

Background: LpxH is a novel pyrophosphate hydrolase in lipid A biosynthesis.
Results: Enzymatic and EPR studies reveal a catalytically important Mn2+ cluster within LpxH.
Conclusion: LpxH is a Mn2+-dependent lipid A enzyme with an active site similar to calcineurin-like phosphatases, not Nudix family hydrolases.
Significance: Unmasking the true nature of LpxH catalysis represents an important step toward structural characterization and development of antibiotics.
Keywords: Electron Paramagnetic Resonance (EPR), Enzymes, Lipids, Lipopolysaccharide (LPS), Metalloenzymes
Abstract
In Escherichia coli and the majority of β- and γ-proteobacteria, the fourth step of lipid A biosynthesis, i.e. cleavage of the pyrophosphate group of UDP-2,3-diacyl-GlcN, is carried out by LpxH. LpxH has been previously suggested to contain signature motifs found in the calcineurin-like phosphoesterase (CLP) family of metalloenzymes; however, it cleaves a pyrophosphate bond instead of a phosphoester bond, and its substrate contains nucleoside diphosphate moieties more common to the Nudix family rather than to the CLP family. Furthermore, the extent of biochemical data fails to demonstrate a significant level of metal activation in enzymatic assays, which is inconsistent with the behavior of a metalloenzyme. Here, we report cloning, purification, and detailed enzymatic characterization of Haemophilus influenzae LpxH (HiLpxH). HiLpxH shows over 600-fold stimulation of hydrolase activity in the presence of Mn2+. EPR studies reveal the presence of a Mn2+ cluster in LpxH. Finally, point mutants of residues in the conserved metal-binding motifs of the CLP family greatly inhibit HiLpxH activity, highlighting their importance in enzyme function. Contrary to previous analyses of LpxH, we find HiLpxH does not obey surface dilution kinetics. Overall, our work unambiguously establishes LpxH as a calcineurin-like phosphoesterase containing a Mn2+ cluster coordinated by conserved residues. These results set the scene for further structural investigation of the enzyme and for design of novel antibiotics targeting lipid A biosynthesis.
Introduction
The outer membrane of Gram-negative bacteria is arranged into an asymmetric bilayer with the inner leaflet consisting of phospholipids such as phosphatidylglycerol and phosphatidylethanolamine and the outer leaflet containing lipopolysaccharide (LPS). LPS serves to shield bacteria from external damage caused by detergents and antibiotics (1) and is anchored into the bacterial outer membrane via a hexacylated saccharolipid called lipid A (1, 2). In addition to serving as a membrane anchor, lipid A also has a critical role in host response to bacterial infection, as it can be recognized by the TLR-4·MD2 complex, leading to activation of the NF-κB pathway, secretion of cytokines, and subsequent clearance of infection (3, 4). However, overactivation of the immune system by lipid A can result in the potentially fatal condition of sepsis (5). Lipid A is required for the growth and survival of most Gram-negative bacteria, making its biosynthesis an attractive target for novel antibiotics (1, 2, 6).
In Escherichia coli, the pathway for lipid A biosynthesis consists of nine enzymes, each of which catalyzes a separate step (1). Nearly all of these nine enzymes are highly conserved throughout Gram-negative bacteria (7). However, an exception to the universal nature of the pathway is the fourth step that involves the hydrolysis of UDP-2,3-diacylglucosamine (UDP-DAGn)2 to yield UMP and 2,3-diacylglucosamine 1-phosphate (more commonly known as lipid X) (Fig. 1). This reaction, thought to be the first membrane-associating step of the pathway, can be carried out by either LpxH (8, 9) or LpxI (10). These two enzymes share no sequence similarity, are never found in the same organism, and attack different phosphates in their catalysis of UDP-DAGn hydrolysis (10). Despite these differences, lpxI can complement an lpxH knock-out in E. coli (10), indicating that LpxI and LpxH have the same function within the context of the lipid A pathway.
FIGURE 1.
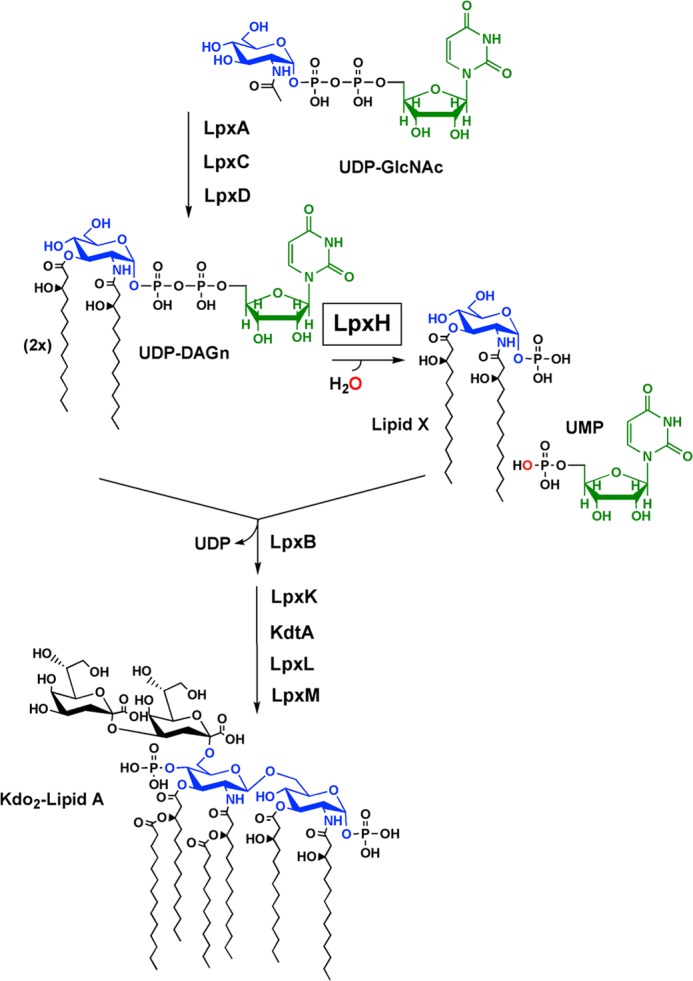
LpxH catalyzes the fourth step in lipid A biosynthesis. UDP-DAGn is synthesized by LpxA, LpxC, and LpxD. Hydrolysis of the molecule by LpxH yields 2,3-diacylglucosamine 1-phosphate (lipid X) and UMP. Lipid X is then condensed with a molecule of UDP-DAGn by LpxB to form disaccharide 1-phosphate and UDP. Following steps of the pathway involve the conversion of disaccharide 1-phosphate to Kdo2-lipid A by LpxK, KdtA, LpxL, and LpxM.
The gene encoding LpxH was first discovered in E. coli, and sequence analysis of LpxH orthologs revealed a DXH(X)∼25GDXXDR(X)∼25GNH(D/E) (where X is any residue) motif that is found in the calcineurin-like phosphoesterase (CLP) superfamily (Pfam00149). These enzymes perform catalysis using an ordered shell of water molecules and divalent or trivalent metal cofactors coordinated by conserved residues of the metallophosphoesterase motif (11, 12). It has been postulated that the metal ions serve to bridge an oxide that performs a nucleophilic attack, to stabilize the formation of a phosphorane intermediate, and to coordinate a nucleophilic hydroxyl group to allow for deprotonation (12, 13). Besides calcineurin, enzymes of the CLP superfamily also include protein serine/threonine phosphatases (14), purple acid phosphatases (15), nucleotidases, sphingomyelin phosphodiesterases (16), exonucleases, and cyclic nucleotide phosphodiesterases (11). These enzymes have broad functions but are thought to have evolved from the same ancestor (17). Compared with other enzymes of this family, LpxH hydrolyzes a unique substrate containing both lipid and nucleotide moieties and does so by breaking a pyrophosphate bond, a type of chemistry that to our knowledge has not been previously reported for CLP enzymes (11). Additionally, no metal dependence of LpxH has been reported (8), adding to the mystery of whether the enzyme functions like other members of the CLP family.
Here, we report cloning, overexpression, and purification of an LpxH ortholog from Haemophilus influenzae (HiLpxH) to near homogeneity (>95%). Using an optimized autoradiographic assay, we have carried out detailed enzymatic characterization of HiLpxH. We show that HiLpxH activity is stimulated over 600-fold by Mn2+, and our EPR studies reveal a Mn2+ cluster that serves to facilitate catalysis. Mutagenesis studies indicate the functional importance of the predicted Mn2+-binding residues in the conserved metallophosphoesterase motif. This work sheds new insights into LpxH catalysis that were previously disguised by enzyme impurity, less-than-optimal assay conditions, and low enzymatic activity. These new observations highlight a strong mechanistic similarity of LpxH to members of the CLP family and set the scene for structural studies of LpxH.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Reagents for bacterial growth included yeast extract, tryptone, and Bacto agar, and were purchased from Difco. The HEPES, phosphate-buffered saline (PBS) components, HCl, salts, ampicillin, isopropyl β-d-thiogalactoside (IPTG), fatty acid-free bovine serum albumin (BSA), EDTA, and DTT were purchased from Sigma. Methanol, chloroform, pyridine, and acetic acid were obtained from EMD Science (Gibbstown, NJ). Radioactive γ-32Pi was purchased from PerkinElmer Life Sciences. The plasmids used in this study were purified using Qiagen Mini-Prep kits (Qiagen, Valencia, CA) according to the manufacturer's protocol. DNA fragments were purified using QIAquick spin kits. Sequencing was carried out by Eton Bioscience, Inc. (Research Triangle Park, NC). Unless otherwise noted, protein concentration was determined either by the bicinchoninic acid assay or Bradford assay (Thermo Scientific, Waltham, MA) depending on compatibility with buffer components. Both of these methods were carried out as described by the manufacturer.
