

Background: mTORC2 integrity is dependent on SIN1 phosphorylation.
Results: mTOR maintains the integrity of mTORC2 by phosphorylation of SIN1 on Ser-260, which is regulated by an ATP-dependent mechanism.
Conclusion: The basal kinase activity of mTORC2, which maintains the constitutive phosphorylation of SIN1, requires a physiological millimolar level of ATP.
Significance: A homeostatic ATP sensor mTOR controls the integrity of mTORC2.
Keywords: ATP, Glucose, mTOR, mTOR Complex (mTORC), Protein Phosphorylation
Abstract
Nutrients are essential for living organisms because they fuel biological processes in cells. Cells monitor nutrient abundance and coordinate a ratio of anabolic and catabolic reactions. Mechanistic target of rapamycin (mTOR) signaling is the essential nutrient-sensing pathway that controls anabolic processes in cells. The central component of this pathway is mTOR, a highly conserved and essential protein kinase that exists in two distinct functional complexes. The nutrient-sensitive mTOR complex 1 (mTORC1) controls cell growth and cell size by phosphorylation of the regulators of protein synthesis S6K1 and 4EBP1, whereas its second complex, mTORC2, regulates cell proliferation by functioning as the regulatory kinase of Akt and other members of the AGC kinase family. The regulation of mTORC2 remains poorly characterized. Our study shows that the cellular ATP balance controls a basal kinase activity of mTORC2 that maintains the integrity of mTORC2 and phosphorylation of Akt on the turn motif Thr-450 site. We found that mTOR stabilizes SIN1 by phosphorylation of its hydrophobic and conserved Ser-260 site to maintain the integrity of mTORC2. The optimal kinase activity of mTORC2 requires a concentration of ATP above 1.2 mm and makes this kinase complex highly sensitive to ATP depletion. We found that not amino acid but glucose deprivation of cells or acute ATP depletion prevented the mTOR-dependent phosphorylation of SIN1 on Ser-260 and Akt on Thr-450. In a low glucose medium, the cells carrying a substitution of SIN1 with its phosphomimetic mutant show an increased rate of cell proliferation related to a higher abundance of mTORC2 and phosphorylation of Akt. Thus, the homeostatic ATP sensor mTOR controls the integrity of mTORC2 and phosphorylation of Akt on the turn motif site.
Introduction
Nutrients are essential entities for living cells. By fueling cellular metabolism, nutrients provide energy for cellular processes and building blocks for anabolic reactions. To cope with a natural inconsistency in the supply of nutrients, eukaryotic cells adapted to regulate the metabolic state according to the abundance of nutrients available to cell. This trait is critical for cell survival in starvation or malnutrition. Each cell continuously monitors levels of the nutrient substances, such as amino acids, glucose, and lipids. The nutrient-sensing pathways carry the critical decision-making mechanisms in cells by controlling the rate of anabolic and catabolic processes (1).
The nutrient-sensing mechanistic target of rapamycin (mTOR)3 pathway has been defined as a major driver of anabolic processes that controls cell growth and proliferation (2, 3). mTOR signaling monitors the abundance of amino acids, and it is also sensitive to depletion of glucose (4–7). The highly conserved protein kinase known as mTOR is the central component of this essential nutrient-sensing pathway (2, 8). mTOR exists in two distinct functional complexes. mTOR complex 1 (mTORC1) is the nutrient-sensitive complex defined by the mTOR-interacting protein raptor. This complex controls cell growth and cell size by regulating protein synthesis and autophagy. Activation of catabolic reactions in cells induced by metabolic stress inhibits mTORC1, which is mediated by activation of TSC and also the AMPK-dependent phosphorylation of raptor on Ser-792 (2, 9). The second complex of mTOR, known as mTORC2, is assembled by binding of its indispensable components rictor and SIN1 (SAPK-interacting protein 1) to mTOR. This complex functions as the regulatory component of growth factor signaling, and it regulates cell proliferation (2, 8). How metabolic stress induced by nutrient deprivation suppresses mTORC2 signaling is not known.
The functional studies identified mTORC2 as the regulatory kinase of the distinct members of the AGC kinase family (where AGC indicates the protein kinase A, G, and C families), including Akt, PKCα, and serum- and glucocorticoid-induced protein kinase (SGK) (10). Phosphorylation of Akt by mTORC2 on the hydrophobic motif Ser-473 site is dependent on the growth factor/phosphatidylinositol 3-kinase (PI3K) signaling. mTORC2 also carries a co-translational phosphorylation of the turn motif Thr-450 site of Akt independent of PI3K. A basal kinase activity of mTORC2 maintains a constitutive phosphorylation of the turn motif site of Akt required for its proper folding (11–13). It has been also shown that the mTOR kinase activity by phosphorylation of SIN1 is required to maintain the integrity of mTORC2 (14). Regulation of a basal kinase activity of mTORC2 has not been addressed previously.
EXPERIMENTAL PROCEDURES
Materials and Cell Culture
Reagents were obtained from the following sources: Dulbecco's modified Eagle's medium (DMEM)/F-12 from Invitrogen; fetal bovine serum (FBS) from Hyclone; Fugene 6 transfection reagent and the complete protease inhibitor mixture from Roche Applied Science; protein G-Sepharose from Pierce; insulin-like growth factor I from Peprotech; the antibodies to Xpress, Myc, and V5 tag from Invitrogen; the antibodies to phospho-Ser-260 SIN1, mTOR, rictor, Akt, phospho-Ser-473, and phospho-Thr-450 Akt from Cell Signaling Technologies; the antibodies to rictor, HRP-labeled anti-rabbit, anti-mouse, anti-goat secondary antibodies, and tubulin from Santa Cruz Biotechnology, Inc.; and the raptor antibody from Bethyl. Lentivaral shRNAs targeting human mTOR were generated and used as described previously. HeLa, MDA-MB-435, and HEK 293T cells were obtained from the American Type Culture Collection and cultured in DMEM/F-12 with 10% FBS, penicillin/streptomycin in 5% CO2 at 37 °C. The wild type and SIN1 null MEF cell lines were kindly provided by Bing Su (Yale University). All of the above cell lines were cultured at a density that allowed cell division throughout the course of the experiment.
Cell Lysis and Immunoblotting
All cells were rinsed with ice-cold PBS before lysis in buffer containing (40 mm HEPES (pH 7.5), 120 mm NaCl, 1 mm EDTA, 10 mm sodium pyrophosphate, 10 mm sodium glycerophosphate, 50 mm NaF, 1% Triton X-100, and protease inhibitor mixture (Roche Applied Science). The scraped lysates were incubated for 20 min at 4 °C to complete lysis. The soluble fractions of cell lysates were isolated by centrifugation at 15,000 × g at 4 °C for 10 min. Samples of the cellular lysates containing an equal amount of proteins were resolved by SDS-PAGE and transferred to PVDF membrane. Proteins were then visualized by immunoblotting and detected with enhanced chemoluminescence (ECL) from the Immobilion Western kit (Millipore).
