

Abstract
We report on the prediction accuracy of ligand-based (2D QSAR) and structure-based (MedusaDock) methods used both independently and in consensus for ranking the congeneric series of ligands binding to three protein targets (UK, ERK2, and CHK1) from the CSAR 2011 benchmark exercise. An ensemble of predictive QSAR models was developed using known binders of these three targets extracted from the publicly-available ChEMBL database. Selected models were used to predict the binding affinity of CSAR compounds towards the corresponding targets and rank them accordingly; the overall ranking accuracy evaluated by Spearman correlation was as high as 0.78 for UK, 0.60 for ERK2, and 0.56 for CHK1, placing our predictions in top-10% among all the participants. In parallel, MedusaDock designed to predict reliable docking poses was also used for ranking the CSAR ligands according to their docking scores; the resulting accuracy (Spearman correlation) for UK, ERK2, and CHK1 were 0.76, 0.31, and 0.26, respectively. In addition, performance of several consensus approaches combining MedusaDock and QSAR predicted ranks altogether has been explored; the best approach yielded Spearman correlation coefficients for UK, ERK2, and CHK1 of 0.82, 0.50, and 0.45, respectively. This study shows that (i) externally validated 2D QSAR models were capable of ranking CSAR ligands at least as accurately as more computationally intensive structure-based approaches used both by us and by other groups and (ii) ligand-based QSAR models can complement structure-based approaches by boosting the prediction performances when used in consensus.
1. Introduction
The 2011 CSAR benchmark exercise provided the scientific community with the opportunity to evaluate and benchmark the reliability of the various computational approaches for predicting protein-ligand interactions. Four targets were considered: UK (UroKinase), ERK2 (Mitogen-activated protein kinase ERK2), CHK1 (Checkpoint Kinase 1), and LPXC (Pseudomonas Aeruginosa UDP-3-O-acyl-GlcNAc deacetylase). The objectives for every participant were to (i) accurately predict the binding pose of each CSAR ligand, and (ii) rank the series of CSAR ligands for different molecular targets according to an assessment of ligands’ binding affinity for each target.
The first objective of the CSAR exercise was clearly thought as a “classical” benchmarking of molecular docking approaches for predicting native-like, accurate binding poses of new ligands towards known targets. Meanwhile, our group especially welcomed the second objective as a unique opportunity to employ ligand-based approaches for ranking the CSAR ligands and compare their overall ranking reliability with that obtained by structure-based approaches used both by other participants and by our group. It is important to underline that CSAR organizers did not put any restrictions on the use of external publicly-available data, methods, and software.1 The participants were even encouraged to use as many sources of any potentially useful information as possible. We also envisioned the possibility of employing both ligand-based2 and structure-based3,4 approaches and exploring some potential complementarities to rank CSAR ligands more accurately.
In this study, we aimed at assessing and ranking the binding affinities of CSAR ligands using a unique consensus approach (see Table 1) that employed two different types of methods: (i) Quantitative Structure-Activity Relationship (QSAR) models built with known inhibitors of UK, ERK2, and CHK1 using two-dimensional molecular descriptors and machine learning techniques; (ii) MedusaDock molecular docking that predicts both the binding poses of CSAR ligands and the corresponding molecular affinities. Beyond the comparison of the prediction power of QSAR models versus structure-based docking5, we pursued the idea of exploring the benefits of using ligand-based and structure-based approaches in a consensus way instead of contrasting them. Similar hybrid strategies have been rarely explored previously6,7 so we took this benchmarking exercise as an opportunity to test such methodology further.
Table 1.
List of approaches used in this study to rank CSAR ligands (see the text for more details).
| Type | Name | ID | Description |
|---|---|---|---|
| Ligand-based (2D) |
QSAR_no_AD | 1 | Consensus QSAR model averaging RF/SiRMS and SVM/Dragon predictions without applicability domain filtering |
| QSAR_AD | 2 | Consensus QSAR model with applicability domain filtering |
|
| Structure- based (3D) |
MEDUSA | 3 | Molecular docking |
| Consensus 2D/3D |
QSAR_no_AD + MEDUSA |
4 | Consensus between models 1 and 3 |
|
QSAR_AD + MEDUSA |
5 | Consensus between models 2 and 3 |
The main goal of this study was to reliably assess the relative ranking of CSAR ligands by predicting their potency towards given kinase targets. To achieve this goal we used cheminformatics approaches to (i) collect and curate chemical data extracted from ChEMBL related to binding towards UK and ERK2, and inhibition towards CHK1 and LPXC; (ii) develop statistically robust, validated, and externally predictive QSAR models to compute CSAR ligands’ activities and rank them accordingly; and (iii) combine QSAR and structure-base docking predictions into consensus relative ranking lists. The results of our studies suggest that ligand-based QSAR approaches can perform similarly or even better than computationally expensive, structure-based approaches. Moreover, we also show potential benefits coming from the synergistic use of both approaches as compared to single method predictions. These benefits mainly relate to the identification and subsequent overriding of activity cliffs (i.e., very similar compounds with dissimilar activities) by enriching the predictions from one structural space (2D or 3D) with the ones from another.