Cloning of H. influenzae lpxH
Amplification of lpxH (HI0375) from H. influenzae genomic DNA (ATCC, Manassas, VA) was accomplished using PCR with primers 50 bp upstream and downstream of lpxH. To facilitate eventual ligation of the gene product into an expression vector, we used primers (Integrated DNA Technologies, Coralville, IA) that allowed for the incorporation of NdeI and XhoI restriction sites to the 5′ and 3′ ends of lpxH and the elimination of the stop codon at the end of the gene. The PCR was carried out using a Mastercycler Gradient Thermocycler (Eppendorf, Hamburg, Germany) with the reagents and protocol of the KOD hot start kit (EMD Chemicals, Gibbstown, NJ); the reaction was additionally supplemented with 3% (w/v) dimethyl sulfoxide. Sequencing of the resulting product using the original primers was used to ensure proper amplification.
To generate an expression vector with a C-terminal His10 tag that was cleavable by tobacco etch virus (TEV) protease, 33 additional nucleotides were added to pET21b (Novagen/EMD Chemicals) to encode for the seven amino acids of the protease site (ENLYFQG) (18) and the four additional histidine residues needed to elongate the affinity tag. This was accomplished using QuikChange (Stratagene, La Jolla, CA) mutagenesis with primers (Integrated DNA Technologies) that were designed to insert the additional nucleotides 3′ to the XhoI restriction site in pET21b. The reaction was carried out using the manufacturer's protocol with an additional supplement of 3% (w/v) dimethyl sulfoxide and 1 m betaine. Following nucleotide insertion, the resulting plasmid, p21t10, was confirmed by sequencing and transformed into chemically competent DH5α cells (Invitrogen). Additionally, p21t10 was transformed into chemically competent E. coli C41(DE3) to create the vector control expression strain VC_T10.
Insertion of amplified H. influenza lpxH into p21t10 was accomplished by digestion of both the PCR fragment and the vector with NdeI and XhoI (New England Biolabs) under conditions described by the manufacturer. After treatment of p21t10 with antarctic phosphatase (New England Biolabs) under conditions described by the manufacturer, digested lpxH was inserted into the plasmid with T4 ligase (Invitrogen) as directed by the manufacturer. The ligation product, pHiHt10, was transformed into chemically competent C41(DE3) E. coli to generate the expression strain HiH_t10. Sequencing with T7 forward and reverse primers confirmed the appropriate insertion of H. influenzae lpxH into p21t10.
Protein Localization
Two samples of cell-free extract from HiH_t10 (obtained as described below with cell debris removed after centrifugation at 10,000 × g) corresponding to 6 mg of total protein were brought to a volume of 1 ml with the appropriate stock solutions to yield final solution concentrations of 200 mm NaCl, 20 mm HEPES, pH 8.0, and either 0 or 1.5% w/v of Triton X-100. These samples were then incubated for 1 h at 4 °C and then subjected to ultracentrifugation at 100,000 × g for 45 min. Resulting supernatants were collected as cytosolic fractions, and pellets were resuspended in 200 mm NaCl and 20 mm HEPES, pH 8.0, to obtain membrane fractions. The fractions were analyzed for the presence of HiLpxH by both SDS-PAGE and the optimized UDP-DAGn hydrolase activity assay, as described below.
Protein Expression and Purification
A typical preparation of HiLpxH began with inoculating an overnight culture of LB with a single colony of HiH_t10. This overnight culture was subsequently used to inoculate 3 liters of LB (10 g of tryptone, 5 g of yeast extract, 10 g of NaCl per liter) supplemented with 100 μg/ml ampicillin to an A600 of 0.02. Cultures were incubated at 30 °C with aeration at 220 rpm until they reached A600 of 0.7–0.8 and then were induced for expression by the addition of 1 mm IPTG and grown for an additional 4–5 h until the A600 reached ∼4. Cells from the growths were pelleted by centrifugation at 5,000 × g, washed with 140 ml of cold PBS, and then stored at −80 °C. For lysis, the frozen pellet was thawed, resuspended in 80 ml of ice-cold 20 mm HEPES, pH 8.0, and passed twice through a French pressure cell (SIM-AMINCO; Spectronic Instruments) at 18,000 p.s.i. The debris from the resulting lysate was removed by centrifugation at 10,000 × g, and the subsequent supernatant was collected as a cell-free extract and stored at −80 °C.
Cell-free extract was thawed and diluted to 5 mg/ml with 20 mm HEPES, pH 8.0. A stock solution of 10% (w/v) Triton X-100 (Thermo Scientific) was used to bring the cell-free extract to a final detergent concentration of 1.5% (w/v). All subsequent steps were carried out at 4 °C. The resulting solution was then mixed by inversion for 1 h and then subjected to ultracentrifugation at 100,000 × g for 45 min. The supernatant was removed and diluted with the appropriate stocks to make a 450-ml solution of ∼3 mg/ml protein, 20 mm HEPES, pH 8.0, 200 mm NaCl, 20 mm imidazole, and 0.5% Triton X-100. This solution was then loaded by gravity onto a packed column of 20 ml of Ni-NTA resin (Qiagen) that had been pre-equilibrated in 20 column volumes of load buffer (20 mm HEPES, pH 8.0, 200 mm NaCl, 20 mm imidazole, 0.1% Triton X-100). The loaded column was next washed with an additional 10 column volumes of load buffer followed by a 15-column volume wash with load buffer supplemented with 30 mm imidazole (final imidazole concentration 50 mm). To exchange HiLpxH out of detergent, the column was subjected to a 50 column volume wash with a solution of 20 mm HEPES, pH 8.0, 200 mm NaCl. Elution of the protein was accomplished by a 7 column volume wash with a solution of 20 mm HEPES, pH 8.0, 200 mm NaCl, and 400 mm imidazole. Samples from each wash of the column were analyzed by SDS-PAGE to ensure proper fractionation.
After elution, a TEV protease reaction was used to cleave the C-terminal His10 tag. The eluted protein was diluted to a volume of 200 ml with the appropriate stock solutions to yield a sample with final concentrations of 2 mm DTT, 2 mm EDTA, 20 mm HEPES, pH 8.0, 200 mm NaCl, 300 mm imidazole, and ∼0.015 mg/ml (∼1:50 of the HiLpxH concentration) of TEV protease. The protease used was prepared as described in the literature (18), and cleavage was monitored using SDS-PAGE. After overnight incubation, the reaction was concentrated to a final volume of 50 ml using 15 ml of Amicon Ultra 10K molecular weight cutoff centrifuge concentrators (Millipore, Billerica, MA) and then dialyzed overnight against 8 liters of 20 mm HEPES, pH 8.0, and 200 mm NaCl. The remaining TEV protease and any uncleaved HiLpxH was removed from the sample by passage over a 5-ml Ni-NTA column pre-equilibrated with 20 column volumes of a solution containing 20 mm HEPES, pH 8.0, 200 mm NaCl, and 20 mm imidazole. The flowthrough was collected and then concentrated using Millipore conical spin concentrations to a final concentration of ∼8 mg/ml. The final protein sample, which contained HiLpxH followed by the residual TEV cleavage site residues of ENLYFQ, was analyzed by SDS-PAGE to confirm purity and stored at −80 °C in 15-μl aliquots.
Generation and Purification of Point Mutants
QuikChange (Stratagene) mutagenesis was used to make alanine point mutants of conserved residues within HiLpxH. The method was carried out as described above, with pHiHt10 serving as template DNA. Specific primers (Integrated DNA Technologies) were created for each desired alanine substitution, and sequencing with T7 forward and reverse primers was used to confirm the mutations. The mutated pHiHt10 vectors were transformed into chemically competent E. coli C41(DE3) to create respective expression strains. These strains, along with wild-type HiH_t10, were grown under conditions described for HiLpxH purification except that the culture volume was 250 ml. Cells from each of the growths were centrifuged at 5,000 × g, and collected pellets were washed with 30 ml of cold PBS and then stored at −80 °C. For lysis, the frozen pellet from each mutant strain and the wild-type strain was thawed, resuspended in 8 ml of ice-cold 20 mm HEPES, pH 8.0, and passed twice through a French pressure cell (Sim-Aminco, Spectronic Instruments) at 18,000 p.s.i. The debris from the resulting lysate was removed by centrifugation at 10,000 × g, and the subsequent supernatant was collected as cell-free extract and stored at −80 °C.
Purification of the protein from these growths was the same as HiLpxH but adjusted to account for the smaller culture size. The cell-free extract was thawed, diluted, and solubilized as described for HiLpxH. Following the centrifugation step after detergent incubation, the supernatant was removed and diluted with the appropriate stocks to make a 20-ml solution of ∼3 mg/ml protein, 20 mm HEPES, pH 8.0, 200 mm NaCl, 20 mm imidazole, and 0.5% Triton X-100. This solution from each strain was then loaded by gravity onto separate columns of 1 ml of Ni-NTA resin (Qiagen) that had been pre-equilibrated in 20 column volumes of the load buffer described above. Washing, detergent exchange, and elution were carried out as outlined for HiLpxH. Samples from each wash of the column were analyzed by SDS-PAGE to ensure proper fractionation. Following elution, the samples were each concentrated to 3 ml using 5 ml of Amicon Ultra 10K molecular weight cutoff centrifuge concentrators (Millipore) and then dialyzed overnight against 1 liter of 20 mm HEPES, pH 8.0, 200 mm NaCl. Resulting protein solutions from each mutant and the wild-type sample were further concentrated to 500 μl and stored in −80 °C.
Synthesis of Radioactive Substrate
32P-Labeled UDP-DAGn was prepared as described previously (10) with some modifications. First, 50 μl of 0.2 m 1H-tetrazole in acetonitrile in the morpholidate reaction was replaced with 20 μl of 0.1 m dicyanoimidazole in acetonitrile (Sigma). Second, after the product of the morpholidate reaction was dried under N2, it was extracted with a single phase Bligh-Dyer (19) with volumes of 0.5 ml of methanol, 0.25 ml of chloroform, and 0.2 ml of PBS. The mixture was centrifuged to pellet debris, and the supernatant was transferred to a 2-ml polypropylene tube and dried under N2. The radioactive substrate was then resuspended in 100–170 μl of 20 mm HEPES, pH 8.0, containing 0.02% Triton X-100 and sonicated for 2 min. One final centrifugation step was used to pellet any insoluble material. The supernatant containing the final [β-32P]UDP-DAGn was transferred to a new tube and stored at −20 °C. These extra extraction steps removed contaminating impurities that interfered with the linearity of our assays.