Immunoprecipitations and Kinase Assays
For immunoprecipitation experiments, the lysis buffer contained 0.3% CHAPS instead of 1% Triton in order to preserve the integrity of the mTOR complexes. One microgram of rictor or raptor antibody was added to the cleared cellular lysates (1 mg of protein content in 700 μl) and incubated with rotation at 4 °C for 90 min. Following a 1-h incubation with 40 μl of a 50% slurry of protein G-agarose, immunoprecipitates captured by protein G-agarose were washed four times with the CHAPS-containing lysis buffer and once with rictor-mTOR kinase buffer (25 mm Hepes, pH 7.5, 100 mm potassium acetate, 2 mm MgCl2). For the kinase reaction, immunoprecipitates were incubated in a final volume of 15 μl at 37 °C for 20 min in the rictor-mTOR kinase buffer containing 500 ng of inactive Akt1-GST and 1 mm ATP. The reaction was stopped by the addition of 200 μl of ice-cold dilution buffer (20 mm MOPS, pH 7.0, 1 mm EDTA, 0.3% CHAPS, 5% glycerol, 0.1% 2-mercaptoethanol, 1 mg/ml BSA). After a quick spin, the supernatant was removed from the protein G-agarose, and a 15-μl portion was analyzed by immunoblotting for phospho-Ser-473 Akt and total Akt level detection. The raptor immunoprecipitates were analyzed for mTORC1 activity as described previously (15). The pelleted protein G-agarose beads were also analyzed by immunoblotting to determine the levels of rictor, mTOR, and raptor in the immunoprecipitates.
Mass Spectrometry
SIN1-V5 protein was resolved on SDS-PAGE by combining 10 V5 immunoprecipitation samples and visualized by Coomassie Blue staining. Excised SIN1 gel bands were washed three times with 50% acetonitrile, 200 mm ammonium bicarbonate. In-gel digestions were conducted according to standard protocols. Reduction was performed with 10 mm DTT in 100 mm ammonium bicarbonate at 60 °C for 30 min, and alkylation was performed with 20 mm iodoacetamide in 100 mm ammonium bicarbonate at room temperature for 30 min in the dark. Digestion with trypsin (1 μg of enzyme added; Promega (Madison, WI)) was performed at 30 °C overnight. Peptides were extracted from the gel bands three times with 60% acetonitrile in 0.1% trifluoroacetic acid at 30 °C for 30 min. The volume was reduced to 10 μl by vacuum centrifugation. Nano-LC/MS/MS was performed on an LTQ linear ion trap mass spectrometer (Thermo Electron Corp.) coupled with an 1100 series nano-LC system (Agilent Technologies). The nano-LC column was a 75-μm inner diameter × 360-μm outer diameter PicoFrit column (New Objective, Woburn, MA) packed with 3-μm Magic C18 resin (Michrom Bioresources, Auburn, CA). Mass spectra were acquired over a 90-min gradient (A, 0.1% formic acid; B, 90% acetonitrile in 0.1% formic acid) by data-dependent acquisition in which the top eight most intense ions per MS scan (mass range of 300–2000 m/z) were selected for collision-induced disassociation MS/MS. Dynamic exclusion was enabled with a repeat count of 2, repeat duration of 0.5 min, and exclusion duration of 1.5 min. MS/MS spectra were analyzed using a combination of Spectrum Mill (Agilent Technologies) and manual interpretation.
SIN1 Mutagenesis, Retroviral Vector, Retroviral Production, and Infection
pMSCV-SIN1-Myc plasmid DNA was obtained from Addgene. The primers for mutagenesis were designed based on the QuikChange primer design program available at Stratagene's Web site. The MSCV plasmid containing the SIN1 fragment has been mutagenized with the QuikChange XLII mutagenesis kit (Stratagene). For stable expression of SIN1 and its phosphomutants, the pMSCV-puro retroviral vector containing the puromycin resistance gene has been used in this study. Plasmids were propagated in and purified from XL-10 Gold bacterial cells. The day prior to transfection, human embryonic kidney (HEK) 293T cells (1.2 × 106) were split into 6-cm dishes in 3 ml of DMEM supplemented with 10% fetal bovine serum. For production of retroviruses, HEK 293T cells were transfected by the calcium phosphate method using 3 μg of transfer vector pMSCV, 0.6 μg of envelope coding plasmid Vsv-g, and 2.4 μg of Gag-pol-expressing plasmid. Retroviruses were harvested 48 h after transfection by centrifugation at 3,000 × g at 4 °C for 15 min. The day prior to infection, cells to be infected were seeded in 6-well dishes. The viral supernatant was added at a ratio of 1:1 to the culture medium in the presence of Polybrene (8 μg/ml), and the cells were centrifuged at 1,800 rpm for 45 min. Cells were incubated with retroviruses for the following 24 h. A second infection was performed following the same protocol the next day. After an additional 24 h of recovery in normal medium, infected cells were passaged and selected with puromycin (2 μg/ml for 2 days).
Purification of the Soluble FLAG-mLST8/Myc-mTOR Heterodimer
The purification of FLAG-mLST8 was carried out according the previous study (16). The FLAG-mLST8 plasmid was co-transfected with Myc-mTOR cDNA in HEK-293T cells. After a 48-h transfection, cells were washed with cold PBS, lysed with 0.3% CHAPS buffer, and incubated with mild agitation for 20 min at 4 °C. The lysate was transferred to the spinning columns for centrifugation for 15 min at 10,000 rpm. After centrifugation, the supernatant was used for the FLAG affinity purification. FLAG M2 affinity resins were washed three times in 0.3% CHAPS lysis buffer and packed into a 10-ml Bio-Rad column. The cellular lysates were applied through the column seven times. After running the lysates, the column was washed with 20 ml of lysis buffer and 10 ml of elution buffer (40 mm HEPES, pH 7.4, 500 mm NaCl, 0.1% CHAPS). The FLAG-mLST8/Myc-mTOR heterodimer was eluted by the elution buffer containing 0.5 mg/ml of the FLAG peptide. The eluted fractions from number 2 to 6 were combined, dialyzed with mTORC2 kinase buffer, and concentrated with Millipore Amicon Ultra-15 centrifugal filter units. The purified FLAG-mLST8/Myc-mTOR heterodimer was analyzed by immunoblotting and applied for the in vitro mTORC2 assembly.
In Vitro mTORC2 Assembly and Kinase Reaction
The assembly of mTORC2 has been performed as described previously (14). Myc-rictor and SIN1-V5 cDNAs were co-transfected in HEK-293T cells. After a 48-h transfection, cells were washed with cold PBS, lysed with 0.3% CHAPS buffer, and subjected to centrifugation for 15 min at 10,000 rpm. The supernatant was applied for immunoprecipitation with anti-V5 antibody. The Myc-rictor/SIN1-V5 immunoprecipitates were incubated with or without the purified soluble FLAG-mLST/Myc-mTOR heterodimer at room temperature for 2 h in mTORC2 kinase buffer with 1 mm ATP. After washing three times with 0.3% CHAPS lysis buffer, the immunoprecipitates were used for the in vitro mTORC2 kinase reaction with wild-type Akt as the substrate.
Tumor Xenografts
The study has been carried out as described previously (17). MEFs constitutively expressing the wild-type, S260D, or S260A SIN1 were transformed by H-Ras overexpression. The MEF cell lines (5 × 106 cells/mouse) were injected subcutaneously into the upper flank region of 6-week-old immunodeficient nude mice (n = 5 for each group). Tumor size was measured after 15 days, and the tumor volume was determined with the standard formula L × W2 × 0.5, where L is the longest length and W is the shortest length in millimeters. The differences in the tumor volume from mice injected with wild-type, S260D, or S260A SIN1-expressing MEFs were compared by one-way analysis of variance. Nude mice were sacrificed, and the tumors were excised.