Here we only report on the results of QSAR modeling and our consensus approach; all results and discussion related to the prediction of CSAR ligand binding poses (and their overall accuracy) by MedusaDock are published in a separate study.8
2. Methods
2.1. Dataset Preparation
2.1.1. Data sources
For each target, we extracted all known associated ligands from the ChEMBL version 12 database.9 For the UK target (CHEMBL3286), a total of 828 binding affinity (Ki) values were retrieved. Approximately 1,450 IC50 values were found for CHK1 (CHEMBL4630), whereas only 91 Ki values were retrieved for ERK2 (CHEMBL4040). For all three sets, we did not consider integrating experimental data coming from qHTS assays, mixing IC50 and Ki values, or adding data from other sources.
The fourth target of the CSAR benchmark exercise, LPXC, was excluded from our study due to the insufficient amount of data available for QSAR modeling. The LPXC-related set extracted from ChEMBL included 53 compounds with exact IC50 values. Among them, only 11 unique compounds had IC50 below 1µM and the overall distribution of pIC50 had a narrow range from 4.2 (inactive compounds) to 6.9 (weakly active compounds) with a strong distribution bias towards inactives. Thus, QSAR analysis of this dataset was not feasible; nevertheless, we have examined whether accurate predictions of LPXC binding affinity for CSAR ligands can be achieved based on global chemical similarity considerations. Indeed, among the 16 new ligands provided by the CSAR organizers, we found a few compounds highly similar to some of the 53 ChEMBL compounds using simple 2D similarity metric (Tanimoto coefficient threshold higher or equal than 0.85). Further examination showed that pairs of highly similar compounds had very similar binding affinities indeed. For instance, only one chlorine group differentiates CSAR_lpxc_11 (pIC50 = 4.7) from ChEMBL107127 (pIC50 = 4.66); CSAR_lpxc_14 (pIC50 = 5.6) was very similar to ChEMBL324440 (pIC50 = 5.80), CHEMBL104577 (pIC50 = 4.51), CHEMBL104043 (pIC50 = 5.92), and CHEMBL107004 (pIC50 = 5.26); CSAR_lpxc_11 (pIC50 = 4.7) was also very similar to CHEMBL104671 (pIC50 = 4.52). This analysis suggests that, even in the absence of sufficiently large amounts of data to enable QSAR modeling, it is still possible to obtain accurate prediction for a subset of LPXC ligands using cheminformatics techniques.
2.1.2. Dataset curation
The compounds retrieved from the ChEMBL database were preprocessed according to a set of guidelines for chemical data curation and standardization that our group published recently.10 Briefly, after the removal of counterions, structures were standardized and converted into canonical tautomeric form with neutral representation and explicit hydrogens. As illustrated in Table 2, only 48 out of 91 compounds remained in the ERK2 dataset after the curation steps including the deletion of stereoisomers and the compounds with uncertain and approximate Ki values. In the end, pKi values for the 48 selected compounds were ranging from 4.60 to 8.70. Similarly, 717 compounds (out of 899 total) were still present in the UK set after curation and their pKi values were ranging from 0.30 to 11. Lastly, 1215 out of 1450 compounds remained in the CHK1 dataset with pIC50 values ranging between 3.68 and 10.
Table 2.
CSAR targets, ligands, and related compounds found in the ChEMBL database.
| Target | Potency measured as |
Number of CSAR ligands to rank |
Number of ChEMBL compounds before curation |
Number of ChEMBL compounds after curation |
|---|---|---|---|---|
| Extracellular signal-regulated kinase (ERK2) |
pKi | 39 | 91 | 48 |
| Urokinase (UK) | pKi | 20 | 828 | 668 |
| Checkpoint kinase (CHK1) |
pIC50 | 45 | 1450 | 1215 |
2.2. Molecular Descriptors
2.2.1. Dragon descriptors
The following types of descriptors were generated using Dragon software (v.5.5, Talete SRL, Milan, Italy): 0D-constitutional descriptors (atom and group counts), 1D-functional groups, 1D-atom centered fragments, 2D-topological descriptors, 2D-walk and path counts, 2D-autocorrelations, 2D-connectivity indices, 2D-information indices, 2D-topological charge indices, 2D-Eigenvalue-based indices, 2D-topological descriptors, 2D-edge adjacency indices, 2D-Burden eigenvalues, and various molecular properties such as octanol-water partition coefficient. Descriptors with low variance (standard deviation lower than 10−4) or missing values were removed. Furthermore, if the correlation coefficient between any two descriptors exceeded 95%, one of them was removed. The remaining descriptors were range-scaled, so that their values were within the interval [0, 1]. Definition and calculation procedures for Dragon descriptors and the related references are given in the Handbook of Molecular Descriptors.11
2.2.2. SiRMS descriptors
HiT QSAR Software12 based on Simplex representation of molecular structure (SiRMS)13,14 was used for generating 2D Simplex descriptors, i.e., number of tetratomic fragments with fixed composition and topological structure. At the 2D level, the connectivity of atoms in simplex, atom type, and bond nature (single, double, triple, or aromatic) have been considered. SiRMS descriptors account not only for the atom type, but also for other atomic characteristics that may impact biological activity of molecules, e.g., partial charge, lipophilicity, refraction, and atom ability for being a donor/acceptor in hydrogen-bond formation (H-bond). For atom characteristics with continuous values (charge, lipophilicity, refraction) the range was converted into several discrete groups. The atoms have been divided into four groups corresponding to their (i) partial charge A≤−0.05<B≤0<C≤0.05<D, (ii) lipophilicity A≤−0.5<B≤0<C≤0.5<D, and (iii) refraction A≤1.5<B≤3<C≤8<D. For atomic H-bond characteristic the atoms have been divided into three groups: A (acceptor of hydrogen in H-bond), D (donor of hydrogen in H-bond), and I (indifferent atom). The usage of sundry variants of differentiation of simplex vertexes (atoms) represents the principal feature of SiRMS approach.15 Detailed description of HiT QSAR based on SiRMS could be found elsewhere.12–14 Constant, low-variance, and correlated (|R| ≥ 0.9) descriptors were excluded prior to modeling. Thus, descriptor pools of 435–889 Simplex descriptors (depending on the dataset) were selected for the statistical processing.
