

Background: Huntington disease is caused by an expanded polyglutamine repeat within the protein huntingtin.
Results: Proteasomal degradation of mutant huntingtin fragments is devoid of polyglutamine peptides as partial cleavage products.
Conclusion: Mammalian proteasomes are capable of entirely degrading expanded polyglutamine sequences.
Significance: Accelerating the mutant huntingtin degradation by the proteasomal pathway obviates toxic species and represents a beneficial therapeutic strategy.
Keywords: Aggregation, Autophagy, Huntington Disease, Polyglutamine, Proteasome
Abstract
Huntington disease is a neurodegenerative disorder caused by an expanded polyglutamine (polyQ) repeat within the protein huntingtin (Htt). N-terminal fragments of the mutant Htt (mHtt) proteins containing the polyQ repeat are aggregation-prone and form intracellular inclusion bodies. Improving the clearance of mHtt fragments by intracellular degradation pathways is relevant to obviate toxic mHtt species and subsequent neurodegeneration. Because the proteasomal degradation pathway has been the subject of controversy regarding the processing of expanded polyQ repeats, we examined whether the proteasome can efficiently degrade Htt-exon1 with an expanded polyQ stretch both in neuronal cells and in vitro. Upon targeting mHtt-exon1 to the proteasome, rapid and complete clearance of mHtt-exon1 was observed. Proteasomal degradation of mHtt-exon1 was devoid of polyQ peptides as partial cleavage products by incomplete proteolysis, indicating that mammalian proteasomes are capable of efficiently degrading expanded polyQ sequences without an inhibitory effect on the proteasomal activity.
Introduction
Huntington disease (HD)3 is a neurodegenerative disorder caused by a polyglutamine (polyQ) repeat expansion in the respective protein huntingtin (Htt) (1, 2). HD occurs when the polyQ tract exceeds a threshold of 35–40 glutamine residues in length with a strong inverse correlation between repeat length and age of onset of disease (3). The polyQ expansion causes neuronal dysfunction through a toxic gain-of-function mechanism in both animal and cellular models independent of its protein context (4–7). N-terminal fragments of mutant Htt (mHtt) protein containing the expanded polyQ tract are highly prone to aggregate and form intracellular inclusion bodies (IBs), as observed in human HD post-mortem brain and in animal or cellular systems (8–12). R6/2 mice expressing exon1 of the human mutant HD gene generate a neurological phenotype similar to human HD with an early onset of symptoms and a fast progression of the disease (13).
The two main intracellular pathways involved in protein degradation are the ubiquitin-proteasome system (UPS) and autophagy. Both pathways play a role in mHtt clearance (14, 15). Although the UPS is active in both the nucleus and the cytoplasm, it is merely capable of degrading unfolded monomeric Htt proteins (16–18). The autophagic pathway is a cytoplasmic degradation machinery and targets soluble and aggregated Htt proteins for lysosomal destruction (19, 20). Interestingly, disappearance of IBs and an amelioration of disease phenotype are observed after shutdown of mHtt expression in a conditional HD mouse model, suggesting that autophagy can remove aggregated mHtt, and HD may be reversible (21). These data are supported by the fact that induction of autophagy decreases both aggregated and soluble mHtt, resulting in reduced toxicity in various models of HD (14). However, cells lack autophagy in the nucleus and do not have the ability to eliminate nuclear aggregates efficiently, which may explain the high frequency of nuclear IBs formed by N-terminal mHtt fragments in human HD post-mortem brain (8). As proteasome activity diminishes with age, Zhou et al. (22) showed that N-terminal mHtt fragments aggregate in the nucleus in association with the age-dependent decrease of proteasome activity in an HD knock-in mouse model. Furthermore, levels of soluble and aggregated mHtt increase upon proteasomal inhibition in cell culture and HD mouse brain material (16–18). Because IBs recruit proteins, including ubiquitin (Ub), many types of chaperones, and whole proteasomes, this suggests that cells attempt to clear the aggregation-prone mHtt protein by the proteasomal pathway (17, 18, 23, 24).
In vitro studies suggest that proteasomes are not capable of cleaving within the expanded polyQ repeat, whereas a partial proteolytic product is released lacking flanking sequences of the polyQ tract (25, 26). However, activating the proteasome by the mutant proteasome activator PA28γ (K188E) appears to improve in vitro degradation of peptides containing 10 glutamines with cleavage after each of the glutamines (27).
