

Background: The epitope and the TNFα inhabitation mechanism of Adalimumab remain unclear.
Results: The crystal structure of the TNFα in complex with Adalimumab is reported at a resolution of 3.1 Å.
Conclusion: The epitope of Adalimumab provided information that Adalimumab may have clinical advantage compared with Infliximab.
Significance: These data reveal the Adalimumab's mechanism of TNFα inhibition and its advantages compared with other TNF inhibitors in clinical practice.
Keywords: Antibodies, Autoimmune Diseases, Crystal Structure, Protein Targeting, Tumor Necrosis Factor (TNF), Adalimumab, Epitope, Mechanism
Abstract
TNFα-targeting therapy with the use of the drugs Etanercept, Infliximab, and Adalimumab is used in the clinical treatment of various inflammatory and immune diseases. Although all of these reagents function to disrupt the interaction between TNFα and its receptors, clinical investigations showed the advantages of Adalimumab treatment compared with Etanercept and Infliximab. However, the underlying molecular mechanism of action of Adalimumab remains unclear. In our previous work, we presented structural data on how Infliximab binds with the E-F loop of TNFα and functions as a TNFα receptor-binding blocker. To further elucidate the variations between TNFα inhibitors, we solved the crystal structure of TNFα in complex with Adalimumab Fab. The structural observation and the mutagenesis analysis provided direct evidence for identifying the Adalimumab epitope on TNFα and revealed the mechanism of Adalimumab inhibition of TNFα by occupying the TNFα receptor-binding site. The larger antigen-antibody interface in TNFα Adalimumab also provided information at a molecular level for further understanding the clinical advantages of Adalimumab therapy compared with Infliximab.
Introduction
TNF is an immunity-modulating cytokine required for immune processes. The unregulated activities of TNFs can lead to the development of inflammatory diseases. Excess amounts of TNFα expressed in cells are associated with the development of immune diseases, including rheumatoid arthritis, Crohn's disease, psoriatic arthritis, and inflammatory bowel disease (1, 2). The function of TNFα requires smooth interaction with its two receptors, TNF receptor 1 (TNFR1)4 and TNF receptor 2 (TNFR2). Blocking the interaction between TNFα and TNFRs has successfully been developed as a therapy in treating inflammatory or autoimmune diseases (3, 4). TNFα neutralization therapies, including the use of a soluble TNFR2-Fc recombinant (Etanercept), a mouse-human chimera mAb (Infliximab), or a human mAb (Adalimumab), have been introduced in the past decades for the management of rheumatoid arthritis and other immune diseases (5).
Although all of these TNFα blockers function by interrupting the TNFα-TNFR interaction, information on whether the different TNFα inhibitors have similar clinical efficacy remains controversial because of the lack of randomized clinical trial meta-analyses. In the early stages of clinical usage of Infliximab, its discontinuation was reported to result in loss of response. This largely affected patients who received long term treatment and later discontinued use (6). Approximately 10% of the patients discontinued the use Infliximab because of the loss of response. The discontinued use caused remissions, representing an additional 13% of patients. This practice pattern still occurs, but the frequency of Infliximab discontinuation in present day clinical practice is considerably lower because of the enhanced understanding that drug holidays may lead to loss of efficacy, attenuation of response, and acute and delayed hypersensitivity reactions through retreatment (7). A longitudinal study involving 93 Crohn's disease patients was performed to compare the effectiveness of Infliximab and Adalimumab. The study suggested that no obvious differences could be found in obtaining and maintaining remission (8). Another study drew a similar conclusion, claiming that the difference between Infliximab and Adalimumab is not obvious because the long term maintenance of the clinical remission between the two antibodies and their effectiveness are similar in both primary and secondary-tertiary centers (9). However, evidence from a Dutch observational study involving 770 patients with rheumatoid arthritis suggested that more patients achieve moderate responses to Adalimumab and Etanercept compared with Infliximab, but the potential baseline biases corrected for in the Dutch study were not specified, and strict response criteria were not included (10). Another study also directly compared the treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with Adalimumab, Etanercept, or Infliximab using the nationwide DANBIO registry, which has been designed to capture operational clinical data as part of routine clinical care. The results showed that Adalimumab had the highest rates in treatment response and disease remission compared with the other two TNFα inhibitors (11). Furthermore, a very recent study implemented a matching adjusted indirect comparison technique, showing that Adalimumab is associated with the higher American College of Rheumatology 70% improvement criteria and Psoriasis Area and Severity Index 50/75/90 response rates compared with Etanercept at week 24 and a higher American College of Rheumatology 70% improvement criteria response rate than that of Infliximab at week 14 (12). These clinical investigations suggested that Adalimumab is more advantageous in TNFα-blocking therapy of autoimmune diseases.
However, the underlying inhibition mechanisms of these TNF blockers with regard to clinical efficacy remain elusive. In particular, no reports have compared the binding epitopes of these drugs that have been widely used, despite the fact that the affinity and the epitope are the crucial elements in evaluating antibody drugs. In a previous work, we reported the crystal structure of the TNFα-Infliximab Fab complex, presenting the inhibition mechanism of Infliximab of the TNFα–TNFR interaction through partial overlap with the TNFα-TNFR interface. We also revealed the pivotal role of the E-F loop of TNFα in Infliximab recognition (5). To further elucidate the inhibition mechanism of Adalimumab, in particular the variation on the mechanism of Infliximab and Adalimumab targeting at the TNFα-TNFR contact, we launched a crystallographic and mutagenesis analysis on the TNFα-Adalimumab Fab complex.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification, and Characterization
Residues 77–233 of human TNFα followed by a His6 tag were expressed in Escherichia coli BL21 (DE3) cells (Novagen) using the pET-22b(+) vector (Novagen). The cells were grown in LB medium at 37 °C until the A600 reached 0.6–0.8, and the expression of the protein was induced with 0.5 mm isopropyl β-d-thiogalactopyranoside for 4 h. The cells were incubated in PBS containing 20 mm phosphate (pH 8.0) and 150 mm NaCl with an additional 1 mg/ml lysozyme, 1 mm PMSF, and 1% Triton X-100 for 20 min on ice and then sonicated. The cell lysate was removed through centrifugation (10,000 × g) and filtration (0.45 μm). Solid ammonium sulfate was added to the supernatant at a 35% final concentration and was immediately mixed and incubated on a roller at 4 °C for 2 h. The precipitant was discarded, and solid ammonium sulfate was continuously added to the supernatant until a final concentration of 60% was achieved. The concentration was immediately mixed and incubated on a roller at 4 °C for 4 h. The precipitated protein was pelleted through centrifugation, and then the supernatant was discarded. The precipitant was dissolved in PBS buffer and separated through gel filtration using a Superdex 75 column (GE Healthcare). The precipitant was desalted to 20 mm Tris-HCl (pH 8.0), and the target fraction was further purified with a 20-column volume linear NaCl gradient elution from a high performance Q-Sepharose (GE Healthcare). The purity was confirmed to be >95% through SDS-PAGE analysis. The bioactivity was measured through a cytotoxicity assay using the TNF-susceptible murine L-929 cell line in the presence of the metabolic inhibitor actinomycin D, as described in Ref. 13.
