

Background: N-Formylated bacterial/mitochondrial peptides in infected/injured tissues are GPCR chemoattractant agonists for neutrophil FPRs.
Results: C-terminal tail FPR phosphopeptides were identified by LC/MS/MS in tryptic digests of FPRs immunopurified from human blood neutrophils.
Conclusion: FPR1 but not FPR2 is monophosphorylated at any one of seven C-terminal tail Ser/Thr residues after fMLF stimulation.
Significance: Decoding of human neutrophil FPR phosphorylation may be important for controlling inflammation.
Keywords: 7-Helix Receptor, Chemokines, Epitope Mapping, Inflammation, Mass Spectrometry (MS), Neutrophil, FPR Phosphorylation, LC/MS/MS, Cytochalasin B, f-Met-Leu-Phe
Abstract
Accumulation, activation, and control of neutrophils at inflammation sites is partly driven by N-formyl peptide chemoattractant receptors (FPRs). Occupancy of these G-protein-coupled receptors by formyl peptides has been shown to induce regulatory phosphorylation of cytoplasmic serine/threonine amino acid residues in heterologously expressed recombinant receptors, but the biochemistry of these modifications in primary human neutrophils remains relatively unstudied. FPR1 and FPR2 were partially immunopurified using antibodies that recognize both receptors (NFPRa) or unphosphorylated FPR1 (NFPRb) in dodecylmaltoside extracts of unstimulated and N-formyl-Met-Leu-Phe (fMLF) + cytochalasin B-stimulated neutrophils or their membrane fractions. After deglycosylation and separation by SDS-PAGE, excised Coomassie Blue-staining bands (∼34,000 Mr) were tryptically digested, and FPR1, phospho-FPR1, and FPR2 content was confirmed by peptide mass spectrometry. C-terminal FPR1 peptides (Leu312–Arg322 and Arg323–Lys350) and extracellular FPR1 peptide (Ile191–Arg201) as well as three similarly placed FPR2 peptides were identified in unstimulated and fMLF + cytochalasin B-stimulated samples. LC/MS/MS identified seven isoforms of Ala323–Lys350 only in the fMLF + cytochalasin B-stimulated sample. These were individually phosphorylated at Thr325, Ser328, Thr329, Thr331, Ser332, Thr334, and Thr339. No phospho-FPR2 peptides were detected. Cytochalasin B treatment of neutrophils decreased the sensitivity of fMLF-dependent NFPRb recognition 2-fold, from EC50 = 33 ± 8 to 74 ± 21 nm. Our results suggest that 1) partial immunopurification, deglycosylation, and SDS-PAGE separation of FPRs is sufficient to identify C-terminal FPR1 Ser/Thr phosphorylations by LC/MS/MS; 2) kinases/phosphatases activated in fMLF/cytochalasin B-stimulated neutrophils produce multiple C-terminal tail FPR1 Ser/Thr phosphorylations but have little effect on corresponding FPR2 sites; and 3) the extent of FPR1 phosphorylation can be monitored with C-terminal tail FPR1-phosphospecific antibodies.
Introduction
Human neutrophils (1) are principal effector cells of the innate immune response and are equipped with an arsenal of antimicrobial activities. They employ a subset of the largest family of cellular receptors, the G-protein-coupled seven-transmembrane domain receptors (GPCRs),2 to seek out and then destroy (2) microbial invaders. By responding to a wide variety of exogenous and endogenous stimulating agents, neutrophils are able to resolve infections but in the process also inflict collateral damage to the neighboring host tissues (3). The GPCRs bind chemotactic agents localized near infected tissue sites and foster the interception of neutrophil traffic in the vasculature by activating β2-integrin adhesion receptors. The adhesins, under the control of the chemotactic receptors (4, 5), help mediate neutrophil passage through the blood vessel walls (diapedesis) and their crawling up concentration gradients through the tissues to sites of infection (6). There, the chemotaxins also contribute to the activation of neutrophil inflammatory responses, including superoxide production and secretion of preformed and newly synthesized degradative enzymes and mediators of inflammation (7).
Neutrophils respond to several chemotactic agents (8, 9), which include bacterial formylated peptides (10); chemokines, such as IL- 8 and MCP-1; and endogenous stimuli, including the complement components C5a and C3a, leukotriene B4 (LTB4), platelet-activating factor, and mitochondrial formylated peptides, among others (10, 11). Inflamed tissue attracts neutrophils using these agents, and as more neutrophils accumulate, autocrine and paracrine events continue to propagate inflammation until this process is turned off by receptor inactivation (12–14), apoptosis (15), resolvin/protectin (16) action, and perhaps other as yet unknown means.
G-protein-coupled activation of human neutrophils by N-formyl-Met-Leu-Phe (fMLF) is mediated not only by FPR1 but also by the lower affinity FPR2, also known as FPRL1, LXA4R, and ALX (17). A third homolog, FPR3, appears to be expressed in cells other than neutrophils, and its function is now under active investigation (18–20). We and others have previously described the molecular associations of fMLF-occupied FPR with G-proteins, the kinetics of FPR internalization, and affinity modulation (21–24). We have shown that it is affinity modulation and complex formation with the cytoskeleton during plasma membrane reorganization prior to internalization that is functionally correlated with response termination (25, 26). Dahlgren and co-workers (27) have extended this work and shown that the response termination process is more complex. Prossnitz and co-workers (28) have shown that FPR phosphorylation is linked to its regulation by the adaptor protein β-arrestin, in a human leukocyte recombinant FPR expression system. The current thinking is that phosphorylation of FPR not only reduces its ability to bind fMLF and G-proteins (28, 29) but also modulates its association with β-arrestin, which competitively inhibits its interaction with G-proteins (29). It is likely that all of the individual steps that modulate activity are linked sequentially and spatially and that the entire system is required to control FPR accessibility to G-proteins in a process that may be important in controlling the inflammatory milieu. Receptor phosphorylation appears to play a role in most of these sensitivity modulation processes (27).
FPR phosphorylation has been implicated as the controlling process for desensitization of FPR action in neutrophils (30). In visual rhodopsin, C-tail phosphorylation is believed to be essential for visual response termination (31), by controlling rhodopsin interactions with arrestin, and similarly phosphorylation in the C-terminal tail of the β-adrenergic receptor is required for down-regulation and desensitization (32). We described two monoclonal antibodies in 2007 that recognized two different regions of the C-terminal tail of FPR1 and FPR2 and showed that these mAbs could differentiate between different states of FPR1 in intact neutrophils, both by immunocytochemistry and flow cytometry. We also showed that the sensitivity of one of the antibodies, NFPRb (also known as NFPR2), to the fMLF-induced activation of recombinant FPR1 expressed in CHO cells was probably due to phosphorylation of its C-terminal tail (33).
In this study, we examine the phosphorylation state of FPR1 in whole cell dodecyl maltoside extracts of control and cytochalasin B-exposed neutrophils, stimulated with fMLF at different concentrations. We show that mAb NFPRb, immunoblotting to a Mr 50,000–70,000 molecular species by SDS-PAGE, depends inversely and saturably on prior exposure of neutrophils to fMLF. In contrast, NFPRa recognizes two species consistent with FPR1 and FPR2 that appear nearly invariant after exposure to fMLF. We also partially immunopurified both FPR1 and FPR2 from whole cell detergent extracts as well as from cell membranes of unstimulated (U) and fMLF + cytochalasin B (CB)-treated (S) cells and analyzed their deglycosylated forms by trypsin digestion followed by mass spectrometry. Our results suggest that phosphorylation occurs individually at seven different sites of the FPR1 C terminus only in the S samples without evidence of any phosphorylation of FPR2. These observations demonstrate the potential for analyzing receptor regulation, association, modification, and potentially, the interplay between the two chemoattractant receptors FPR1 and FPR2 in their native stimulated and unstimulated cellular environments.
EXPERIMENTAL PROCEDURES
Reagents
Octyl β-d-glucopyranoside (OG), PMSF, and dithiothreitol (DTT) were purchased from Calbiochem; n-dodecyl-β-d-maltopyranoside (DDM) was from Anatrace; Centricon concentrators were purchased from Millipore; and the 50 Sonic Dismembrator probe sonicator was obtained from Fisher. The epitope-mimicking peptides for mAb NFPRa, NH2-GQDFRERLIH-COOH (peptide C), and NH2-KHDFRSRLA-COOH (peptide D), for mAb NFPRb NH2-STLPSAEVELQAK-OH (peptide A), and NH2-STLPSIEVASYWR-OH (peptide B) were obtained from Global Peptides and Pi Proteomics. Econo-Pac 10 DG desalting columns (30 × 10 ml) were from Bio-Rad. Goat anti-mouse IgG (H+L) DyLight 800-conjugated antibody and the Pierce SDS-PAGE Sample Prep Kit were from Thermo Scientific, and Odyssey Infrared Imaging Blocking Buffer was from LI-COR. All other reagents were obtained from Sigma-Aldrich.
Buffers
Cell resuspension and stimulation buffers were as follows: DPBS(+) (136 mm NaCl, 2.7 mm KCl, 1.5 mm KH2PO4, Na2HPO4, supplemented with 1 mm MgCl2, 0.9 mm CaCl2, and 0.1% dextrose) and RPMI without phenyl red made as directed by the manufacturer (Sigma-Aldrich). Relaxation buffer (Relax) contained 10 mm Hepes, 100 mm KCl, 10 mm NaCl, pH 7.4; Relax(+) contained 10 mm Hepes, 100 mm KCl, 10 mm NaCl, 1 mm EDTA, 0.1 mm DTT, fresh 1 mm PMSF, 1:1000 diluted Sigma P8340 protease inhibitor mixture, and 1:1000 Sigma phosphatase inhibitor mixtures 2 and 3, pH 7.4. Phosphate-buffered saline (PBS) contained 137 mm NaCl, 3 mm KCl, 1.5 mm KH2PO4, 8 mm Na2HPO4, pH 7.4. Cell lysis buffer was as follows: Relax(+) plus 2 mm MgCl2, 10 μg/ml chymostatin, 2.5 μm latrunculin, 1 unit/ml benzoase, with or without the nonionic detergent, 2% DDM. SDS denaturation buffer (TS buffer) contained 0.2 m Tris-HCl, 2% SDS, pH 8. 2× SDS-PAGE sample buffer (non-reducing, SB) contained 0.4% SDS, 0.12 m Tris-HCl, pH 6.8, 20% glycerol without bromphenol blue for IR fluorescence assays. Special FPR electrotransfer buffer contained 0.19 m sodium/glycine, 20% methanol, 25 mm Tris-base, 0.02% SDS, pH 8.5.
Neutrophil Preparations
Human neutrophils were prepared as described by Henson and Oades (34) and by the low LPS dextran method described by DeLeo et al. (35). The latter cells were used for preparation of unstimulated cells because they were unprimed and minimally perturbed. Maximal stimulation was carried out on the former primed cell preparation essentially as described previously by Parkos et al. (37) with minor modifications. Briefly, after resuspension in DPBS(+) containing +10 μm CB, 80 μg/ml catalase, and 50 units/ml superoxide dismutase, a 7-min preincubation at 37 °C was followed by the addition of 1 μm fMLF and continued incubation for 10 min with brief gentle mixing. The stimulated cells were then diluted 5-fold with ice-cold DPBS(+) and placed on ice followed by resuspension, maintaining the cells at 0–4 °C in homogenization buffer for preparation of membranes or in Relax(+) buffer for direct solubilization.