2.3. QSAR Modeling
In this study, we developed a series of QSAR models following the workflow and other guidelines we published elsewhere.10,16 The QSAR modeling workflow can be divided into three major steps2,16: (i) data curation, preparation, and analysis, (ii) model building, and (iii) model validation/selection. Here we followed a 5-fold external cross-validation procedure: for each CSAR target, the full set of compounds with known experimental activity was randomly split into five modeling (80% of the full set) and external validation sets (remaining 20%). Models were built using the modeling set compounds only, and it is important to emphasize that the external set compounds were never taken into account to build and/or select the models. Briefly, each modeling set was split into many training and test sets for SVM method and plethora of training and out-of-bag set for RF approach; then the models were built using the compounds belonging to each training set and applied to test set compounds for assessing their properties. Pearson’s correlation coefficient (R2), Root Mean Square Error (RMSE), and Spearman’s rank correlation coefficient (ρ) were used to assess the prediction performances of developed models.
Best models were identified and selected according to estimated R2 values for test set (SVM) or out-of-bag set (RF). Then, selected models were applied to the external set compounds to predict their experimental properties. This overall procedure is repeated five times to ensure that every compound from the full set is present once (and only once) in the external test set. While compounds were present in the external test sets, they have never been used to derive, bias, or select the models; thus, the entire procedure gives more or less fair estimation of the true predictivity of the models. In addition, 1000 rounds of Y-randomization were performed for each selected model in order to avoid chance correlations.
Model’s Applicability Domain (AD) aims to determine whether the given model is capable of predicting the activity of a query compound within a reasonable error.16 In this study, we defined the AD of SVM models as a threshold distance DT between a query compound and its nearest neighbors in the training set. If the distance of the test compound from any of its k nearest neighbors in the training set exceeds the threshold, the prediction is considered unreliable. For RF models the AD was estimated using the local (Tree) approach that was described by Artemenko et al.17
2.4. Random Forest (RF)
Random Forest models were constructed according to the original RF algorithm18 implemented by Polischuk et al.19 RF is an ensemble of single decision trees. Outputs of all trees are aggregated to obtain one final prediction. Each tree has been grown as follows: (i) a bootstrap sample was produced from the whole set of N compounds to form a training set for the current tree. Compounds which are not in this current tree training set are placed in the out-of-bag (OOB) set (OOB set size is ~ N/3); (ii) the best split by CART algorithm20 among the m randomly selected descriptors from the entire pool in each node is chosen. Value of m is just one tuning parameter for which RF models are sensitive; (iii) each tree is then grown to the largest possible extent. There is no pruning. Prediction of out-of-bag set is made but each tree predicts values only for compounds which are not included into training set of that tree (for OOB set only). Since RF possesses its own reliable statistical characteristics (based on OOB set prediction) which could be used for validation and model selection, no cross-validation has been performed18. Thus, the final model is chosen by lowest error for prediction of OOB set and only after that resulting model was applied for blind prediction of external test set/fold compounds.
2.5. Support Vector Machines (SVM)
The description of the original SVM algorithm could be found in many publications.21 Briefly, molecular descriptors are first mapped onto a high dimensional feature space using various kernel functions. Then, SVM finds a separating hyperplane with the maximal margin in this high dimensional space in order to separate compounds with different activities. Models built with this machine learning technique allow the prediction of a target property using a set of descriptors solely calculated from the structure of a given compound. In this study, we used the WinSVM program developed in our group on the basis of the open-source libSVM package. The WinSVM program provides users with a graphical interface to prepare input data; split datasets into training and test sets; set up parameters for SVM grid calculations, including iterative and simultaneous grid optimization of SVM parameters; launch and follow calculation progress via a powerful graphical interface; select models with the best prediction accuracy for both training and internal test sets; and to apply them to the external evaluation set as an ensemble consensus model. The program also allows one to visualize molecular structures and various plots, making the use of SVM easier and more appropriate for QSAR modeling in order to obtain robust and predictive models and apply them to virtual libraries. WinSVM is freely available for academic laboratories from the following web site: http://www.unc.edu/~fourches/.