RESULTS
Glucose, but Not Amino Acids, Is Required for the Akt Phosphorylation on Thr-450, SIN1 Protein Stability, and Integrity of mTORC2
We hypothesized that if mTOR is a central component of the nutrient-sensing pathway (2), then a basal kinase activity of mTORC2 is nutrient-dependent. How nutrients regulate mTORC2 is poorly characterized, and we studied the nutrient depletion effects on mTORC2 in HeLa and MDA-MB-435 human cancer cell lines. We detected a decrease in the protein levels of SIN1 and rictor following glucose deprivation for 20 h (Fig. 1A). Most likely, these effects on the mTORC2 components are related to the metabolic stress response but not cell death, because we detected a decrease in cell number only following 42 h of glucose deprivation (data not shown). Amino acid deprivation of cells for a similar period of time did not indicate detectable changes in the abundance of SIN1 or rictor. We also observed that the glucose deprivation also caused inhibition of a basal kinase activity of mTORC2, as indicated by decreased phosphorylation of Akt on the turn motif Thr-450 site. These data indicate that the basal kinase activity of mTORC2 is sensitive to glucose but not amino acid deprivation, and its low activity is associated with a decrease in abundance of SIN1 and rictor.
FIGURE 1.

The mTORC2 integrity and Akt phosphorylation on Thr-450 are sensitive to ATP depletion. A, decreased SIN1 and rictor protein abundance under glucose starvation is associated with reduced basal activity of mTORC2, as detected by Akt phosphorylation (p-Akt) on Thr-450. MDA-MB-435 and HeLa cells were incubated with glucose-free or amino acid (AA)-free medium for 20 h. Immunoblotting was used to detect phosphorylation of Akt on Thr-450 and the abundance of the indicated proteins in cell lysates. B, integrity of mTORC1 and mTORC2 is glucose-dependent. MDA-MB-435 cells were incubated with glucose-free medium for 10, 16, and 20 h or with amino acid-free medium for 20 h, respectively. The mTOR immunoprecipitates were prepared from cell lysates to examine the complex integrity of mTORC1 and mTORC2. Immunoblotting was used to detect phosphorylation of raptor on Ser-792 and the abundance of the indicated proteins in the immunoprecipitates and cell lysates. C, detection of cellular ATP levels under glucose starvation and acute ATP depletion. MDA-MB-435 cells were incubated with glucose-free medium for 0, 3, 6, 9, 12, or 15 h and collected for quantitative detection of ATP by a luciferase-driven bioluminescence assay (Roche Applied Science ATP Bioluminescence Assay Kit CLS II). For acute ATP depletion, MDA-MB-435 cells were incubated with 5 mm 2-DG and 10 μm rotenone in glucose-free medium for 1, 2, or 4 h and collected for quantitative detection of ATP. Three independent experiments were shown for each condition. D, acute ATP depletion leads to a low abundance of SIN1. MDA-MB-435 cells were incubated with 5 mm 2-DG and 10 μm rotenone in glucose-free medium for 1, 1.5, and 2 h. Immunoblotting was used to detect abundance of the indicated proteins in the cell lysates. Error bars, S.E.
To follow up our initial finding, we addressed the question of whether the integrity of the mTOR complexes is sensitive to nutrient deprivation. By analyzing the mTOR immunoprecipitates, we found that the integrity of mTORC1 and mTORC2 is not sensitive to amino acid deprivation, as detected by the similar levels of raptor, rictor, and SIN1 bound to mTOR purified from the cells incubated with or without amino acids (Fig. 1B). On the contrary, we found that the integrity of both mTOR complexes was affected by glucose deprivation. The glucose-dependent decrease in abundance of mTORC1 and mTORC2 that was initially detected at 16 h became more evident following glucose starvation for 20 h. We observed that the disintegration of mTORC1 has been associated with the AMP-activated protein kinase-dependent phosphorylation of raptor on Ser-792 (9). The initial weak phosphorylation that was detected at 10 h gradually increased and became intense at 20 h of glucose deprivation. The glucose-dependent posttranslational modification of raptor altered its mobility to a slower migratory form on a gel and decreased its binding to mTOR, but the abundance of raptor has not been altered, as detected in the cell lysates. We found that the disintegration of mTORC2 was carried out by a different mechanism because the abundance of its essential components SIN1 and rictor was substantially decreased, as detected in the cell lysate following glucose starvation for 20 h. A role of AMP-activated protein kinase in the regulation of SIN1 and rictor has not been described previously, but mTOR kinase-dependent regulation of SIN1 turnover and the integrity of mTORC2 has been reported (14). A low basal kinase activity of mTORC2 under glucose starvation is supported by a weak phosphorylation of Akt on Thr-450, known as a constitutively phosphorylated site, by mTORC2. Because the mTOR kinase activity is required to maintain the integrity of mTORC2 by phosphorylation of SIN1 (14), we proposed that under glucose deprivation, the disintegration of mTORC2 might be linked to the mTOR-dependent regulation of SIN1.
Glucose Deprivation Effects on mTORC2 Signaling Are Linked to Cellular ATP Level
Glucose is a major source of energy in mammalian cells, and a lack of glucose causes ATP depletion in cells (18). Within 15 h of the glucose deprivation, we detected less than 15% of the intracellular ATP level compared with control cells (Fig. 1C). The glucose-dependent decline in ATP level correlates well with the disintegration of mTORC2. To determine whether a cellular ATP level controls basal mTORC2 activity and SIN1 abundance, we induced the acute depletion of ATP in cells by simultaneous inhibition of glycolysis (2-DG) and mitochondrial oxidative phosphorylation (rotenone) (19). We observed that incubation of cells with both drugs for 2 h caused a decreased phosphorylation of Akt on the turn motif Thr-450 site that associated with a substantial decrease of SIN1 protein (Fig. 1D) and alteration of its mobility in a gel, indicating dephosphorylation of the protein as described previously (14). This observation suggests that cellular ATP levels control the integrity of mTORC2 by regulation of expression of SIN1. We found that ATP depletion inhibited the basal kinase activity of mTORC2, as detected by Akt phosphorylation on Thr-450 and also dephosphorylation of SIN1.
A High Level of ATP Is Required for Optimal Kinase Activity of mTORC2 and mTORC1
How might cellular ATP level regulate the kinase activity of mTORC2? This regulation may be carried out by a regulation of the kinase component of this complex mTOR that has been identified previously as the homeostatic ATP sensor (20). It has been reported that the optimal kinase activity of mTOR requires an ATP concentration above 1 mm, whereas most of the kinases require much lower ATP and are effective at concentrations below 0.1 mm. To address the sensitivity of mTORC2 to ATP levels, we examined the question of what is an optimal concentration of ATP for the functional activity of this kinase complex. The ATP titration study determined that mTORC2 requires an ATP concentration above 1.2 mm for its effective kinase activity (Fig. 2A). This finding shows that mTORC2 operates efficiently as the kinase at a high (millimolar) ATP concentration, and it is consistent with the initial finding indicating a hypersensitivity of the mTOR kinase activity to the ATP level (20). We also performed the analogous functional study of mTORC1 and identified that it also carried the optimal activity to phosphorylate its substrate S6K1 on Thr-389 when the ATP concentration was above 1.2 mm (Fig. 2B). We interpret that mTOR as the kinase, with its distinctive threshold for ATP level above 1 mm, determines a high sensitivity of both mTOR complexes to cellular ATP levels. In the case of mTORC2, an ATP-dependent regulatory mechanism controls not only its functional activity but also the kinase complex integrity carried by the mTOR-dependent phosphorylation of SIN1. This critical phosphorylation site of SIN1 has not been identified.