Optimized in Vitro Assay for UDP-DAGn Hydrolase Activity
General autoradiographic assays for hydrolase activity were similar to that previously described (8) with optimized modifications. Reactions mixtures were a final volume of 15 μl in 0.6-ml polypropylene tubes and contained 20 mm HEPES, pH 8.0, 0.5% (w/v) BSA, 0.05% (w/v) Triton X-100, 1 mm MnCl2, 100 μm UDP-DAGn (prepared as described previously (20)), 1,000 cpm/μl [β-32P]UDP-DAGn, and enzyme. Previous studies of E. coli LpxH reported a small enhancement in enzyme stability and activity when the protein was purified with MnCl2 (8); therefore, MnCl2 was incorporated into the assay. All reaction components besides the enzyme were mixed to a volume of 12 μl and equilibrated at 30 °C for 10 min, after which 3 μl of enzyme was added to start the reaction. Unless otherwise noted, enzyme samples were diluted in a buffer identical to the assay mixture but lacking any lipid substrate. Aliquots of 1.5 μl were taken from the reactions at various time intervals and spotted onto 20 × 20-cm glass-backed silica gel thin layer chromatography (TLC) plates (EMD Chemicals, Darmstadt, Germany). These plates were developed in a 25:15:4:2 chloroform/methanol/water/acetic acid tank system, dried, exposed to phosphoscreens, scanned, and quantified as described previously (8).
Comparison of in Vitro Assays
To assess the effectiveness of the optimized in vitro assay, E. coli LpxH (EcLpxH) cell-free extract, HiLpxH cell-free extract, and purified HiLpxH were analyzed for activity under both the original and the improved assay conditions. To prepare the EcLpxH cell-free extract, E. coli C41(DE3) was transformed with pKJB2 (8), a pET21a+ plasmid containing E. coli lpxH. A 250-ml culture of the resulting strain was grown at 30 °C in LB media and induced for expression with IPTG at mid-log phase. After 5 h, cells were harvested and lysed to generate cell-free extract using the method described above. The same procedure was carried out in parallel with strain HiH_t10 to prepare the HiLpxH cell-free extract. Purified HiLpxH was isolated by the aforementioned technique. Activity assessment under original conditions was conducted as reported previously with the final protein concentrations in the assay for EcLpxH cell-free extract, HiLpxH cell-free extract, and HiLpxH being 1.8, 4.5, and 8.5 μg/ml respectively. Samples were also tested under the optimized in vitro conditions with protein concentrations of 45 ng/ml EcLpxH cell-free extract, 110 ng/ml HiLpxH cell-free extract, and 1.7 ng/ml HiLpxH.
Kinetic Parameters, pH Optimum, and Detergent Dependence of HiLpxH
To determine the Km and Vmax values of HiLpxH with respect to UDP-DAGn, purified HiLpxH was assayed under standard conditions except the concentration of UDP-DAGn was varied from 20 to 400 μm. The velocities from each of these reactions were plotted against substrate concentration, and using KaleidaGraph (Synergy Software, Reading PA), the resulting curve was fit to the Michaelis-Menten equation. To maintain linear conversion at various substrate concentrations, the HiLpxH in the assay was varied from 6 to 63 pm.
To assess the effect of pH on HiLpxH activity, purified protein was assayed under standard conditions except 20 mm HEPES, pH 8.0, was replaced with a triple buffer system containing 100 mm sodium acetate, 50 mm bis(2-hydroxyethyl)iminotris-(hydroxymethyl)hexane, and 50 mm Tris at a pH from 4.0 to 9.0. KaleidaGraph was used to fit a single-limb pKa curve (Equation 1) to the resulting data. The enzyme concentration of HiLpxH needed to see linear activity in the assay at different pH values ranged from 30 pm to 372 nm.
 |
To monitor the detergent dependence of HiLpxH, the Triton X-100 concentration in the standard assay was varied from 0 to 18 mm (1.2% w/v). Purified HiLpxH was diluted in the standard buffer lacking Triton X-100, added to the reactions covering the mentioned range of detergent concentrations, and assayed for activity. Reported total detergent concentrations accounted for the small amount of detergent (0.01 mm, 0.0007% w/v) present in both the [β-32P]UDP-DAGn and the nonradioactive UDP-DAGn. Linear activity could be observed at final enzyme concentrations of 30 and 6 pm.
Metal Dependence of HiLpxH
To analyze the metal dependence of HiLpxH, a modified autoradiographic assay was employed. First, the enzyme sample was diluted with standard reaction buffer that replaced 1 mm MnCl2 with 2 mm EDTA, incubated on ice for 20 min, then diluted again into standard reaction buffer with no MnCl2, and incubated on ice for an additional 10 min. This EDTA-treated enzyme was next added to reaction mixtures in which 1 mm MnCl2 was replaced by no di- or trivalent metal ion, 2 mm NaCl, or each of the following chloride salts at 1 mm: Ca2+, Co2+, Cu2+, Fe3+, Mg2+, Mn2+, Ni2+, and Zn2+. To ensure linear activity under each condition, the concentration of HiLpxH in the assays were varied from 35 to 178 nm.
Another form of the modified autoradiographic assay was used to assess HiLpxH dependence on MnCl2 in which the protein was diluted in standard reaction buffer containing no MnCl2 and then diluted into reactions in which the MnCl2 concentration ranged from 0 to 5 mm. Protein concentration was 980 pm in the 0 mm assay condition and 10 pm in all others.
EPR Spectroscopy
X-band EPR spectra were recorded using Bruker Biospin (Billerica, MA) Bruker E-500 spectrometer equipped with super high Q cavity providing conventional perpendicular EPR mode (i.e. microwave magnetic field B1 is perpendicular to the static magnetic field B0). All room temperature spectra were recorded at the same experimental conditions: 10 G modulation amplitude at 100 kHz, 2-milliwatt incident microwave power at 9.86 GHz frequency. For EPR measurements at cryogenic temperatures, the spectrometer was outfitted with ESR 910 cryostat and ITC-4 temperature controller (Oxford Instruments, Concord, MA). Temperature at the sample cavity was calibrated using a CernoxTM thin film sensor (Lake Shore Cryotronics Inc., Westerville, OH). Incident microwave power was kept at levels to avoid saturation, and modulation amplitude was set to about ⅓ to ¼ of the narrowest peak-to-peak line width.
Manganese Titration with EPR Detection at Room Temperature
To assess binding of Mn2+ to HiLpxH, 80 μl of a solution containing 100 μm purified HiLpxH, 20 mm HEPES, pH 8.0, and 200 mm NaCl was titrated in successive additions with 1 μl of 1.645 mm MnCl2 stock containing the same buffer as the protein solution. After each addition of manganese stock, the sample was mixed and incubated for >5 min at room temperature. Subsequently, ∼20 μl of the titrated solution was drawn into a quartz capillary with 0.70-mm inner diameter and 0.87-mm outer diameter (VitroCom, Mountain Lakes, NJ). One end of the capillary was sealed with Critoseal® (McCormick Scientific, St. Louis, MO) and inserted into a 3-mm inner diameter quartz EPR tube (Wilmad-LabGlass, Vineland, NJ). The tube was then fixed inside the EPR resonator so that the section of the capillary containing the aqueous sample was covering the entire sensitivity region of the EPR resonator, and the Critoseal® plug was located outside. After taking an EPR spectrum, the protein solution was recovered into the vial containing the rest of the sample, and a new portion of MnCl2 was added. The same quartz tube was used for all the protein-containing samples. When changing the sample, solution was drawn in and out of the capillary twice to ensure complete replacement of the previous sample. For a control calibration of EPR signal intensity versus Mn2+ concentration, the same procedure was used except that a buffer-only solution was used.
After collection of spectra, the concentration of free Mn2+ ion present in solutions was evaluated by either least squares fitting of the high field hyperfine component of the six-line Mn2+ EPR spectrum using EWVoigt program (21) or by measuring peak-to-peak intensity of that component as the peak line width did not change. Both measurements yielded identical results. The determined free Mn2+ concentrations were analyzed based on a two-site binding model (Equation 2),
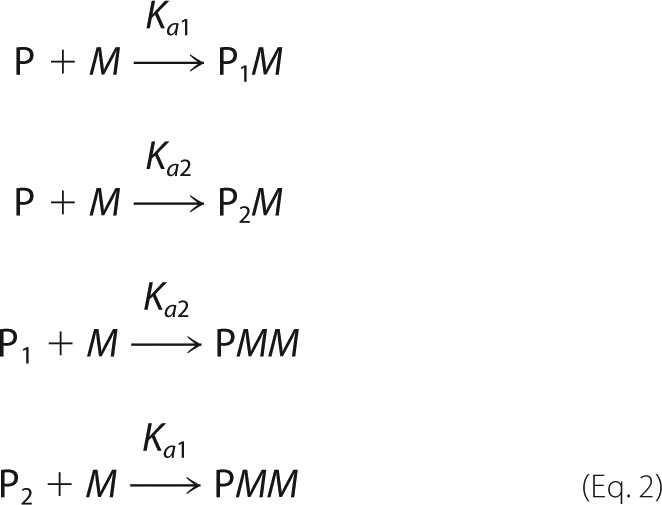 |
where P is the protein with only the first (P1M), and the second (P2M), or both metal-binding sites occupied by metal ion M, and Ka1 and Ka2 are the corresponding association constants. All experimental spectra were corrected for a small background signal from the resonator that was recorded using a capillary containing only protein solution in buffer. Least squares fitting of experimental data were accomplished using the Levenberg-Marquardt optimization, and parameter uncertainties were determined by standard covariance matrix method using SigmaPlot (Systat Software, San Jose, CA).