2.6. MedusaDock
The MedusaDock software22 was used to generate and score all ligand-receptor binding poses for the different CSAR targets. MedusaDock performs conformational sampling of both ligand and receptor side chains simultaneously and synergistically. Details of the docking method can be found elsewhere.8,23 Briefly, a library of ligand rotamers is generated in a stochastic manner “on the fly”: ligand conformations are explored by random variation of ligands’ rotatable angles and excluding those conformations that feature atomic clashes. The docking protocol involves two steps. First, a representative set of ligand conformations is generated by clustering the stochastic library of rotamers. Each representative conformation is rapidly fitted into a “smoothed” receptor pocket by disabling the van der Waals repulsion between the ligand and the receptor side chains and subsequent rigid-body docking. Second, fine-docking is performed from each of the coarsely-docked poses, where the binding pose is minimized by iterative repacking of the rotamers of ligand and receptor side chains as well as ligand rigid-body minimization. In the second fine-docking step, the van der Waals repulsions between ligand and receptor side chains are included. The MedusaScore scoring function was utilized to guide the docking.23
3. Results and Discussion
3.1. Presence of CSAR duplicates in the ChEMBL modeling set
First, for each target, we used ISIDA/Duplicates software to search for structural duplicates between CSAR ligands and the compounds retrieved from the ChEMBL database. We were not expecting to find any duplicate compounds assuming that none of the “blind set” CSAR ligands were supposed to be in the public domain already. Surprisingly, we identified several CSAR ligands that were indeed present in the ChEMBL sets with known experimental affinities for the targets of interest. Results are summarized in Table 3.
Table 3.
List of CSAR compounds with identified structural duplicates retrieved in the ChEMBL database and their reported activities.
| CSAR compounds (experimental activity) |
ChEMBL compounds identical to CSAR compounds |
|---|---|
|
CSAR_chk1_1 (CSAR pIC50=7.60) |
CHEMBL401274; pIC50=7.60 4-(6,7-dimethoxy-2,4-dihydro-indeno[1,2- c]pyrazol-3-yl)-phenol; |
|
CSAR_chk1_3 (CSAR pIC50=8.30) |
CHEMBL248396; pIC50=8.30 4-(6,7-dimethoxy-2,4-dihydro-indeno[1,2- c]pyrazol-3-ylethynyl)-2-methoxy-phenol; |
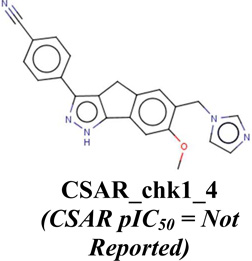 |
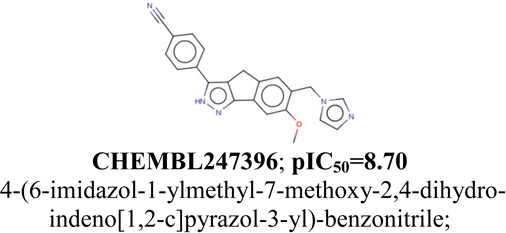 |
|
CSAR_chk1_6 (CSAR pIC50=8.80) |
CHEMBL245796; pIC50=8.80 4'-(6,7-dimethoxy-1,4-dihydro-indeno[1,2- c]pyrazol-3-yl)-biphenyl-4-ol; |
|
CSAR_chk1_13 (CSAR pIC50=7.64) |
CHEMBL248010; pIC50=7.64 4'-{6-[2-(5-ethyl-pyridin-2-yl)-ethoxy]-7-methoxy- 2,4-dihydro-indeno[1,2-c]pyrazol-3-yl}-biphenyl- 4-ol; |
|
CSAR_chk1_20 (CSAR pIC50=4.80) |
CHEMBL396034; pIC50=4.77 8-chloro-5,10-dihydro-dibenzo[b,e][1,4]diazepin- 11-one; |
|
CSAR_uk_18 (CSAR pKi=6.30) |
CHEMBL319264; pKi=6.35 8-Amino-naphthalene-2-carboxamidine; |
|
CSAR_erk2_30 (CSAR pKi=7.10) |
CHEMBL220320; pKi=7.07 N-benzyl-4-(4-(3-chlorophenyl)-1H-pyrazol-3-yl)- 1H-pyrrole-2-carboxamide; |
In total, six duplicates were found in the CHK1 dataset: 5 out of 6 compounds were CHK1 inhibitors with pIC50 ranging from 7.60 (ChEMBL401274) to 8.80 (ChEMBL245796). The sixth compound (ChEMBL396034) was annotated as being inactive (pIC50 = 4.77). Only one duplicated structure was identified for both the UK dataset (CSAR_UK_18 with ChEMBL319264) and the ERK2 dataset (CSAR_ERK2_30 with ChEMBL220320).
When submitting our prediction results to CSAR organizers, we enclosed this list of structural duplicates and a letter underlining that (i) some CSAR compounds and their supposedly unknown experimental activities were indeed present in the public domain and thus could potentially bias the results of the overall benchmark, (ii) simple methods such as similarity search can easily identify them, and (iii) our group honestly acknowledged that we knew the experimental values for those duplicate compounds and thus we advocated for the removal of those compounds from the CSAR ligand set in order to calculate unbiased statistics between the different participants. Although these ligands were present in the training sets we used to develop models, our group submitted only the values obtained from the 5-fold external cross-validation when these compounds were blindly predicted.
Later when the experimental activities of all CSAR ligands were revealed, we indeed observed their perfect agreement with the values retrieved from the ChEMBL database (see Table 3).
3.2. Prediction performance of QSAR models
QSAR modeling results are given in Table 4. Models built using the SiRMS fragment descriptors and Random Forests afforded reasonable prediction performances evaluated by Spearman rank correlation ρ ranging from 0.78 (CHK1) to 0.85 (UK). When considering models' applicability domains, the reliability of RF predictions increased (up to ρ=0.89 for UK) but the coverage decreased, i.e., ca. 25% of the compounds had to be excluded due to the model applicability domain.
Table 4.