Adalimumab was cloned, expressed, and purified following reported procedures. EcoRV and XbaI sites were added to the 5′-end of the heavy chain variable region gene (VH), and an NheI site was added to the 3′-end. The PCR product was cloned into the pGEM-T vector, and the sequence of the product was confirmed through DNA sequencing. VH was excised through EcoRV and NheI digestion and then inserted into the EcoRV/NheI sites of the pAH4604 vector containing the human gamma-1 constant region gene (CH). The resultant pAH4604-VH vector was cleaved with XbaI and BamHI. The 3.3-kb fragment containing the human antibody heavy chain gene was cloned into the pcDNA3.1(−) vector (Invitrogen), which was digested with the same restriction enzymes and yielded the heavy chain expression vector pcDNA3.1(−)VHCH. The human κ chain constant cDNA (CL) was obtained as a 0.3-kb PCR product derived from pAG4622. The light chain variable region gene (VL) of Adalimumab was fused to the 5′-end of CL through the overlapping PCR method. The resultant human light chain gene (VLCL), which has a HindIII site upstream of the start codon and an EcoRI site downstream of the stop codon, was cloned into the pGEM-T vector. The sequence of the chain was then verified. VLCL was excised through HindIII and EcoRI digestion and was ligated into pcDNA3.1 The Zeo(+) vector (Invitrogen) cleaved with the same restriction enzymes yielded the chimeric light chain expression vector pcDNA3.1 Zeo(+)VLCL. The light and heavy chain expression vectors were co-transfected into Chinese hamster ovary K1 cells using Lipofectamine 2000 (Invitrogen). The stable transfectants were isolated through limiting dilution in the presence of 600 μg/ml G418 and 300 μg/ml Zeocin. The culture supernatants from the individual cell clones were analyzed for antibody production through a sandwich enzyme-linked immunosorbent assay. The assay used goat anti-human IgG Fc (KPL, Gaithersburg, MD) as the capture antibody and goat anti-human κ-HRP (Southern Biotechnology Associates, Birmingham, AL) as the detecting antibody. Purified human IgG1/κ (Sigma) was used as the standard control. The clones producing the highest amount of recombinant antibody were selected and grown in serum-free medium. The recombinant antibodies were purified through protein A affinity chromatography from the serum-free culture supernatant. The antibody concentrations were determined by absorbance at 280 nm, and the purity was confirmed through SDS-PAGE analysis. Bioactivity was measured in a cytotoxicity antagonist assay using the TNF-susceptible murine L-929 cell line in the presence of the metabolic inhibitors actinomycin D and TNFα.
The Fab fragment of Adalimumab for the crystallographic investigation was obtained through papain digestion of the antibody. The digested protein sample was loaded onto a Protein A-Sepharose 4 FF column (GE Healthcare). The Fab fragment eluted in the flow through was separated from the Fc fragment and further purified through ion exchange chromatography using a Q-Sepharose FF column (GE Healthcare). The protein sample was concentrated to ∼10 mg/ml and then exchanged to a stock buffer containing 10 mm Tris-HCl (pH 8.0) and 100 mm NaCl. TNFα was subsequently mixed with an excess of Adalimumab Fab, and the complex was purified through gel filtration chromatography (GE Healthcare). This complex was dialyzed against 50 mm Tris-HCl (pH 8.0) and 150 mm NaCl and was concentrated to 30 mg/ml.
Crystallization
Crystallization was performed at 291 K through the hanging drop vapor diffusion technique. The crystals were obtained by mixing 1 μl of the protein solution with an equal volume of a reservoir solution. The mixture drop was equilibrated against 500 μl of the reservoir solution. The crystals were obtained with a reservoir solution containing 30% PEG 400, 0.1 m sodium acetate trihydrate (pH 4.6), and 0.1 m cadmium chloride hydrate, which reached the final dimensions of 100 × 100 × 100 μm3 with the best diffraction within 1 week. The crystals were then cryo-protected through soaking in a cryo-protectant composed of the reservoir solution and 5% glycol. The cryo-protected crystals were subsequently flash-cooled in liquid nitrogen and then transferred into a dry nitrogen stream at 100 K for x-ray data collection.
X-ray Data Collection, Processing, and Structure Determination
The diffraction data for the TNFα-Adalimumab Fab complex were collected at Beamline BL17A (Photon Factory) with a resolution of 3.1 Å. The data were processed, integrated, and scaled using the HKL2000 package (14). The crystals belong to space group I213 with cell parameters a = b = c = 161.8 Å, α = β = γ = 90°. The statistics of all data collections and structure refinements are summarized in Table 1.
TABLE 1.
Data collection and refinement statistics
| Parameters | TNFα-Humira Fab complex |
|---|---|
| Data collection statistics | |
| Cell parameters | a = b = c = 161.8 ′, α = β = γ = 90° |
| Space group | I213 |
| Wavelength used (Å) | 1.0000 |
| Resolution (Å) | 50.0 (3.2)-3.1c |
| No. of all reflections | 226,799 (11,466) |
| No. of unique reflections | 24,923 (1274) |
| Completeness (%) | 100.0 (100.0) |
| Average I/σ(I) | 8.0 (5.1) |
| Rmerge (%)a | 17.2 (64.3) |
| Refinement statistics | |
| No. of reflections used (σ(F) > 0) | 12,943 |
| Rwork (%)b | 18.67 |
| Rfree (%)b | 27.50 |
| Root mean square deviation bond distance (Å) | 0.010 |
| Root mean square deviation bond angle (°) | 1.412 |
| Average overall B value (Å2) | 48.2 |
| Ramachandran plot (excluding Pro and Gly) | |
| Residues in most favored regions | 504 (88.3%) |
| Residues in additionally allowed regions | 42 (7.4%) |
a Rmerge = ΣhΣl|Iih−<Ih>|/ΣhΣI<Ih>, where <Ih> is the mean of multiple observations, Iih, of a given reflection h.
b Rwork = Σ||Fp(obs) −|Fp(calc)||/Σ|Fp(obs)|; Rfree is an R factor for a selected subset (5%) of reflections that was not included in prior refinement calculations.
c The numbers in parentheses are corresponding values for the highest resolution shell (2.5–2.4 ′).
The TNFα-Adalimumab Fab structure was solved through the molecular replacement method, which employs the crystal structures of apo TNFα (Protein Data Bank code 1TNF) and GA101 Fab (Protein Data Bank code 3PP3) as the initial searching model using the program PHASER (15). The clear solutions in both the rotation and translation functions indicated the presence of one complex molecule, including one TNFα and one Adalimumab Fab molecule, in one asymmetric unit. This result is consistent with the Matthews coefficient and solvent content (16). The inconsistent residues were manually rebuilt in the program Coot (17) under the guidance of the Fo − Fc and 2Fo − Fc electron density maps. The residues were refined in PHENIX (18), and the respective working Rfactor and Rfree values decreased from 0.42 and 0.48 to 0.19 and 0.28, respectively, for all data from 50.0 to 3.1 Å. The refinement was monitored by calculating Rfree based on a subset containing 5% of the total reflections. Model geometry was verified using the program PROCHECK (19). The data collection and refinement statistics are presented in detail in Table 1. All structure figures were prepared using PyMOL (20).