Manganese Titration with EPR Detection at Cryogenic Temperatures
Low temperature EPR experiments were employed for further assessment of the Mn2+-protein interaction and for the existence of a Mn2+ cluster. For these experiments, samples containing 20 mm HEPES, pH 8.0, 200 mm NaCl, 10% (v/v) glycerol, and 234 μm of HiLpxH were incubated with molar equivalents of MnCl2 ranging from 0 to 2.0. After assembly, these samples were stored at −80 °C. Just before an EPR measurement, a sample was thawed and drawn into 3 -mm inner diameter precision quartz EPR tubes (Wilmad-LabGlass) and then frozen again by immersing the tube into liquid nitrogen before being placed in a pre-cooled EPR cryostat.
RESULTS
Expression and Purification of HiLpxH
The gene encoding LpxH from H. influenzae was amplified from genomic DNA and cloned into a modified pET21b expression vector designed to yield a protein product that was tagged at the C terminus with an eight-residue TEV protease cleavage site followed by 10 histidine residues. Transformation of this plasmid into E. coli C41 resulted in strain HiH_t10. When HiH_t10 was grown at 30 °C in the presence of IPTG, the overexpression of a 27-kDa protein matching the molecular weight of the predicted protein product was evident when compared with a vector control strain VC_t10 via SDS-PAGE (Fig. 2A).
FIGURE 2.
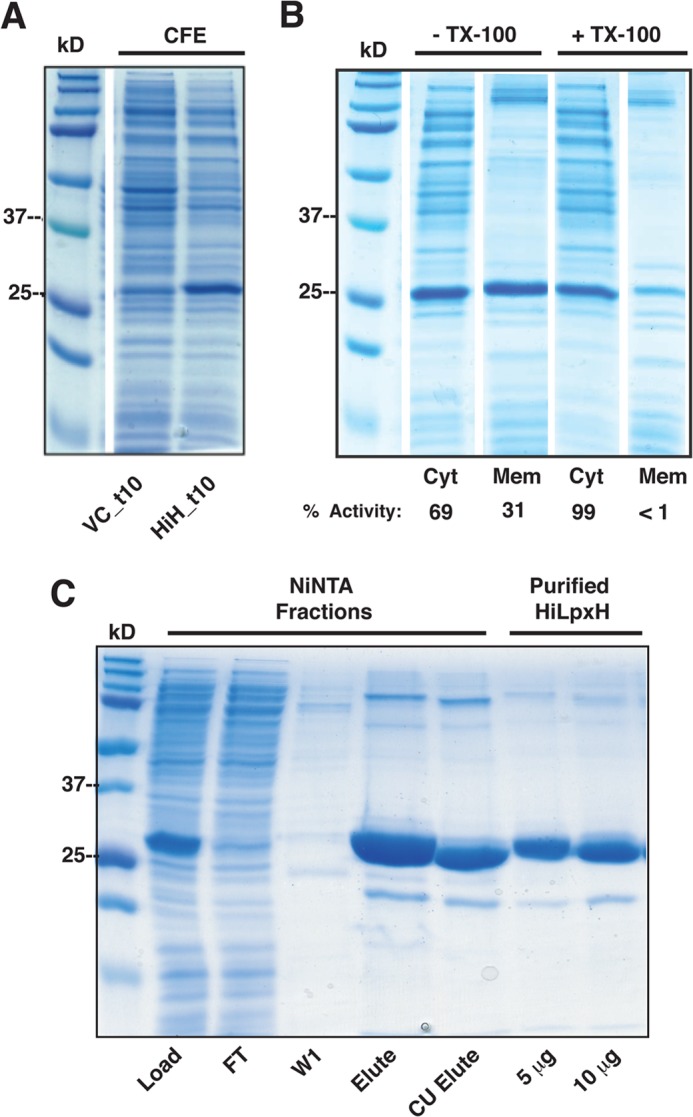
SDS-PAGE analysis of overexpression and purification of HiLpxH. A, cell-free extract (CFE) of VC_t10 and HiH_t10 prepared as described under “Experimental Procedures.” The 27-kDa band that is present in the HiH_t10 lane and not in the VC_t10 lane corresponds to overexpressed HiLpxH. B, cytosolic (Cyt) and membrane (Mem) fractions of HiH_t10 cell-free extracts incubated in the absence or presence of Triton X-100 (TX-100). The activity of each fraction, as determined by autoradiographic assay, is listed below the lanes as a percentage of total LpxH activity observed in the corresponding cell-free extract. C, analysis of fractions from HiLpxH purification outlined under “Experimental Procedures.” Fractions from the Ni-NTA purification include the Load, denoting the solubilized cell-free extract from HiH10_t that was loaded onto the column, FT representing the flow-through from the column, W1 signifying the 50 mm imidazole wash, and Elute indicating the tagged HiLpxH obtained from the 400 mm imidazole wash. The CU elute lane shows protein that flowed through the second clean-up Ni-NTA column after TEV protease digestion. The purified HiLpxH fractions depict the final sample of purified HiLpxH that was used in autoradiographic assays and EPR experiments. The labels 5 μg and 10 μg indicate how much protein was loaded in each lane.
When overexpressed in E. coli C41, HiLpxH partitioned to both the membrane and the cytosolic fractions, as indicated by SDS-PAGE and activity assay (Fig. 2B). This membrane association of the protein is similar to the behavior reported for EcLpxH (8). To increase the yield of protein for purification, the cell-free extract from HiH_t10 was incubated with Triton X-100 to solubilize any HiLpxH that may be associated with the membrane, resulting in recovery of nearly all the LpxH activity to the soluble fraction (Fig. 2B).
Following detergent treatment, Ni-NTA chromatography was used to isolate the His10-tagged HiLpxH. Any remaining detergent from this initial solubilization step was removed with an extensive wash in buffer lacking detergent or imidazole after the protein was loaded onto the Ni-NTA column. The initial Ni-NTA step resulted in a 10-fold increase in specific activity (Table 1) and yielded protein that was ∼90% pure by SDS-PAGE (Fig. 2C). Following dialysis to remove imidazole and cleavage of the His tag and subsequent clean-up with another Ni-NTA column, the final protein sample showed marginal increase in purity by gel but a 6-fold enhancement in specific activity. This corresponded to a dramatic increase in total active units (480%, Table 1), suggesting that although the inclusion of an affinity tag facilitated protein purification, its presence inadvertently impaired the enzyme activity. Overall, we were able to consistently recover ∼30 mg of HiLpxH per 1 liter of HiH_t10 culture. This final protein sample was ∼95% pure (Fig. 2C) and exhibited a 60-fold increase in specific activity (Table 1).
TABLE 1.
Purification of HiLpxH from E. coli strain HiH_t10
Specific activities were determined under optimized assay conditions as described under “Experimental Procedures.”
| Step | Protein | Volume | Units | Specific activity | Yield | Purification |
|---|---|---|---|---|---|---|
| mg | ml | mmol/min | mmol min−1 mg−1 | % | -fold | |
| Ni-NTA load | 1,400 | 400 | 182 | 0.13 | 100 | 1 |
| Ni-NTA elution | 210 | 140 | 273 | 1.3 | 150 | 10 |
| Final sample | 112 | 16 | 874 | 7.8 | 480 | 60 |
HiLpxH Activity in Optimized Autoradiographic Assay
LpxH activity can be monitored with a TLC-based assay utilizing a [β-32P]UDP-DAGn substrate that retains the radioactive label upon hydrolysis to lipid X, making it possible to quantify substrate conversion using autoradiography. The previously published assay conditions contained 25 mm HEPES, pH 8.0, 100 μm UDP-DAGn, and 1,000 cpm/μl [β-32P]UDP-DAGn (8). Based on the observations of increased stability of the E. coli ortholog in the presence of MnCl2 and slight activation of the enzyme upon preincubation with the metal (8), we have added 1 mm MnCl2 to the assay dilution buffer as well as to the reaction. Additionally, we supplemented the reaction with 0.05 mg/ml BSA and 0.05% Triton X-100 to aid in protein stability and substrate solubility. When purified HiLpxH was assayed with these modifications, we were able to track conversion of [β-32P]UDP-DAGn to 32P-lipid X (Fig. 3A) and calculate an activity that was 1,000-fold higher than that calculated when HiLpxH was assayed under previously reported conditions (Fig. 3B). This increase in activity under optimized conditions was also observed when the cell-free extract of strains overexpressing HiLpxH (HiH CFE) and EcLpxH (EcH CFE) were tested for lipid X formation. The LpxH expression from both of these cell-free extract samples appeared equal by SDS-PAGE (data not shown) and corresponded to similar levels of activity under both conditions, indicating the activation was not ortholog-specific. Furthermore, activity in the optimized system is linear with both time (Fig. 3C) and protein concentration (Fig. 3D).
FIGURE 3.

Optimized assay shows enhancement in LpxH activity and is linear with time and protein concentration. A, scan of phosphoscreen exposed to silica TLC plate from a representative HiLpxH assay using 10 pm enzyme. Product conversion is calculated by quantifying intensity of lipid X as a percentage of the total intensity of each lane and then subtracting the background conversion calculated from the no enzyme (no rxn) control. B, specific activity of various protein samples under original assay conditions compared with optimized assay conditions. Cell-free extract samples overproducing EcLpxH (EcH CFE) and HiLpxH (HiH CFE) were prepared in parallel and exhibit similar levels of protein expression. C, percent of 100 μm UDP-DAGn converted to lipid X at specific time points under standard assay conditions with 10 pm HiLpxH. Data were fit to a linear curve using KaleidaGraph and showed an R2 value of 0.998. D, percent of 100 μm UDP-DAGn converted to lipid X at various HiLpxH concentrations after 10 min under standard reaction conditions. KaleidaGraph was once again used to fit data to a linear curve with an R2 value of 0.995. Plots generated in C and D are the result of the average of three independent data sets.