Quantitative FPR1 and FPR2 Immunoblotting
For analysis of cellular FPR1 phosphorylation by immunoblotting with NFPRb, neutrophils were resuspended in RPMI at a density of 1.1 × 107/ml and divided into 500-μl aliquots into individual 1.5–2-ml polyethylene tubes. These suspensions were pre-exposed to vehicle or cytochalasin B for 10 min and mixed by agitation occasionally, after which volume of the appropriately diluted stock fMLF/vehicle was added to each tube. After 10 min, the reaction was quenched on ice, after the addition of 1 ml of iced RPMI. Cells were then pelleted at 500 × g for 2 min at 2 °C and carefully aspirated, discarding the supernatants. 300 μl of DDM lysis buffer (plus PMSF, protease inhibitor mixture, and phosphatase inhibitors) was then added and mixed by vortexing to remove the pellets from the side of the tubes. The tubes were capped and tumbled for 45 min at 4 °C and then centrifuged in a table top centrifuge for 30 min at 17,000 rpm at 4 °C. 200 μl of the supernatant was placed into a new tube, avoiding the pellet. FPR1 and FPR2 in the pellet as measured by NFPRa binding was negligible. This sample was then reduced and alkylated as follows: 1) the addition of 200 μl of 18 mm DTT (final concentration 9 mm DTT) in TS buffer, mixed and incubated at 60 °C for 5 min and then cooled for 5 min at room temperature; 2) the addition of 100 μl of 300 mm N-ethylmaleimide (final concentration 60 mm NEM) in TS buffer, mixed and incubated at room temperature for 10 min; 3) the addition of 500 μl of 2-fold concentrated SB followed by mixing, incubation at room temperature for 10 min, and then freezing at −20 °C. After 24–72 h, the sample was thawed, and 40 μl/lane containing ∼1.5 × 105 cell equivalents/lane was loaded and then separated on a 9% polyacrylamide gel containing 28 lanes. The gels were run as described previously (33) and then electroeluted onto Immobilon PVDF membranes in special FPR electrotransfer buffer (see above). The transfer was carried out at 100 V to 0.2 V-h at a maximum of 0.5 A for ∼2 h. The Immobilon was then washed in milliQ deionized water for 15 min and then dried overnight between two sheets of Whatman filter paper. The next day, the PVDF was re-wet for 15 s in 100% methanol (HPLC grade) and then rinsed twice in deionized water, followed by soaking in PBS for ∼2 min. The blot was blocked by soaking in undiluted Odyssey blocking buffer at room temperature for 1 h on a gently oscillating rocker. The blot was subsequently transferred into the NFPRa or NFPRb primary antibody, diluted to 1.2 and 3.0 μg/ml, respectively, in Odyssey blocking buffer with 0.2% Tween nonionic detergent, and incubated at 37 °C for 1 h with rocking. The blot was then removed from the primary mAb and washed five times for 10 min each in PBS plus 0.1% Tween with rocking. Incubation of the washed blot in secondary mAb was accomplished at room temperature with gentle rocking in Odyssey blocking buffer containing 0.2% Tween, 0.01% SDS, with goat anti-mouse IgG (H + L) DyLight 800-conjugated antibody at a dilution of 1:10,000. The incubation was shielded from light by Al foil. This incubation was followed by five washes of 10-min duration in PBS plus 0.1% Tween while shielded from ambient light. The blots were scanned in a LI-COR Odyssey infrared fluorescence digital scanner using the 780-nm excitation while wet and subsequently stored dry between two sheets of Whatman filter paper, protected from light.
Scans were quantified in two ways. For routine assays, 8-bit tif images from Odyssey scans of wet transfers were saved to files and analyzed using GE Life Sciences ImageQuant by inverting images, rotating 90°, and analyzing individual bands in a 28-lane gel as a single virtual lane in ImageQuant, allowing up and down quantitation for any chosen molecular weight range for all conditions of the experiment. The integrated signals minus background and corresponding ratios of background-subtracted signals for FPR1, measured with NFPRb and NFPRa, were calculated and plotted, after normalizing for maximum range, or against the control as indicated. The normalized data were then analyzed by GraphPad Prism version 3.03 for Windows (GraphPad Software, San Diego, CA) using a one- or two-site nonlinear regression fit and plotted as sigmoidal inhibition curves on a logarithmic abscissa, as shown, for example, in Fig. 2.
FIGURE 2.
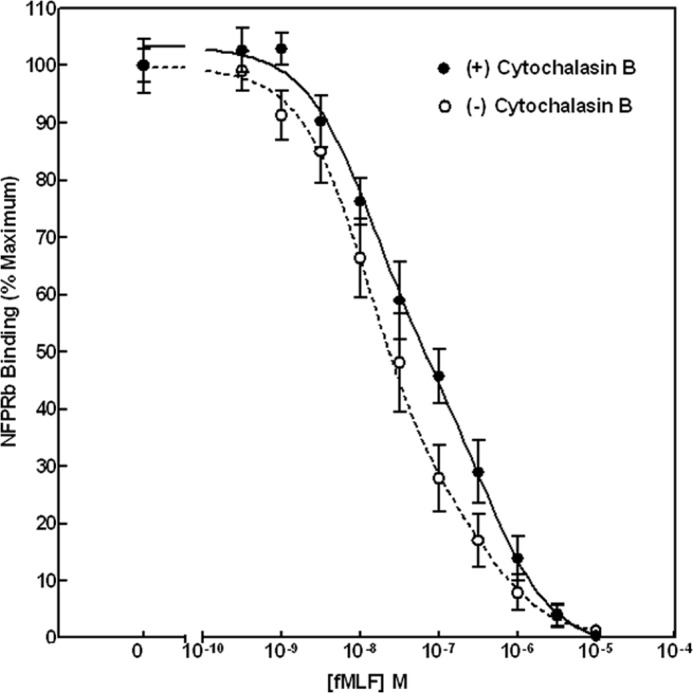
Effect of cytochalasin B on fMLF-stimulated neutrophil FPR1 phosphorylation. Digitized scans of IR fluorescent antibody-bound NFPRa and NFPRb on PVDF immunoblots shown in Fig. 1A were quantified, as described under “Experimental Procedures.” The figure represents the normalized full-scale changes in NFPRb/NFPRa ratios. Using a one-site fit to the data, the EC50 values (EC50 = 73 ± 21 nm (+CB; n = 6) and 33 ± 8 nm (−CB; n = 6)) were determined from nonlinear regression analysis for six paired experiments. The differences were significant with a two-tailed p value of 0.029 derived in GraphPad Prism version 3.0. A two-site fit is shown in the figure (see text). Error bars show S.E.
Extraction of FPR Directly from Cells
Pellets containing up to 1–3 × 109 S or U neutrophils, prepared as above, were resuspended in at least 5–15 ml of lysis buffer without detergent, followed by 1:1 dilution in the same buffer with 2% DDM, and then tumbled at room temperature for 45 min. The cell lysates were centrifuged at 17,000 × g for 30 min in a Beckman Ti-60 rotor at 22 °C. The soluble extract or supernatant was used for further purification of FPR except for two 50-μl aliquots used for quantitation. To clear the extract of Sepharose-binding components and flavocytochrome b, needed for other purposes, the extract was mixed with a 500-μl wet-packed bead volume of CNBr-Sepharose covalently coupled to mAb CS9 that recognizes p22phox (36). The matrix was pre-equilibrated with Relax buffer containing 1% DDM. Any IgG1 mAb, however, would achieve the clearing, and other mAbs can be substituted. The suspension was tumbled in a rotator (∼4 rpm) at room temperature for 40 min and then poured into a plastic fritted column, drained, and washed once with 1 ml of Relax buffer with 1% DDM, which was added to the cleared extract (CS9 postbind).
FPR Extraction from Neutrophil Membranes
FPR1 and FPR2 were also extracted and purified from membranes prepared from U neutrophils and S cells (pretreated with 10 μm cytochalasin B for 7 min at 37 °C followed by 1 μm fMLF for 10 min as described above). The latter membranes were prepared by N2 cavitation, exactly as described in Parkos et al. (37), but with protease and phosphatase inhibitor mixtures at dilutions of 1:1000 and 1:100, respectively. The membranes from unstimulated cells were separated from primary granules by sedimentation through a 40% sucrose cushion, as described by Klotz et al. (38). Briefly, neutrophil N2 cavitates, from cellular suspensions made at a cell density of 1 × 108 cells/ml in Relax(+), were collected and centrifuged at 1000 × g to remove pellets and debris. Any actin-rich foam was rehomogenized with the low speed pellet in a Dounce homogenizer and added back to the low speed supernatant to form the 1KS fraction. 10 ml of the 1KS fraction was underlaid with 6.5 ml of 45% sucrose in Relax(+) buffer in thick walled Ti-60 polycarbonate tubes and centrifuged for 120 min at 45,000 rpm. 9 ml of the supernatant layer above the interface was removed and discarded, followed by separate removal of the interface plus all but 0.5–1 ml of the sucrose layer, to leave the pellet undisturbed. This second fraction, the 100KS1, was diluted 1:1 in Relax(+) buffer and recentrifuged for 1 h at 45,000 rpm in a Ti-60 Beckman rotor. The supernatants were carefully removed, and the pellets were resuspended to the original volume in Relax(+) buffer plus 1 m NaCl by light sonication or with a Dounce homogenizer, followed by one cycle of freeze/thaw in liquid N2/water at ambient temperature, and recentrifuged. This second high speed pellet was similarly resuspended in Relax(+) to a cell equivalent density of 5 × 108/ml and brought to 20% glycerol, frozen, and stored at −70 °C. After thawing, both types of membranes were washed in 1 m NaCl, prior to resuspension in the extraction buffer. Subsequent steps were essentially identical.
Thawed membranes at room temperature were brought to 1% DDM by dilution from a 20% stock in water and were extracted for 1 h with continuous tumbling. The extract was clarified by centrifugation at room temperature in a TLA 100.2 Beckman rotor at 55,000 rpm for 30 min. The supernatant was separated from the pellet, and the latter was re-extracted for 1 h in one-half the volume of the original extraction after resuspension with light sonication. This extract was pooled and recentrifuged, and the supernatants were combined. Recovery of NFPRa binding activity for FPR was ∼50–70%. This extract was then precleared of Sepharose and irrelevant IgG1 binding activities by exposure to the CS9 affinity matrix, as described above.
Serial Immunoaffinity Capture of FPR1 and FPR2
The flavocytochrome b-cleared extract (CS9 postbind extract) was then mixed with a 300-μl wet-packed bead volume of CNBr-Sepharose that was covalently coupled to mAb NFPRb and pre-equilibrated with Relax buffer containing 1% DDM. NFPRb recognizes the C terminus of human neutrophil FPR1 (33). The suspension was allowed to tumble at room temperature for 1–2 h, after which the beads were drained as described above for the CS9 matrix, washed with 1 ml of Relax/DDM that was added to the NFPRb postbind. The NFPRb postbind was then added to a 300-μl wet-packed bead volume of CNBr-Sepharose that was covalently coupled to mAb NFPRa. NFPRa binds the juxtamembrane segment of the FPR C-terminal tail (33). This suspension was tumbled with NFPRa-coupled beads for 1–2 h at room temperature.
Prior to the addition of the NFPRb postbind to the NFPRa beads, the drained 1× washed NFPRb beads, were washed a second time with Relax/DDM (1%), followed by two washes with 5 ml of Relax buffer, but now containing 1% OG. After the last wash, the drained beads in a plastic fritted column were tumbled overnight with 1 ml of Relax/OG (1%) containing a 200 μm concentration of the two (A and B) NFPRb-specific peptides plus 0.02% NaN3 at 4 °C. The same was done to the NFPRa affinity matrix, except that the final wash and subsequent elution step was carried out using a 200 μm concentration of the C + D NFPRa-specific peptide mix. After overnight exposure to each of the respective peptide-containing buffers, the matrices were again serially eluted four additional times with 1 ml of their respective peptide-containing buffers after 10 min of tumbling incubation at room temperature. After the last peptide elution, the matrices were washed first with 500 μl of 100 mm sodium glycine, 100 mm KCl, pH 2.2, plus 1% OG, followed again by three additional serial low pH elutions, each followed by immediate neutralization with 12.5 μl of 2 m Tris. Following these steps, the drained matrix was washed with 10 ml of phosphate-buffered saline (PBS), pH 7.2, without any detergent, followed by two elutions with 500 μl of PBS containing 1% SDS after 10 min of equilibration. The drained beads were then solubilized in TS buffer plus 18 mm DTT). The peptide and low pH elutions were separately pooled and concentrated to 300 μl. All of the final eluted fractions were supplemented with 80% glycerol to a final concentration of 20% glycerol and then stored at −70 °C.
Deglycosylation of FPR
To prepare and maximize the affinity-purified material for mass identification, after tryptic digestion, all of the peptide and low pH eluates from NFPRb and NFPRa of the unstimulated sample (and likewise the stimulated samples) were pooled (approximately 30–50% of the starting 1–1.5 × 109 cell equivalents) of extracted FPR1. The initial volumes ranged from ∼8 to 10 ml each and were then concentrated 10-fold on an Amicon Ultracel 15 with 30 kDa cut-off. This material was then brought to 0.1 m K2HPO4, pH 8, 1% SDS, 1% OG, and (5 μg/ml) recombinant His-CBM-PNGase F (Mr 48,000), as prepared by Kwan et al. (39). In some cases, when the purity of the 34-kDa deglycosylated FPR was not paramount, 5 units of commercially available N-glycosidase F (Sigma-Aldrich; Mr 34,000) was used and treated identically. The deglycosylation proved to be critical for improving yields and avoiding the suppression of the digested FPR peptide signals by contaminating keratins in the 60,000–70,000 molecular weight range. After this incubation, the sample was further concentrated and buffer-exchanged using a Pierce SDS-PAGE Sample Prep Kit, following the manufacturer's instructions. Two 50-μl elutions of the Pierce resin were carried out, and the samples were run separately on a preparative Bio-Rad precast 8% Precise Protein Gel, subsequently stained with Gel Code Blue Safe Protein Stain and destained as per the manufacturer's instructions. Bands were excised and prepared for mass analysis, as described below.