In this study, we investigated whether the proteasome can degrade mHtt-exon1 with an expanded polyQ stretch, both in vitro and in neuronal cells, by targeting mHtt-exon1 exclusively to the proteasome. We show that mHtt-exon1 is subsequently fully degraded by the cellular UPS, thereby preventing mHtt-exon1 accumulation and aggregation. The proteasomal degradation of soluble mHtt-exon1 does not lead to the release of a polyQ peptide as partial cleavage product by incomplete proteolysis. Furthermore, mammalian proteasomes are capable of degrading expanded polyQ sequences, and proteasomal activity is not affected by the presence of mHtt-exon1.
EXPERIMENTAL PROCEDURES
Constructs
Htt-exon1-97Q constructs with C-terminal sequences encoding the CL1 degron and the C terminus of the mouse ornithine decarboxylase (MODC), respectively, were generated by introducing dsDNA oligonucleotides with a final stop codon. For the generation of the Ub-R-KK-Htt-exon1-97Q construct, the N-terminal degron signal sequence was cut out as a XhoI/NcoI fragment, followed by Klenow fill from the plasmid R15Q (kindly provided by M. D. Kaytor (Emory University, Atlanta, GA)) (28) and cloned into a 5′ XhoI site after blunt ending the vector containing an Htt-exon1-97Q sequence. The N-terminal degron signal sequence from the lacI gene encodes a ubiquitin followed by a stretch of 40 amino acids consisting of two lysines for polyubiquitination as a target signal for the proteasomal degradation (29). The N-terminal ubiquitin is removed by cellular ubiquitinases with a subsequent exposure of a stabilizing amino acid (arginine). Ub-R-KK-Htt-exon1-97Q-C4 was generated by introducing a tetracysteine (C4) tag sequence (FLNCCPGCCMEP) with a stop codon in a 3′ BamHI site. Htt-exon1-25/97Q-H4 constructs were generated by cloning the Htt-exon1-25/97Q sequence with a 5′ XhoI and 3′ BamHI site into a vector encoding a C-terminal H4 tag (HA-His-His-HA, kindly provided by J. Steffan (University of California)). The Htt-exon1-25Q-GFP construct was kindly provided by R. Kopito (Stanford University). GFP-Ub, GFP-Ub-Q112, and Ub-Q112 were generated as described previously (23). Ub-G76V-GFP was a kindly provided by N. Dantuma (Karolinska Institute, Stockholm).
Cell Culture, Transfection, and Electroporation
Atg5+/+ mouse embryonic fibroblast (MEF) and Atg5−/− MEF cells (kindly provided by N. Mizushima (Tokyo Medical and Dental University)), TppII−/− MEF cells (kindly provided by K. Rock (University of Massachusetts Medical School)), and Neuro-2a cells were maintained in DMEM (Invitrogen) supplemented with 10% fetal calf serum, 1 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator with 5% atmospheric CO2. Neuro-2a cells were transfected with PEI according to the manufacturer's instructions (Polysciences Europe). Atg5+/+ MEF, Atg5−/− MEF, and TPPII−/− MEF cells were electroporated with DNA constructs using the Neon Electroporation System (Invitrogen) according to the manufacturer's instructions. For inhibitor studies, transfected or electroporated cells were incubated for 24 h and treated for the last 16 h with various inhibitors: 5 mm 3-methyladenine (3-MA), 50 nm epoxomicin, 0.5 μm bestatin, or 100 μm 1,10-phenanthroline (all purchased from Sigma-Aldrich) or 1 μm PAQ22 (SOPACHEM). 200 nm bafilomycin A1 (Sigma-Aldrich) was added to transfected cells for the last 4 h, whereas treatment with DMSO served as a control. Loss of Neuro-2a cell membrane integrity was detected by uptake of propidium iodide (PI) (Sigma-Aldrich). Transfected cells were cultured for 48 h in total, and for the last 4 h, they were treated with 500 nm staurosporine (Sigma-Aldrich) prior to staining cells with PI (5 μg/ml) and Hoechst 33342 (5 μg/ml; Invitrogen). Quantification of the percentage of PI-positive cells was performed by fluorescent microscopy, where a minimum of three fields per condition were counted.