FIGURE 2.
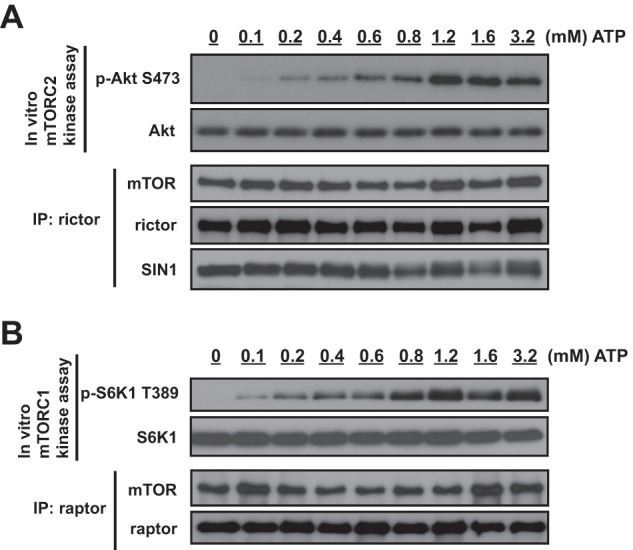
Both mTOR complexes require a millimolar level of ATP for their optimal kinase activities. A, for the mTORC2 kinase reaction, rictor immunoprecipitates (IP) prepared from lysates of MDA-MB-435 cells growing in cell culture medium containing 10% serum were used for in vitro kinase assays with full-length WT Akt1 as the substrate and different concentrations of ATP (0, 0.1, 0.2, 0.4, 0.6, 0.8, 1.2, 1.6, and 3.2 mm, respectively). B, for the mTORC1 kinase reaction, raptor immunoprecipitates prepared from lysates of MDA-MB-435 cells growing in cell culture medium containing 10% serum were used for in vitro kinase assays with full-length WT S6K1 as the substrate and different concentrations of ATP (0, 0.1, 0.2, 0.4, 0.6, 0.8, 1.2, 1.6, and 3.2 mm, respectively).
Phosphorylation of SIN1 on Ser-260 by mTOR Is Sensitive to ATP Depletion
To identify the mTOR-dependent phosphorylation of SIN1, we analyzed SIN1 phosphorylation following the reconstitution of mTORC2 with the wild-type or kinase-dead form of mTOR. Co-expression of the four essential components of mTORC2 carrying distinct tags (Myc-mTOR, Xp-rictor, V5-SIN1, and HA-mLST8) has been sufficient to assemble the functional kinase complex (14). By immunoprecipitation of V5-SIN1, we isolated mTORC2, as detected by co-purification of mTOR and rictor (Fig. 3A). Mass spectrometry analysis of the protein band corresponding to SIN1 revealed its phosphorylation on Ser-260 when mTORC2 was assembled with the functional wild-type mTOR but not with its kinase-dead form (Fig. 3B). Based on this analysis, the Ser-260 residue of SIN1 has been identified as the mTOR-dependent phosphorylation site. This finding marks mTOR as a potential Ser-260 kinase of SIN1, which is also supported by the hydrophobic nature of this site (Fig. 3C). It is surrounded by hydrophobic amino acids and resembles the known hydrophobic motif sites of mTOR (10, 21). To determine whether mTOR functions as the Ser-260 kinase of SIN1, we have performed the functional study by assembly of mTORC2 in vitro, as described previously (14). We assembled the functional mTORC2 kinase complex by co-incubation of the soluble mLST8/mTOR heterodimer with the immobilized rictor/SIN1 heterodimer, as detected by the phosphorylation of its substrate Akt on Ser-473 (Fig. 3D). Importantly, following the mTORC2 assembly in vitro, we also detected phosphorylation of SIN1 on Ser-260 by the phosphospecific antibody. The specificity of the phospho-Ser-260 SIN1 antibody has been initially validated by detection of the wild-type SIN1 but not its S260A phosphomutant (data not shown). In addition, the Ser-260 site is localized within the most conserved SIN1 sequence, designated as Box1 (residues 225–267) of the CRIM (conserved region in the middle) domain (22), as illustrated in Fig. 3C, and therefore might carry a critical regulatory role. Thus, our study identified that mTOR is the Ser-260 kinase of SIN1.
FIGURE 3.

Phosphorylation of SIN1 on the hydrophobic and conservative Ser-260 site by mTOR is sensitive to glucose starvation and ATP depletion. A, to reconstitute mTORC2, Xpress-rictor, SIN1-V5, and HA-mLST8 constructs were co-transfected with Myc-mTOR (wt) or Myc-mTOR (KD) in HEK-293T cells, respectively. Immunoprecipitates (IP) prepared from lysates of HEK-293T cells with anti-V5 antibody were resolved in SDS-PAGE and visualized by Coomassie Blue staining. The SIN1 protein bands were excised and analyzed by mass spectrometry. B, phosphorylation of Ser-260 in SIN1 identified by MS/MS. SDS-PAGE purification followed by enzymatic digestion and LC/MS/MS was used to isolate peptide containing a phosphorylated serine residue in position 4 (S*). Expected masses for the y and b ions are listed above and below the peptide sequence. Ions that were positively identified are highlighted in blue and red. y ions at 940.5 and 773.5 define the phosphoserine residue at position 4. The abundant ion at 597.5 is the neutral loss of 98 Da from the doubly charged parent ion. C, Ser-260 is a conserved and hydrophobic site of SIN1. SIN1 protein domains are as follows. CRIM, conserved region in the middle; RBD, Raf-like Ras-binding domain; PH, pleckstrin homology. Box1 (amino acids 225–267) is the highly conserved region located within the CRIM domain. The mTOR kinase-dependent SIN1 site (Ser-260) resides in Box1. The Ser-260 residue of SIN1 and the known mTORC2 phosphorylation sites of Akt, SGK1, and PKCα are surrounded by hydrophobic amino acids. *, hydrophobic amino acids. D, mTOR is the Ser-260 kinase of SIN1. In vitro mTORC2 assembly was performed by incubation of two heterodimers, rictor/SIN1 (in an immobilized form) and mTOR/mLST8 (in a soluble form). The assembled mTORC2 complex in vitro has been examined by the in vitro mTORC2 kinase reaction and also by phosphorylation of SIN1 on Ser-260. E, glucose starvation results in dephosphorylation of SIN1 on Ser-260. MDA-MB-435 cells were incubated in the medium with or without glucose for 10 and 16 h. As controls, the cells were also incubated in 10% serum medium or amino acid-free medium for 16 h. The SIN1 protein was immunopurified, and immunoblotting was used to detect the abundance of SIN1 and its phosphorylation on Ser-260. F, acute ATP depletion leads to dephosphorylation of SIN1 on Ser-260 and mTORC2 complex dissociation. MDA-MB-435 cells were incubated with 5 mm 2-DG and 10 μm rotenone in glucose-free medium for 1 h. The SIN1 immunoprecipitates were prepared from cell lysates to examine the complex formation of mTORC2. Immunoblotting was used to detect phosphorylation of SIN1 on Ser-260 and the abundance of the indicated proteins in the immunoprecipitates and cell lysates.