Statistical characteristics of QSAR models for CSAR datasets assessed by 5-fold external validation.
| Method | Descriptor | Target | NO AD | WITH AD | |||||
|---|---|---|---|---|---|---|---|---|---|
| R2 | RMSE | Spearman ρ |
R2 | RMSE | Spearman ρ |
Coverage | |||
| RF | SiRMS |
CHK1 (n = 1215) |
0.64 | 0.77 | 0.78 | 0.72 | 0.66 | 0.85 | 75% |
| SVM | Dragon | 0.64 | 0.76 | 0.77 | 0.65 | 0.73 | 0.81 | 72% | |
| QSAR_CONSENSUS | 0.67 | 0.74 | 0.79 | 0.68 | 0.71 | 0.83 | 87% | ||
| RF | SiRMS |
ERK2 (n = 48) |
0.71 | 0.62 | 0.80 | 0.69 | 0.56 | 0.79 | 75% |
| SVM | Dragon | 0.62 | 0.68 | 0.77 | 0.59 | 0.65 | 0.72 | 73% | |
| QSAR_CONSENSUS | 0.69 | 0.62 | 0.79 | 0.67 | 0.59 | 0.76 | 90% | ||
| RF | SiRMS |
UK (n = 668) |
0.69 | 0.66 | 0.85 | 0.77 | 0.53 | 0.89 | 75% |
| SVM | Dragon | 0.68 | 0.72 | 0.84 | 0.70 | 0.64 | 0.86 | 71% | |
| QSAR_CONSENSUS | 0.71 | 0.68 | 0.87 | 0.73 | 0.61 | 0.88 | 88% | ||
The prediction power of SVM models based on Dragon descriptors was slightly lower than that of RF models with ρ ranging from 0.77 (CHK1) to 0.84 (UK). In particular, ERK2 predictions were less accurate with R2 going down from 0.71 (RF) to 0.62 (SVM). With applicability domain, ranking accuracy of SVM models were ranging from ρ= 0.72 (ERK2) to 0.86 (UK).
In addition to the individual RF and SVM models, we also explored the predictive power of the simple consensus prediction where activities for external compounds were predicted as simple arithmetic means of predictions made with RF and SVM models. Obtained results showed that in most cases, with or without taking into account models’ applicability domain, the consensus model was consistently achieving higher reliability compared to any of the individual QSAR models. For instance, the modeling results obtained for UK were as follows: without applicability domain filtering, RF models afforded very good performance (ρ = 0.85, R2 = 0.69) as well as SVM models (ρ = 0.84, R2 = 0.68). The consensus model improved the accuracy reaching up to ρ = 0.87 and even ρ = 0.88 taking into account the applicability domain. Importantly, the coverage of the consensus is significantly boosted from 71–75% (individual SVM and RF models) up to 88%. This result means that more compounds were predicted correctly compared to individual QSAR models.
3.3. Application of QSAR models to CSAR Ligands
The results of activity prediction for CSAR ligands are given in Table 5. To match the ranking metric used by the organizers, ranking performance was evaluated using the Spearman correlation coefficient ρ expressing the ranking accuracy of ligands by comparing the ligands’ rank orders based on model's predicted potency (pKi or pIC50 depending on the target) with the actual experimental rank provided by the CSAR organizers.1 As discussed below, model predictive accuracy was evaluated both for all ligands as was stipulated by the CSAR challenge organizers as well for ligands found within the AD of QSAR models only to follow our standard modeling workflow (see Methods).
Table 5.
Spearman correlation coefficient (ρ) between experimental and predicted ranks of CSAR ligands.
| Type | Ranking Methods | ρ | ||
|---|---|---|---|---|
| UK (n=20) |
ERK2 (n=39) |
CHK1 (n=45) |
||
| 2D | QSAR_no_AD | 0.77 | 0.60 | 0.55 |
| QSAR_AD | 0.78 | 0.59 | 0.55 | |
| 3D | MEDUSA | 0.76 | 0.31 | 0.26 |
| 2D/3D |
QSAR_no_AD + MEDUSA |
0.79 | 0.48 | 0.45 |
|
QSAR_AD + MEDUSA |
0.82 | 0.50 | 0.45 | |
When predicting all ligands in the absence of any AD, the QSAR models afforded relatively high accuracy for UK ligands (ρ= 0.77, n = 20) and lower accuracies for ERK2 (ρ= 0.60, n = 39) and CHK1 (ρ= 0.55, n = 45) ligands (Table 5; QSAR_no_AD). Independently, we have employed an ad hoc scheme to make predictions for all compounds when using the respective AD thresholds for individual RF and SVM models. Thus, for compounds found either within or outside of the AD of the individual models the predicted activities were averaged whereas for compounds found within the AD of only one model the activity predicted by that model was used. As shown in Table 5, the prediction accuracy for this QSAR_AD model was similar to that for the QSAR_no_AD model.
In addition, we also made predictions for ligands within the models’ AD only, i.e., with reduced coverage of the CSAR datasets. Indeed, many CSAR ligands were found to be outside of the respective AD of either SVM or RF models. Certain ligands were even outside of the AD of both models: 14 compounds for UK, 8 compounds for ERK2, and 13 compounds for CHK1. Only six out of 20 UK ligands were found to be within the AD of one of the models making it non-sensible to evaluate model prediction accuracy in this case. After removing compounds outside of the AD, the Spearman correlation coefficients between experimental and predicted ranks (QSAR_AD model) for the remaining CSAR compounds increased to 0.64 for both ERK2 (n=31, coverage = 79.5%) and CHK1 (n=32, coverage = 71.1%) datasets as compared to 0.59 and 0.55, respectively, when all compounds were considered (see Table 5). Thus, the effect of AD on prediction accuracy of QSAR models is dataset dependent. In one of the considered cases (UK), the default AD appears over-restrictive whereas in two other cases the use of AD slightly improves model accuracy but at the expense of reduced data coverage, which is typical for QSAR-based predictions.