Kinetics and Affinity Assay of TNFα Mutants
Site-directed mutants (TNFP20A, TNFQ21A, TNFE23A, TNFK65A, TNFQ67A TNFT72A, TNFK90A, TNFV91A, TNFN92A, TNFE110A, TNFP113A, TNFE135A, TNFI136A, and TNFE146A) were created through PCR. The mutants were expressed and purified as recommended for wild-type proteins. Adalimumab Fab was immobilized on the surface of a CM-5 sensor chip (GE Healthcare) through amine coupling following the manufacturer's instructions. The maximal electrostatic interaction was obtained with 10 mm sodium acetate (pH 5.0). We regularly obtained Adalimumab Fab immobilization levels ranging from ∼1000 to 1500 resonance units. The BIAcore T100 (GE Healthcare) instrument for the binding experiments was operated at 25 °C, and the assay buffer was PBS. The contact time (the period during which the analyte consisting of TNFα mutants 4 and 11 was perfused over the chip) was limited to 300 s. The flow rate was set at 30 μl/min. A 10 mm glycine (pH 2.0) solution was used to dissociate the bound TNF at the end of each experiment while retaining the surface integrity for chip surface regeneration.
RESULTS
Structure of the TNFα-Adalimumab Fab Complex
The TNFα-Adalimumab Fab complex was crystallized, and the structure was determined and refined to 3.1 Å resolution with a final Rwork value of 19.7% (Rfree = 28.5%) (Table 1). One TNFα-Adalimumab Fab complex molecule in the asymmetric unit with a Matthews coefficient of 3.5 Å3/Da exists, corresponding to 64% of the solvent content (21). The central TNFα trimer is bound by three symmetrically arranged Adalimumab Fab molecules related through a crystallographic 3-fold axis (Fig. 1), which is analogous to the structures of TNFα-TNFR2 (22), TNFβ-TNFR1 (23), and TNFα in complex with other binding partners (5, 24).
FIGURE 1.
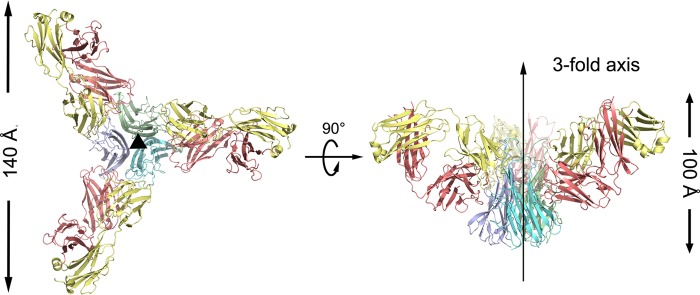
Overall structure of the TNFα-Adalimumab Fab complex. The TNFα-Adalimumab Fab complex is shown as ribbon diagrams in two orientations: top view, looking down the crystallographic 3-fold symmetry axis; side view, with the crystallographic 3-fold axis vertical (middle). The central TNFα molecules are colored green, blue, and cyan, respectively; the light chain and heavy chain of the Adalimumab Fab are colored yellow and red, respectively.
Superimposing TNFα in the TNFα-Adalimumab Fab complex with its free form yielded a root mean square deviation of 0.9 Å for all of the Cα atoms, indicating that no significant overall structural difference occurred, except for several key residues on the antibody-antigen interface (Fig. 2). Residues TNFLeu-29, TNFArg-31, TNFSer-52, and TNFTyr-56, which are crucial for TNFα cytotoxicity and TNFR interaction (25), were confirmed to have the correct conformation through a cytotoxicity assay.
FIGURE 2.

Structural variations of TNFα in free form and complex forms. A single subunit of the TNFα trimer in the free form or complex forms of the TNFα-Infliximab Fab, the Adalimumab Fab, and TNFR2 are shown. The free state of the TNFα molecule is colored orange, whereas complex states with Infliximab Fab, Adalimumab Fab, and TNFR2 are colored pale green, red, and light blue, respectively.
The Adalimumab Fab presents a canonical β-sandwich immunoglobulin fold with the heavy chain folding into the VH and CH domains and the light chain folding into the VL and CL domains. The elbow angle, defined as the subtended angle by two pseudo 2-fold axes relating VH to VL and CH to CL of Adalimumab Fab, was ∼140°. The complementarity-determining region (CDR) loops L1, L2, L3, H1, and H2 of Adalimumab belong to the Chothia canonical classes (26) 2, 1, 1, 1, and 3, respectively. The CDR loops L1, L2, L3, H1, H2, and H3 of Adalimumab form a large deep pocket to accommodate the epitope (Fig. 3), whereas not all of the CDR loops of Infliximab participate in the interaction with TNFα (5).
FIGURE 3.
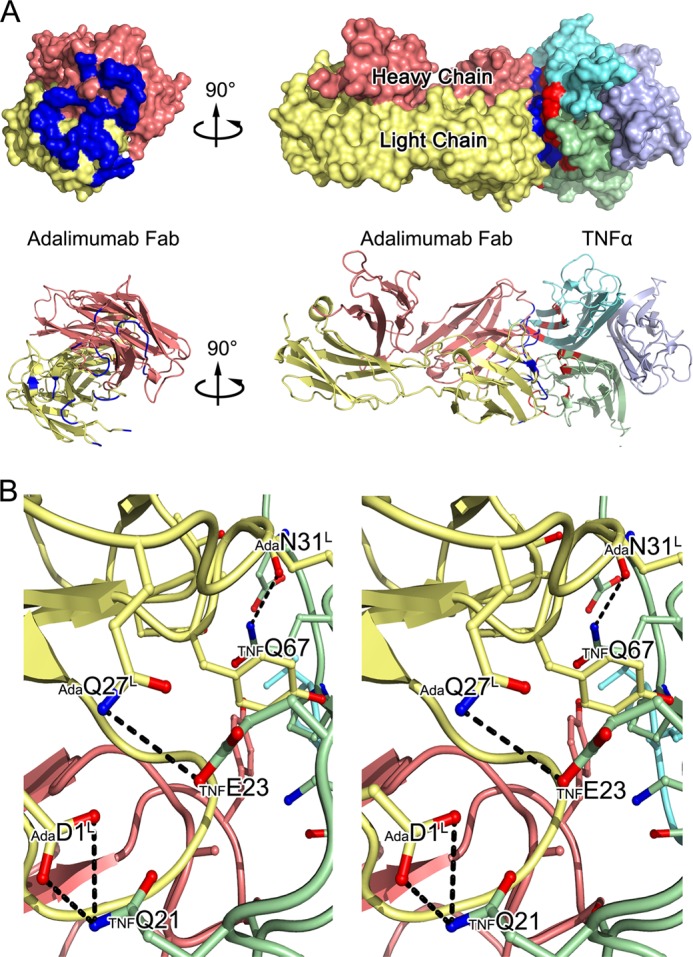
The TNFα-Adalimumab Fab interface. A, surface representations of the Adalimumab Fab (left) and the TNFα-Adalimumab Fab complex (right) and ribbon diagrams corresponding to the surfaces shown above with the same color scheme. The light chain and heavy chains of the Adalimumab Fab are colored yellow and red, respectively, whereas TNFα molecules are colored green, blue, and cyan. Contact surfaces are highlighted in blue on Adalimumab and red on TNFα. B, stereo view of the detailed TNFα-Adalimumab Fab interface. The residues that are involved in the intermolecular interaction are shown as colored sticks with the same scheme as the surface representation above; the Adalimumab Fab and TNFα molecules are presented as ribbon diagrams. Dashed lines denote hydrogen bonds.