Protein Composition of Peptide-eluted Fractions
10-μl aliquots of samples eluted directly from the antibody affinity matrices were denatured (7 m urea, 2 m thiourea, 3% CHAPS, 1% ASB-14, 20 mm Tris-HCl, 8.4), reduced with 5 mm tributylphospine for 1 h at 4 °C, alkylated with 10 mm 4-vinylpyridine for 2 h at 4 °C, and labeled with 600 pmol of Z-dye Blue2 (81–85) for 30 min on ice in the dark. Excess dye was quenched with 1 μl of 100 mm lysine. Samples were diluted 1:2 in Laemmli sample buffer (Bio-Rad) and separated on 10% SDS-PAGE. The gels were imaged using 488-nm excitation on a Typhoon Trio (GE Healthcare) fluorescence imager. Protein bands were excised manually and trypsinized “in gel,” as described below. Peptide composition was determined by LC/MS/MS mass spectrometry, as described below, and searched against NCBInr database using the MASCOT (Matrix Science, London, UK) search engine.
In-gel Tryptic Digestion and Protein Identification by Mass Spectrometry
Protein bands were excised from the gels, washed, in-gel reduced, and S-alkylated and were digested with porcine trypsin (Promega) overnight at 37 °C following Maaty et al. (40). The solutions containing peptides released during in-gel digestion were transferred to sample analysis tubes prior to mass analysis. LC/MS/MS measurements were performed using an Agilent 1100 HPLC system coupled to a 6520 Accurate-Mass Quadrupole Time-of-Flight LC/MS (Q-TOF) (Agilent Technologies), controlled with Agilent MassHunter Work station software (version B.04.00). Injected samples were first trapped and desalted on a Zorbax 300SB-C18 Agilent HPLC-Chip enrichment column (40-nl volume) for 3 min with 0.1% formic acid delivered by the auxiliary pump at 4 μl/min. The peptides were then reverse eluted and loaded onto the analytical capillary column (150 mm × 75-μm inner diameter, also packed with 5-μm Zorbax 300SB-C18 particles) connected in-line to the mass spectrometer, with a flow of 0.6 μl/min. Peptides were eluted with a 5–50% acetonitrile gradient over 40 min. Data-dependent acquisition of collision-induced dissociation tandem mass spectrometry (MS/MS) was utilized. Parent ion scans were run over the m/z range of 300–1,700 at 2 and 1 spectra/s for MS and MS/MS, respectively. Peptide mass analysis by MALDI was conducted using a Bruker Autoflex 3 TOF/TOF. Data were visualized using MassHunter Qualitative Analysis software (Agilent Technologies) and MGF and mzData compound list files were used to query an in-house database, using MASCOT (Matrix Science, London, UK) and Peptide shaker (X!tandem and OMSSA) software using 25 ppm for MS and MS/MS ion mass tolerance.
RESULTS
The high affinity N-formyl peptide receptor of human neutrophils (or FPR1) is in relatively low abundance and has, until fairly recently, been difficult to detect immunologically as a resolved molecular entity. In 2007, we described a pair of mAbs that could detect the unphosphorylated C terminus of FPR1 (mAb NFPRb) and the juxtamembrane region of the C-terminal tail of the receptor (mAb NFPRa). We were able to show that these mAbs could function together as probes of the phosphorylation state of the FPR C terminus because binding of NFPRb to immunoblots of lysates of FPR1-expressing CHO cells 1) depended inversely on prior exposure of the cells to fMLF and 2) could be regenerated after treatment of alkali-stripped membranes with alkaline phosphatase (33). Although we showed that fixed and permeabilized neutrophils bound NFPRb, in an fMLF-sensitive fashion, the molecular species responsible for its binding was only identified in isolated membranes of stimulated neutrophils, and its dependence on fMLF exposure was not quantitatively examined.
fMLF Stimulation of Neutrophils Decreases NFPRb Binding to FPR1
To show that NFPRa and NFPRb bind to receptor in extracts of whole neutrophils with fMLF sensitivity, suggestive of phosphoregulation, we examined immunoblotting of the NFPRa/NFPRb mAb pair to FPR1 of neutrophils as a function of neutrophil exposure to fMLF. Because mAb NFPRb binds the Ser/Thr-rich C-terminal region of FPR (33) and mAb NFPRa binds a juxtamembrane region without Ser/Thr, the expectation was for NFPRa to be insensitive to neutrophil fMLF exposure, whereas NFPRb would be sensitive (33). Indeed, in the two groups of lanes on the right-hand side of Fig. 1, immunoblots of NFPRa (bottom) and NFPRb (top) binding to SDS-PAGE-separated and electrotransferred FPR show such activity. The samples examined derived from DDM extracts of suspensions of whole neutrophils pre-exposed at 37 °C to concentrations of fMLF ranging from 0 to 10 μm (from left to right). In the top blot on the right, the decline in binding of NFPRb, as a function of prior exposure of the cells to fMLF, is visually evident from the intensity of the signal of a molecular species with a relative molecular weight of 50,000–65,000 (termed the 60K or 50–65K region) that decreases with increasing concentration of exposure to fMLF. From quantitation of these blots, shown in Fig. 2, it is clear that the decline in sensitivity was saturable with an EC50 = (3.3 ± 0.8) × 10−8 m fMLF, which closely approximates the low affinity (G-protein-unassociated) value of the fMLF Kd to neutrophil membranes of 2.5 × 10−8 m (41). A stable minimum of binding was achieved in 10 min with a t½ of ∼15 s.3
FIGURE 1.

fMLF-sensitive C-terminal phosphoregulation of FPR1 in suspensions of human neutrophils. A, immunoblots of neutrophil extracts with NFPRa and NFPRb as primary mAbs. Suspension neutrophils were incubated with logarithmically increasing (triangle) concentrations of fMLF at 0, 3 × 10−10, 1 × 10−9, 3 × 10−9, 1 × 10−8, 3 × 10−8,1 × 10−7, 3 × 10−7, 1 × 10−6, 3 × 10−6, and 1 × 10−5 m for 10 min at 37 °C in the presence (left set of lanes) and absence (right set of lanes) of 10 μm cytochalasin B. Aliquots were diluted in ice-cold buffer and centrifuged at low speed for 2 min, and the pellets were solubilized in 1% DDM, cleared by centrifugation, denatured in SDS-PAGE sample buffer, reduced and alkylated, and immunoblotted at ∼1 × 105 cell equivalents/lane, as described in detail under “Experimental Procedures.” Blotting was carried out with either NFPRa (bottom panels) or NFPRb (top panels) as the primary antibodies and infrared fluorescing secondary antibodies. The blots were developed for IR fluorescence detection using LI-COR Dylight 800-conjugated secondary antibody and measured on a LI-COR infrared fluorescence imager.
Importantly, the corresponding bottom right-hand blot of Fig. 1 shows that NFPRa recognition of the 50–65K region of the blot does not change more than 10–15% over this fMLF concentration range. Because the amount of signal in the pellet fraction of the DDM extraction was negligible (not shown), this observation suggests that the 50–65K species was not being selectively removed from the DDM extract by a change in solubility properties due to its regulation in the cell under conditions of this assay. Also of interest is another narrower band below the 50–65K band, between the Mr 39,000 and 51,000 markers (45K species), which we previously assigned to the “low affinity” fMLF-binding species FPRL1 (now called FPR2). FPR2 carries the same epitope recognized by NFPRa on FPR1, and the binding of NFPRa to the 45K species also remains effectively constant through the time course (not shown) and concentration range of fMLF tested.
Although NFPRa specifically binds the putative FPR1 (60K) and FPR2 (45K) bands in immunoblots, it also strongly cross-reacts with two bands in the 35,000 and 30,000 molecular weight range that are derived from the cytosol. Fig. 3 shows the enrichment of the 60K and 45K bands from equivalent cell amounts of low speed supernatants and high speed pellets of nitrogen cavitates of unstimulated whole neutrophils. These are clearly bound by NFPRa in both fractions, although an apparent suppressive effect of excess neutrophil protein is removed by the removal of the cytosol after centrifugation of the low speed supernatants at 100,000 × g. Removal of the cytosolic proteins by sedimentation also results in the loss of the two prominent cross-reactive bands at Mr 35,000 and 30,000 observed in the bottom panel of Fig. 1. This result clearly confirms the recognition of the broad 60K and 45K receptor bands in the particulate membrane fractions and shows that the unknown lower molecular weight protein species are not sedimentable at 100,000 × g but are recoverable in the supernatant fraction (not shown). These bands are also not bound by either antibody in non-denaturing DDM extracts of whole neutrophils because they are not retained by immunoaffinity matrices of NFPRa or NFPRb (see below and Fig. 4). We conclude that the 35,000/30,000 bands reveal binding sites for NFPRa only when denatured and bound by nitrocellulose or PVDF and do not impede FPR immunopurification as described below. It would be of interest, however, to identify these proteins because they may have some regulatory function related to the latent FPR-like C-terminal tail structures.
FIGURE 3.
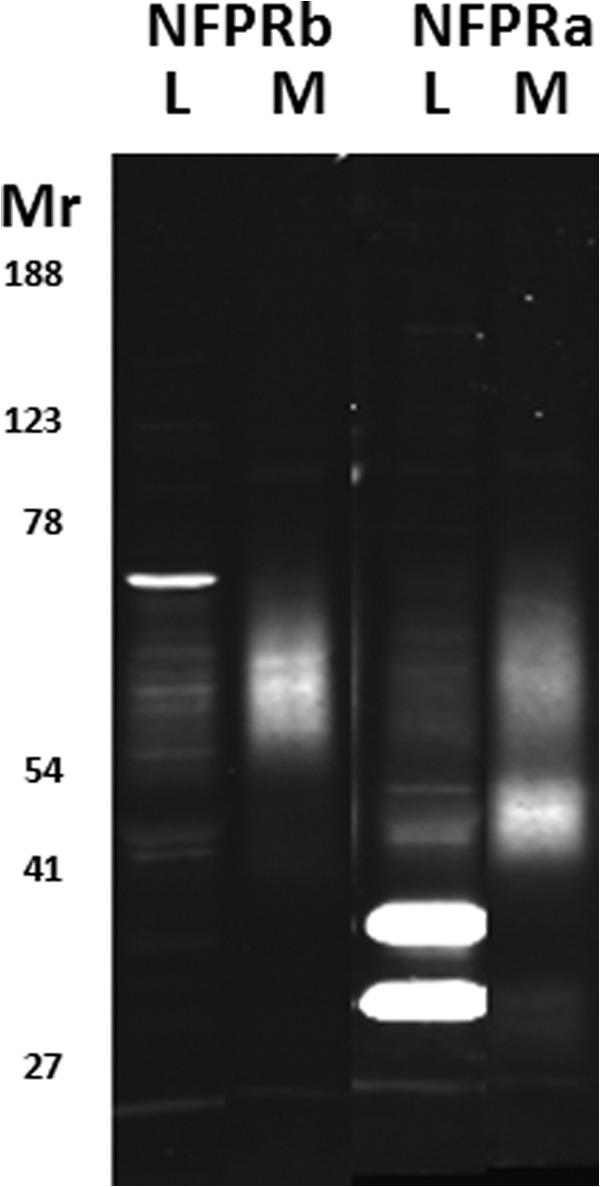
Recognition of FPR1 and FPR2 in unstimulated neutrophil homogenates and membranes. SDS-PAGE-separated and immunoblotted whole cell lysates (L) and salt-washed membranes (M) from N2 cavitates of unstimulated neutrophils, as described under “Experimental Procedures,” were developed with the NFPRb (left two lanes) and NFPRa (right two lanes) primary mAbs and visualized by infrared fluorescence scanning with a LI-COR Odyssey scanner. Each lane contains 2 × 105 cell equivalents. The broadly staining band between Mr ∼50,000 and 65,000 (also known as 60K) is putatively FPR1 and is recognized by both mAbs. The band between Mr ∼41,000 and 50,000 (also known as 45K) comigrates with FPR2 and is only recognized by NFPRa. The more intense bands at ∼30,000 and ∼36,000 are soluble cytosolic species and are lost in the particulate fraction.
FIGURE 4.