Apparent Kinetic Parameters, pH Rate Profile, and Detergent Dependence of HiLpxH
Although the enzymatic properties of EcLpxH had been reported previously (8), these observations were obscured by sample impurity, less-than-optimal assay conditions, and low enzymatic activity. Therefore, it is critical to revisit the characterization of LpxH using a highly purified protein sample and our newly optimized assay conditions. The specific activity of purified HiLpxH was determined at varied concentrations of UDP-DAGn to calculate the basic kinetic parameters of the enzyme (Fig. 4A). When this information was fit to a Michaelis-Menten model, it resulted in an apparent Km of 79.4 ± 11.0 μm for the lipid substrate and an apparent Vmax of 18.1 ± 0.9 mmol min−1 mg−1.
FIGURE 4.
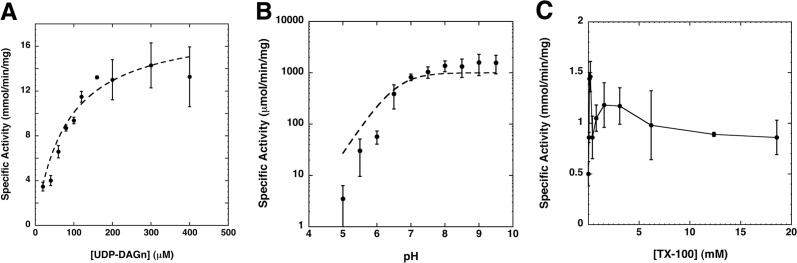
Enzymatic analysis of HiLpxH. A, HiLpxH activity under standard conditions is dependent upon UDP-DAGn concentration. The apparent Km was 79.4 ± 11.0 μm and the apparent Vmax was 18.1 ± 0.9 mmol min−1 mg−1, both calculated by fitting the data using KaleidaGraph. B, changes in pH alter the in vitro activity of HiLpxH. The enzyme exhibits a dramatic increase in activity as the pH of the reaction reaches 7 but very little change upon progressively more alkaline conditions. Data were fit to Equation 1 using KaleidaGraph, yielding a pKa of 6.56 ± 0.38. C, effect of Triton X-100 concentration on HiLpxH-specific activity at 100 μm UDP-DAGn. Surface dilution kinetics are not apparent as activity does not appear to have a strong correlation with detergent amount, and the enzyme displays detectable activity even at very low Triton X-100 (TX-100) concentrations. All plots shown were each generated from the average of three separate experiments.
The high specific activity of HiLpxH also enabled us to determine the pH dependence of LpxH catalysis. LpxH is most active at slightly alkaline pH values and exhibits a sharp decrease in its catalytic activity at low pH. It is important to note that such a drop in LpxH activity at acidic pH is not due to enzyme instability as preincubation of HiLpxH at low pH does not alter the apparent enzyme activity at the standard condition of pH 8.0 (data not shown). These data were fit to a single lobed pH curve described by Equation 1 with a pKa = 6.6 ± 0.4 (Fig. 4B).
Finally, the activity of HiLpxH was assessed at a range of Triton X-100 concentrations from 0 to 18 mm. As shown in Fig. 4C, the enzyme is active without detergent and demonstrates stimulation in hydrolysis as the detergent concentration approaches the critical micelle concentration (0.19 mm). At detergent amounts above the critical micelle concentration, the protein initially shows a decrease in hydrolytic ability, after which activity appears to be unaffected by the detergent concentration in the assay. Such behavior is not indicative of surface dilution kinetics, suggesting that although the enzyme likely associates with the surface of the detergent-substrate micelles in vitro during catalysis, it is not directly embedded in a detergent micelle.
Metal Dependence of HiLpxH
Although Mn2+ has been previously suggested to play a role in maintaining enzyme structure, facilitating membrane association, or contributing to the formation of the active site of EcLpxH (8), the exact function of metal ions in LpxH remains to be elucidated as inclusion of Mn2+ in enzymatic assays using partially purified EcLpxH only has a marginal effect on the enzyme activity. To determine how metal ions altered the capability of LpxH to hydrolyze UDP-DAGn, we first incubated highly purified HiLpxH with a reaction buffer in which the MnCl2 was replaced with 2 mm EDTA to remove any co-purifying metal ions and then diluted it into standard assay conditions containing 1 mm of the following chloride salts: Ca2+, Co2+, Cu2+, Fe3+, Mg2+, Mn2+, Ni2+, and Zn2+. Assay conditions where the di- or trivalent metal salt was replaced by 2 mm NaCl or no metal additive were also included as controls. Results were compared with the activity of HiLpxH in the presence of no metal and are shown in Fig. 5A. The Mn2+ assay condition showed activity that was 670-fold greater than the condition with no metal. The Ni2+, Co2+, and Fe3+ conditions also enhanced activity; however, the increase (10–50-fold) was substantially lower than that seen when Mn2+ was present. The activity of the enzyme was negligible in NaCl, indicating that the ionic strength of the reaction was not responsible for any stimulation observed. The enzyme did show residual activity in the absence of metal, albeit only visible at enzyme concentrations 1,000-fold greater than that usually used in the assay.
FIGURE 5.

Metal dependence of HiLpxH. A, HiLpxH treated with EDTA was assayed under standard conditions supplemented with either metal, NaCl, or no metal. Resulting specific activity was calculated and compared with the specific activity of the no metal control. Data shown are the means of three separate experiments. B, UDP-DAGn hydrolysis activity of HiLpxH was determined as a function of MnCl2 concentration in the assay. The data were fit to a Michaelis-Menten model with KaleidaGraph to obtain a Km of 487 ± 113 μm for Mn2+.
To further investigate Mn2+-dependent stimulation, HiLpxH activity was determined at a range of Mn2+ concentrations (Fig. 5B). Enzyme activity increased with Mn2+ concentrations up to 2.5 mm, an ∼108-fold molar excess compared with the HiLpxH concentration in the assay. After this point, there seemed to be no further stimulation in product hydrolysis. This pattern was indicative of Michaelis-Menten saturation kinetics, suggesting the metal acts as a pseudo-substrate for HiLpxH. Consequently, the data were fit to determine an apparent Km of 487 ± 113 μm for Mn2+. This term was designated the KMetal for the enzyme.
Evidence for Two Mn2+-binding Sites in HiLpxH
To further probe the interaction of Mn2+ with HiLpxH, we generated a Mn2+ titration curve using EPR spectroscopy, a technique that has been used to characterize the Mn2+-binding site in another CLP family enzyme, bacteriophage λ protein phosphatase (22). This approach relies on the fact that a Mn2+ ion coordinated to a protein-binding site exhibits very broad EPR lines caused by large zero-field splitting effects, making it undetectable at room temperature, whereas free Mn2+ yields a distinct EPR signal under the same condition. Representative room temperature EPR spectra from the titration of HiLpxH with manganese are shown in Fig. 6A. For samples with Mn2+-to-protein ratio above 1:1, an EPR spectrum revealed a six-line hyperfine pattern (nuclear spin I = 5/2) with a splitting of ∼95 G positioned at g ≈2.0 that is characteristic of free (hexoaqua) Mn2+ ion. The intensity of the spectrum varied, increasing concomitantly with the manganese-to-protein ratio (Fig. 6A). Spectra obtained from samples with Mn2+-to-protein ratio of ≤1 demonstrated no detectable six-line component characteristic of the free Mn2+ ion, indicating that the free metal concentration was below the EPR detection limit (<1 μm) and that nearly all the Mn2+ ions in these samples are bound to the protein.
FIGURE 6.

Room temperature EPR analysis of Mn2+ binding. A, representative room temperature continuous wave X-band EPR spectra from samples containing 100 μm of HiLpxH and the appropriate concentration of MnCl2 to yield 1 eq of Mn2+, 1.4 eq of Mn2+, and 2.0 eq of Mn2+. For a comparison, an EPR spectrum from a control sample containing only buffer and MnCl2 at the same concentration as 0.6 Mn2+ eq. B, double integrated intensity of free (hexoaqua) Mn2+ EPR signal as a function of Mn2+ concentration for the room temperature titration experiment (circles) and a control titration experiment without the protein present (squares). Line represents least squares fit using a two-site binding model.
The spectra were further analyzed to assess the binding of the metal ion to LpxH. As the intensity of the six-line hyperfine component correlates to the amount of free Mn2+ in the sample tube, the concentration of unassociated ion could be determined by comparing the double integral of the high field hyperfine component of the HiLpxH-MnCl2 titration spectra with that from a control titration containing only MnCl2. Fig. 6B shows the intensity of EPR spectra of free hexoaqua Mn2+ ion as a function of total [Mn2+] for samples containing either MnCl2 in buffer (squares) or MnCl2 and HiLpxH (circles). Above ∼200 μm Mn2+, which represents [Mn2+]/[HiLpxH] ratios above 2, the slopes of both plots are approximately the same, indicating that concentration of protein-bound Mn2+ species plateaus and that each HiLpxH binds two Mn2+ ions.
Based on this observation, the titration curve in Fig. 6B was fit with a two-site binding model (Equation 2). Although the least squares fit was of exceptionally high quality (Rsqr = 0.998) for Ka2, yielding a value of 5.8 ± 0.6 × 105 m−1 (or Kd2 = 1.7 μm), it was found to be insensitive to Ka1 as long as Ka1 >50 × 106 m−1 (or Kd1 < 20 nm). Indeed, no free Mn2+ was detectable in EPR spectra when the metal-to-protein ratio was below 1, consistent with the notion that the binding affinity for the first metal is very tight, well below the detection limit of the EPR instrument (∼1 μm).