Interactions between TNFα and Adalimumab
The Adalimumab Fab binds to TNFα through a large and highly complementary interface, with a total buried surface area of 2,540 Å2 (Fig. 3A), which is consistent with the high avid association between Adalimumab and TNFα (27). The Adalimumab epitope on TNFα is composed of a number of discontinuous segments, including residues TNFPro-19, TNFGln-20, TNFGlu-23, TNFLys-65 to TNFGln-67, TNFGlu-10 to TNFPro-113, TNFTyr-141, and TNFAla-145, to TNFGlu-146 and TNFThr-71, TNFHis-72, TNFThr-77, TNFThr-79, TNFSer-81, TNFLys-89 to TNFAsn-91, and TNFGlu-135 to TNFAsn-137 of an adjacent TNFα protomer (Figs. 3 and 4). Both the heavy and light chains of Adalimumab participated in the interaction with TNFα, with all of the contacts coming from CDRs. CDRs L2 and H2 contribute to the majority of the interactions with the antigen. Additional contributions are given from CDRs L1, L3, H1, and H3. Over 20 pairs of hydrogen bonds or salt bridges stabilized the TNFα-Adalimumab Fab complex (Table 2), which indicates a strong and stable interface within this antigen-antibody pair.
FIGURE 4.
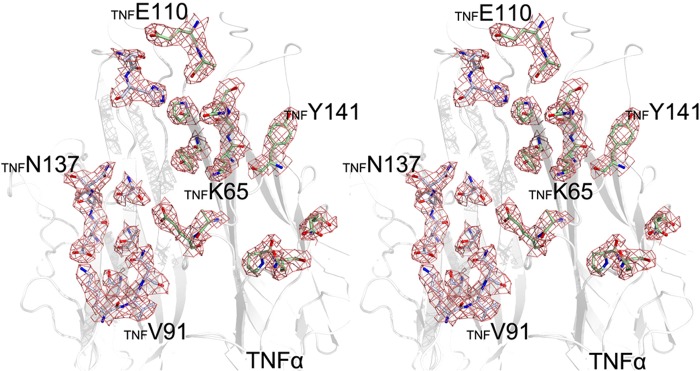
A stereo view of the epitope density map in the TNFα-Adalimumab Fab binding interface. The omit map of the epitope on the TNFα polypeptide is contoured at 1.0 σ. The TNFα molecule is shown as a white cartoon, whereas the epitope is represented as colored sticks.
TABLE 2.
Complete list of interactions between TNFα and the Adalimumab Fab (≤3.6 ′)
| TNFα |
Adalimumab Fab |
Distance | |||
|---|---|---|---|---|---|
| Residue | Atom | Residue | Atom | CDR loop | |
| Å | |||||
| Gln-67 | Cδ | Ile-56H | Cδ1 | H2 | 3.04 |
| Oϵ1 | Ile-56H | Cδ1 | 2.82 | ||
| Nϵ2 | Ile-56H | Cδ1 | 3.18 | ||
| Nϵ2 | Ser-53H | Oγ | 3.58 | ||
| Nϵ2 | Ser-55H | Oγ | 3.44 | ||
| Pro-70 | Cβ | Ser-105H | Oγ | H3 | 3.46 |
| Cβ | Tyr-50L | OH | L2 | 3.45 | |
| Cβ | Ser-105H | Cβ | H3 | 3.57 | |
| Cγ | Tyr-103H | O | H3 | 3.03 | |
| Ser-71 | Cβ | Tyr-50L | Cϵ1 | L2 | 3.25 |
| Cβ | Cζ | L2 | 3.34 | ||
| His-73 | Cβ | Tyr-50L | Cδ2 | L2 | 3.50 |
| Thr-105 | O | Tyr-102H | OH | H3 | 2.86 |
| Glu-107 | N | Tyr-102H | OH | H3 | 3.58 |
| Cα | OH | H3 | 3.53 | ||
| Cβ | OH | H3 | 3.51 | ||
| Ala-109 | O | Tyr-103H | OH | H3 | 3.22 |
| Glu-110 | Cβ | Asn-31H | Nδ2 | H2 | 3.53 |
| Asn-137 | O | Trp-94L | N | L3 | 2.92 |
| Ser-93L | Cα | 3.30 | |||
| Ser-93L | Oγ | 3.55 | |||
| Asn-137 | Cβ | Ser-93L | Oγ | L3 | 3.48 |
| Cγ | His-92L | O | 3.58 | ||
| Nδ2 | His-92L | O | 2.81 | ||
| Nδ2 | His-92L | Cϵ1 | 3.54 | ||
| Nδ2 | His-92L | Nϵ2 | 3.43 | ||
| Arg-138 | Cδ | His-92L | O | L3 | 3.28 |
| NH1 | Ser-91L | O | 3.44 | ||
| Asp-140 | Oδ1 | Trp-94L | Cβ | L3 | 3.52 |
| Oδ1 | Trp-94L | Cγ | L3 | 3.56 | |
| Oδ2 | Arg-52H | Cζ | H2 | 3.58 | |
| Oδ2 | Arg-52H | Nθ1 | H2 | 3.27 | |
| Oδ2 | Arg-52H | Nθ2 | H2 | 3.00 | |
| Tyr-141 | Cϵ1 | Arg-52H | Nθ1 | H2 | 3.43 |
| OH | Trp-33H | Cθ2 | H1 | 3.35 | |
The light chain of Adalimumab interacts with strands A and C, as well as the A-A′ and the E-F loops of TNFα. Residues AdaAsp-1 and AdaArg-93 of CDR L3 form three hydrogen bonds and a salt bridge with TNFPro-20, TNFGln-21, and TNFGlu-23 at the beginning of strand A and the A-A′ loop of TNFα. An extensive network of intermolecular side chain hydrogen bonds between CDR L1 and strand C of TNFα contributes to most of the light chain interactions and positions the side chain of AdaArg-30, AdaAsn-31, and AdaTyr-32 of CDR L1 interacting with TNFLys-65 and TNFGln-67 of TNFα. CDR L2 additionally contributes to the antigen-antibody communication through the hydrogen bond formed by the side chain of AdaThr-53 with the residue TNFAla-111 on the E-F loop of TNFα.