To study the role of SIN1 phosphorylation, we first assessed whether the Ser-260 phosphosite is nutrient-dependent. We analyzed the phosphorylation of SIN1 immunopurified from cells deprived of amino acids or glucose. We observed that phosphorylation of SIN1 on Ser-260 was decreased following glucose but not amino acid starvation for 16 h (Fig. 3E). This time point correlates well with ATP depletion caused by glucose starvation (Fig. 1C). To link the regulation of SIN1 phosphorylation with the cellular ATP level, we induced acute ATP depletion in cells as described in Fig. 1D. We found that ATP depletion has been effective at causing dephosphorylation of SIN1 on Ser-260 within 1 h (Fig. 3F). This dephosphorylation event has also been associated with a decrease in abundance of mTORC2, as detected by the level of mTOR co-purified with SIN1. This implies that a cellular ATP depletion within 1 h led to disintegration of mTORC2. Our study indicates that SIN1 phosphorylation on Ser-260 is sensitive to ATP depletion, and this finding reflects the millimolar ATP level threshold for the basal activity of its kinase.
mTOR-dependent Phosphorylation of SIN1 on Ser-260 Controls the Stability of SIN1 and Integrity of mTORC2
The mTOR kinase activity is required for SIN1 protein stability and integrity of mTORC2. We found that mTOR phosphorylated SIN1 on Ser-260, and this phosphorylation was sensitive to ATP depletion. We hypothesized that if Ser-260 is a major mTOR-dependent regulatory site of SIN1, then the mutation of this site, mimicking or preventing phosphorylation, will affect SIN1 protein stability and mTORC2 integrity. We generated the SIN1 phosphomutant cDNAs by a substitution of Ser-260 to alanine (S260A) (to prevent phosphorylation) or to aspartic acid (S260D) (to mimic phosphorylation). The reconstitution of mTORC2 by co-expressing its four essential components (14) indicated an uneven expression of SIN1 and its phosphomutants (Fig. 4A). Consistent with the previous study (14), we observed a low expression of the wild-type SIN1 when it was co-expressed with the kinase-dead form of mTOR. Importantly, we found that mTOR kinase-dependent expression of SIN1 was related to its phosphorylation on Ser-260. A low expression of the S260A SIN1 mutant was detected when it was co-expressed with either the wild-type or kinase-dead form of mTOR. On the contrary, expression of the S260D SIN1 mutant was not dependent on the kinase activity of mTOR, because we detected a relatively high abundance of this mutant when it was co-expressed with the wild type or kinase-dead form of mTOR. This finding indicates a critical role of the Ser-260 phosphorylation site in regulation of expression of SIN1. The S260D mutation, by mimicking the mTOR-dependent phosphorylation, leads to elevated expression of SIN1, whereas the S260A mutation, by preventing phosphorylation, results in a low expression. This regulation of protein expression was carried out by an accelerated lysosomal protein degradation of SIN1, because inhibition of lysosomal protein degradation by chloroquine was effective in the recovery of the S260A SIN1 mutant expressed transiently (Fig. 4B) or in the stable cell line (Fig. 4C). This observation agrees with the previous finding indicating that mTOR, by phosphorylation of SIN1, prevents its lysosomal degradation (14). Thus, mTOR, by phosphorylation of SIN1 on Ser-260, regulates the protein turnover of SIN1 and implicates this phosphorylation site in the regulation of mTORC2 integrity.
FIGURE 4.
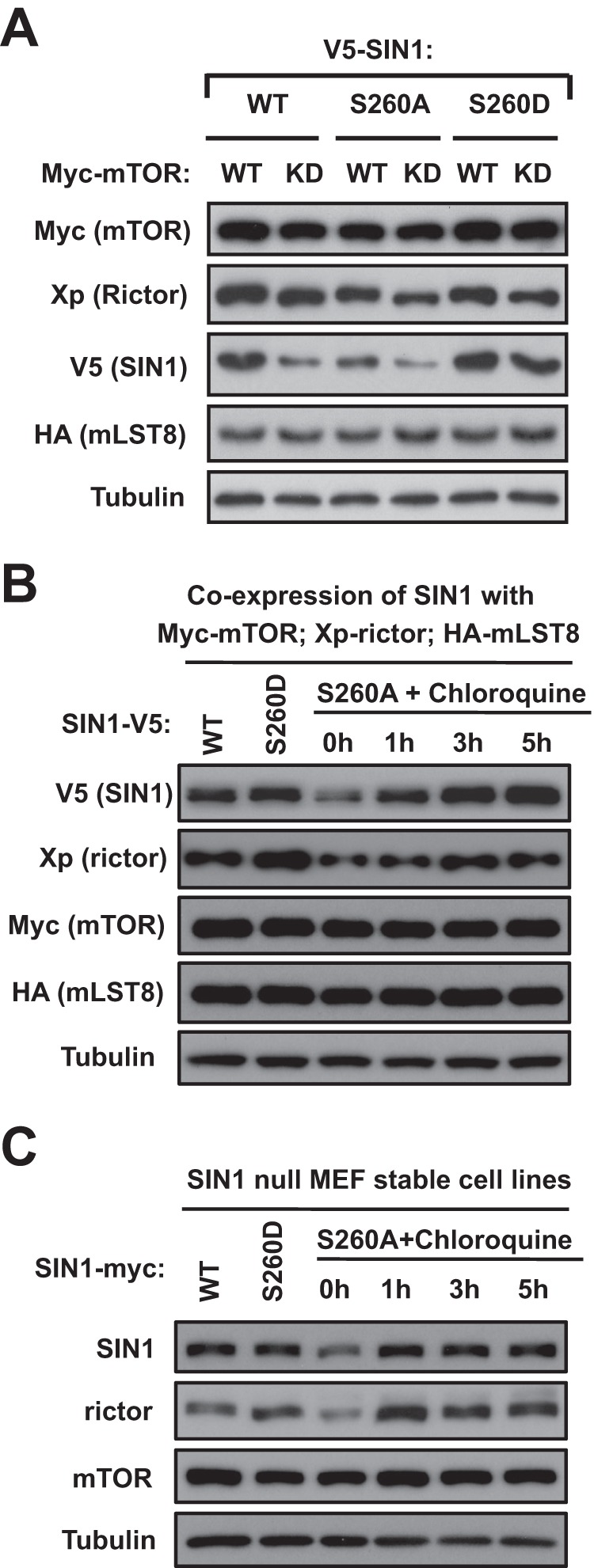
SIN1 protein stability is dependent on its phosphorylation on Ser-260. A, the SIN1 Ser-260 phosphorylation site is required for protein stability. The V5-tagged wild-type or phosphospecific SIN1 recombinant proteins (S260D and S260A) were co-expressed with Myc-mTOR (wt) or Myc-mTOR (KD), respectively, and another two mTORC2 components (Xp-rictor and HA-mLST8) in HEK-293T cells. The total cell lysates were analyzed by Western blotting with the indicated tag antibodies. B and C, the Ser-260 phosphorylation site of SIN1 prevents its lysosomal degradation. SIN1-V5 WT, S260D, or S260A construct was co-transfected with Myc-mTOR WT, Xpress-rictor, and HA-mLST8 constructs into HEK-293T cells (B). After a 48-h transfection, cells expressing SIN1-V5 S260A cDNA were treated with 100 μm chloroquine for 1, 3, and 5 h. SIN1-null MEFs stably expressing SIN1 Ser-260A phosphomutant were treated with 100 μm chloroquine for 1, 3, and 5 h (C). The total cell lysates were analyzed by Western blotting with the indicated antibodies.