Indication of di-Mn2+ Cluster Formation in HiLpxH
Observing that two metal ions seem to associate with HiLpxH and knowing that other members of the CLP family have been identified to contain metal centers, the possibility of such cluster formation in HiLpxH was investigated using cryogenic EPR spectroscopy. The presence of a close pair of Mn2+ ions can be monitored through the detection of spin exchange coupling in the EPR spectra, as noted with both di-manganese cluster bridged by oxygen atoms (23) and other CLP enzymes (11, 22). Fig. 7A shows representative experimental continuous wave EPR spectra measured at 28 K from a buffered protein solution in the absence of MnCl2 (top) and from protein samples with varying Mn2+-to-protein ratios. It should be noted that the EPR spectrum of HiLpxH in the absence of Mn2+ revealed small but easily observable features at g = 4.3 (Fig. 7A, marked by a dashed line) that were assigned to a low symmetry Fe3+ ion nonspecifically bound to protein. The g = 4.3 signal was used as an internal EPR intensity marker, as the protein concentration was kept constant in all samples.
FIGURE 7.

Cryogenic EPR spectra of HiLpxH in the presence of Mn2+. A, representative continuous wave X-band EPR spectra measured at 28 K. From top to bottom are background signal from an EPR resonator outfitted with a low temperature quartz insert; signal from samples a range of [Mn2+]/[HiLpxH] ratios; and signal from a Mn2+ only solution. Position of the g = 4.3 marker originating from a low symmetry Fe3+ ion nonspecifically bound to the protein is shown by a dashed line. The areas of the spectra indicating the presence of a di-Mn2+ cluster (g = 14, 7.7, 3.1, and 2.48) are denoted by arrows. B, spectra from A enhanced in the range 500–2,500 G to highlight the increase in hyperfine splitting at g = 7.7 for greater [Mn2+]/[HiLpxH] ratios.
The cryogenic experiments revealed that an increase in the [Mn2+]/[HiLpxH] ratio resulted in systematic changes in EPR spectra at this temperature as indicated by the progressive appearance of new features upon further addition of MnCl2 to the samples. Some of these features are attributed to Mn2+ ions bound to the protein, although others likely arise from nonassociated hexoaqua Mn2+, as such features are also obtained from a control experiment using a MnCl2 solution supplemented with 10% (v/v) glycerol (Fig. 7A, Mn only spectrum). The control spectrum is easily identified by a sharp six-line hyperfine pattern at g = 2.04 (Fig. 7A, marked with arrow) arising from electronic spin S = ±1/2 transition that is split into six lines with a splitting of about 95 G by the Mn2+ nuclear spin I = 5/2.
Patterns indicative of a symmetric exchange-coupled pair of Mn2+ ions are evident in the cryogenic EPR spectra of HiLpxH and MnCl2. Specifically, at g ≈7.7, we observed a component with a multiline pattern characterized by an effective hyperfine splitting of ∼40.2 G. Such a pattern appears first in the spectrum from the HiLpxH sample containing 1.25 eq of Mn2+, and the intensity of this pattern increased with greater metal-to-protein ratios (Fig. 7B). The hyperfine splitting constants for binuclear Mn2+ complexes reported in the literature range from 40 to 47 G (23–32). Thus, the splitting of 40.2 G we observed for the HiLpxH/Mn2+ sample is consistent with the existence of binuclear manganese center in the protein.
In support of the spectral feature at g ≈7.7, other spectral features similar to those detected for a binuclear Mn2[(OPPh2)2N]4 complex (33) were observed at g ≈14 in HiLpxH samples with more than 1 Mn2+ eq (Fig. 7B). The signals at g ≈3.1 and 2.48 (Fig. 7B) also can be attributed to a binuclear cluster (34), although they might exhibit a substantial overlap with the signal originating from a singly occupied HiLpxH Mn2+ site as well as the free Mn2+ signal. Although none of these components show resolved hyperfine patterns with splitting characteristic of the binuclear Mn2+ center, such a loss in resolution is not uncommon for many binuclear Mn2+ complexes (24–26, 34, 35).
Point Mutagenesis of Conserved Residues
After establishing that LpxH possesses a binuclear Mn2+ metal cluster, we proceeded to examine the functional role of conserved residues in catalysis. Sequence alignment of LpxH orthologs reveals three areas of amino acid conservation (Fig. 8) that show homology to the motifs characteristic of the CLP family. The most conserved CLP family motif, DXH(X)∼25GDXXDR(X)∼25GNH(D/E), where X indicates any amino acid (the underlined histidine is not completely conserved), is clustered at the N-terminal region of LpxH (Fig. 8, motif 1, green). The middle region of the protein contains a motif of a highly conserved histidine followed by an aspartate (Fig. 8, motif 2, cyan) that is less ubiquitous in the CLP family and not always detectable from sequence alignment (13). Finally, at the C terminus of LpxH, there is a UUXGHXH motif (Fig. 8, motif 3, orange), with U representing any hydrophobic residue, which was first characterized in a sub-class of CLP enzymes that are involved in DNA repair and polymerization (36, 37).
FIGURE 8.

Sequence alignment of LpxH orthologs highlighting conserved residues. Motifs characteristic of the calcineurin-like phosphatases are boxed in color as follows: green represents motif 1, the most conserved residues across the family (DXHX∼25GDXXDRX∼25GNH(D/E)); cyan denotes a lesser conserved motif, motif 2, not always clearly detectable through sequence alignment (usually a highly conserved His followed by Asp); and orange depicts motif 3, an additional motif first identified in a subset of enzymes that are involved in DNA repair (UUXGHXH where U is a hydrophobic amino acid). Residues that were mutated to alanine are italicized and underlined. Alignment was generated using the ClustalW2 tool on the EBI website. Sequences were obtained from the NCBI server.
To assess the importance of these areas of conservation, we made the following alanine point mutants of amino acids (Fig. 8, underlined) from the motif in each region: D9A, H11A, D42A, R81A in motif 1; H115A and D117A in motif 2; and H196A in motif 3. These point mutants, along with a wild-type HiLpxH control, were expressed in E. coli with a C-terminal His10 tag and purified using Ni-NTA chromatography. The mutant proteins overexpressed at levels comparable with that of wild type, and the final purities of all protein forms were similar (data not shown). The isolated protein samples were analyzed for UDP-DAGn hydrolase activity using the radioactive assay, and their resulting activities were compared with that of the wild-type control (Table 2). All of the point mutants showed a decrease in specific activity; however, the residues of the motif in the N-terminal region displayed the most significant change. The D9A mutant exhibited a 20,000-fold inhibition of hydrolysis, 10-fold over any of the other mutants. The other mutants of the N-terminal region, as well as the H115A mutation in the middle region and the H196A mutation in the C-terminal region, showed similar levels of hydrolysis inactivation. This is in contrast to the D117A mutant from the middle region that was only slightly impaired compared with wild type.
TABLE 2.
Activity of purified HiLpxH point mutants compared with wild-type HiLpxH
Samples were analyzed for activity using optimized assay conditions as outlined under “Experimental Procedures.”
| Conserved region | Mutation | Specific activity | decrease compared with WT |
|---|---|---|---|
| μmol/min/mg | -fold | ||
| WT | 6,900 ± 700 | 1 | |
| Motif 1 | D9A | 0.03 ± 0.004 | 200,000 |
| H11A | 0.41 ± 0.02 | 20,000 | |
| D42A | 0.09 ± 0.01 | 80,000 | |
| R81A | 0.93 ± 0.04 | 7,000 | |
| Motif 2 | H115A | 0.57 ± 0.008 | 12,000 |
| D117A | 3,020 ± 90 | 2 | |
| Motif 3 | H196A | 1.4 ± 1 | 5,000 |
DISCUSSION
Optimized Assay Conditions Unmask the Genuine Nature of LpxH Catalysis
Previous studies of E. coli LpxH identified it as a peripheral membrane protein with conserved sequences found in the CLP family of phosphoesterases, but the enzyme properties of EcLpxH have been obscured by limited sample purity (60%) and low activity under assay conditions (8). To reveal the true nature of LpxH catalysis, we cloned and purified the H. influenzae ortholog of LpxH to near homogeneity and identified optimized assay conditions with 1,000-fold enrichment in LpxH-specific activity. These developments facilitated enzymatic reassessment of LpxH, leading to new insights into the protein's properties. Specifically, we noted the apparent Vmax value of HiLpxH, 18.1 mmol min−1 mg−1, is ∼1,000-fold greater than the corresponding E. coli value, 17.2 μmol min−1 mg−1 (8), which was determined in the absence of BSA, Triton X-100, and MnCl2. Additionally, the previously reported pH-rate profile showed small variation of activities within the same order of magnitude over the entire range of tested pH values (8), whereas that for HiLpxH reveals a 1,000-fold increase in activity over three pH units. The mono-limbic shape of the pH-rate profile for HiLpxH is similar to that reported for LpxI (10).
Perhaps the most notable difference between previous assessments of LpxH and those reported here is the activity of the enzyme in the presence of detergent. Although EcLpxH demonstrated surface dilution kinetics, with the enzyme showing inactivation at high levels of detergent (8), HiLpxH retains a relatively consistent level of activity over a wide range of detergent concentrations. The low purity of the EcLpxH protein and unsatisfactory assay conditions may have masked enzymatic activity in a micellar system. Because HiLpxH does not obey surface dilution kinetics, it is likely this enzyme is not embedded within the membrane but partitions between the cytosol and the inner membrane surface to perform catalysis, much like the model suggested for LpxI (10). This similarity in membrane interaction is consistent with the idea that LpxI and LpxH are functionally interchangeable within the lipid A pathway.
LpxH Contains a Mn2+ Cluster for Catalysis
LpxH is classified by sequence to belong to the CLP family that employs metal ions for catalysis, but its metal dependence has never been thoroughly investigated. Our enzymatic analyses reveal Mn2+ is the most likely candidate for HiLpxH's cofactor. The calculated KMetal of the enzyme for Mn2+ (487 μm) is near the estimated concentration of Mn2+ in E. coli (38), further supporting the plausibility of the metal as a physiological cofactor. Although CLPs have been shown to contain two different metals in their binuclear active sites, those containing Mn2+ usually have this ion occupying both sites (11).