Second, we analyzed the results obtained by using the MedusaDock scores for ranking the CSAR ligands. MedusaDock was as accurate as QSAR models for UK. However, the QSAR models were found to have twice as high predictive power than MedusaDock for ERK2 (ρ= 0.60 versus ρ= 0.31) and CHK1 (ρ= 0.55 versus ρ= 0.26).
Overall, we could make the following observations: (i) the “true” accuracy of QSAR models and MedusaDock for ranking CSAR ligands is slightly (UK) or significantly (CHK1) worse than the one found at the modeling and validation stages, (ii) ligand-based QSAR models performed better than computationally expensive molecular docking, and (iii) QSAR models’ applicability domains in their current form do not significantly improve the overall prediction accuracy for the remaining compounds.
3.4. Consensus scoring using QSAR predictions and MedusaDock
As part of the exercise, we considered another type of consensus models including the predictions coming from both QSAR models and Medusa docking. Ranks for CSAR ligands predicted by the QSAR models (e.g., QSAR_no_AD) were added to the ranks predicted by Medusa docking. Then, the ligands were re-ranked based on these summed QSAR/Medusa ranks. The overall results are shown in Table 5.
The accuracy of QSAR/Medusa consensus predictions was higher than the accuracy reached by Medusa predictions for all three targets. This remark is particularly true for CHK1 and ERK2 for which QSAR/Medusa consensus model was found to be almost twice more predictive than Medusa alone: in the case of ERK2 for instance, ρ=0.48 compared to ρ=0.31 respectively.
Also we noticed that the QSAR/Medusa consensus predictions afforded higher ranking accuracy than individual QSAR models and MedusaDock only in the case of UK (ρ=0.79–0.82 versus ρ=0.76–0.78). Due to the higher ranking accuracy obtained by QSAR models over Medusa for both ERK2 and CHK1, this result was expected.
3.5. Half success or half failure?
The analysis of the results revealed the overall reliability of QSAR models to rank CSAR ligands from the most active to the most inactive, especially for UK. However, there is a significant portion of ligands that have been mispredicted by both QSAR and docking. Among them, some compounds predicted to be active were confirmed as being weak active or inactive. In this section, we are giving some examples and some clues to improve our current approach based on what we learned in this exercise.
First, the results tend to contradict the general principle commonly trusted by the QSAR community posing that the bigger the modeling set is, the more predictive the model will be. In this CSAR benchmark, our largest modeling set included 1,215 compounds for the CHK1 target. Although QSAR models developed using this large dataset afforded reasonable prediction power in the 5-fold external cross-validation procedure (Table 4), the set of 45 CSAR ligands tested towards CHK1 was the most difficult to annotate as shown by the results: Spearman ρ = 0.55 for QSAR models as compared to ρ = 0.59–0.78 for UK and ERK2, ρ = 0.26 for docking as compared to ρ = 0.31–0.76 for UK and ERK2. Besides the ranking accuracy per se, the consensus QSAR model was indeed able to correctly predict 17 out of 21 CHK1 actives and 10 out of 24 inactive compounds, but missed 14 false positives and 4 false negatives (sensitivity = 0.81, specificity = 0.42, and balanced accuracy = 0.61 considering the activity threshold of pIC50 = 7). Out of these 18 mispredicted compounds, we should underline that 8 compounds have their experimental pIC50 ranging from 6.5 to 7.7, which is very close to the activity threshold we used to separate active from inactive compounds.
The smallest modeling set (ERK2) included 48 compounds only. Nevertheless, QSAR models built for this small modeling set afforded relatively good prediction performance at the 5-fold cross-validation stage (R2=0.69, RMSE=0.62, and ρ=0.79) and reasonable reliability on CSAR ligands (ρ=0.60). As illustrated on Figure 1, the balanced accuracy is reaching 0.77 for the 39 CSAR ligands tested towards ERK2 using an affinity threshold of pKi = 7 to distinguish active from inactive compounds. These results demonstrate once again the importance of the cross-validation procedures in the QSAR modeling workflow but also the fact that such procedures must involve the building and selection of QSAR models using the modeling sets only and a truly external validation with the test sets.
Figure 1.

Experimental versus predicted binding affinities (pKi) for ERK2 CSAR ligands (n = 39) based on QSAR_no_AD model’s predictions. Correctly predicted ERK2 binders (pKi ≥ 7) are colored in green, whereas correctly predicted non-binders are colored in red (balanced accuracy = 0.77). Mispredicted compounds are colored in black.
Second, the overall accuracy of 2D QSAR models was affected by the presence of large activity cliffs in both the modeling and the external sets of ligands. To illustrate this point, let’s consider again the example of ERK2 ligands and more precisely the CSAR_ERK2_1 compound. As shown in Figure 2, very similar structures to CSAR_ERK2_1 found in our modeling set are annotated as strong binders with pKi equal to 8.4 and above. It is thus not surprising that our QSAR models computed CSAR_ERK2_1’s affinity towards ERK2 to be approximately pKi = 6.9. However the experimental binding affinity has been determined to be 4.8 only. This perfectly corresponds to the case of activity cliffs.24
Figure 2.