Soluble and Insoluble Fractionation
Cells were harvested and lysed in 1× TEX buffer (70 mm Tris/HCl, pH 6.8, 1.5% SDS, 20% glycerol). After sonification (Soniprep150, Sanyo) 50 mm DTT was added fresh, and samples were centrifuged at 14,000 rpm at room temperature. The pelleted SDS-insoluble fraction was incubated with 100% formic acid at 37 °C for 40 min, followed by evaporation of the formic acid using a SpeedVac system (Eppendorf). 1× TEX buffer supplemented with 0.05% bromphenol blue was added to the pellet, and the soluble and insoluble fractions were loaded on an SDS-polyacrylamide gel.
Proteasome Activity Labeling
Cells were harvested in TSDG buffer (10 mm Tris/HCl, pH 7.5, 25 mm KCl, 10 mm NaCl, 1.1 mm MgCl2, 0.1 mm EDTA, and 8% glycerol) and lysed by three consecutive freeze-thaw cycles with liquid nitrogen. After high speed centrifugation (15 min at 14,000 rpm and 4 °C), the total protein concentration of the clarified lysate was determined by a Bradford protein assay. Proteasomes were labeled in the cell lysate with 0.5 μm activity-based probe BODIPY-epoxomicin for 1 h at 37 °C (kindly provided by H. Overkleeft (Institute of Chemistry, Leiden)) (30). Sample loading buffer was added to 10 μg of lysate, and the samples were boiled for 3 min and loaded onto a 12.5% SDS gel with subsequent in-gel fluorescence imaging using a Typhoon imager (GE Healthcare) with the 580 BP 30 filter to detect the BODIPY-epoxomicin probe.
Western Blot Analysis and Filter Trap Assay
Cells were harvested in lysis buffer (50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 20 mm N-ethylmaleimide, supplemented with complete miniprotease inhibitor mixture (Roche Applied Science)). Total cell lysates were boiled for 10 min at 99 °C with 1× Laemmli sample loading buffer (350 mm Tris/HCl, pH 6.8, 10% SDS, 30% glycerol, 6% β-mercaptoethanol, bromphenol blue) fractionated by SDS-PAGE, and transferred to a PVDF membrane (0.45-μm pore size; Schleicher & Schuell). Western blot membranes were blocked with 5% milk; incubated with primary antibodies anti-Htt 1C2 (1:1000; MAB1574, Millipore), anti-polyQ (1:1000; 3B5H10, Sigma-Aldrich), anti-Htt N18 (1:1000; BML-PW0595-0100, Enzo), anti-HA (1:1000; H3663, Sigma-Aldrich), polyclonal rabbit anti-GFP (1:1000; kindly provided by J. Neefjes, Nederlands Kanker Instituut), anti-β-actin (1:1000; SC-130656, Santa Cruz Biotechnology, Inc.), anti-α2 (1:1000; ab22666, Abcam), anti-p62 (1:500; Clonagen), anti-LC3 (1:500; 48394, Abcam), and anti-ubiquitin (1:100; U5379, Sigma-Aldrich); and subsequently incubated with secondary antibodies IRDye 680 or IRDye 800 (1:10.000; LI-COR). Infrared signal was detected using the Odyssey imaging system (LI-COR). A filter trap assay was performed with the pellet obtained after high speed centrifugation of the cell lysate (15 min at 14,000 rpm at 4 °C). Pellet with aggregates was resuspended in Benzonase buffer (1 mm MgCl2, 50 mm Tris/HCl, pH 8.0) and incubated for 1 h at 37 °C with 125 units of Benzonase (Merck). The reaction was stopped by adding 2× termination buffer (40 mm EDTA, 4% SDS, 100 mm DTT fresh). Samples with 50 μg of protein extract diluted in 2% SDS buffer were filtered through a 0.2-μm pore size cellulose acetate membrane (Schleicher & Schuell), pre-equilibrated in 2% SDS wash buffer (2% SDS, 150 mm NaCl, 10 mm Tris/HCl, pH 8.0), and spotted on the membrane in doublets. Filters were washed twice with 0.1% SDS buffer (0.1% SDS, 150 mm NaCl, 10 mm Tris, pH 8.0) and blocked with 5% milk for further treatment like Western blot membranes.