The interface between the heavy chain of Adalimumab and TNFα is primarily composed of the residues in the G-H loop, several amino acids in strand D, and the D-E loop of an adjacent TNFα protomer. AdaHis-57 in CDR H2 and AdaSer-103 and AdaThr-104 in CDR H3 make hydrogen bonds with TNFAla-145 to TNFSer-147 in the G-H loop. Moreover, the residues AdaAsn-54 and AdaGly-56 of CDR H2 contact TNFThr-79, TNFSer-81, and TNFAsn-92 in strands D and E and TNFGlu-135 in strand G of another neighboring TNFα polypeptide.
Comparison of the Interfaces between TNFα and TNFRs and the Infliximab Fab and the Adalimumab Fab
A comparison of the interfaces in TNFα-TNFRs, TNFα-Infliximab, and TNFα-Adalimumab Fab provides a better understanding of the mechanism of TNFα inhibition by blocking the communication with TNFRs, which is what allows Adalimumab to effectively inhibit the TNFα function more directly compared with Infliximab (Fig. 5).
FIGURE 5.
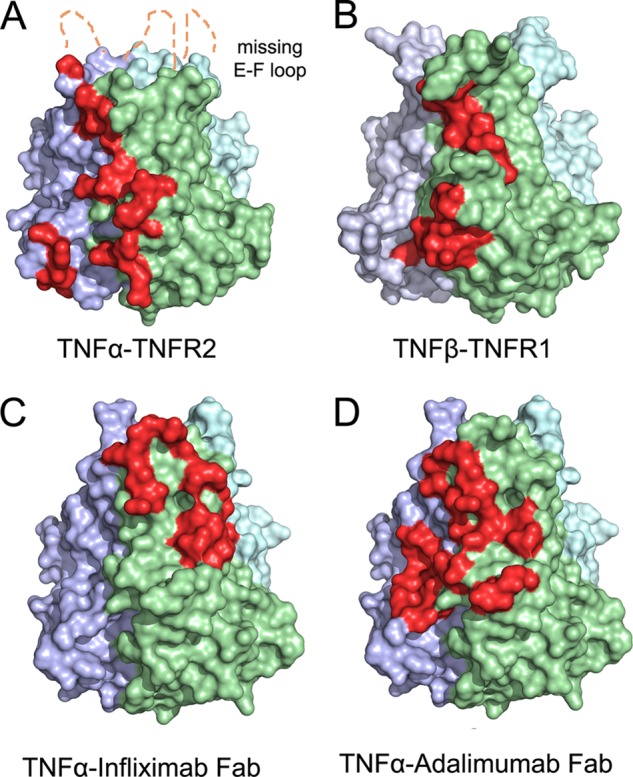
A comparison of the interface between TNFα and receptors and Infliximab/Adalimumab Fab complexes. A comparison of the interface between TNFα and receptors and mAbs is shown. TNFα from the complex structures is represented as a colored surface with TNFR2 and the mAb Fabs interface highlighted in red at one of three interfaces on the TNFα trimer. The E-F loop region, which is missing in the TNFα-TNFR2 (A) complex because of a lack of interaction, is labeled. The TNFβ from the TNFβ-TNFR1 (B) complex structure is shown as a colored surface with one of the TNFR1-binding sites highlighted in red. The TNFα-Infliximab Fab (C) and the TNFα-Adalimumab Fab (D) interfaces are shown as colored surfaces with TNFα-binding sites highlighted in red.
In the TNFα-TNFR2 complex, one TNFR2 molecule interacts with two adjacent TNFα protomers, such as in the TNFα-Adalimumab complex. By contrast, the antigen-antibody interaction only involves one TNFα molecule in the TNFα-Infliximab complex (5). The structures of the extracellular domains of the TNFR superfamily are composed of cysteine-rich domains (CRDs) that typically contain six cysteine residues that form three disulfide bonds (23). In the TNFα-TNFR2 complex, the CRD2 and CRD3 of TNFR2 play important roles in the cytokine-receptor interaction. CRD2 and CRD3 contact strand A, the A-A′ loop, and the G-H loop of one TNFα protomer and generate several hydrogen bonds with strand D, the D-E loop, E-F loop, and the G-H loop of a neighboring TNFα molecule. These contacts greatly overlap with the TNFα-Adalimumab interface, in which strand D, strand E, strand G, the E-F loop, and the G-H loop of TNFα have important roles. By contrast, a different subset of the secondary structures of TNFα, including strand C, strand D, strand G, the C-D loop and E-F loop, interacts with Infliximab.
Residues TNFRAsp-58, TNFRSer-59, TNFRGln-63, TNFRTrp-67, TNFRGlu-70, TNFRCys-71, TNFRCys-74, TNFRSer-76, TNFRArg-77 of CRD2, TNFRTyr-103, TNFRGln-109, and TNFRArg-113 of CRD3 in TNFR2, as well as TNFGln-21, TNFGln-23, TNFArg-32, TNFAla-33, TNFHis-73, TNFSer-86, TNFTyr-87, TNFPro-113, TNFTyr-115, TNFAsp-143, TNFPhe-144, TNFAla-145, TNFGlu-146, and TNFGln-149 of TNFα, participate and greatly contribute to these interactions in the cytokine-receptor interface. Among them, TNFGln-21 in strand A, TNFGln-23 in the A-A′ loop, TNFHis-73 in strand D, TNFPro-113 and TNFTyr-115 in the E-F loop, and TNFAla-145 and TNFGln-146 in the G-H loop have considerably important roles in both the interface of the TNFα-TNFR2 complex and the TNFα-Adalimumab complex, whereas none of these residues can be observed in the TNFα-Infliximab structure. Additionally, the Infliximab paratope consists of five CDRs, namely L2, L3, H1, H2, and H3, whereas the Adalimumab paratope is composed of all six CDRs (Fig. 6).
FIGURE 6.

Sequence comparison between two TNFα therapeutic antibodies (Infliximab and Adalimumab). The CDRs are highlighted by black frames and labeled. The residues that play crucial roles in the antibody-antigen interaction are framed with blue frames. The residue numbers (top) refer to those in Infliximab.
These structural features reveal that the Adalimumab epitope directly overlaps the TNFR binding area with a larger area of the antigen-antibody interface of TNFα-Adalimumab (2,340 Å2), whereas the Infliximab epitope is distant from the receptor-binding sites with less interacting surface (1,977 Å2).
Structure-based Mutagenesis Study on the Antigen-Antibody Interface
We identified 14 selected TNFα residues for mutagenesis analysis, including TNFPro-20, TNFGln-21, TNFGlu-23, TNFLys-65, TNFGln-67, TNFLys-72, TNFLys-90, TNFVal-91, TNFAsn-92, TNFGlu-110, TNFPro-113, TNFGlu-135, TNFIle-136 and TNFGlu-146 (Table 2), according to the structural information of the TNFα-Adalimumab Fab. We substituted each residue with alanine and measured the binding affinities with Adalimumab through surface plasmon resonance to study the effects of these residues on the TNFα-Adalimumab interaction (Table 3).
TABLE 3.