We took advantage of the paramagnetic properties of Mn2+ to probe the binding of the metal to the HiLpxH with EPR spectroscopy. Results from room temperature EPR titrations suggest that two tight-binding Mn2+ ions are interacting with the protein with varying affinities. This observation is common for CLP enzymes, as binding at one site can be as much as 100 times stronger than that at the second site (11, 13). Subsequent cryogenic EPR experiments showed evidence of strong spin exchange coupling, making it likely that the two metal-binding sites in HiLpxH are in close proximity and form a cluster. Cluster formation is beneficial for the catalysis of reactions, as the proximity of the metal ions in an active site allows for a symmetric delocalization of charge that is not possible with only one metal (39). This delocalized distribution over two metals permits them to act like a unit in stabilizing transition states of concerted mechanisms that facilitate the concurrent anionic ligand addition and anionic ligand dissociation. In the case of hydrolases, this stabilization of the transition state lowers the activation barrier for both nucleophilic attack by the activated water and phosphoester bond cleavage. Additionally, it has been reported that having two close Mn2+ ions in an active site decreases the strength of bridging-ligand field potential and increases the Lewis acidity of the metals, allowing them to have a higher affinity for ligands like H2O and facilitating the formation of a hydroxide anion that performs the nucleophilic attack in a hydrolysis reaction (39).
Intriguingly, although both our enzymatic assay and the EPR measurements establish LpxH as a metalloenzyme, the two techniques yielded very different affinities for Mn2+ binding, with the enzymatic KMetal term being 2 orders of magnitude greater than the EPR-determined Kd values. The disparity in metal affinity calculations may be indicative of three Mn2+-binding sites in LpxH: two sites in the binuclear cluster revealed in the EPR studies with affinities in the low nanomolar and micromolar ranges, and another weak-binding Mn2+ that facilitates the catalysis but only binds LpxH in the presence of the substrate UDP-DAGn, yielding an apparent KMetal in the high micromolar to sub-millimolar range. Supporting this notion, members of the protein phosphate M family have been shown to contain a third Mn2+ cofactor in proximity of the di-metal cluster found in their active sites (40). This third metal has been shown to have significantly weaker affinity (high micromolar to sub-millimolar) than the ions in metal cluster (40) and has been suggested to directly take part in catalysis (41) or have a regulatory role important in substrate recognition (42).
Conserved Residues in the Metal-binding Motifs Define LpxH as a CLP, Not Nudix Family Hydrolase
The hydrolase reaction catalyzed by LpxH is more commonly carried out by enzymes in the Nudix family, which cleave the pyrophosphate bond of nucleoside diphosphates linked to another moiety (43). Although these enzymes also use metals and activated water molecules in catalysis, they have a characteristic sequence motif (“Nudix box”) of GX5EX7REUXEEXGU, with glutamate residues in the conserved motif positioning the cofactors. The metal cofactors in Nudix enzymes are almost always Mg2+ (43), making them much less diverse that those of the CLP family.
In the CLP family, metal chelation is achieved by use of the residues in the Asp-rich metallophosphoesterase motif, highlighted in Fig. 9A (11, 13). The functional groups of these amino acids directly serve as ligands to the ions and perform secondary coordination of waters and amino acids that in turn coordinate the metals, as observed in the structure of bacteriophage λ protein phosphatase (Fig. 9B). The two metal cofactors are often bridged by a carboxylate (D49, Fig. 9B); such an orientation is a common structural motif in binuclear enzymes (11, 39).
FIGURE 9.
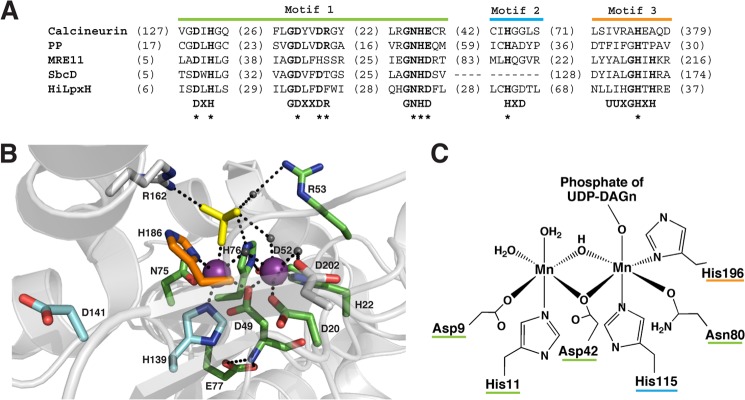
HiLpxH's active site is likely structured in a way similar to other CLP enzymes. A, alignment of conserved CLP motifs in various family members. The most conserved portion of the motif is labeled motif 1. Motifs 2 and 3 indicate portions of the motif less ubiquitous throughout the family. B, active site of bacteriophage λ protein phosphatase modified from Protein Data Bank code 1G5B (45). Residues marked in green, cyan, and orange correspond to those that are part of motifs 1–3, respectively, and their location in the motifs are denoted by * in A. Residues marked in gray are not part of the CLP motif but have roles in active site coordination. Mn2+ ions are represented by purple spheres, water molecules are indicated by dark gray spheres, and the SO4 substrate mimic is shown in yellow. Black dashed lines indicate possible modes of coordination as suggested by intramolecular distances. C, proposed Mn2+ coordination of HiLpxH based on similarity to the enzymes of the CLP family. Metal-chelating residues are underlined in color to indicate their location in the conserved CLP motifs.
The investigated alanine point mutants of conserved residues in the HiLxpH metallophosphoesterase motifs have shown a significant decrease in activity, supporting the similarity of the protein to the CLP family. These residues in HiLpxH are likely to participate in metal coordination, similar to the corresponding residues in other CLP enzymes (13, 44–47) (seen with the positions of Asp-20, His-22, Asp-49, His-139, and His-186 in the structure of bacteriophage λ protein phosphatase in Fig. 8B), and their mutations impair hydrolysis by abrogating metal binding. The D117A mutation only had a slight effect on HiLpxH activity suggesting it has a negligible role in catalysis. Support for this hypothesis comes in the position of D117's corresponding residue in bacteriophage λ protein phosphatase, which lies distant from the enzyme's binuclear site.
Taken together, the mutagenesis data from HiLpxH support an active site structure similar to that found in other CLP enzymes, with the residues of the conserved CLP motif in HiLpxH coordinating a high affinity binuclear Mn2+ site (Fig. 8C). This strongly suggests that despite the fact LpxH cleaves the pyrophosphate bond of a substrate containing a nucleoside di-phosphate linked to a lipid moiety, it is a unique member of the CLP family rather than the Nudix family (11).
Conclusion
Through development of a robust assay system and utilization of a nearly homogeneous purified protein sample, we have been able to unmask the genuine features of LpxH catalysis. Our studies reveal that the LpxH is a membrane-associating Mn2+-dependent enzyme that is more similar to LpxI in terms of pH and detergent profiles than previously reported. Furthermore, it shows strong similarities to the enzymes of the CLP family, making it possible to propose an active site structure with a binuclear site coordinated by residues of the conserved CLP motifs. These findings pave the way for structural studies of LpxH to understand the unique features of this enzyme for processing lipid substrate. Because of the importance of lipid A in bacterial pathogenesis and growth, detailed enzyme kinetic information of LpxH gained from our studies also help set the scene for further exploration of its inhibition as a method of antibiotic development.
Acknowledgments
We thank the late Dr. Christian R. H. Raetz for his meaningful contributions to the foundations of this work as well as supportive guidance. Additionally, we thank Dr. Louis Metzger and the members of the Raetz laboratory for helpful discussions. The EPR experiments at North Carolina State University were supported by National Institutes of Health Grant DE-FG02-02ER15354 (to A. I. S.) and National Science Foundation Grant MCB-0451510 (to T. I. S.).
This work was supported, in whole or in part, by National Institutes of Health Grants GM-51310 (to C. R. H. R. and P. Z.) and AI-055588 (to P. Z.).