To assess if a specific SIN1 phosphorylation regulates the integrity of mTORC2, we have optimized expression levels of SIN1 and its phosphomutants (Fig. 5A). Reconstitution of mTORC2 with the wild type of mTOR showed a low expression of the S260A SIN1 mutant, and we attained a similar level of expression by applying only 30% of the cDNA amount encoding the wild type or S260D mutant. Following optimization of the SIN1 expression, we detected a low abundance of mTORC2 assembled with the SIN1 S260A mutant, as observed by a decreased level of the mutant co-purified with mTOR. This observation indicates that prevention of the SIN1 phosphorylation by mTOR interferes with the integrity of mTORC2. Consistent with this finding, a replacement of mTOR with its kinase-dead form shows deficiency in mTORC2 integrity not only with the S260A SIN1 mutant but also with its wild type form. Importantly, the integrity of mTORC2 remained insensitive to the mTOR kinase activity when the complex was assembled with the SIN1 mutant mimicking constitutive phosphorylation on Ser-260 (S260D). Thus, based on our reconstitution study, we find that a single phosphorylation site of SIN1 regulates integrity of mTORC2.
FIGURE 5.

Phosphorylation of SIN1 on Ser-260 controls the integrity of mTORC2. A, the V5-tagged wild-type or phosphospecific SIN1 (S260D and S260A) cDNAs were co-expressed with Myc-mTOR (wt) or Myc-mTOR (KD), respectively, and the other two mTORC2 components (Xp-rictor and HA-mLST8) in HEK-293T cells. To optimize the SIN1 expression, the Myc-mTOR WT was co-transfected with a cDNA amount containing 30% V5-SIN1 WT, 30% V5-SIN1 S260D, or 100% V5-SIN1 S260A. The Myc-mTOR-KD was co-transfected with a cDNA amount containing 100% V5-SIN1 WT, 30% V5-SIN1 S260D, or 100% V5-SIN1 S260A. The Myc-mTOR immunoprecipitates (IP) were prepared from cell lysates to examine the complex integrity of mTORC2. Immunoblotting was used to detect the amount of the indicated proteins in the immunoprecipitates and cell lysates. B, stable expression of the SIN1 S260D phosphomutant associates with a higher phosphorylation of Akt and abundance of mTORC2. SIN1-null MEFs carrying a stable expression of WT SIN1 or its phosphomutants were grown in 10% serum-containing medium. SIN1 immunoprecipitates from stable MEFs were used to examine mTORC2 integrity. Immunoblotting was performed to detect the indicated proteins in the immunoprecipitates and cell lysates.
SIN1 Phosphorylation on Ser-260 Regulates Cell Proliferation and Glucose Sensitivity
To examine the biological role of the mTOR-mediated SIN1 phosphorylation on Ser-260, we generated stable cell lines by restoring expression of the wild-type SIN1 or its phosphomutants in the SIN1 null MEFs. We detected a higher basal phosphorylation of Akt on Ser-473 in cells expressing the SIN1 S260D mutant compared with those expressing the wild-type SIN1 or its S260A mutant (Fig. 5B). Phosphorylation of Akt on Ser-473 in these cell lines correlated well with the differences in abundance of mTORC2. By pulling down SIN1 and detecting the levels of mTOR, we found that cells expressing the S260D mutant maintained a high abundance of mTORC2 compared with cells expressing the wild-type SIN1, whereas the lowest abundance of mTORC2 was detected in cells expressing the S260A mutant (Fig. 5B). Similar to the mTORC2 reconstitution study by transient expression (Fig. 5A), the stable expression of SIN1 also indicates an important role of its Ser-260 site in controlling the integrity of mTORC2. Interestingly, regulation of the mTORC2 integrity by SIN1 phosphorylation on Ser-260 is independent of its binding partner rictor (23–25), because the SIN1 phosphomutants did not show any differences in binding to rictor. We also found that the abundance of mTORC2 induced by mimicking the mTOR-dependent phosphorylation of SIN1 carried out its functional activity not only by the elevated basal phosphorylation of Akt but also by a higher cell proliferation rate (Fig. 6A). Analysis of cell number following incubation of MEFs in growth medium containing serum for 48 h shows that restoration of the wild-type SIN1 expression increased the proliferation rate up to 63%. A similar analysis carried out by expression of the S260D mutant led to a substantial increase in cell proliferation to 89%, whereas expression of the S260A mutant showed only a 27% increase. This study outlines the biological relevance of SIN1 phosphorylation by mTOR, indicating a potent regulatory role in cell proliferation by altering its rate at least three times. To extend our finding to an in vivo model, we analyzed the effects of the SIN1 phosphomutants on subcutaneous tumor growth. Initially, to establish tumor growth, the SIN1 null MEFs reconstituted with the wild type SIN1 or its phosphomutants were transformed by overexpressing an equal amount of H-Ras (data not shown). We observed that cells expressing the wild type of SIN1 or its phosphomimetic S260D mutant grew into large tumors, whereas the wild-type SIN1 expression yielded slightly bigger tumors. Most importantly, mice injected with cells expressing the SIN1 S260A mutant, preventing phosphorylation of this site, developed much smaller tumors than those injected with either wild-type SIN1 or its S260D mutant-expressing cells (Figs. 6, B and C). These results suggest that mTOR-dependent phosphorylation of SIN1 on Ser-260 by controlling the integrity of mTORC2 is important in regulation of cell proliferation and tumor growth.
FIGURE 6.

Phosphorylation of SIN1 on Ser-260 regulates cell proliferation and tumor growth. A, cell proliferation assay; the SIN1 null cells carrying a stable expression of SIN1 and its phosphomutants shown in Fig. 5B were analyzed by counting cells at 48 and 72 h after incubation in the medium containing 10% FBS. The ratio of the proliferation rate was graphed with GraphPad Prism 5 software. *, p < 0.001 for all pair-wise comparisons; one-way analysis of variance, post hoc intergroup comparisons with the Holm-Sidat test. B and C, SIN1 phosphorylation on Ser-260 regulates tumor growth. MEFs described in A were transformed by overexpression of the oncogenic form of H-Ras and injected into 6-week-old immunodeficient nude mice (n = 5 for each group; 3 × 106 cells/mouse). Tumor size was measured after 3, 4, and 5 weeks; the tumor sizes were calculated, and the volumes are shown by a line graph (B). *, p < 0.04 for the 4-week time point; **, p < 0.015 for the 5-week time point; one-way analysis of variance, post hoc intergroup comparisons with the Holm-Sidat test; p values are indicated. Mice injected with MEFs expressing each form of SIN1 were sacrificed, and representative images of the excised tumors are shown in C. Error bars, S.E.