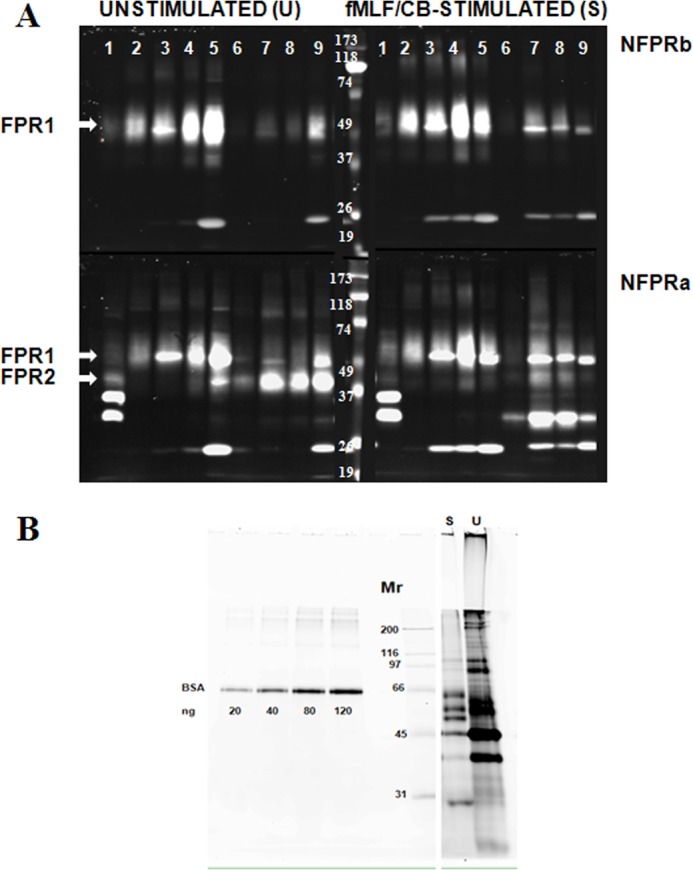
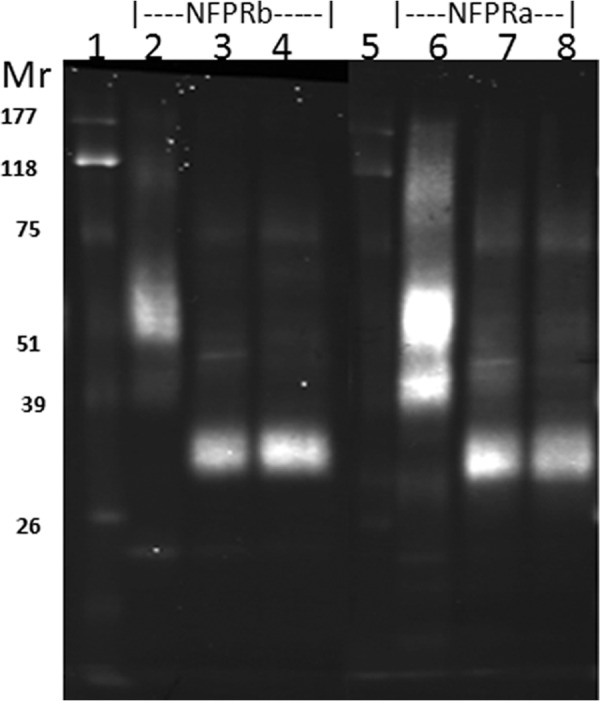
Partial purification of FPR1 and FPR2 from DDM lysates of unstimulated and fMLF/cytochalasin B-stimulated neutrophils by serial immunoaffinity capture using NFPRb- and NFPRa-Sepharose. A, serial affinity purification of FPR1 and FPR2. U and S neutrophils prepared as described under “Experimental Procedures” were lysed in DDM-containing lysis buffer at 2 × 108 cells/ml, precleared of debris antibody binding components first by centrifugation at 17,000 × g and then by passage over an isotype-matched mAb CS9 affinity matrix. The extracts (lane 1) were then serially exposed to NFPRb-Sepharose followed by exposure of the flow-through to an NFPRa-Sepharose matrix. The NFPRb matrix (lanes 2–5) and the NFPRa matrix (lanes 6–9) were eluted with first epitope mimetic peptide A&B buffer (lane 2) and then a low pH buffer (lane 3) and 1% SDS-containing buffer (lane 4). The NFPRa matrix was similarly eluted with mimetic peptide C&D buffer (lane 6), followed by a low pH buffer (lane 7) and 1% SDS-containing buffer (lane 8). The washed matrix beads were then put into SDS-PAGE sample buffer, heated, centrifuged, and run in lanes 5 (NFPRb-beads) and 9 (NFPRa-beads). Mr markers are shown in the center panel of the bottom row. Only the peak fractions are shown. B, protein composition of NFPRb peptide eluates. 10-μl samples of each NFPRb eluate U and fMLF/CB were labeled with 600 pmol of fluorescence protein Z dye Blue 2 (see “Experimental Procedures”) as described previously using similarly labeled bovine serum albumin as a standard. Major bands were tryptically digested and identified by Agilent ESI quadrupole-TOF mass analysis in Table 1.
Effect of CB on fMLF-induced Decrease in NFPRb Binding to FPR1
Enhanced activation of neutrophils results if they are stimulated with fMLF in the presence of CB (42, 43). The enhancement has been attributed to perturbation of cytoskeletal organization of the cell through multiple mechanisms, including augmented protein kinase activation in the cell as well as release of FPR1 from a tight cytoskeletal association and laterally segregated, G-protein-poor membrane domains (25, 26, 44–46). To examine the effect of CB on FPR1 phosphorylation in the enhanced response, we repeated the examination of the NFPRb binding to FPR1 as a function of neutrophil exposure to fMLF but in the presence of 10 μm CB. The effect CB on putative fMLF-stimulated FPR1 phosphorylation is shown in Fig. 1 with quantitation in Fig. 2. The results indicate that there is a modest but significant change (p = 0.029) in the concentration dependence of NFPRb binding to the 60K species on prior neutrophil fMLF exposure, shifting the EC50 by a factor of ∼2 to 7.4 ± 2.2 × 10−8 m. Two-site analysis suggests two populations of receptors with EC50 values of 13 and 420 nm, where the CB-exposed population has fewer higher affinity receptors (53% versus 75%). This result suggests that the aggregate effect of enhancing the activation state of the cell by stimulating it with fMLF in the presence of cytochalasin B may be to cause decreased phosphorylation of FPR1. This result could be interpreted as arising from lower FPR1 kinase activity (i.e. GRK2 or GRK3) or higher FPR1 phosphatase activity. The shift is consistent with the generation of less phosphorylated (i.e. less inactivated) receptor and hence a higher response promoted by cellular CB exposure during fMLF stimulation. It is consistent with a slower but still saturable FPR1 phosphorylation at high fMLF, reflected in superoxide production rates that subside, more slowly (42, 47).
Immunoaffinity Purification of FPR1 and FPR2
Although the immunoblotting characteristics of the NFPR mAbs are consistent with recognition of both FPRs in their respective C-terminal regions, independent supporting evidence is required to conclude that NFPRa and NFPRb bind neutrophil FPR1. To show that the species identified by NFPRa and NFPRb immunochemically are indeed FPR1 and FPR2, we needed to positively identify both species in immunoaffinity isolates by tryptic digestion and mass analysis. To provide sufficient material for identification by mass spectrometry, we devised a method for isolating FPR from detergent extracts of membranes and whole neutrophils based, in part, on our experience with the immunopurification of flavocytochrome b from human neutrophils (36, 49). Such a method might also be potentially useful for identifying molecular partners of FPR and for providing material for phosphorylation analysis of this important GPCR system of human neutrophils. Briefly, we partially purified both FPR1 and FPR2 from DDM extracts of whole cells or membranes by serial immunoaffinity purification on NFPRb matrices, followed by a second stage of immunoaffinity purification on NFPRa. This serial procedure was carried out successfully on whole cell DDM extracts of unstimulated human neutrophils and works equally well on extracts of membranes made from fMLF-stimulated or unstimulated cells.
Because NFPRb does not appear to efficiently bind the 45K or 60K species on immunoblots from maximally fMLF-stimulated cells (see Fig. 1), we attempted to first separate the unmodified 60K species by binding the 1% DDM extracts of U or S whole neutrophils (prepared as described under “Experimental Procedures”) to the NFPRb matrix first for 1–2 h at room temperature. The unbound material or flow-through volume was then incubated with the NFPRa matrix in a similar way, washed, and eluted overnight in the cold with the respective antigen mimetic peptides. These specific elutions were then followed by shorter low pH and 1% SDS elutions at room temperature the next day (see “Experimental Procedures”). Fig. 4 presents a composite immunoblot of an analysis of the U and S samples arranged horizontally so that the left column shows the blots obtained from the U cells, and the right column shows blots from the S cells. Because each type of blot was probed with the two antibodies, the top row of the composite shows the blots developed using NFPRb as the primary antibody (sensitive to FPR1 modification), whereas the bottom row of blots represents those developed with NFPRa as the primary mAb (which binds both FPR1 and FPR2 and is relatively insensitive to exposure of the cells to fMLF). Each of the four panels in Fig. 4 show the CS9-precleared starting material in lane 1, followed in lanes 2–5 by samples from the peak elution fractions of the first NFPRb column and in lanes 6–9 by peak fractions from the second NFPRa column. The elution fractions are the antigen mimetic peptide eluates (lane 2, NFPRb peptide; lane 6, NFPRa peptide); the pH 2.3 eluates (lanes 3 and 7); the 1% SDS eluates (lanes 4 and 8); and the residual activity left on the beads when directly solubilized and heated in SDS-PAGE sample buffer (lanes 5 and 9). The recovery of receptor based on quantitative immunoblotting ranged between 50 and 70%. The relatively sharply focused species at approximately Mr 50,000 and 25,000 are leaked antibody heavy and light chain, respectively, and were also detected when the primary antibody is omitted (not shown). The minor species at Mr ∼100,000 was variably present in different preparations and probably represents incomplete reduction and alkylation prior to SDS-PAGE.
Examination of the 60K and 45K bands, corresponding to the U and S conditions, reveals interesting information. First, from an evaluation of the top row of NFPRb blots in Fig. 4, it is clear that the NFPRb matrix (lanes 2–5) retains most (∼80–90%) of the species recognized by NFPRb for both stimulus conditions. Because the U and S samples represent the same cell equivalent load, this result is somewhat surprising and contrasts with what is observed in Fig. 1. The receptor amount retained by NFPRb in fractions 2–5 in the stimulated S samples does not appear to be significantly different from that retained by the corresponding fractions in the U samples, although there is variance in the relative amounts of activity retained by the columns sensitive to the different elution conditions. This result is similar to that found if neutrophils are stimulated with fMLF alone, without CB (not shown), and thus does not appear to be the result of events unique to cells under the influence of this fungal agent. Retention of the 60K band by the column matrix is also relatively strong, compared with our experience with flavocytochrome b (36) and requires additional low pH or SDS help to achieve full recovery.
Examination of the bottom tier of the blots shows the retention of the 60K and 45K species by both matrices as developed the NFPRa antibody. Despite the contaminating immunoglobulin band at ∼50,000 and ∼25,000, the distribution of the broader underlying bands suggests that nearly all of the 45K species (putatively FPR2) was retained by the second (NFPRa) column (lanes 6–9) in the case of the U samples. The S samples show that slightly more 60K species was retained on the NFPRa matrix and, surprisingly, a new species at about 30,000 that was also retained by the second column, suggesting a major shift in the migration of either the former 60K or 45K species. In separate analytical gels run to compare the cytosolic bands (30,000/35,000) mentioned above in reference to Figs. 1 and 3, the new 30,000 species was found only in the S samples and was resolved from the cytosolic bands (not shown). Additionally, it is a very tightly associated membrane protein that is not stripped from S membranes even after treatment with 10 mm NaOH (33); nor was it observed in membranes derived from cells stimulated with fMLF only (not shown). Because there is no evidence of a new 30,000 species in the NFPRa blots of the fMLF/CB or fMLF only samples (Fig. 1), we believe that this alteration of the migration pattern of the FPRs may result from partially digested FPR proteolyzed by an endogenous protease during the extended purification required for this analysis. Similar changes in apparent molecular weight have been observed when papain is used to deglycosylate FPR1 by digestion of intact neutrophils (50).
To assess the purity of these fractions, we attempted to directly develop equivalent gels using conventional silver or Coomassie Blue stains after SDS-PAGE. However, only an abundant (Mr ∼45,000) protein (β-actin) was visualized. To obtain more sensitivity, we also fluorescently labeled the peptide eluate fractions from the NFPRb matrix with the protein amino group-reactive Z dye Blue 2 (see “Experimental Procedures”) and examined their compositions by SDS-PAGE and fluorescence imaging on a slab gel imager. An image of this analysis is shown in grayscale in Fig. 4B for the U and S samples. The FPR1 region between the Mr 50,000 and 65,000 markers in the both cases showed 2–3 sharp bands over a background of fainter density. Multiple attempts at mass analysis, using analogously prepared fractions from whole cells or membranes of this region for FPR1 peptides by in-gel tryptic digestion were negative for all of the fractions shown in Fig. 4A. Instead there was a strong signal for several cytokeratin peptides, as shown in Table 1. Further analysis of these fractions allowed us to identify the origin of the following bands with their relative molecular weights (also summarized in Table 1): lactoferrin (80,000), serum albumin (65,000), β-actin (45,000), myeloperoxidase (55–60K), IgG heavy chain (50,000), and glyceraldehyde-3-phosphate dehydrogenase (40,000). The S fractions tended to be less contaminated with these bands because such cells have been degranulated (37, 51). Based on immunoblotting, the 25,000 band of the S sample is probably column immunoglobulin-derived, supported by mass analysis of other preparations. Attempting similar purifications using 1 m NaCl-washed membranes (37) or NaOH-washed membranes (52) resulted in less contamination by neutrophil-soluble or granule proteins, but still not enough to sufficiently diminish the ion-suppressing effect of the cytokeratins in the mass spectrometer. However, after pooling and concentrating these fractions from multiple cell preparations, several statistically significant identifications of the same FPR1 peptides shown in Tables 2 and 3 for the deglycosylated receptor preparations analyzed below were made without deglycosylation of FPR1 (not shown).