Kinetics and binding affinity of TNFα mutants with the Adalimumab Fab
Kinetics and affinity of TNFα mutant and the Adalimumab Fab were analyzed using a BIAcore T100. Wild type TNFα and the mutants were passed over the immobilized Adalimumab Fab surface, and the data were globally analyzed using a simultaneous fit for both dissociation (kd) and association (ka). The value for KD was calculated as kd/ka.
| ka | kd | KD | |
|---|---|---|---|
| m−1·s−1 | s−1 | m | |
| WT TNFα | 4.784 ± 0.717 × 105 | 5.512 ± 0.826 × 10−5 | 1.152 ± 0.172 × 10−10 |
| TNFP20A | 3.785 ± 0.567 × 105 | 8.491 ± 1.273 × 10−5 | 2.244 ± 0.336 × 10−10 |
| TNFQ21A | 2.286 ± 0.342 × 104 | 5.338 ± 0.801 × 10−4 | 2.335 ± 0.350 × 10−8 |
| TNFE23A | 2.665 ± 0.399 × 105 | 3.886 ± 0.582 × 10−4 | 1.458 ± 0.218 × 10−9 |
| TNFK65A | 3.211 ± 0.481 × 104 | 4.339 ± 0.650 × 10−4 | 1.351 ± 0.202 × 10−8 |
| TNFQ67A | 2.557 ± 3.383 × 104 | 5.087 ± 0.763 × 10−4 | 1.989 ± 0.298 × 10−8 |
| TNFT72A | 4.778 ± 0.716 × 105 | 5.033 ± 0.755 × 10−5 | 1.053 ± 0.157 × 10−10 |
| TNFK90A | 3.956 ± 0.593 × 105 | 3.893 ± 0.583 × 10−4 | 9.841 ± 1.476 × 10−10 |
| TNFV91A | 1.159 ± 0.173 × 105 | 8.496 ± 1.274 × 10−5 | 5.593 ± 0.838 × 10−10 |
| TNFN92A | 2.554 ± 0.383 × 105 | 5.223 ± 0.783 × 10−4 | 2.045 ± 0.306 × 10−9 |
| TNFE110A | 2.144 ± 0.321 × 105 | 2.967 ± 0.445 × 10−5 | 1.384 ± 0.207 × 10−10 |
| TNFP113A | 1.187 ± 0.178 × 106 | 1.394 ± 0.209 × 103 | 1.175 ± 0.176 × 10−9 |
| TNFE135A | 2.493 ± 0.373 × 104 | 2.948 ± 0.442 × 10−4 | 1.183 ± 0.177 × 10−8 |
| TNFI136A | 4.666 ± 0.699 × 105 | 5.133 ± 0.769 × 10−5 | 1.100 ± 0.165 × 10−10 |
| TNFE146A | 2.337 ± 0.350 × 104 | 4.223 ± 0.633 × 10−4 | 1.808 ± 0.271 × 10−8 |
The replacement of TNFPro-21, TNFThr-72, TNFLys-90, TNFVal-91, TNFGlu-110, and TNFIle-136 with alanine residues did not obviously affect the binding capacity of TNFα with Adalimumab, whereas the substitutions on TNFGlu-23, TNFAsn-92, and TNFPro-113 showed 5–10-fold decreases in binding. Notably, the mutant TNFQ21A presented a sharp decrease in the binding to Adalimumab with a 200-fold lower binding affinity. The same phenomenon was observed in the TNFK65A, TNFQ67A, TNFE135A, and TNFE146A mutations. All of these mutants resulted in a 100–200-fold affinity decrease. The TNFGln-21 of strand A, TNFLys-65 and TNFGln-67 of strand C, as well as TNFGlu-135 and TNFGlu-146 of the G-H loop, which are crucial for TNFα-Adalimumab interaction, also play key roles in TNFα-TNFR2 communication (22).
DISCUSSION
Etanercept, Infliximab, and Adalimumab have remarkably enhanced the treatment of immune diseases after they were successfully developed. A number of clinical investigations have studied the current use of these TNFα inhibitors and revealed that Adalimumab has an advantage in therapeutic treatment. However, the cause for this distinct efficacy remains elusive, although all of these TNFα inhibitors function as blockers that interrupt TNFα-TNFR communication.
Because Etanercept is a soluble TNFR2-Fc recombinant, the structure of TNFα-TNFR2 explains the mechanism by which Etanercept blocks the TNFα-TNFR interaction by occupying the receptor binding site on TNFα (22). One Etanercept/TNFR2 molecule interacted with two TNFα molecules, and the majority of the interface was made up of CRD2 and CRD3 regions of Etanercept/TNFR2 and the interface of two adjacent TNFα protomers, with a buried surface of 2,500 Å2 (22) (Fig. 5A). The epitope of Infliximab on TNFα is primarily composed of C-D and E-F loop residues and several key residues in strands C and D of the TNFα molecule, with a total buried surface of 1,977 Å2 (Fig. 5C). Interestingly, one Infliximab Fab contacts only one TNFα protomer, whereas two adjacent TNFα protomers both contribute to the Etanercept/TNFR2 contact. Although both Etanercept and Infliximab bind to TNFα, their affinity for binding to TNFα is controversial. Scallon et al. (28) suggested that Infliximab has a slightly lower KD value of 4.5 × 10−10 m compared with 1.15 × 10−9 m for Etanercept. However, Smith et al. (29) showed greater affinity of Etanercept with a KD of 2.35 × 10−11 m compared with the lower KD value of 1.17 × 10−10 m of Infliximab. The larger affinity displayed by Infliximab is believed to be a consequence of the greater stability of the TNFα-Infliximab complex (28), whereas the greater affinity of Etanercept was attributed to the faster rate of ligand binding (29). The area of buried surfaces shown in the structural information is likely consistent with the greater KD value of Etanercept.
Although the binding affinities displayed by Etanercept and Infliximab are debated, Adalimumab has been reported to bind TNFα with a relatively higher affinity than Etanercept and Infliximab, with KD values ranging from 7.05 × 10−11 m (30) to 1.0 × 10−10 m (31). The buried surface of one Adalimumab Fab with trimeric TNFα is consistently 2,536 Å2, which is larger than those of one Infliximab Fab and one Etanercept molecule with trimeric TNFα. The structural comparisons of the TNFα-Adalimumab Fab with the TNFα-Infliximab and TNFα-TNFR2 complex reveal that the Adalimumab epitope extensively overlaps with the TNFα-TNFR2 interface, whereas Infliximab only partially occupies the TNFα-TNFR2 binding area and is mainly targeted at the E-F loop of TNFα and spatially obstructs the correct interaction with TNFRs. Furthermore, a total of 7 of 21 TNFα residues involved in TNFR2 binding also participate in contacting the Adalimumab Fab. The three regions, including residues TNFGln-21, TNFGlu-23, TNFAla-145, and TNFGlu-146 that form two negatively charged surface patches, residues TNFPro-113 and TNFTyr-115 that form a hydrophobic surface patch and residues TNFVal-85–TNFTyr-87 and TNFThr-89 of an adjacent TNFα that also form a negatively charged surface patch, are shared by both TNFR2 and Adalimumab Fab. Notably, although TNFGln-21, TNFGlu-23, TNFHis-73, TNFPro-113, TNFTyr-115, TNFAla-145, and TNFGln-146 play essential roles in the TNFR2 and Adalimumab interaction, they are irrelevant for TNFα-Infliximab recognition. In the previous investigations, random mutagenesis studies on TNFα were performed to produce inactive molecules that lost their cytotoxic activity. Interestingly, the residues that were identified to lose most cytotoxic activity are involved in both TNFR2 and Adalimumab interactions. For example, the activity of TNFα dropped when alterations were introduced in TNFGlu-23, TNFPro-113, and TNFTyr-115 without marked changes in immunoreactivity or physicochemical characteristics, as well as TNFQ146K mutation, which causes a nearly complete loss of the cytotoxicity for TNFα (32). Moreover, TNFAsp-143, TNFGln-149, and TNFGlu-24, which are important residues for the TNFR2 recognition not only in the structure analysis but also in previous mutagenesis studies (22, 32, 33), are involved in the TNFα-Adalimumab interface.