Our initial observation indicated a dependence of mTORC2 integrity on glucose deprivation linked to ATP depletion. The mTORC2 kinase activity is highly sensitive to cellular ATP level, and the metabolic stress causing depletion of ATP inhibits a basal kinase activity of mTORC2 that accelerates breakdown of the kinase complex by preventing phosphorylation of SIN1 on Ser-260 by mTOR. Based on our finding, we proposed that cells expressing the phosphomimetic S260D mutant of SIN1 will be more resilient to glucose starvation. To address further the functional role of SIN1 phosphorylation, we studied SIN1 null cell lines carrying a stable expression of the wild-type SIN1 or its phosphomutants in the medium containing insulin-like growth factor 1 and regular (5.5 mm) or low (0.55 mm) glucose concentration. Following incubation of MEFs for 48 h in cell culture medium with a regular glucose concentration, we detected a similar basal phosphorylation of Akt on Ser-473 in cells expressing the wild-type SIN1 and its S260D mutant. A low Akt phosphorylation has been observed in cells expressing the S260A mutant (Fig. 7A). The phosphorylation of Akt on its regulatory mTORC2-dependent site correlated well with the cell proliferation rates of these cell lines (Fig. 7B). The SIN1 null cells incubated in the medium containing a regular glucose concentration and insulin-like growth factor 1 did not proliferate, as indicated by cell number. Restoring the expression of the wild-type SIN1 or its S260D mutant in SIN null cells induced proliferation in the range of 60%, and expression of its S260A mutant was 2-fold less effective, inducing proliferation only about 30%. Prevention of the mTOR-dependent phosphorylation of SIN1 caused a low activity of mTORC2 and slow proliferation rate. Instead, cells expressing the wild type of SIN1 or its phosphomimetic mutant showed a similar higher activity of mTORC2 and rate of proliferation when cultured in the serum-free medium containing a regular glucose concentration and supplemented with insulin-like growth factor 1. This similarity might also explain a comparable tumor growth rate induced by these two cell lines detected in the xenograft study (Fig. 6). Our data suggest that the wild type of SIN1 and its phosphomimetic mutant show similar phenotypes when expressed in cells incubated in a medium containing glucose, which is required to maintain a physiological level of ATP.
FIGURE 7.

Sensitivity to glucose depletion depends on phosphorylation of SIN1 on Ser-260. A, SIN1-null MEFs stably expressing wild type SIN1 and its phosphomutants were grown without serum in medium containing 5.5 or 0.55 mm glucose and 100 ng/ml IGF-1 for 48 h. Immunoblotting was used to detect the indicated proteins and Akt phosphorylation in cell lysates. B and C, cell proliferation was performed in the cell lines analyzed in A. Analysis of the cells incubated in medium containing 5.5 or 0.55 mm glucose is presented in B and C, respectively. The ratio of proliferation rate was graphed with GraphPad Prism 5 software. *, p < 0.001 for all pair-wise comparisons; one-way analysis of variance; post hoc intergroup comparisons with Holm-Sidat test. Error bars, S.E.
Switching these cell lines to the medium containing low glucose indicated the sensitivity of the wild-type SIN1 but not its phosphomimetic S260D mutant to a lack of glucose (Fig. 7C). In a low glucose medium, the cells carrying expression of the wild-type SIN1 indicated a substantial decrease in Akt phosphorylation and abundance of SIN1 that has also been reflected in a decreased rate of cell proliferation (27%). On the contrary, we found that cells expressing the S260D mutant remained insensitive to a low glucose concentration, as detected by the phosphorylation of Akt, abundance of SIN1, and also higher a cell proliferation rate (53%). Thus, our study indicates that a glucose-dependent sensitivity of the mTORC2 signaling is mediated by mTOR phosphorylation of SIN1 on Ser-260 required to maintain the integrity of mTORC2.
DISCUSSION
Our results show that the cellular ATP level regulates the basal kinase activity of mTORC2 because the optimal kinase activity of this complex requires ATP concentration at the physiological range above 1.2 mm. This observation is consistent with the original finding that mTOR as the homeostatic ATP sensor requires a millimolar concentration of ATP for its kinase activity, whereas a 0.1 mm ATP concentration is optimal for most protein kinases (20). We found that the ATP-dependent basal kinase activity of mTORC2 carries the autoregulatory mechanism controlling integrity of the kinase complex, where mTOR stabilizes SIN1 by phosphorylation of its Ser-260 site and maintains the integrity of mTORC2. By a similar mechanism, mTORC2 controls a proper folding of its downstream effector Akt by its phosphorylation on the turn motif Thr-450 site. Our study shows that mTOR, a key regulator of anabolic processes in cells, also restrains its own signaling by the autoregulatory mechanism if cellular energy supplies get low.
mTORC2 regulates Akt by controlling phosphorylation of its two distinct sites. Phosphorylation of Akt on the Ser-473 site by mTORC2 is dependent on growth factor/PI3K signaling, whereas mTORC2, together with PDK1, by phosphorylation of Akt on the regulatory Ser-473 and catalytic Thr-308 sites couples activation of Akt to growth factor signaling (10). It has been reported that the PI3K-dependent activation of mTORC2 is sensitive to endoplasmic reticulum stress (17) and takes place in association with ribosomes (26). mTORC2 also carries out the PI3K-independent regulation of Akt by phosphorylation of the Thr-450 turn motif site. This phosphorylation as a co-translational modification of Akt is required for its proper folding. It has been defined as a constitutively phosphorylated site that facilitates the carboxyl-terminal folding and stabilizes newly synthesized Akt. Dephosphorylation of Akt on Thr-450 caused by a loss of mTORC2 renders Akt in an unstable form that requires assistance from the heat shock protein HSP90 to sustain its expression (11–13). It is remarkable that the cellular cues controlling regulation of the Thr-450 site have not been identified. Our study shows that phosphorylation of Akt on the turn motif is regulated by a cellular energy state, because its kinase mTORC2 requires a physiological millimolar level of ATP for its basal activity and is not effective if the abundance of ATP is decreased under metabolic stress. This finding indicates that Akt phosphorylation on Thr-450 may serve as a marker of cellular ATP level by reflecting a basal kinase activity of mTORC2. Thus, the homeostatic ATP sensor mTOR controls a proper folding of its critical downstream effector Akt by phosphorylation of its turn motif site.
The basal kinase activity of mTORC2 is sufficient to maintain a constitutive phosphorylation of Akt on the turn motif site. Regulation of the basal kinase activity of mTORC2 has not been addressed previously. We found that the basal kinase activity of mTORC2 is dependent on glucose but not amino acid deprivation. Glucose starvation, by causing a decrease of ATP and inhibition of the kinase activity of mTORC2, leads to the phosphorylation-dependent destabilization of its essential components SIN1 and rictor. These effects are not related to a glucose-sensing mechanism, because they are observed following long term glucose deprivation (20 h), when a substantial decrease of cellular ATP level has occurred. This critical role of ATP level in regulation of mTORC2 has been further supported by the observation that an acute ATP depletion (within 2 h) has been effective in causing similar effects on mTORC2. Our data indicate that mTORC2 requires a millimolar physiological ATP concentration for its optimal kinase activity, and the basal kinase activity of mTORC2 is sensitive to the metabolic stress associated with a cellular ATP depletion. We interpret that mTOR as the kinase component and a homeostatic ATP sensor of mTORC2 becomes inactive under metabolic stress because of a substantial decrease in cellular ATP level.