ReAsH Staining
For imaging, cells were seeded on coverslips and transfected with the plasmids. Cells were cultured for 24 h, rinsed with prewarmed 1× PBS buffer, and labeled for 30 min with prewarmed DMEM containing 1 μm ReAsH (kindly provided by H. Overkleeft (Leiden University)) and 10 μm 1,2-ethanedithiol (Sigma-Aldrich) at 37 °C. After staining, cells were washed five times for 15 min each in prewarmed DMEM containing 1 mm 1,2-ethanedithiol and 10% FCS. Finally, cells were washed twice with 1× PBS, fixed with 4% paraformaldehyde solution, and mounted on objective glasses using Vectashield (Vector Laboratories).
Fluorescence Microscopy
Cells were transfected or electroporated with the indicated DNA constructs 24–48 h prior to imaging, and fluorescent cells were scored for the percentage of cells with aggregates and the percentage of PI-positive cells using an inverted fluorescence microscope (Leica DMR). For imaging, transfected cells were fixed on coverslips, and images were obtained using a confocal microscope equipped with an argon/krypton laser and a 63× objective (Leica TCS SP2).
Htt Protein and 20 S Proteasome Purification
Neuro-2a cells were transfected with the Htt-exon1-25/97Q-H4 construct, and 48 h after expression, they were harvested in lysis buffer (50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 20 mm N-ethylmaleimide, supplemented with complete miniprotease inhibitor mixture (Roche Applied Science). Centrifugation at 14,000 rpm at 4 °C for 15 min was performed to remove cell debris and protein aggregates. Cell lysates were immunoprecipitated for 2 h using EZview Red Anti-HA Affinity Gel (Sigma-Aldrich) to purify Htt-exon1-25/97Q-H4. Proteins were eluted for 5 min at room temperature with 0.1 m glycine, pH 2.5, and immediately neutralized with 1.5 m Tris/HCl, pH 8.8, to avoid deamidation of proteins. The protein eluate was dialyzed against 50 mm Tris/HCl, pH 7.5, for 2 h at 4 °C with an additional exchange of buffer after 1 h. 20 S proteasomes (kindly provided by B. Dahlmann (Charité, Berlin, Germany)) were purified from human erythrocytes as described previously (31).
In Vitro Degradation Assays
To analyze the in vitro proteasomal degradation of purified Htt-exon1-25/97Q-H4 protein by the 20 S mammalian proteasome, 50 pmol of purified Htt protein was incubated with 1.4 pmol of mammalian 20 S proteasomes in 1× 20 S buffer (10 mm Tris/HCl, pH 7.4, 30 mm NaCl, 1 mm MgCl2, 400 μm fresh DTT) in a total volume of 15 μl treated with or without 40 μm MG132 at 37 °C for the indicated times. Reactions were stopped by adding 6× sample loading buffer. Aggregated Htt was spun down at 14,000 rpm for 15 min and treated as an insoluble fraction. To assess the effect of the inhibitors 3-MA and epoxomicin on the enzymatic activity of the proteasome in Neuro-2a cells, 5 mm 3-MA, 50 nm epoxomicin, and DMSO as control were added to living Neuro-2a cells, respectively; cells were incubated for 16 h and subsequent lysed in KMH buffer (110 mm KAc, 2 mm MgAc, and 20 mm HEPES, pH 7.2) containing 100 μm digitonin. The cytoplasmic fraction was obtained after centrifugation (15 min at 14,000 rpm), and protein concentration was determined by the Bradford protein assay. The assay was performed with 30 μg of total cytoplasmic protein and fluorogenic substrate with an end concentration of 100 μm Suc-LLVY-AMC, 150 μm Ac-RLR-AMC, and 150 μm Ac-GPLD-AMC (all purchased from Enzo). To assess the effects of purified Htt proteins with a normal and an expanded polyQ stretch on the enzymatic activity of purified mammalian 20 S proteasome in vitro, chymotrypsin-like activity of the proteasome was monitored by Suc-LLVY-AMC digestion after incubation of 1.4 pmol of proteasomes with 50 pmol of purified wtHtt or mHtt treated with or without 40 μm MG132 for 16 h at 37 °C. Experiments were performed in triplicates, and generation of free AMC was measured at 37 °C with the spectrophotometric plate reader FLUOstar Optima (BMG Labtec).