TABLE 1.
Proteins identified in peptide eluted fractions from NFPRb affinity matrix
Peptide-eluted fractions from the NFPRb matrix shown in Fig. 4A were labeled with the protein amino group reactive Z dye Blue 2 (see “Experimental Procedures”) and separated by SDS-PAGE. Migrating protein bands were detected by fluorescence imaging on a Typhoon (GE Healthcare) slab gel fluorescence imager. An image of this analysis is shown in grayscale in Fig. 4B. Individual bands were excised and tryptically digested, and peptides were identified by electrospray ionization-quadrupole-TOF and MASCOT analysis. The proteins identified by this analysis are shown in the table with accession numbers, protein names, MASCOT scores, numbers of peptides detected, percentage coverage of the protein, and the expected molecular weights.
| Accession number | Protein | MASCOT score | Unique peptides | Coverage | Expected Mr |
|---|---|---|---|---|---|
| % | thousands | ||||
| CAA37116 | Lactoferrin | 181 | 10 | 16 | 78 |
| CAA23754 | Serum albumin | 211 | 10 | 17 | 69 |
| P04264 | Keratin 1 | 595 | 13 | 24 | 66 |
| P35908 | Keratin 2 | 393 | 9 | 14 | 66 |
| CAA32649 | Keratin 10 | 545 | 12 | 25 | 60 |
| CAA33438 | Myeloperoxidase | 251 | 10 | 14 | 84 (observed 55–60) |
| AAB28159 | IgG heavy chain | 163 | 4 | 10 | 51 |
| NP_001092 | β-Actin | 448 | 11 | 32 | 42 |
| CAA25833 | Glyceraldehyde-3-phosphate dehydrogenase | 175 | 6 | 23 | 36 |
TABLE 2.
FPR1 and FPR2 peptides with modifications from stimulated and unstimulated neutrophils
The protein bands shown in Fig. 6 were excised from gels, digested with porcine trypsin, and subjected to LC/MS/MS mass analysis as described under “Experimental Procedures.” m/z data were analyzed by MASCOT and Peptide Shaker using 25 ppm for MS and MS/MS ion mass tolerance. The left column shows the identified peptides with residue modifications underlined. The upper set is from FPR1, and the lower set is from FPR2. Each subsequent column shows the residue sequence numbers, type of modification, calculated molecular weights, measured experimental m/z ratios, charge, ppm mass error, p values, fractions from which the data were derived, and the analysis program used.
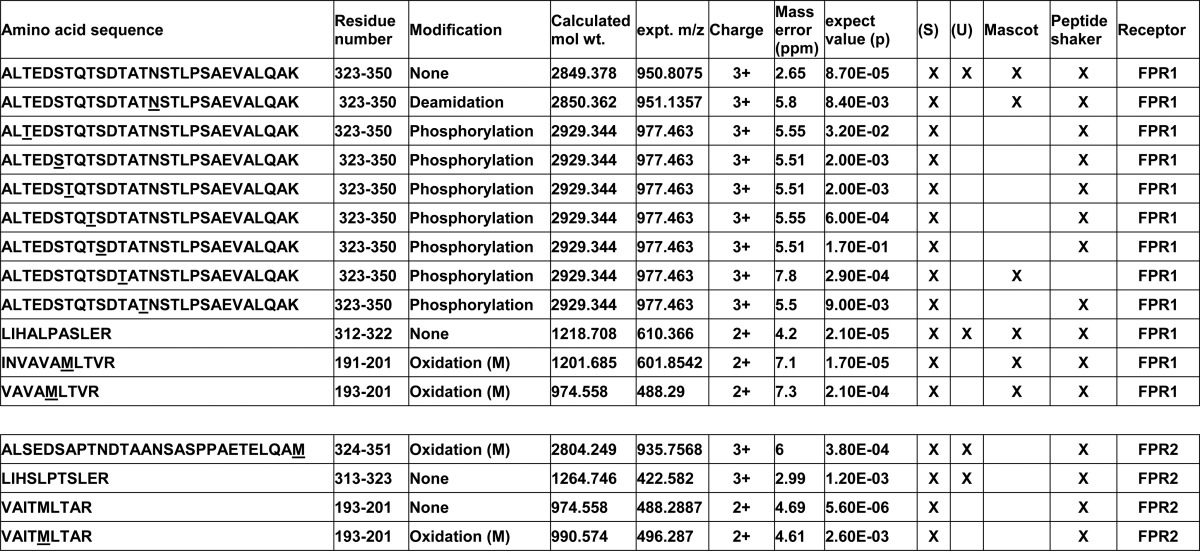
TABLE 3.
Alignment of FPR1 and FPR2 showing underlined and shaded identified peptide regions
The LALIGN (Swissprot EXPASy) output for FPR1 and FPR2 are shown to allow comparison of the sequence peptides of the two GPCRs. The FPR1 (gi|119592446|gb|EAW72040.1) and FPR2 (gi|4503781|ref|NP_001453.1) sequences were aligned by application of Pearson's LALIGN program as implemented by the Huang and Miller algorithm of a linear protein sequence alignment (80). The C-terminal Lys and Met were added to the alignment sequences. They show the insertion of Phe273 in the sequence for FPR2, explaining its total length of 351 amino acid residues. The underlined sequence regions were identified by mass analysis described under “Experimental Procedures.” The gray highlight differentiates individual peptides of the C-terminal portion of the extracellular loop 2 (E2) and the N-terminal portion of the C-terminal tail of both proteins.

Mass Analysis of Unstimulated and fMLF/CB-stimulated FPR
High confidence identification, in the absence of the strong cytokeratin signals in the same mass spectra, was finally achieved by pooling all of the eluate fractions of similarly prepared membrane extracts of neutrophils from both mAbs and deglycosylating them to their core peptide chain molecular weights (calculated Mr for FPR1 = 38,446; FPR2 = 38,964). This deglycosylation procedure was carried out by overnight exposure to recombinant CBM-PNGase F, which is a His-tagged fusion protein of a carbohydrate binding module of Chryseobacterium meningosepticum and PNGase F deglycosylating enzyme. This fusion protein, having a molecular weight of 48,000 (39), was significantly shifted from the apparent relative SDS-PAGE molecular weight of deglycosylated FPR1 and FPR2 at ∼31,000–36,000 and the molecular weight of the native PNGase F (Mr 36,000) measured by SDS-PAGE. Fig. 5 shows the result of such manipulation on a pooled preparation of mAb eluates from U cells. Lanes 2 and 6 represent the pooled column-purified eluates from whole cell lysates. The deglycosylated products detected by NFPRb are shown in lanes 3 and 4, and products detected by NFPRa are shown in lanes 7 and 8. Clearly, both 60K and 45K species appear to have very similar polypeptide molecular weights after exposure to both types of PNGases and do not appear to be resolvable by SDS-PAGE. However, deglycosylation induced a shift to a region of the gel with a lower keratin content and thus provided an additional important purification step. This shift, combined with pooling of the NFPRa and NFPRb eluates, allowed the visualization of the Coomassie-stained deglycosylated FPR bands in preparative gels, shown in Fig. 6 side by side with immunoblots of corresponding more lightly loaded samples, run on other lanes of the gel.
FIGURE 5.
Deglycosylation of FPR by His-CBM-PNGase F and PNGase F. Pooled (0.5 ml) of the 1% SDS eluates from both the NFPRa and NFPRb affinity matrices representing ∼5 × 107 cell equivalents from unstimulated cells were divided into two aliquots, and each was deglycosylated as described under “Experimental Procedures” using recombinant His-CBM-PNGase F (39) (lanes 3 and 7) or commercial PNGase F (Sigma) (lanes 4 and 8). The deglycosylated samples showed a shift of both FPR1 and FPR2 bands to Mr ∼34,000, the previously reported deglycosylated SDS-PAGE apparent molecular weight of FPR1. The calculated peptide molecular weight of FPR1 and FPR2 is ∼38,000, supporting their indistinguishability by SDS-PAGE. The glycosylated starting material is shown in lanes 2 and 6. Lanes 2–4 were developed with NFPRb. Lanes 6–8 were developed with NFPRa. The FPR1 and FPR2 signals represent about 1 × 106 cell equivalents/lane.
FIGURE 6.
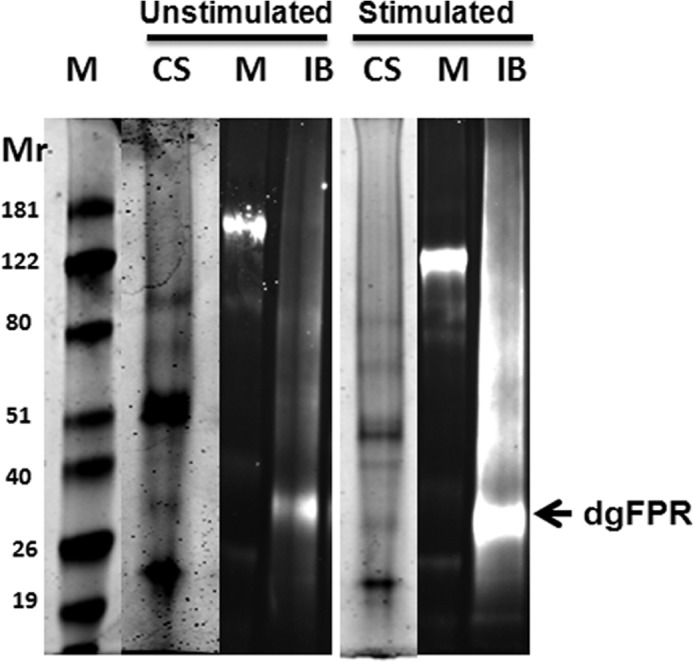
Preparative SDS-PAGE for mass analysis of deglycosylated FPR (dgFPR) from S and U human neutrophils. All fractions from both NFPRa and NFPRb purifications from five separate preparations were pooled, concentrated, deglycosylated with His-PNGase F (39), and desalted into SDS-PAGE-compatible buffer of 100-μl final volume. The Coomassie-stained (CS) lanes contain the U sample, representing the yield from 2 × 109 cells, and the S sample from 6 × 109 cells. The relative molecular weight markers (M) are to the left of the immunoblotted samples (IB) and between the Coomassie-stained samples used for excision and mass analysis. The location of the deglycosylated FPR band is indicated by an arrow.
The Coomassie-stained bands in these gels were excised and subjected to in-gel trypsinolysis followed by LC/MS/MS analysis. This analysis allowed the unequivocal identification of several tryptic peptides from both the U and S bands, shown in Fig. 6. The peptides, identified with their m/z values, signal strengths, and significance values, are shown in Tables 2 and 3. Two adjacent peptides, 312LIHALPASLER322 and 323ALTEDSTQTSDTATNSTLPSAEVALQAK350, account for 83% of the predicted C-terminal tail (Met304–Lys350) of FPR1, as can be clearly seen with shaded circles in the “snake” diagram of FPR1 shown in Fig. 7. The third peptide, 191INVAVAMLTVR201, is near the C terminus of the predicted second extracellular loop and was accompanied by an internal fragment 193VAVAMLTVR201. This latter interesting result reflects the fact that our preparations were made from neutrophils from a pooled mixed population of blood donors and supports the presence of an amino acid polymorphism at N192K (17), validating the sensitivity of our method. In addition to the FPR1 peptides, we also found the analogous peptides from similar regions in FPR2. These peptides were the two C-terminal regions 313LIHSLPTSLER323 and 324ALSEDSAPTNDTAANSASPPAETELQAM351 and the corresponding extracellular loop 2 region, 193VAITMLTAR201. The alignment of FPR1 and FPR2, showing the relative positions of the identified peptides, is shown in Table 3. Overall, these mass spectrometry results clearly support the partial immunopurification of FPR1 and FPR2 and the presence of both species in the deglycosylated extracts used in this analysis. These results also support their unequivocal recognition by NFPRa and NFPRb antibodies.
FIGURE 7.

