs nascent gag polypeptides to a pericentriolar region of the cell. Traffic. 2003;4:660–670. doi: 10.1034/j.1600-0854.2003.00125.x. [DOI] [PubMed] [Google Scholar]
- Shehu-Xhilaga M, Crowe SM, Mak J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J Virol. 2001;75:1834–1841. doi: 10.1128/JVI.75.4.1834-1841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Srinivasakumar N, Hammarskjold ML, Rekosh D. Requirements for incorporation of Pr160Gag-Pol from human-immunodeficiency-virus type-1 into virus-like particles. J Virol. 1993;67:2266–2275. doi: 10.1128/jvi.67.4.2266-2275.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerfelt MA, Petteway SR, Dreyer GB, Hunter E. Effect of retroviral proteinase-inhibitors on Mason-Pfizer monkey virus maturation and transmembrane glycoprotein cleavage. J Virol. 1992;66:4220–4227. doi: 10.1128/jvi.66.7.4220-4227.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CS, Hunter E. Variable sensitivity to substitutions in the N-terminal heptad repeat of Mason-Pfizer monkey virus transmembrane protein. J Virol. 2003;77:7779–7785. doi: 10.1128/JVI.77.14.7779-7785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonigo P, Barker C, Hunter E, Wainhobson S. Nucleotide-sequence of Mason-Pfizer monkey virus — an immunosuppressive D-type retrovirus. Cell. 1986;45:375–385. doi: 10.1016/0092-8674(86)90323-5. [DOI] [PubMed] [Google Scholar]
- Stansell E, Tytler E, Walter MR, Hunter E. An early stage of Mason-Pfizer monkey virus budding is regulated by the hydrophobicity of the gag matrix domain core. J Virol. 2004;78:5023–5031. doi: 10.1128/JVI.78.10.5023-5031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlach J, Lipov J, Rumlova M, Veverka V, Lang J, Srb P, Knejzlik Z, Pichova I, Hunter E, Hrabal R, Ruml T. D-retrovirus morphogenetic switch driven by the targeting signal accessibility to Tctex-1 of dynein. Proc Natl Acad Sci USA. 2008;105:10565–10570. doi: 10.1073/pnas.0801765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaka Y, Katoh I, Copeland TD, Oroszlan S. Murine leukemia-virus protease is encoded by the gag-pol gene and is synthesized through suppression of an amber termination codon. Proc Natl Acad Sci USA. 1985;82:1618–1622. doi: 10.1073/pnas.82.6.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]