In Vitro Transcription and Translation
Htt proteins were transcribed and translated in vitro with a TNT T7-coupled reticulocyte lysate system according to the manufacturer's instructions (Promega). The in vitro protein expression level and protein degradation by the proteasome were determined by adding 100 μm MG132 to reticulocyte cell lysate prior to protein expression and subsequent Western blot analysis.
Proteinase K Treatment
Purified mHtt protein (1 μg) was incubated with 100 μg/ml proteinase K (Invitrogen) at 37 °C in 50 mm Tris/HCl, pH 7.4, reaction buffer for the indicated times. After digest, aggregates were captured by centrifugation (14,000 rpm for 15 min) and solubilized by formic acid (insoluble fraction). The proteinase K digest was stopped by adding 6× sample loading buffer, and proteins were subjected to SDS-PAGE.
Mass Spectrometry
Reactions of purified Htt-exon1-25/97Q-H4 with mammalian 20 S proteasomes after an incubation time of 16 h at 37 °C were analyzed for Htt peptide generation. As a control, Htt-exon1-25/97Q-H4 only and mammalian 20 S proteasomes only were analyzed. A <3-kDa peptide pool fraction was separated using an Amicon Ultra 0.5-ml Centrifugal Filter 3K device (Millipore) for subsequent mass spectrometry analysis. After adjusting the pH of the <3-kDa fractions to 3.0 (using 10% trifluoroacetic acid (TFA)), samples were desalted via C18-Stage Tip purification as described (32). Peptide samples were analyzed by B. Florea (Leiden University) on a Surveyor nano-LC system (Thermo) connected to an LTQ-Orbitrap mass spectrometer (Thermo). The column was packed with BioSphere C18 5-μm 120-Å particles from Nanoseparations (Nieuwkoop, The Netherlands). Instrument settings and measuring parameters were used as described by Florea et al. (30). The resulting raw data were analyzed by MaxQuant software (version 1.2.2.5) (33, 34) using the Andromeda search algorithm against a manually generated fasta-file containing the sequence of Htt-exon1-25/97Q-H4. Search parameters were as follows: Enzyme, any cleavage allowed; Max. miss cleavages, 40. To determine Htt-exon1-97Q-H4 and 20 S proteasome co-precipitated proteins, the 3-kDa filter supernatant (≥3-kDa fraction) was diluted in 100 μl of 50 mm NH4HCO3 (ABC) buffer and trypsin-digested at 37 °C overnight. Digests were stopped by adjusting the pH with 10% TFA to 3.0. Samples were analyzed using the same protocol as for the <3-kDa fractions. A search was performed instead against the ipi.HUMAN.v3.68 database. Search parameters were as follows: Enzyme, trypsin; Max. miss cleavages, 2.
Statistical Analysis
All values were obtained from three independent repeated experiments and expressed as mean ± S.D. Statistical analysis was performed using Student's t test. p < 0.05 was considered statistically significant.
RESULTS
mHtt-exon1 Is Degraded by Macroautophagy and the Proteasome
To determine whether macroautophagy and the proteasome are involved in mHtt-exon1 degradation, mHtt-exon1-97Q was expressed for 24 h in Neuro-2a cells treated with the proteasomal inhibitor epoxomicin or the autophagy inhibitor 3-MA for the last 16 h of expression. Cells were harvested for soluble and insoluble fractionation, and a filter trap assay was used to evaluate both monomeric and aggregated mHtt-exon1 levels using the 1C2 antibody, which recognizes polyQ sequences. Whereas autophagy inhibition increased the level of mHtt-exon1 aggregates detected in the insoluble fraction (arrow) and on the filter trap (Fig. 1A), proteasome inhibition showed no obvious effect on the amount of monomeric and aggregated mHtt-exon1 in Neuro-2a cells. Furthermore, the protein level of wtHtt-exon1-25Q expressed in Neuro-2a cells was elevated upon inhibition of the autophagic degradation by 3-MA (Fig. 1B). To verify the effect of 3-MA as an inhibitor of macroautophagy, Neuro-2a cells were treated for 16 h with 3-MA in combination with the inhibitor bafilomycin A1 or DMSO as a control. Western blot analysis clearly shows an effect of 3-MA on the endogenous LC3-II level, indicating an inhibition of the formation of LC3-II-positive autophagosomes by 3-MA (Fig. 1C). Assessing the effect of epoxomicin and 3-MA on the proteasomal catalytic activity in treated Neuro-2a cells, endogenous proteasomes were labeled in the cell lysates with a proteasome activity-based probe, where the catalytic activities were visualized by in-gel