FPR1 predicted transmembrane structure showing identified peptides and Ser/Thr phosphorylation sites. A “snake” diagram of the predicted transmembrane structure of FPR1 is shown. The tryptic peptides identified by mass spectrometry are shaded gray, and Ser/Thr phosphorylation sites are identified by seven darkly shaded star borders around amino acid residue circles. The extracellular loops are marked E1, E2, and E3, and the cytoplasmic loops are marked C1, C2, and C3. The predicted α-helical regions are shown as stacks of closely packed circles representing each type of amino acid residue, with arrows depicting the direction along which the sequences should be read. The predicted transmembrane region is boxed for helices I–VII. The predicted amphipathic helix VIII (67) is also shown. A putative disulfide bond between Cys98 and Cys176 is shown as an arc connecting the two residues. Underlined residues represent positions in the sequence for which normal SNP variants exist (17). These polymorphisms are I11T, V47L, L97M, L101V, R190W, N192K, and A346E. The phage display-mapped epitopes for each mAb are marked by a rounded rectangle for NFPRb and an oval for NFPRa showing specific C-terminal tail residues identified by the mapping. This model is based on a structural alignment of the FPR1 sequence on a template derived from superposition of several known x-ray crystal structures of GPCRs (human A2a adenosine receptor (73), human M2 muscarinic acetylcholine receptor (PDB code 3UON) (74), CXCR4 chemokine receptor (PDB code 3ODU) (75), human histamine H1 receptor (PDB code 3RZE) (76), neurotensin nts1 receptor (PDB code 4GRV) (77), human dopamine receptor (PDB code 3PBL) (78), metarhodopsin 2 (PBD code 3PQR) (79), and the β-adrenergic receptor (PBD code 3SN6) (48)), using the homology modeling functions of Accelrys Discover version 3.5.
LC/MS/MS Shows FPR1 Phosphorylation in fMLF/CB-stimulated Samples
Because NFPRb binding to FPR1 appeared to be sensitive to the neutrophil stimulation conditions used to prepare the S samples, we were interested to know if receptor phosphorylation could be detected. Searching of LC/MS/MS data for phosphorylation of serine or threonine led to the identification of several phosphorylated forms of the peptides from the extreme C-terminal fragment of only FPR1 in the stimulated population of cells (Table 2). The representative LC/MS/MS spectrum of the C-terminal unphosphorylated peptide is shown in Fig. 8A. The phosphorylated forms are shown in Fig. 8B with phospho-Thr325 and Fig. 8C with phospho-Thr331 identified within red highlight boxes. Of particular interest was the observation that the seven identified phospho-peptides were derived only from FPR1 and found only in the S sample prepared from fMLF/CB-stimulated neutrophils. Moreover, each peptide was phosphorylated at only one of the following Ser/Thr loci: Thr325, Ser328, Thr329, Thr331, Ser332, Thr334, and Thr339. Also significant was the absence of phosphorylated peptides from FPR2. Because Thr339 resides in the mapped epitope of NFPRb, the phosphorylation of this site would be sufficient to explain the diminished binding of NFPRb to phosphorylated FPR1. However, given that the alternative sites were unique phosphorylation sites on peptides from other FPR1 molecules, the result also suggests that the binding of NFPRb to FPR1 may be diminished by those modifications or that significant dephosphorylation during preparation or measurement had occurred.
FIGURE 8.

LC-MS/MS analysis of tryptic peptides from FPR1. A, fragmentation of the C-terminal FPR1 peptide ALTEDSTQTSDTATNSTLPSAEVALQAK. The 3+ charge state (950.807 m/z) was fragmented using collision-induced disassociation. b and y fragment ions are labeled and shown above bars going up (y) or down (b) to indicate sequence coverage. B, MS/MS spectrum of fragment ions from the Thr325-phosphorylated form of the peptide in A. The 3+ precursor ion (977.463 m/z) was used for collision-induced disassociation. C, MS/MS spectrum of fragment ions from the Thr331-phosphorylated form of the peptide in A. The triply charged ion was selected for collision-induced disassociation. Data are from the PeptideShaker proteomics search engine version 0.19.3.
DISCUSSION
The phosphorylation of rhodopsin following receptor activation was detected in light-activated rods long before G-proteins were even implicated in rhodopsin-mediated signal transduction (53). We now know that such covalent modification of GPCRs serves as a universal regulator of this superfamily of receptors (54). After more than 2 decades of intense research on the phosphorylation of hundreds of non-olfactory GPCR, primarily in recombinant systems, the role of G-protein receptor kinases (GRKs) and arrestins in this process has been firmly established. The field is now turning toward analysis of the receptors in their physiological context (55). Through understanding of the sequential steps and kinetics of the phosphorylation of the FPRs in leukocytes, the consequences of inappropriate FPR regulation and its possible relationship to disease may be revealed.
Because the regulation of neutrophil responses is of critical importance to the host and some of this regulation is mediated through GPCR, understanding of a neutrophil GPCR prototype is of both basic and medical interest. An extreme example of how neutrophils must be tightly regulated is found at the intestinal locus, which serves as a barrier to enormous numbers of luminal bacteria (56). Although neutrophils are equipped to respond to bacterial protein degradation products at infinitesimal concentrations using N-formyl peptide receptors (57), these responses are tightly controlled at the intestinal lumen/epithelial interface. Indeed, direct measurement of formyl-Met-Leu-Phe concentrations in the colonic lumen suggests that the concentration of this peptide alone is ∼1 μm (58). Given that nascent bacterial proteins have N-formyl methionines at their N termini, it is reasonable to suppose that this concentration represents only a lower limit. The published affinity of the N-formyl peptide receptors in neutrophils ranges widely. G-protein-coupled and uncoupled forms of FPR1 demonstrate affinities between 5.2 × 10−10 and 2.5 × 10−8 m, respectively. In the “high affinity” cytoskeletal/arrestin FPR-associated complex, the affinity for fMLF is even higher, exhibiting non-dissociable properties (44, 59). This high affinity may help explain the high sensitivity to formyl peptides in neutrophils in the range below its lower EC50 for chemotaxis. Clearly, for neutrophils to efficiently migrate up concentration gradients, spanning concentrations from the tens of picomolar to the tens of micromolar, additional regulatory mechanisms must be present and functional. Furthermore, given the deleterious consequences of chemotactic overstimulation in a directional gradient of neutrophils (60–62), important adaptive regulatory controls for this receptor system would be expected.
In human neutrophils, Ye and co-workers (63) showed that cytosolic GRK2 and, less efficiently, GRK3, when reconstituted in vitro, phosphorylated the C-terminal 47-amino acid fragment of FPR1, expressed as a fusion protein with glutathione S-transferase in vitro. They found that the phosphorylation occurred hierarchically, showing significant dependence of C-terminal Ser/Thr phosphorylations of the C terminus on N-terminal Ser/Thr phosphorylations of the same segment. These C-terminal phosphorylations are critical to FPR1 internalization, G-protein, and arrestin interaction (29) and are thus implicated in the important mechanisms of receptor-initiated signal transduction and affinity control. In heterologous expression systems, the phosphorylation of the sequence 328DSTQTST339 affects β-arrestin affinity for the FPR, whereas the affinity of the FPR1·β-arrestin complex for its fMLF analogs is dependent on phosphorylation in the 333TATNST339 region (28, 64). It is believed that multiple phosphorylations of FPR, in the cytoplasmic face, will also act to finely tune the sensitivity of neutrophils during adaptive migration, as is postulated for rhodopsin for light (31) and Dictyostelium discoidium for cAMP (65).
To obtain site-specific information about FPR1 phosphorylation in its native neutrophil context, we applied mass spectrometry-based peptide sequencing to NFPRa/NFPRb-immunopurified, deglycosylated FPR1 and FPR2. The NFPRb and NFPRa mAbs had been raised against histidine-tagged recombinant FPR1, expressed in sF9 cells, and affinity-purified from their lysophosphatidylglycerol-solubilized membranes (33). Both antibodies were epitope-mapped, using affinity-selected members of the J404 random nonapeptide phage display library (66), and the identified loci were confirmed by site-directed mutagenesis of recombinant FPR1 expressed in the CHO cell system (67). These results suggested that the antibodies recognized the recombinant forms of FPR1 and probably recognized the corresponding native forms in neutrophils, both in SDS-PAGE immunoblots and by immunofluorescence of fixed neutrophil suspensions or glass-adherent cells prepared by cytospin (33). Further experiments suggested that the rFPR1 was phosphorylated because NFPRb binding to the receptor was restored after treatment of rFPR1-CHO membranes with alkaline phosphatase at pH7 (33). Because Ser338, Thr339, and Ser342 were located within the NFPRb epitope (see Fig. 7) and Ser338, Thr336, and Thr334 are adjacent to it, we had inferred that that the phosphorylation of these C-terminal residues of FPR1 was interfering with the binding of the mAb NFPRb to its epitope and was responsible for its sensitivity to fMLF stimulation.
In the current work, using phospho-sensitive and insensitive anti-C-terminal FPR mAbs, we show that the molecular species they recognize in neutrophils are indeed FPR1 and FPR2. We also examined the nature of the modifications to the C terminus occurring after prolonged (10-min) activation of neutrophils by fMLF in a highly stimulative condition (+CB). Although attempts were made to examine “highly purified” FPR, this was technically difficult because of the low abundance of the receptors relative to highly basic and adhesive neutrophil granule proteins. Our preparations also had a significant presence of keratins in the same molecular weight range as FPR1 and, in certain preparations, a number of neutrophil cytoplasmic proteins, which confounded earlier attempts to identify the FPRs in our preparations. These problems were, however, circumvented by pooling multiple preparations and deglycosylating the partial SDS-denatured immunopurified receptor-enriched fractions and then separating the FPRs from the bulk keratins by SDS-PAGE. In so doing, we were able to identify peptides by LC/MS/MS analysis with more than 80% coverage of the C-terminal tails of FPR1 and FPR2 as well as coverage of a C-terminal region of the predicted second extracellular loop. The peptides generated also confirmed the presence of the N192K amino acid polymorphism in our population of blood donors, because the recovered fragments of the second extracellular loop of FPR1 corresponded to two polymorphic forms. One contained Asn192 in the 191INVAVAMLTVR201 fragment, whereas the other had to have been cleaved by trypsin at Lys192 of the polymorphic FPR1 form, leaving the 193VAVAMLTVR201 fragment.
The excellent coverage of the FPR1 C terminus sequence in the mass spectra and the differential binding of the NFPR mAbs after neutrophil stimulation suggested that some phosphorylated peptide forms might be present. Indeed, our LC/MS/MS mass analysis of the partially purified FPR mixtures resulted in the identification of seven C-terminal FPR1 peptides that were each phosphorylated on only one Ser or Thr shown in Table 2 and Fig. 7. Phosphorylation was observed only in the FPR1 C-terminal tryptic peptide and only found in extracts obtained from fMLF/CB-stimulated neutrophils. This result suggests that FPR1 from fMLF/CB-stimulated neutrophils was monophosphorylated at Thr325, Ser328, Thr329, Thr331, Ser332, Thr334, or Thr339 on seven different receptor molecules. No peptides with multiple phosphorylations were detected. We cannot rule out, however, that multiply phosphorylated peptides are present but more difficult to detect with the LC/MS/MS conditions that were used or that they are dephosphorylated faster than isolation allowed. Nevertheless, our studies represent novel identification of GPCR phosphorylation sites in primary cells and allow evaluation of past studies made on rFPR1 expressed in heterologous systems.
It is noteworthy that no phosphorylations were detected on peptides derived from FPR2 from either U or S neutrophils. The concentration of fMLF used to stimulate the neutrophils for FPR purification in this study was 1 μm. The Kd for fMLF binding to FPR1 versus FPR2 differs by at least an order of magnitude (∼50 nm versus ∼500 nm (17)), and CB appears to shift the EC50 of FPR1 phosphorylation by 2–4-fold higher values. Thus, one might expect, by analogy to FPR1 for cells treated with CB, that the Kd for FPR2 might also be shifted, resulting in only 30–40% maximal FPR2 phosphorylation. Furthermore, Dalli et al. (68) showed that FPR2 function is quite specific and high affinity for the natural N-terminal domain of annexin 1 ligand (AnxA1(2–50); 4 nm) as well as for lipoxin A4 (2 nm (69)) and orders of magnitude less sensitive to peptide fragments, such as fMLF, suggesting that the efficiency of fMLF-activated FPR2 phosphorylation is probably even lower than suggested by Kd arguments. Together, these considerations may explain the lack of detectable FPR2 phosphorylation.
In their comprehensive combinatorial mutational analysis of the regulation of rFPR1 expression and function in U937 cells, Potter et al. (29) restricted phosphorylation to groups of sites on the ΔST mutant in which all C-terminal serines and threonines with the exception of Ser319 and Thr325 were substituted by alanine or glycine. They concluded that Ser328, Ser332, and Ser338 were individually critical and sufficient for internalization, desensitization, and arrestin binding and that other sites had modulating influences. Although we detected phosphorylation at Ser328 and Ser332, we did not detect phosphorylated Ser338 but instead found that Thr339 as well as Ser325 are phosphorylated. Additionally, because we found receptor peptides only individually phosphorylated at one site, it is difficult to explain how a combinatorial code of regulation might be operative without invoking additional mechanisms. Of course, it is important to note that the fMLF stimulation was carried out on CB-treated neutrophils, which places FPR1 in a different cellular context and significantly alters desensitization and receptor processing. Nevertheless, the ability to individually identify phosphorylation sites from neutrophil fractions suggests that FPR1 phospho-decoding may now be feasible and allow understanding the FPR1 functional state in different stimulated contexts, especially in those relevant to inflammation.