Structural neighbors of the CSAR_ERK2_1 compound retrieved in the ChEMBL database.
In Figure 2, we showed CSAR_ERK2_1 as well as two other compounds CSAR_ERK2_6 and CSAR_ERK2_9 that have not been assessed correctly by our QSAR models. Despite the fact that the QSAR model succeeded to correctly predict the increasing activity trend CSAR_ERK2_1 (pKi = 4.8) < CSAR_ERK2_6 (pKi = 6.1) < CSAR_ERK2_9 (pKi = 6.8), the model did calculate their binding affinities with ΔpKipred-exp > 1.5 log units. Interestingly, the docking score obtained for CSAR_ERK2_1 is relatively high (−42.2) meaning that the binding of the compound is predicted to be unfavorable. To provide the necessary context, MedusaDock scores were ranging from −59.2 (very favorable docking) to −36.4 (unfavorable docking) for the ERK2 CSAR ligands. As a result, this observation opens the way for defining new strategies to calculate consensus predictions between QSAR models and docking scores as well as identifying potential activity cliffs such as CSAR_ERK2_1. Simply summing up the ranks from QSAR models and docking scores as we did in this exercise does not seem to be the optimal workflow. On the contrary, using docking score thresholds to automatically discard some compounds from the predicted actives is more likely to avoid the prediction of false-positives such as CSAR_ERK2_1.
Third, based on the results presented in this study, there are some additional evidences how to complement structure-based predictions from ligand-based predictions. On Figures 3 and 4, we plotted the MedusaDock scores versus QSAR_no_AD predictions. The 2D/3D correlation reached R2 = 0.67 for UK and only 0.42 for CHK1. These values are indeed important to analyze because they measure the level of concordance between the two different modeling approaches for the CSAR ligands and can be computed without the knowledge of the experimental values of the compounds. The challenge is thus to find new ways to use these correlation plots for establishing rules to calculate a new type of 2D/3D consensus. Also, it seems logical that one way to assess the potential benefit of the 2D/3D consensus requires the calculation of their correlation coefficient for modeling set compounds (and thus there is a need for docking the modeling set compounds as well).
Figure 3.

MedusaDock scores plotted versus QSAR_no_AD pKi predictions for the 20 CSAR ligands towards UK (R2 = 0.67). UK binders (pKi ≥ 7) are colored in green, whereas non-binders are colored in red.
Figure 4.

MedusaDock scores plotted versus QSAR_no_AD pKi predictions for the 45 CSAR ligands towards CHK1 (R2 = 0.42). CHK1 inhibitors (pIC50 ≥ 7) are colored in green, whereas inactive compounds are colored in red.
Fourth, compared to the other research teams who participated in the CSAR benchmarking, the reasonable prediction performances obtained by our QSAR models ranked our group in the top-10%. Moreover, our QSAR models occupied top-2 and top-3 positions for ranking both CHK1 actives and inactives. Our models were ranked fifth for ERK 2 prediction reliability. We have processed only three targets and the overall performance among all the targets cannot be estimated for our models, but based on the results for separate targets we can expect that our group was ranked among top-3 research teams.
Overall, structure-based approaches may not be viewed as intuitively better or more predictive than ligand-based QSAR models and this CSAR benchmarking exercise serves to illustrate this point. It is well-known that correlation between docking scoring functions and experimental binding affinities is typically low25 or moderate26. Furthermore, as shown by our collaborators at UNC for the same CSAR sets8, structure-based approaches (and MedusaDock especially) are accurate in generating native-like poses. However, as this study shows, the docking scores for those native-like poses do not correlate with experimental binding affinities well (Table 5) and thus do not allow a correct ranking. This observation highlights a known fact that different scoring functions are needed for predicting ligand poses versus predicting binding affinities. Lastly, we should stress that unlike universal scoring functions used in docking studies, QSAR models are specifically trained and selected towards a given target using a set of respective ligands with experimental activities. Thus, it may be underappreciated but not necessarily surprising that ligand-based QSAR models can, in fact, have better accuracy than most of the structure-based docking approaches in prognosticating target-specific ligand binding affinities.
4. Conclusions
In this study, both structure-based (molecular docking) and ligand-based (QSAR models) approaches were used both independently and in the form of a 2D/3D consensus model to rank untested ligands based on their predicted potency. In this exercise of blind predictions, QSAR models developed with publicly-available experimental data extracted from the ChEMBL database were shown to outperform predictions obtained by several molecular docking approaches. These results confirmed that when QSAR models are rigorously derived using curated chemical datasets and statistically relevant procedures for model selection and validation, then their prediction power can be at least as accurate as computationally expensive structure-based docking. Our results also emphasized the validity of QSAR models as a critical component of a virtual screening platform. Moreover, we showed the potential benefits of using both QSAR and docking predictions altogether to assess and eventually override the presence of activity cliffs in the sets of ligands. However, in this particular CSAR benchmark we did not notice a dramatic boost in predictions’ accuracy using the current implementation of our QSAR/docking consensus model. We believe the CSAR benchmark represents a great initiative to honestly benchmark (i) structure-based scoring functions and docking software with each other as well as with (ii) ligand-based cheminformatics methods, whose prediction accuracy will continue to rise along with the increasing number of experimental data available in online repositories.