fluorescence (Fig. 1D). In addition, the degradation of fluorogenic proteasomal AMC substrates added to Neuro-2a cell lysates was analyzed (Fig. 1E). Whereas epoxomicin reduces the caspase-like, trypsin-like, and chymotrypsin-like proteasomal activities, referred to as β1, β2, and β5, 3-MA had no effect on the proteasomal activity nor on the level of polyubiquitinated material. To exclude the possibility that 3-MA treatment inhibits the proteasomal degradation of substrates at a level upstream of the proteasome catalytic activity, the degradation of the proteasomal reporter Ub-G76V-GFP in Neuro-2a cells was analyzed. Epoxomicin but not 3-MA increased the protein level of the proteasomal reporter substrate, showing that 3-MA inhibits autophagic but not proteasomal degradation of proteins (Fig. 1F). Although proteasomal inhibition did not lead to an increase of Htt-exon1 protein levels, the impairment of the ubiquitin proteasome system might be compensated by an up-regulation of autophagy (reviewed in Ref. 35). To examine the role of the proteasome in mHtt-exon1 degradation in more detail, Htt-exon1-97Q-C4 was expressed in WT and Atg5-deficient MEFs (36) and treated with epoxomicin. The short C4 tag with 12 amino acids binds the biarsenical dye ReAsH, and the Htt protein becomes fluorescent (41). The Atg5-Atg12 protein conjugation is essential for the formation of LC3-II-positive autophagosomes, with LC3-II being a key marker for autophagosomes (37). Atg5−/− MEFs deficient in LC3-II-positive autophagosomes showed an increase of mHtt-exon1 aggregates after proteasomal inhibition compared with WT MEFs, indicating that soluble mHtt-exon1 can indeed be targeted by the proteasome when autophagy is impeded (Fig. 2, A and B). To exclude the possibility that Atg5−/− MEFs compensate with an increase of proteasomal catalytic activity compared with WT MEFs, cellular proteasomes were labeled in the cell lysates with a proteasome activity-based probe, where the catalytic activities were visualized by in-gel fluorescence (Fig. 2C). Together, these data indicate that mHtt-exon1 can be degraded by both macroautophagy and the proteasome.
FIGURE 1.

Intracellular degradation of Htt-exon1. A, soluble and insoluble fractionation of Neuro-2a cell lysate after transient transfection of Htt-exon1-97Q or an empty vector as control. Six hours after transfection, cells were treated for 16 h with DMSO, epoxomicin, or 3-MA and harvested. Soluble Htt and formic acid-dissolved and non-dissolved (arrow) Htt aggregates were detected on Western blot by the 1C2 antibody. SDS-insoluble Htt aggregates were analyzed by a filter trap assay in doublets. β-Actin was used as a loading control. Shown is a quantification of soluble and insoluble mHtt protein levels; *, p < 0.05 (n = 3). B, Western blot analysis of Neuro-2a cells after transient transfection of Htt-exon1-25Q or an empty vector as control. Six hours after transfection, cells were treated for 16 h with DMSO, epoxomicin, or 3-MA and harvested. Htt was detected on Western blot by the 1C2 antibody. β-Actin was used as a loading control. Shown is a quantification of soluble wtHtt protein levels; *, p < 0.05 (n = 3). C, Neuro-2a cells were treated for 16 h with 3-MA in combination with bafilomycin A1 or DMSO as a control (last 4 h) before harvest. Endogenous LC3-I and -II levels were detected by the LC3 antibody. β-Actin was used as a loading control. D, Neuro-2a cells were treated for 16 h with DMSO, epoxomicin, or 3-MA before harvest. The three proteasomal catalytic sites (β1, β2, and β5) were labeled in the Neuro-2a cell lysate with activity probe. As a control for the inhibitor treatment, the level of polyubiquitinated proteins was detected by a ubiquitin antibody. E, 3-MA has no inhibitory effect on proteasome catalytic activity. Neuro-2a cells were treated for 16 h with DMSO, epoxomicin, or 3-MA before harvest. Cells were lysed, and the chymotrypsin-like, trypsin-like, and caspase-like activities of the proteasomes were monitored by the substrates Suc-LLVY-AMC, Ac-RLR-AMC, and Ac-GPLD-AMC, respectively; ***, p < 0.001; **, p < 0.01 (n = 3). F, Western blot with a GFP antibody after expression of the proteasomal reporter Ub-G76V-GFP in Neuro-2a cells. Six hours after transfection, cells were treated for 16 h with DMSO, epoxomicin, or 3-MA and harvested. β-Actin was used as a loading control; *, p < 0.05 (n = 3). Error bars, S.D.