- UDP-DAGn
- UDP-2,3-diacylglucosamine
- CLP
- calcineurin-like phosphoesterase
- EcLpxH
- E. coli LpxH
- HiLpxH
- H. influenzae LpxH
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- Lipid X
- 2,3-diacylglucosamine 1-phosphate
- TEV protease
- tobacco etch virus protease
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Raetz C. R., Whitfield C. (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raetz C. R., Reynolds C. M., Trent M. S., Bishop R. E. (2007) Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Park B. S., Song D. H., Kim H. M., Choi B. S., Lee H., Lee J. O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 [DOI] [PubMed] [Google Scholar]
- 4. Rietschel E. T., Kirikae T., Schade F. U., Mamat U., Schmidt G., Loppnow H., Ulmer A. J., Zähringer U., Seydel U., Di Padova F. (1994) Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 8, 217–225 [DOI] [PubMed] [Google Scholar]
- 5. Russell J. A. (2006) Management of sepsis. N. Engl. J. Med. 355, 1699–1713 [DOI] [PubMed] [Google Scholar]
- 6. Barb A. W., Zhou P. (2008) Mechanism and inhibition of LpxC: an essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr. Pharm. Biotechnol. 9, 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Opiyo S. O., Pardy R. L., Moriyama H., Moriyama E. N. (2010) Evolution of the Kdo2-lipid A biosynthesis in bacteria. BMC Evol. Biol. 10, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Babinski K. J., Ribeiro A. A., Raetz C. R. (2002) The Escherichia coli gene encoding the UDP-2,3-diacylglucosamine pyrophosphatase of lipid A biosynthesis. J. Biol. Chem. 277, 25937–25946 [DOI] [PubMed] [Google Scholar]
- 9. Babinski K. J., Kanjilal S. J., Raetz C. R. (2002) Accumulation of the lipid A precursor UDP-2,3-diacylglucosamine in an Escherichia coli mutant lacking the lpxH gene. J. Biol. Chem. 277, 25947–25956 [DOI] [PubMed] [Google Scholar]
- 10. Metzger L. E., 4th, Raetz C. R. (2010) An alternative route for UDP-diacylglucosamine hydrolysis in bacterial lipid A biosynthesis. Biochemistry 49, 6715–6726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitić N., Smith S. J., Neves A., Guddat L. W., Gahan L. R., Schenk G. (2006) The catalytic mechanisms of binuclear metallohydrolases. Chem. Rev. 106, 3338–3363 [DOI] [PubMed] [Google Scholar]
- 12. Cleland W. W., Hengge A. C. (2006) Enzymatic mechanisms of phosphate and sulfate transfer. Chem. Rev. 106, 3252–3278 [DOI] [PubMed] [Google Scholar]
- 13. White D. J., Reiter N. J., Sikkink R. A., Yu L., Rusnak F. (2001) Identification of the high affinity Mn2+-binding site of bacteriophage λ phosphoprotein phosphatase: effects of metal ligand mutations on electron paramagnetic resonance spectra and phosphatase activities. Biochemistry 40, 8918–8929 [DOI] [PubMed] [Google Scholar]
- 14. Zhuo S., Clemens J. C., Stone R. L., Dixon J. E. (1994) Mutational analysis of a Ser/Thr phosphatase. Identification of residues important in phosphoesterase substrate binding and catalysis. J. Biol. Chem. 269, 26234–26238 [PubMed] [Google Scholar]
- 15. Battistuzzi G., Dietrich M., Löcke R., Witzel H. (1997) Evidence for a conserved binding motif of the dinuclear metal site in mammalian and plant purple acid phosphatases: 1H NMR studies of the di-iron derivative of the Fe(III)Zn(II) enzyme from kidney bean. Biochem. J. 323, 593–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ago H., Oda M., Takahashi M., Tsuge H., Ochi S., Katunuma N., Miyano M., Sakurai J. (2006) Structural basis of the sphingomyelin phosphodiesterase activity in neutral sphingomyelinase from Bacillus cereus. J. Biol. Chem. 281, 16157–16167 [DOI] [PubMed] [Google Scholar]
- 17. Koonin E. V. (1994) Conserved sequence pattern in a wide variety of phosphoesterases. Protein Sci. 3, 356–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lucast L. J., Batey R. T., Doudna J. A. (2001) Large-scale purification of a stable form of recombinant tobacco etch virus protease. BioTechniques 30, 544–546 [DOI] [PubMed] [Google Scholar]
- 19. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 20. Radika K., Raetz C. R. (1988) Purification and properties of lipid A disaccharide synthase of Escherichia coli. J. Biol. Chem. 263, 14859–14867 [PubMed] [Google Scholar]
- 21. Belford A. I., Belford R. L. (1995) Rapid quantitation from inhomogeneously broadened EPR spectra by a fast convolution algorithm. J. Magn. Reson. A 113, 65–73 [Google Scholar]
- 22. Rusnak F., Yu L., Todorovic S., Mertz P. (1999) Interaction of bacteriophage λ protein phosphatase with Mn(II): evidence for the formation of a [Mn(II)]2 cluster. Biochemistry 38, 6943–6952 [DOI] [PubMed] [Google Scholar]
- 23. Hayden J. A., Hendrich M. P. (2010) EPR spectroscopy and catalase activity of manganese-bound DNA-binding protein from nutrient starved cells. J. Biol. Inorg. Chem. 15, 729–736 [DOI] [PubMed] [Google Scholar]
- 24. Blanchard S., Blain G., Rivière E., Nierlich M., Blondin G. (2003) Temperature dependence of X- and Q-band EPR spectra of the dinuclear manganese(II) complex [(NO2Bpmp)Mn2(mu-OAc)2]+: determination of the exchange constant and of the spin parameters for the S = 1, 2, and 3 spin states. Chemistry 9, 4260–4268 [DOI] [PubMed] [Google Scholar]
- 25. Howard T., Telser J., DeRose V. J. (2000) An electron paramagnetic resonance study of Mn2(H2O)(OAc)4(tmeda)2 (tmeda = N,N,N′,N′-tetramethylethylenediamine): a model for dinuclear manganese enzyme active sites. Inorg. Chem. 39, 3379–3385 [DOI] [PubMed] [Google Scholar]
- 26. Mathur P., Crowder M., Dismukes G. C. (1987) Dimanganese(II) complexes of a septadentate ligand. Functional analogs of the manganese pseudocatalase. J. Am. Chem. Soc. 109, 5227–5233 [Google Scholar]
- 27. Blanchard S., Blondin G., Rivière E., Nierlich M., Girerd J. J. (2003) X- and Q-band EPR studies of the dinuclear Mn(II) complex [(Bpmp)Mn2(mu-OAc)2]+. Determination of the spin parameters for the S = 1 and S = 2 spin states. Inorg. Chem. 42, 4568–4578 [DOI] [PubMed] [Google Scholar]
- 28. Chakraborty P., Chandra S. K. (1994) Synthesis, structure, EPR, and electrochemical studies of a μ2-phenoxo bridged manganese(II) dimer afforded by a binucleating macrocyclic ligand. Polyhedron 13, 683–687 [Google Scholar]
- 29. Kessissoglou D. P., Li X., Butler W. M., Pecoraro V. L. (1987) Mononuclear manganese(IV) complexes of hydroxyl-rich Schiff base ligands. Inorg. Chem. 26, 2487–2492 [Google Scholar]
- 30. Mabad B., Cassoux P., Tuchagues J. P., Hendrickson D. N. (1986) Manganese(II) complexes of polydentate Schiff bases. 1. Synthesis, characterization, magnetic properties, and molecular structure. Inorg. Chem. 25, 1420–1431 [Google Scholar]
- 31. Mathur P., Dismukes G. C. (1983) Models for the photosynthetic water oxidizing enzyme. 2. Electronic, magnetic, and EPR characterization of a binuclear manganese(II) semiquinone complex. J. Am. Chem. Soc. 105, 7093–7098 [Google Scholar]
- 32. Gelasco A., Kirk M. L., Kampf J. W., Pecoraro V. L. (1997) The [Mn(2)(2-OHsalpn)(2)](2-,-,0,+) system: Synthesis, structure, spectroscopy, and magnetism of the first structurally characterized dinuclear manganese series containing four distinct oxidation states. Inorg. Chem. 36, 1829–1837 [DOI] [PubMed] [Google Scholar]
- 33. Tzima T. D., Ferentinos E., Maganas D., Melissas V. S., Sanakis Y., Kyritsis P. (2013) Electronic and magnetic properties of the binuclear [Mn2{(OPPh2)2N}4] complex, as revealed by magnetometry, EPR and density functional broken-symmetry studies. Polyhedron 52, 706–712 [Google Scholar]
- 34. Golombek A. P., Hendrich M. P. (2003) Quantitative analysis of dinuclear manganese(II) EPR spectra. J. Magn. Reson. 165, 33–48 [DOI] [PubMed] [Google Scholar]
- 35. Epel B., Schäfer K. O., Quentmeier A., Friedrich C., Lubitz W. (2005) Multifrequency EPR analysis of the dimanganese cluster of the putative sulfate thiohydrolase SoxB of Paracoccus pantotrophus. J. Biol. Inorg. Chem. 10, 636–642 [DOI] [PubMed] [Google Scholar]
- 36. Aravind L., Koonin E. V. (1998) Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 26, 3746–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sharples G. J., Leach D. R. (1995) Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol. Microbiol. 17, 1215–1217 [DOI] [PubMed] [Google Scholar]
- 38. Outten C. E., O'Halloran T. V. (2001) Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science 292, 2488–2492 [DOI] [PubMed] [Google Scholar]
- 39. Dismukes G. C. (1996) Manganese enzymes with binuclear active sites. Chem. Rev. 96, 2909–2926 [DOI] [PubMed] [Google Scholar]
- 40. Wehenkel A., Bellinzoni M., Schaeffer F., Villarino A., Alzari P. M. (2007) Structural and binding studies of the three-metal center in two mycobacterial PPM Ser/Thr protein phosphatases. J. Mol. Biol. 374, 890–898 [DOI] [PubMed] [Google Scholar]
- 41. Su J., Schlicker C., Forchhammer K. (2011) A third metal is required for catalytic activity of the signal-transducing protein phosphatase M tPphA. J. Biol. Chem. 286, 13481–13488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rantanen M. K., Lehtiö L., Rajagopal L., Rubens C. E., Goldman A. (2007) Structure of Streptococcus agalactiae serine/threonine phosphatase. The subdomain conformation is coupled to the binding of a third metal ion. FEBS J. 274, 3128–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mildvan A. S., Xia Z., Azurmendi H. F., Saraswat V., Legler P. M., Massiah M. A., Gabelli S. B., Bianchet M. A., Kang L. W., Amzel L. M. (2005) Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 433, 129–143 [DOI] [PubMed] [Google Scholar]
- 44. Goldberg J., Huang H. B., Kwon Y. G., Greengard P., Nairn A. C., Kuriyan J. (1995) Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 376, 745–753 [DOI] [PubMed] [Google Scholar]
- 45. Voegtli W. C., White D. J., Reiter N. J., Rusnak F., Rosenzweig A. C. (2000) Structure of the bacteriophage λ Ser/Thr protein phosphatase with sulfate ion bound in two coordination modes. Biochemistry 39, 15365–15374 [DOI] [PubMed] [Google Scholar]
- 46. Schenk G., Carrington L. E., Hamilton S. E., de Jersey J., Guddat L. W. (1999) Crystallization and preliminary x-ray diffraction data for a purple acid phosphatase from sweet potato. Acta Crystallogr. D Biol. Crystallogr. 55, 2051–2052 [DOI] [PubMed] [Google Scholar]
- 47. Hopfner K. P., Karcher A., Craig L., Woo T. T., Carney J. P., Tainer J. A. (2001) Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 105, 473–485 [DOI] [PubMed] [Google Scholar]