In early stage usage of Infliximab, a number of patients discontinued the use of Infliximab because of the loss of response (6). It was proposed that Infliximab is a chimeric mAb, and its usage in humans could lead to the production of “antibodies to Infliximab” in a small subset of patients (34–37). Using more human sequence content by grafting murine CDRs may be crucial for the integral capacity of antigen binding and should be retained during humanization (38). Although this practice pattern still occurs, in present day clinical practice, the frequency of Infliximab discontinuation for this reason is low (7). However, the efficacy of Adalimumab-based TNF-blocking therapy in autoimmune diseases is higher than that of Etanercept and Infliximab. The structure shown here illustrates how Adalimumab prevents ligands from binding to TNFR2, inhibits ligand-receptor binding, and blocks TNFR activation. The primary consequence of Adalimumab binding to TNFα is the steric blocking of TNFα and the prevention of ligand binding to the receptor. The solvent-accessible surface contributed by the ligand-receptor interaction covers over 60% of the total interface between TNFα and the Adalimumab Fab, which indicates a straightforward overlap between the TNFα receptor binding sites and the Adalimumab epitope. Moreover, several residues that are crucial for TNFα receptor binding also participate in the TNFα-Adalimumab Fab interface, especially the groove between the two associated TNFα molecules in the TNFα trimer. These results may partly explain the clinical data regarding the more significant effectiveness of the treatment compared with placebo in inducing remission in patients with Crohn's disease who are intolerant of or have lost response to Infliximab. Therefore, binding Adalimumab to TNFα can efficiently compete with binding TNFRs to TNFα. The interface between TNFα and TNFRs is blocked in the presence of a sufficient amount of Adalimumab, which prevents the function of TNFα in the pathological process. Taken together, these data indicate that Adalimumab occupies the binding site and competitively inhibits the binding of TNFR to TNFα. Combined with the structural information of TNFα-Infliximab, these structures provide information to improve the interface complementarity between antibody and antigen, to strengthen the interaction, and to thereby enhance the binding affinity through altering the paratope of the antibody in targeted therapy and antibody engineering. Moreover, this information may also be useful to evaluate the antibody in the early stages of use. A therapeutic antibody whose epitope directly occupies the receptor binding position and competitively inhibits TNFR-TNFα communication, such as Adalimumab, may have better and more predictable clinical effects than an antibody that has a more distant epitope and uses the steric properties, such as Infliximab. Once a new antibody is identified as having promising results in preclinical studies, a crystallography study to determine its precise epitope may help us in making strategic decisions before proceeding with costly clinical drug trials.
In summary, our findings structurally explain the varying clinical observations for the anti-TNF antibodies. An evaluation of the effectiveness showed that Adalimumab had better outcomes than Infliximab from a molecular view. This finding, which agrees with another clinical report (10), highlights an opportunity for therapeutic improvement despite the significant advances in the past decade. The precise epitope revealed by our complex structure provides useful information for structure-based improvements for current TNFα mAbs and highlights the importance of identifying the predictors of a beneficial outcome through the structural and biological characteristics of the drug.
Acknowledgments
We thank the staffs of Photon Factory, Japan Synchrotron Radiation Facility, and Beijing Synchrotron Radiation Facility for generous help in collecting x-ray data.
This work was supported by 973 Project Grants 2010CB833600, 2013CB911103, 2012CB724500, and 2010CB735605; National Natural Science Foundation of China Grants 31170678 and 31000332; Key Projects in the Tianjin Science and Technology Pillar Program Grant 10ZCKFSY08800; and 2011 Science and Technology Innovation Fund Grant 11C26211203971.
The atomic coordinates and structure factors (code 3WD5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TNFR1
- TNF receptor 1
- CRD
- cysteine-rich domain
- CDR
- complementarity-determining region.
REFERENCES
- 1. Shealy D. J., Visvanathan S. (2008) Anti-TNF antibodies. Lessons from the past, roadmap for the future. Handb. Exp. Pharmacol. 181, 101–129 [DOI] [PubMed] [Google Scholar]
- 2. Palladino M. A., Bahjat F. R., Theodorakis E. A., Moldawer L. L. (2003) Anti-TNF-α therapies: the next generation. Nat. Rev. Drug Discov. 2, 736–746 [DOI] [PubMed] [Google Scholar]
- 3. Aggarwal B. B. (2003) Signalling pathways of the TNF superfamily. A double-edged sword. Nat. Rev. Immunol. 3, 745–756 [DOI] [PubMed] [Google Scholar]
- 4. Tansey M. G., Szymkowski D. E. (2009) The TNF superfamily in 2009. New pathways, new indications, and new drugs. Drug Discov. Today 14, 1082–1088 [DOI] [PubMed] [Google Scholar]
- 5. Liang S., Dai J., Hou S., Su L., Zhang D., Guo H., Hu S., Wang H., Rao Z., Guo Y., Lou Z. (2013) Structural basis for treating tumor necrosis factor α (TNFα)-associated diseases with the therapeutic antibody Infliximab. J. Biol. Chem. 288, 13799–13807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schnitzler F., Fidder H., Ferrante M., Noman M., Arijs I., Van Assche G., Hoffman I., Van Steen K., Vermeire S., Rutgeerts P. (2009) Long-term outcome of treatment with Infliximab in 614 patients with Crohn's disease. Results from a single-centre cohort. Gut 58, 492–500 [DOI] [PubMed] [Google Scholar]
- 7. Seminerio J. L., Loftus E. V., Jr., Colombel J. F., Thapa P., Sandborn W. J. (2013) Infliximab for Crohn's disease. The first 500 patients followed up through 2009. Dig. Dis. Sci. 58, 797–806 [DOI] [PubMed] [Google Scholar]
- 8. Zorzi F., Zuzzi S., Onali S., Calabrese E., Condino G., Petruzziello C., Ascolani M., Pallone F., Biancone L. (2012) Efficacy and safety of Infliximab and Adalimumab in Crohn's disease. A single centre study. Aliment. Pharmacol. Ther. 35, 1397–1407 [DOI] [PubMed] [Google Scholar]