Acknowledgements
The authors gratefully thank the help of Drs. Ashutosh Tripathi (UNC) and Regina Politi (UNC) for fruitful discussions. The authors also acknowledge the financial support of NIH (grant GM66940 to A.T. and R01GM080742 to N.V.D.) and EPA (RD 83382501 and R832720).
References
- 1.Damm-Ganamet KL, Smith RD, Dunbar JB, Stuckey JA, Carlson HA. Journal of chemical information and modeling. 2013 doi: 10.1021/ci400025f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tropsha A. Molecular Informatics. 2010;29:476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 3.Bursulaya BD, Totrov M, Abagyan R, Brooks CL. Journal of computer-aided molecular design. 2003;17:755–63. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y, Shoichet BK. Nature chemical biology. 2009;5:358–64. doi: 10.1038/nchembio.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain SV, Ghate M, Bhadoriya KS, Bari SB, Chaudhari A, Borse JS. Organic and medicinal chemistry letters. 2012;2:22. doi: 10.1186/2191-2858-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsieh J-H, Yin S, Liu S, Sedykh A, Dokholyan NV, Tropsha A. Journal of chemical information and modeling. 2011;51:2027–35. doi: 10.1021/ci200146e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scotti L, Jaime Bezerra Mendonca F, Junior, Rodrigo Magalhaes Moreira D, Sobral da Silva M, R Pitta I, Tullius Scotti M. Current topics in medicinal chemistry. 2012;12:2785–809. doi: 10.2174/1568026611212240007. [DOI] [PubMed] [Google Scholar]
- 8.Ding F, Dokholyan NV. Journal of chemical information and modeling. 2012 [Google Scholar]
- 9.ChEMBL Database. [accessed Mar 13, 2013]; https://www.ebi.ac.uk/chembl/
- 10.Fourches D, Muratov E, Tropsha A. Journal of Chemical Information and Modeling. 2010;50:1189–1204. doi: 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Todeschini R, Consonni V. In: Handbook of Molecular Descriptors. Todeschini R, Consonni V, editors. Vol. 11. Wiley-VCH Verlag GmbH; Weinheim, Germany: 2000. p. 667. [Google Scholar]
- 12.Kuz’min VE, Artemenko AG, Muratov EN. Journal of computer-aided molecular design. 2008;22:403–421. doi: 10.1007/s10822-008-9179-6. [DOI] [PubMed] [Google Scholar]
- 13.Muratov EN, Artemenko AG, Varlamova EV, Polischuk PG, Lozitsky VP, Fedchuk AS, Lozitska RL, Gridina TL, Koroleva LS, Sil’nikov VN, Galabov AS, Makarov Va, Riabova OB, Wutzler P, Schmidtke M, Kuz’min VE. Future medicinal chemistry. 2010;2:1205–1226. doi: 10.4155/fmc.10.194. [DOI] [PubMed] [Google Scholar]
- 14.Kuz’min VE, Artemenko AG, Muratov EN, Volineckaya IL, Makarov Va, Riabova OB, Wutzler P, Schmidtke M. Journal of medicinal chemistry. 2007;50:4205–4213. doi: 10.1021/jm0704806. [DOI] [PubMed] [Google Scholar]
- 15.Artemenko A, Muratov E, Kuz’min V, Kovdienko N, Hromov A, Makarov V, Riabova O, Wutzler P, Schmidtke M. The Journal of antimicrobial chemotherapy. 2007;60:68–77. doi: 10.1093/jac/dkm172. [DOI] [PubMed] [Google Scholar]
- 16.Tropsha A, Golbraikh A. Current pharmaceutical design. 2007;13:3494–3504. doi: 10.2174/138161207782794257. [DOI] [PubMed] [Google Scholar]
- 17.Artemenko AG, Muratov EN, Kuz’min VE, Muratov NN, Varlamova EV, Kuz’mina AV, Gorb LG, Golius A, Hill FC, Leszczynski J, Tropsha A. SAR and QSAR in environmental research. 2011;22:575–601. doi: 10.1080/1062936X.2011.569950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breiman L. Machine Learning. 2001;45:5–32. [Google Scholar]
- 19.Polishchuk PG, Muratov EN, Artemenko AG, Kolumbin OG, Muratov NN, Kuz’min VE. Journal of chemical information and modeling. 2009;49:2481–8. doi: 10.1021/ci900203n. [DOI] [PubMed] [Google Scholar]
- 20.Breiman L, Friedman JH, Olshen RA, Stone CJ. Classification and Regression Trees. Belmont: Wadsworth Publishing; 1984. p. 358. [Google Scholar]
- 21.Vapnik VN. The Nature of Statistical Learning Theory. New York: Springer-Verlag; 1995. [Google Scholar]
- 22.Ding F, Yin S, Dokholyan NV. Journal of chemical information and modeling. 2010;50:1623–1632. doi: 10.1021/ci100218t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin S, Biedermannova L, Vondrasek J, Dokholyan NV. J Chem Inf Model. 2008;48:1656–1662. doi: 10.1021/ci8001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maggiora GM. Journal of chemical information and modeling. 2006;46:1535. doi: 10.1021/ci060117s. [DOI] [PubMed] [Google Scholar]
- 25.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. J.Med.Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 26.Biesiada J, Porollo A, Velayutham P, Kouril M, Meller J. Human genomics. 2011;5:497–505. doi: 10.1186/1479-7364-5-5-497. [DOI] [PMC free article] [PubMed] [Google Scholar]