It should also be noted that in addition to sites on the C-terminal tail, FPR1 has 11 other predicted cytoplasmic Ser/Thr amino acid residues (threonines 56, 58, 60, 61, 133, 138, and 226 and serines 63, 140, 236, and 237; see also Fig. 8) located on the first, second, and third predicted intracellular loops. Kang et al. (70) mutated LXA4R/FPR2 Ser236 and Ser237 and showed that in heterologous systems, the expressed mutants displayed altered responses and phosphoprotein immunoblotting patterns consistent with receptor phosphorylation. Table 3 shows that these FPR2 potential phosphorylation sites are conserved in FPR1. These additional potential phosphorylation sites were not evident in our tryptic digests, precluding conclusion about the phosphorylation state of these amino acid residues. Cleavage of the purified proteins with additional proteases may increase sequence coverage by our methods and requires further study.
On another analytical level, it is clear that only one FPR1 phosphorylation site is within the phage display-mapped epitope recognized by NFPRb (see Fig. 7). Because the remaining sites are at most 13 residues distant from the beginning of the NFPRb epitope at Ser338 and because 28 Å is the maximum average dimension of the antibody-protein antigen interface (71) even an extended peptide conformation might have steric/charge effects at such distances. Thus, with appropriate partial renaturation after SDS-PAGE and transfer onto PVDF membranes, phosphorylation at sites other than Ser339 could potentially interfere with the NFPRb binding to FPR1 observed in immunoblots. We therefore conclude that, at least qualitatively, the differential recognition of FPR1 by the two antibodies arises from phosphorylation of the C-terminal tail in or near the region of the FPR1 epitope recognized by NFPRb and that the NFPRa mAb must have independent access to the protein not at all influenced by the C-terminal phosphorylation sites. Indirectly, this understanding suggests that such remote influences may also play a role in the specific binding of regulatory proteins to FPR1.
Last, we speculate that unique individual and scattered phosphorylation of the C terminus of FPR1 could have unique functions in regulation of FPR1. Possibilities include biochemical or spatial sequestration of populations of FPR by regulatory systems of the cell. Thus, a more systematic study of the kinetics of phosphorylation with fMLF alone, with cytochalasin B, or with pro- and anti-inflammatory cytokines might help to clarify the relevance of the detected phosphorylation pattern. Ultimately, such a diversity of regulatory sites on a regulatory protein under the influence of cellular kinases and phosphatases may demand a systems biology approach (72) to understanding the global influence of FPR “phospho-coding” on the innate immune response.
In summary, our results report a convenient assay of activated and phosphorylated FPR1 and thus present a way to follow this form of FPR1 regulation in the native human neutrophil cellular milieu. Our results also report the first identification of the neutrophil FPR1 Ser/Thr sites that are phosphorylated after fMLF stimulation of suspension neutrophils. We believe that the method could be adapted to allow kinetic evaluation of the coding and hierarchical phosphorylation hypotheses of FPR1 regulation. Furthermore, these results suggest that examination of phosphorylation patterns of leukocyte receptors under different conditions in their primary cells obtained from human blood is within reach and should provide a fruitful and significant analysis of normal and aberrant receptor regulation.
Acknowledgments
We thank Drs. Emily Kwan and R. Antony Warren for kindly providing the CBM-PNGase F plasmid, which significantly increased yields of deglycosylated FPR1 free of cytokeratin contamination. We are also thankful to Dr. William Nauseef for reading the manuscript and for providing helpful suggestions, especially on early versions. The Montana State University mass spectrometry facility is supported by Murdock Charitable Trust Grant 2009084 and National Institutes of Health Grants P20RR-020185, 1R42RR-025311, P20GM-103394-05 and P20RR-024237 from the COBRE Program of the National Center for Research Resources.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-AI22735-19 (to A. J. J.), 5R01AI-064107 (to E. A. D. and T. L.), 2R42RR-021790 (to E. A. D. and G. K.-A.), 2R42RR-021740 (to E. A. D.), and R01 AI081961-01A1 (to B. B.). This work was also supported by a Crohn's and Colitis Foundation Helmsley Scholar award (to A. J. J.).
M Riesselman and A. Jesaitis, unpublished results.
- GPCR
- G-protein-coupled receptor
- fMLF
- N-formyl-Met-Leu-Phe
- CB
- cytochalasin B
- CBM
- carbohydrate binding module
- U
- unstimulated
- S
- fMLF + CB-treated
- OG
- octyl β-d-glucopyranoside
- DDM
- n-dodecyl-β-d-maltopyranoside
- FPR
- N-formyl peptide chemoattractant receptor
- PNGase F
- N-glycosidase F
- GRK
- G-protein receptor kinase
- 60K or 50–65K
- molecular species with a relative molecular weight of 50,000–65,000
- 45K
- narrow band below 60K, between the Mr 39,000 and 51,000 markers.
REFERENCES
- 1. Dale D. C., Boxer L., Liles W. C. (2008) The phagocytes. Neutrophils and monocytes. Blood 112, 935–945 [DOI] [PubMed] [Google Scholar]
- 2. Nauseef W. M. (2007) How human neutrophils kill and degrade microbes. An integrated view. Immunol. Rev. 219, 88–102 [DOI] [PubMed] [Google Scholar]
- 3. McDonald B., Kubes P. (2012) Neutrophils and intravascular immunity in the liver during infection and sterile inflammation. Toxicol. Pathol. 40, 157–165 [DOI] [PubMed] [Google Scholar]
- 4. Powner D. J., Pettitt T. R., Anderson R., Nash G. B., Wakelam M. J. (2007) Stable adhesion and migration of human neutrophils requires phospholipase D-mediated activation of the integrin CD11b/CD18. Mol. Immunol. 44, 3211–3221 [DOI] [PubMed] [Google Scholar]
- 5. Springer T. A. (1990) Adhesion receptors of the immune system. Nature 346, 425–434 [DOI] [PubMed] [Google Scholar]
- 6. Pick R., Brechtefeld D., Walzog B. (2013) Intraluminal crawling versus interstitial neutrophil migration during inflammation. Mol. Immunol. 55, 70–75 [DOI] [PubMed] [Google Scholar]
- 7. Swirski F. K., Nahrendorf M. (2013) Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 339, 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williams M. R., Azcutia V., Newton G., Alcaide P., Luscinskas F. W. (2011) Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 32, 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sadik C. D., Kim N. D., Luster A. D. (2011) Neutrophils cascading their way to inflammation. Trends Immunol. 32, 452–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marasco W. A., Phan S. H., Krutzsch H., Showell H. J., Feltner D. E., Nairn R., Becker E. L., Ward P. A. (1984) Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J. Biol. Chem. 259, 5430–5439 [PubMed] [Google Scholar]
- 11. Carp H. (1982) Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med. 155, 264–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bünemann M., Hosey M. M. (1999) G-protein coupled receptor kinases as modulators of G-protein signalling. J. Physiol. 517, 5–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kovacs J. J., Hara M. R., Davenport C. L., Kim J., Lefkowitz R. J. (2009) Arrestin development. Emerging roles for β-arrestins in developmental signaling pathways. Dev. Cell 17, 443–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reiter E., Lefkowitz R. J. (2006) GRKs and β-arrestins. Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165 [DOI] [PubMed] [Google Scholar]
- 15. Simon H. U. (2003) Neutrophil apoptosis pathways and their modifications in inflammation. Immunol. Rev. 193, 101–110 [DOI] [PubMed] [Google Scholar]
- 16. Ariel A., Serhan C. N. (2007) Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 28, 176–183 [DOI] [PubMed] [Google Scholar]
- 17. Ye R. D., Boulay F., Wang J. M., Dahlgren C., Gerard C., Parmentier M., Serhan C. N., Murphy P. M. (2009) International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 61, 119–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S. D., Kim J. M., Jo S. H., Lee H. Y., Lee S. Y., Shim J. W., Seo S. K., Yun J., Bae Y. S. (2009) Functional expression of formyl peptide receptor family in human NK cells. J. Immunol. 183, 5511–5517 [DOI] [PubMed] [Google Scholar]
- 19. Prevete N., Salzano F. A., Rossi F. W., Rivellese F., Dellepiane M., Guastini L., Mora R., Marone G., Salami A., De Paulis A. (2011) Role(s) of formyl-peptide receptors expressed in nasal epithelial cells. J. Biol. Regul. Homeost. Agents 25, 553–564 [PubMed] [Google Scholar]
- 20. Rabiet M. J., Macari L., Dahlgren C., Boulay F. (2011) N-Formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J. Biol. Chem. 286, 26718–26731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bommakanti R. K., Bokoch G. M., Tolley J. O., Schreiber R. E., Siemsen D. W., Klotz K.-N., Jesaitis A. J. (1992) Reconstitution of a physical complex between the N-formyl chemotactic peptide receptor and G protein. Inhibition by pertussis toxin-catalyzed ADP ribosylation. J. Biol. Chem. 267, 7576–7581 [PubMed] [Google Scholar]
- 22. Jesaitis A. J., Naemura J. R., Painter R. G., Schmitt M., Sklar L. A., Cochrane C. G. (1982) The fate of the N-formyl-chemotactic peptide receptor in stimulated human granulocytes. Subcellular fractionation studies. J. Cell. Biochem. 20, 177–191 [DOI] [PubMed] [Google Scholar]
- 23. Sklar L. A., Finney D. A., Oades Z. G., Jesaitis A. J., Painter R. G., Cochrane C. G. (1984) The dynamics of ligand-receptor interactions. Real-time analysis of association, dissociation, and internalization of an N-formyl peptide and its receptors on the human neutrophil. J. Biol. Chem. 259, 5661–5669 [PubMed] [Google Scholar]
- 24. Sklar L. A., Bokoch G. M., Button D., Smolen J. E. (1987) Regulation of ligand-receptor dynamics by guanine nucleotides. Real-time analysis of interconverting states for the neutrophil formyl peptide receptor. J. Biol. Chem. 262, 135–139 [PubMed] [Google Scholar]
- 25. Jesaitis A. J., Tolley J. O., Allen R. A. (1986) Receptor-cytoskeleton interactions and membrane traffic may regulate chemoattractant-induced superoxide production in human granulocytes. J. Biol. Chem. 261, 13662–13669 [PubMed] [Google Scholar]
- 26. Jesaitis A. J., Tolley J. O., Bokoch G. M., Allen R. A. (1989) Regulation of chemoattractant receptor interaction with transducing proteins by organizational control in the plasma membrane of human neutrophils. J. Cell Biol. 109, 2783–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu H., Karlsson J., Bylund J., Movitz C., Karlsson A., Dahlgren C. (2006) Ligand recognition and activation of formyl peptide receptors in neutrophils. J. Leukoc. Biol. 79, 247–256 [DOI] [PubMed] [Google Scholar]
- 28. Key T. A., Foutz T. D., Gurevich V. V., Sklar L. A., Prossnitz E. R. (2003) N-Formyl peptide receptor phosphorylation domains differentially regulate arrestin and agonist affinity. J. Biol. Chem. 278, 4041–4047 [DOI] [PubMed] [Google Scholar]
- 29. Potter R. M., Maestas D. C., Cimino D. F., Prossnitz E. R. (2006) Regulation of N-formyl peptide receptor signaling and trafficking by individual carboxyl-terminal serine and threonine residues. J. Immunol. 176, 5418–5425 [DOI] [PubMed] [Google Scholar]
- 30. Ali H., Richardson R. M., Tomhave E. D., Didsbury J. R., Snyderman R. (1993) Differences in phosphorylation of formylpeptide and C5a chemoattractant receptors correlate with differences in desensitization. J. Biol. Chem. 268, 24247–24254 [PubMed] [Google Scholar]
- 31. Arshavsky V. Y. (2002) Rhodopsin phosphorylation. From terminating single photon responses to photoreceptor dark adaptation. Trends Neurosci. 25, 124–126 [DOI] [PubMed] [Google Scholar]
- 32. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riesselman M., Miettinen H. M., Gripentrog J. M., Lord C. I., Mumey B., Dratz E. A., Stie J., Taylor R. M., Jesaitis A. J. (2007) C-terminal tail phosphorylation of N-formyl peptide receptor. Differential recognition of two neutrophil chemoattractant receptors by monoclonal antibodies NFPR1 and NFPR2. J. Immunol. 179, 2520–2531 [DOI] [PubMed] [Google Scholar]