It is known that SIN1 is an unstable protein without its binding partner rictor (24, 25). By forming a heterodimer, SIN1 and rictor interact with another heterodimer, formed by mTOR and mLST8, to assemble the functional kinase complex of mTORC2. Within the complex, the kinase activity of mTOR by phosphorylation of SIN1 maintains the integrity of mTORC2 (14). Our study is consistent with the previous findings and indicates that the ATP-dependent basal kinase activity of mTORC2 carries the autoregulatory mechanism controlling the integrity of the kinase complex; mTOR stabilizes SIN1 by phosphorylation of its hydrophobic and conserved Ser-260 site and maintains the integrity of mTORC2. This specific phosphorylation site controls the turnover of SIN1 by preventing its lysosomal degradation. How a single phosphorylation site on SIN1 controls the integrity of a large multiprotein complex is an interesting question. It is conceivable that phosphorylation of the Ser-260 site on SIN1 by mTOR hinders a recognition site responsible for the disintegration of mTORC2 and lysosomal degradation of SIN1. It has been recently reported that a proper protein folding is critical in the assembly of mTORC2 and that a single amino acid substitution on rictor has precluded the assembly of mTORC2 (23). Because the endoplasmic reticulum has been identified as the main localization site of mTORC2 (27), this phosphorylation-dependent degradation pathway might be linked to proper protein folding monitoring by endoplasmic reticulum quality control that tags mTORC2 containing the non-phosphorylated form of SIN1 for disintegration and lysosomal degradation. A disintegration step in this process is supported by the observation that the abundance of SIN1 and rictor but not of mTOR and mLST8 is highly sensitive to phosphorylation of SIN1 on Ser-260. Thus, our study indicates that a specific phosphorylation of SIN1 on Ser-260 is a critical site controlling the integrity of mTORC2 by an ATP-dependent autoregulatory mechanism.
Acknowledgments
We thank Dr. Bing Su for providing SIN1 null MEFs and David Sabatini and Timothy Peterson for providing the mTORC1 substrate HA-GST-S6K1 protein. We also thank Cell Signaling Technology (Danvers, MA) for development of the phospho-SIN1 Ser-260 antibody. We gratefully acknowledge Professors B.Z. Abdraimov and E.B. Sydykov (L. N. Gumilyov Eurasian National University) for active support of the Ph.D. training program.
This work was supported, in whole or in part, by National Institutes of Health Grant CA 133522 (to D. D. S.) This work was also supported by the M. D. Anderson Trust Fellow Fund.
- mTOR
- mechanistic target of rapamycin
- mTORC1
- mTOR complex 1
- mTORC2
- mTOR complex 2
- SIN1
- SAPK-interacting protein 1
- SAPK
- (stress-activated protein kinases)
- rictor
- rapamycin-insensitive companion of mTOR
- raptor
- regulatory associated protein of mTOR
- 2-DG
- 2-deoxy-d-glucose
- HEK
- human embryonic kidney.
REFERENCES
- 1. Lindsley J. E., Rutter J. (2004) Nutrient sensing and metabolic decisions. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 139, 543–559 [DOI] [PubMed] [Google Scholar]
- 2. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR. From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 4. Han J. M., Jeong S. J., Park M. C., Kim G., Kwon N. H., Kim H. K., Ha S. H., Ryu S. H., Kim S. (2012) Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149, 410–424 [DOI] [PubMed] [Google Scholar]
- 5. Hara K., Yonezawa K., Weng Q. P., Kozlowski M. T., Belham C., Avruch J. (1998) Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 273, 14484–14494 [DOI] [PubMed] [Google Scholar]
- 6. Avruch J., Long X., Ortiz-Vega S., Rapley J., Papageorgiou A., Dai N. (2009) Amino acid regulation of TOR complex 1. Am. J. Physiol. Endocrinol. Metab. 296, E592–E602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 [DOI] [PubMed] [Google Scholar]
- 8. Sarbassov D. D., Ali S. M., Sabatini D. M. (2005) Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 17, 596–603 [DOI] [PubMed] [Google Scholar]
- 9. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pearce L. R., Komander D., Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 11. Facchinetti V., Ouyang W., Wei H., Soto N., Lazorchak A., Gould C., Lowry C., Newton A. C., Mao Y., Miao R. Q., Sessa W. C., Qin J., Zhang P., Su B., Jacinto E. (2008) The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 27, 1932–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ikenoue T., Inoki K., Yang Q., Zhou X., Guan K. L. (2008) Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 27, 1919–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oh W. J., Wu C. C., Kim S. J., Facchinetti V., Julien L. A., Finlan M., Roux P. P., Su B., Jacinto E. (2010) mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 29, 3939–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen C. H., Sarbassov dos D. (2011) The mTOR (mammalian target of rapamycin) kinase maintains integrity of mTOR complex 2. J. Biol. Chem. 286, 40386–40394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sancak Y., Thoreen C. C., Peterson T. R., Lindquist R. A., Kang S. A., Spooner E., Carr S. A., Sabatini D. M. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 [DOI] [PubMed] [Google Scholar]
- 16. Sarbassov dos D., Bulgakova O., Bersimbaev R. I., Shaiken T. (2012) Isolation of the mTOR complexes by affinity purification. Methods Mol. Biol. 821, 59–74 [DOI] [PubMed] [Google Scholar]
- 17. Chen C. H., Shaikenov T., Peterson T. R., Aimbetov R., Bissenbaev A. K., Lee S. W., Wu J., Lin H. K., Sarbassov dos D. (2011) ER stress inhibits mTORC2 and Akt signaling through GSK-3β-mediated phosphorylation of rictor. Sci. Signal. 4, ra10. [DOI] [PubMed] [Google Scholar]
- 18. Rolland F., Winderickx J., Thevelein J. M. (2001) Glucose-sensing mechanisms in eukaryotic cells. Trends Biochem. Sci. 26, 310–317 [DOI] [PubMed] [Google Scholar]
- 19. Meriin A. B., Yaglom J. A., Gabai V. L., Zon L., Ganiatsas S., Mosser D. D., Zon L., Sherman M. Y. (1999) Protein-damaging stresses activate c-Jun N-terminal kinase via inhibition of its dephosphorylation. A novel pathway controlled by HSP72. Mol. Cell. Biol. 19, 2547–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dennis P. B., Jaeschke A., Saitoh M., Fowler B., Kozma S. C., Thomas G. (2001) Mammalian TOR. A homeostatic ATP sensor. Science 294, 1102–1105 [DOI] [PubMed] [Google Scholar]
- 21. Ali S. M., Sabatini D. M. (2005) Structure of S6 kinase 1 determines whether raptor-mTOR or rictor-mTOR phosphorylates its hydrophobic motif site. J. Biol. Chem. 280, 19445–19448 [DOI] [PubMed] [Google Scholar]
- 22. Schroder W. A., Buck M., Cloonan N., Hancock J. F., Suhrbier A., Sculley T., Bushell G. (2007) Human Sin1 contains Ras-binding and pleckstrin homology domains and suppresses Ras signalling. Cell. Signal. 19, 1279–1289 [DOI] [PubMed] [Google Scholar]
- 23. Aimbetov R., Chen C. H., Bulgakova O., Abetov D., Bissenbaev A. K., Bersimbaev R. I., Sarbassov D. D. (2012) Integrity of mTORC2 is dependent on the rictor Gly-934 site. Oncogene 31, 2115–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frias M. A., Thoreen C. C., Jaffe J. D., Schroder W., Sculley T., Carr S. A., Sabatini D. M. (2006) mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 16, 1865–1870 [DOI] [PubMed] [Google Scholar]
- 25. Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., Su B. (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 [DOI] [PubMed] [Google Scholar]
- 26. Zinzalla V., Stracka D., Oppliger W., Hall M. N. (2011) Activation of mTORC2 by association with the ribosome. Cell 144, 757–768 [DOI] [PubMed] [Google Scholar]
- 27. Boulbés D. R., Shaiken T., Sarbassov dos D. (2011) Endoplasmic reticulum is a main localization site of mTORC2. Biochem. Biophys. Res. Commun. 413, 46–52 [DOI] [PMC free article] [PubMed] [Google Scholar]