- 9. Tursi A., Elisei W., Picchio M., Penna A. (2013) Letter. Are Infliximab and Adalimumab similar for Crohn's disease in clinical practice? Aliment. Pharmacol. Ther. 37, 763–764 [DOI] [PubMed] [Google Scholar]
- 10. Kievit W., Adang E. M., Fransen J., Kuper H. H., van de Laar M. A., Jansen T. L., De Gendt C. M., De Rooij D. J., Brus H. L., Van Oijen P. C., Van Riel P. C. (2008) The effectiveness and medication costs of three anti-tumour necrosis factor α agents in the treatment of rheumatoid arthritis from prospective clinical practice data. Ann. Rheum. Dis. 67, 1229–1234 [DOI] [PubMed] [Google Scholar]
- 11. Hetland M. L., Christensen I. J., Tarp U., Dreyer L., Hansen A., Hansen I. T., Kollerup G., Linde L., Lindegaard H. M., Poulsen U. E., Schlemmer A., Jensen D. V., Jensen S., Hostenkamp G., Østergaard M. (2010) Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with Adalimumab, Etanercept, or Infliximab. Results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum. 62, 22–32 [DOI] [PubMed] [Google Scholar]
- 12. Kirson N. Y., Rao S., Birnbaum H. G., Kantor E., Wei R. S., Cifaldi M. (2013) Matching-adjusted indirect comparison of Adalimumab vs. Etanercept and Infliximab for the treatment of psoriatic arthritis. J. Med. Econ. 16, 479–489 [DOI] [PubMed] [Google Scholar]
- 13. Matthews N., Neale M. L. (1987) Lymphokines and Interferons: A Practical Approach, p. 296, IRL Press, London [Google Scholar]
- 14. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode, in Macromolecular Crystallography: Part A (Carter C. W., Jr., Sweet R. M., eds) pp. 307–326, Academic Press, Orlando, FL: [DOI] [PubMed] [Google Scholar]
- 15. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 16. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 17. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 18. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX. Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 19. Laskowski R., MacArthur M., Moss D., Thornton J. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 20. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Schrödinger, LLC, New York [Google Scholar]
- 21. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 22. Mukai Y., Nakamura T., Yoshikawa M., Yoshioka Y., Tsunoda S., Nakagawa S., Yamagata Y., Tsutsumi Y. (2010) Solution of the structure of the TNF-TNFR2 complex. Sci. Signal. 3, ra83. [DOI] [PubMed] [Google Scholar]
- 23. Banner D. W., D'Arcy A., Janes W., Gentz R., Schoenfeld H. J., Broger C., Loetscher H., Lesslauer W. (1993) Crystal structure of the soluble human 55 kd TNF receptor-human TNFβ complex. Implications for TNF receptor activation. Cell 73, 431–445 [DOI] [PubMed] [Google Scholar]
- 24. Yang Z., West A. P., Jr., Bjorkman P. J. (2009) Crystal structure of TNFα complexed with a poxvirus MHC-related TNF binding protein. Nat. Struct. Mol. Biol. 16, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Idriss H. T., Naismith J. H. (2000) TNFα and the TNF receptor superfamily. Structure-function relationship(s). Microsc. Res. Tech. 50, 184–195 [DOI] [PubMed] [Google Scholar]
- 26. Al-Lazikani B., Lesk A. M., Chothia C. (1997) Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 273, 927–948 [DOI] [PubMed] [Google Scholar]
- 27. Kaymakcalan Z., Sakorafas P., Bose S., Scesney S., Xiong L., Hanzatian D. K., Salfeld J., Sasso E. H. (2009) Comparisons of affinities, avidities, and complement activation of Adalimumab, Infliximab, and Etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol. 131, 308–316 [DOI] [PubMed] [Google Scholar]
- 28. Scallon B., Cai A., Solowski N., Rosenberg A., Song X. Y., Shealy D., Wagner C. (2002) Binding and functional comparisons of two types of tumor necrosis factor antagonists. J. Pharmacol. Exp. Ther. 301, 418–426 [DOI] [PubMed] [Google Scholar]
- 29. Davis T., Friend D., Smith C. A. (2002) Comparative TNF binding characteristics of etanercept (Enbrel) and infliximab (Remicade). Ann. Rheum. Dis. 61, 184 [Google Scholar]
- 30. Granneman R. G., Zhang Y., Noertersheuser P. A., Velagapudi R. B., Awni W. M., Locke C. S. (2003) Pharmacokinetic/pharmacodynamic (PK/PD) relationships of adalimumab (HumiraTM) in rheumatoid arthritis (RA) patients during phase II/III clinical trials. Arthritis Rheum. 48, S140 [Google Scholar]
- 31. Santora L. C., Kaymakcalan Z., Sakorafas P., Krull I. S., Grant K. (2001) Characterization of noncovalent complexes of recombinant human monoclonal antibody and antigen using cation exchange, size exclusion chromatography, and BIAcore. Anal. Biochem. 299, 119–129 [DOI] [PubMed] [Google Scholar]
- 32. Van Ostade X., Tavernier J., Fiers W. (1994) Structure-activity studies of human tumour necrosis factors. Protein Eng. 7, 5–22 [DOI] [PubMed] [Google Scholar]
- 33. Mukai Y., Shibata H., Nakamura T., Yoshioka Y., Abe Y., Nomura T., Taniai M., Ohta T., Ikemizu S., Nakagawa S., Tsunoda S., Kamada H., Yamagata Y., Tsutsumi Y. (2009) Structure-function relationship of tumor necrosis factor (TNF) and its receptor interaction based on 3D structural analysis of a fully active TNFR1-selective TNF mutant. J. Mol. Biol. 385, 1221–1229 [DOI] [PubMed] [Google Scholar]
- 34. Bachmann F., Nast A., Sterry W., Philipp S. (2010) Safety and efficacy of the tumor necrosis factor antagonists. Semin. Cutan. Med. Surg. 29, 35–47 [DOI] [PubMed] [Google Scholar]
- 35. Alonso-Ruiz A., Pijoan J. I., Ansuategui E., Urkaregi A., Calabozo M., Quintana A. (2008) Tumor necrosis factor α drugs in rheumatoid arthritis. Systematic review and metaanalysis of efficacy and safety. BMC Musculoskelet. Disord. 9, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wiens A., Venson R., Correr C. J., Otuki M. F., Pontarolo R. (2010) Meta-analysis of the efficacy and safety of Adalimumab, Etanercept, and Infliximab for the treatment of rheumatoid arthritis. Pharmacotherapy 30, 339–353 [DOI] [PubMed] [Google Scholar]
- 37. Baidoo L., Lichtenstein G. R. (2005) What next after Infliximab? Am. J. Gastroenterol. 100, 80–83 [DOI] [PubMed] [Google Scholar]
- 38. Bernett M. J., Karki S., Moore G. L., Leung I. W., Chen H., Pong E., Nguyen D. H., Jacinto J., Zalevsky J., Muchhal U. S., Desjarlais J. R., Lazar G. A. (2010) Engineering fully human monoclonal antibodies from murine variable regions. J. Mol. Biol. 396, 1474–1490 [DOI] [PubMed] [Google Scholar]