- 34. Henson P. M., Oades Z. G. (1975) Stimulation of human neutrophils by soluble and insoluble immunoglobulin aggregates. J. Clin. Invest. 56, 1053–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DeLeo F. R., Renee J., McCormick S., Nakamura M., Apicella M., Weiss J. P., Nauseef W. M. (1998) Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J. Clin. Invest. 101, 455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taylor R. M., Burritt J. B., Foubert T. R., Snodgrass M. A., Stone K. C., Baniulis D., Gripentrog J. M., Lord C., Jesaitis A. J. (2003) Single-step immunoaffinity purification and characterization of dodecylmaltoside-solubilized human neutrophil flavocytochrome b. Biochim. Biophys. Acta 1612, 65–75 [DOI] [PubMed] [Google Scholar]
- 37. Parkos C. A., Allen R. A., Cochrane C. G., Jesaitis A. J. (1987) Purified cytochrome b from human granulocyte plasma membrane is composed of two polypeptides with relative molecular weights of 91,000 and 22,000. J. Clin. Invest. 80, 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klotz K. N., Jesaitis A. J. (1994) The interaction of N-formyl peptide chemoattractant receptors with the membrane skeleton is energy-dependent. Cell. Signal. 6, 943–947 [DOI] [PubMed] [Google Scholar]
- 39. Kwan E. M., Boraston A. B., McLean B. W., Kilburn D. G., Warren R. A. (2005) N-Glycosidase-carbohydrate-binding module fusion proteins as immobilized enzymes for protein deglycosylation. Protein Eng. Des. Sel. 18, 497–501 [DOI] [PubMed] [Google Scholar]
- 40. Maaty W. S., Ortmann A. C., Dlakić M., Schulstad K., Hilmer J. K., Liepold L., Weidenheft B., Khayat R., Douglas T., Young M. J., Bothner B. (2006) Characterization of the archaeal thermophile Sulfolobus turreted icosahedral virus validates an evolutionary link among double-stranded DNA viruses from all domains of life. J. Virol. 80, 7625–7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koo C., Lefkowitz R. J., Snyderman R. (1983) Guanine nucleotides modulate the binding affinity of the oligopeptide chemoattractant receptor on human polymorphonuclear leukocytes. J. Clin. Invest. 72, 748–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jesaitis A. J., Tolley J. O., Painter R. G., Sklar L. A., Cochrane C. G. (1985) Membrane-cytoskeleton interactions and the regulation of chemotactic peptide-induced activation of human granulocytes. The effects of dihydrocytochalasin B. J. Cell. Biochem. 27, 241–253 [DOI] [PubMed] [Google Scholar]
- 43. Liu L., Harbecke O., Elwing H., Follin P., Karlsson A., Dahlgren C. (1998) Desensitization of formyl peptide receptors is abolished in calcium ionophore-primed neutrophils. An association of the ligand-receptor complex to the cytoskeleton is not required for a rapid termination of the NADPH-oxidase response. J. Immunol. 160, 2463–2468 [PubMed] [Google Scholar]
- 44. Jesaitis A. J., Naemura J. R., Sklar L. A., Cochrane C. G., Painter R. G. (1984) Rapid modulation of N-formyl chemotactic peptide receptors on the surface of human granulocytes. Formation of high affinity ligand-receptor complexes in transient association with cytoskeleton. J. Cell Biol. 98, 1378–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harbecke O., Liu L., Karlsson A., Dahlgren C. (1997) Desensitization of the fMLP-induced NADPH-oxidase response in human neutrophils is lacking in okadaic acid-treated cells. J. Leukoc. Biol. 61, 753–758 [DOI] [PubMed] [Google Scholar]
- 46. Forsman H., Önnheim K., Andréasson E., Christenson K., Karlsson A., Bylund J., Dahlgren C. (2013) Reactivation of desensitized formyl peptide receptors by platelet activating factor. A novel receptor cross talk mechanism regulating neutrophil superoxide anion production. PLoS One 8, e60169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bengtsson T., Dahlgren C., Stendahl O., Andersson T. (1991) Actin assembly and regulation of neutrophil function. Effects of cytochalasin B and tetracaine on chemotactic peptide-induced O2⨪ production and degranulation. J. Leukocyte Biol. 49, 236–244 [DOI] [PubMed] [Google Scholar]
- 48. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lord C. I., Riesselman M. H., Gripentrog J. M., Burritt J. B., Jesaitis A. J., Taylor R. M. (2008) Single-step immunoaffinity purification and functional reconstitution of human phagocyte flavocytochrome b. J. Immunol. Methods. 329, 201–207 [DOI] [PubMed] [Google Scholar]
- 50. Dolmatch B., Niedel J. (1983) Formyl peptide chemotactic receptor. Evidence for an active proteolytic fragment. J. Biol. Chem. 258, 7570–7577 [PubMed] [Google Scholar]
- 51. Mukherjee G., Quinn M. T., Linner J. G., Jesaitis A. J. (1994) Remodeling of the plasma membrane after stimulation of neutrophils with f-Met-Leu-Phe and dihydrocytochalasin B. Identification of membrane subdomains containing NADPH oxidase activity. J. Leukocyte Biol. 55, 685–694 [DOI] [PubMed] [Google Scholar]
- 52. Jesaitis A. J., Erickson R. W., Klotz K.-N., Bommakanti R. K., Siemsen D. W. (1993) Functional molecular complexes of human N-formyl chemoattractant receptors and actin. J. Immunol. 151, 5653–5665 [PubMed] [Google Scholar]
- 53. Kühn H., Dreyer W. J. (1972) Light dependent phosphorylation of rhodopsin by ATP. FEBS Lett. 20, 1–6 [DOI] [PubMed] [Google Scholar]
- 54. Premont R. T., Inglese J., Lefkowitz R. J. (1995) Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J. 9, 175–182 [DOI] [PubMed] [Google Scholar]
- 55. Butcher A. J., Kong K. C., Prihandoko R., Tobin A. B. (2012) Physiological role of G-protein coupled receptor phosphorylation. Handb. Exp. Pharmacol. 79–94 [DOI] [PubMed] [Google Scholar]
- 56. Fournier B. M., Parkos C. A. (2012) The role of neutrophils during intestinal inflammation. Mucosal. Immunol. 5, 354–366 [DOI] [PubMed] [Google Scholar]
- 57. Rabiet M. J., Huet E., Boulay F. (2007) The N-formyl peptide receptors and the anaphylatoxin C5a receptors. An overview. Biochimie 89, 1089–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roberts E. C., Hobson C. H., Anderson R. P., Chadwick V. S. (1990) Radio-immunoassay for formyl methionyl leucyl phenylalanine. II. Demonstration of an enterohepatic circulation of immunoreactive bacterial chemotactic peptides in man. J. Gastroenterol. Hepatol. 5, 38–43 [DOI] [PubMed] [Google Scholar]
- 59. Key T. A., Bennett T. A., Foutz T. D., Gurevich V. V., Sklar L. A., Prossnitz E. R. (2001) Regulation of formyl peptide receptor agonist affinity by reconstitution with arrestins and heterotrimeric G proteins. J. Biol. Chem. 276, 49204–49212 [DOI] [PubMed] [Google Scholar]
- 60. Huber-Lang M. S., Riedeman N. C., Sarma J. V., Younkin E. M., McGuire S. R., Laudes I. J., Lu K. T., Guo R. F., Neff T. A., Padgaonkar V. A., Lambris J. D., Spruce L., Mastellos D., Zetoune F. S., Ward P. A. (2002) Protection of innate immunity by C5aR antagonist in septic mice. FASEB J. 16, 1567–1574 [DOI] [PubMed] [Google Scholar]
- 61. Huber-Lang M. S., Younkin E. M., Sarma J. V., McGuire S. R., Lu K. T., Guo R. F., Padgaonkar V. A., Curnutte J. T., Erickson R., Ward P. A. (2002) Complement-induced impairment of innate immunity during sepsis. J. Immunol. 169, 3223–3231 [DOI] [PubMed] [Google Scholar]
- 62. Huber-Lang M. S., Sarma J. V., McGuire S. R., Lu K. T., Guo R. F., Padgaonkar V. A., Younkin E. M., Laudes I. J., Riedemann N. C., Younger J. G., Ward P. A. (2001) Protective effects of anti-C5a peptide antibodies in experimental sepsis. FASEB J. 15, 568–570 [DOI] [PubMed] [Google Scholar]
- 63. Prossnitz E. R., Kim C. M., Benovic J. L., Ye R. D. (1995) Phosphorylation of the N-formyl peptide receptor carboxyl terminus by the G protein-coupled receptor kinase, GRK2. J. Biol. Chem. 270, 1130–1137 [DOI] [PubMed] [Google Scholar]
- 64. Key T. A., Vines C. M., Wagener B. M., Gurevich V. V., Sklar L. A., Prossnitz E. R. (2005) Inhibition of chemoattractant N-formyl peptide receptor trafficking by active arrestins. Traffic 6, 87–99 [DOI] [PubMed] [Google Scholar]
- 65. Zhang M., Goswami M., Hereld D. (2005) Constitutively active G protein-coupled receptor mutants block dictyostelium development. Mol. Biol. Cell 16, 562–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Burritt J. B., Bond C. W., Doss K. W., Jesaitis A. J. (1996) Filamentous phage display of oligopeptide libraries. Anal. Biochem. 238, 1–13 [DOI] [PubMed] [Google Scholar]
- 67. Suvorova E. S., Gripentrog J. M., Jesaitis A. J., Miettinen H. M. (2009) Agonist-dependent phosphorylation of the formyl peptide receptor is regulated by the membrane proximal region of the cytoplasmic tail. Biochim. Biophys. Acta 1793, 406–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dalli J., Consalvo A. P., Ray V., Di Filippo C., D'Amico M., Mehta N., Perretti M. (2013) Proresolving and tissue-protective actions of annexin A1-based cleavage-resistant peptides are mediated by formyl peptide receptor 2/lipoxin a4 receptor. J. Immunol. 190, 6478–6487 [DOI] [PubMed] [Google Scholar]
- 69. Chiang N., Serhan C. N., Dahlén S. E., Drazen J. M., Hay D. W., Rovati G. E., Shimizu T., Yokomizo T., Brink C. (2006) The lipoxin receptor ALX. Potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 58, 463–487 [DOI] [PubMed] [Google Scholar]
- 70. Kang Y., Taddeo B., Varai G., Varga J., Fiore S. (2000) Mutations of serine 236–237 and tyrosine 302 residues in the human lipoxin A4 receptor intracellular domains result in sustained signaling. Biochemistry 39, 13551–13557 [DOI] [PubMed] [Google Scholar]
- 71. Ramaraj T., Angel T., Dratz E. A., Jesaitis A. J., Mumey B. (2012) Antigen-antibody interface properties. Composition, residue interactions, and features of 53 non-redundant structures. Biochim. Biophys. Acta 1824, 520–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kobayashi S. D., Deleo F. R. (2013) Systems biology and innate immunity. J. Innate Immun. 5, 97–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hino T., Arakawa T., Iwanari H., Yurugi-Kobayashi T., Ikeda-Suno C., Nakada-Nakura Y., Kusano-Arai O., Weyand S., Shimamura T., Nomura N., Cameron A. D., Kobayashi T., Hamakubo T., Iwata S., Murata T. (2012) G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 482, 237–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Haga K., Kruse A. C., Asada H., Yurugi-Kobayashi T., Shiroishi M., Zhang C., Weis W. I., Okada T., Kobilka B. K., Haga T., Kobayashi T. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482, 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shimamura T., Shiroishi M., Weyand S., Tsujimoto H., Winter G., Katritch V., Abagyan R., Cherezov V., Liu W., Han G. W., Kobayashi T., Stevens R. C., Iwata S. (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. White J. F., Noinaj N., Shibata Y., Love J., Kloss B., Xu F., Gvozdenovic-Jeremic J., Shah P., Shiloach J., Tate C. G., Grisshammer R. (2012) Structure of the agonist-bound neurotensin receptor. Nature 490, 508–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chien E. Y., Liu W., Zhao Q., Katritch V., Han G. W., Hanson M. A., Shi L., Newman A. H., Javitch J. A., Cherezov V., Stevens R. C. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Choe H. W., Kim Y. J., Park J. H., Morizumi T., Pai E. F., Krauss N., Hofmann K. P., Scheerer P., Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655 [DOI] [PubMed] [Google Scholar]
- 80. Huang X., Miller W. (1991) A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 12, 337–357 [Google Scholar]
- 81. Dratz E. A., Grieco P. A. (January 20, 2004) Novel Zwitterionic Dyes for Labeling in Proteomics and Other Biological Analyses, United States Patent 7,582,260B2
- 82. Dratz E. A., Grieco P. A. (June 22, 2007) Novel Zwitterionic Dyes for Labeling in Proteomics and Other Biological Analyses, United States Patent 7,833,799B2
- 83. Dratz E. A., Grieco P. A. (June 22, 2007) Novel Zwitterionic Dyes for Labeling in Proteomics and Other Biological Analyses, United States Patent 8,197,758B2
- 84. Dratz E. A., Grieco P. A. (August 31, 2009) Novel Zwitterionic Dyes for Labeling in Proteomics and Other Biological Analyses, United States Patent 8,197,759B2
- 85. Dratz E. A., Grieco P. A. (January 8, 2009) Novel Zwitterionic Dyes for Labeling in Proteomics and Other Biological Analyses, United States Patent Novel Optical Labeling Molecules in Proteomics and Other Biological Analyses, World Patent, WO 2009/005871-A2