FIGURE 2.
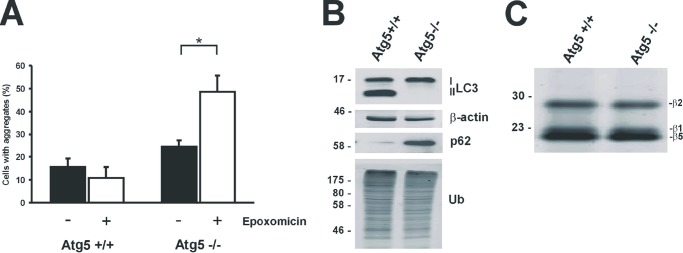
Proteasomal degradation of mHtt-exon1 in Atg5-deficient cells. A, quantification of Htt aggregates after expression of Htt-exon1-97Q-C4 in Atg5+/+ and Atg5−/− MEF cells. Six hours after electroporation, cells were treated for 16 h with DMSO or epoxomicin and fixed on coverslips for staining and aggregate scoring; *, p < 0.05 (n = 3). B, Western blot analysis for detection of endogenous LC3-I and LC3-II levels in electroporated Atg5+/+ and Atg5−/− MEF cells. C, the proteasomal catalytic sites (β1, β2, and β5) were labeled in Atg5+/+ and Atg5−/− MEF cell lysate with activity probe. Error bars, S.D.
Targeting mHtt-exon1 to the Proteasome
Previous studies showed inefficient in vitro proteasomal degradation of fusion proteins containing an expanded polyQ stretch, leading to the conclusion that eukaryotic proteasomes fail to cleave within expanded polyQ sequences (25, 26). However, it remained unclear whether expanded polyQ stretches embedded in native polyQ protein sequences like mHtt-exon1 are also inefficiently degraded by proteasomes in living cells. In the case of remaining polyQ stretches as partial cleavage products of incomplete proteolysis, these fragments would accumulate within the cell and start to aggregate, as previously shown in living cells expressing pure expanded polyQ peptides without flanking sequences (23).
To examine whether intracellular proteasomes are able to fully degrade mHtt and also cleave within the expanded polyQ tract, we prevented degradation of mHtt by the autophagic pathway and targeted the mHtt-exon1 protein with a specific degradation signal to the proteasomal pathway. To generate a mHtt-exon1 protein, which is exclusively degraded via the proteasomal degradation pathway and independent of macroautophagic clearance within the cell, degron signals for the ubiquitin-dependent and ubiquitin-independent proteasomal destruction were fused to the mHtt-exon1 protein. C-terminal degrons, such as the 16-amino acid-long ubiquitin-dependent CL1 degron and the ubiquitin-independent C-terminal PEST sequence from MODC are known to reduce the half-life of GFP by introduction of these additional proteolytic signals (24, 38, 39). These degron signals were fused to the C terminus of mHtt-exon1 (Fig. 3A). To evaluate the effect of the specific C-terminal degrons on mHtt-exon1 degradation and protein aggregation, Neuro-2a cells were transfected with the Htt-exon1-97Q constructs, expressed for 24 h, and treated with the inhibitors epoxomicin and 3-MA. Despite these degron signals, mHtt-exon1 with a CL1 or a MODC degron was still a target for macroautophagy, similar to mHtt-exon1 without an additional degron signal (Fig. 3B). Only the autophagy inhibitor 3-MA increased the amount of aggregated mHtt-exon1 in the SDS-insoluble fraction detected by the filter trap analysis, whereas neither the CL1 degron nor the MODC degron targeted mHtt-exon1 protein exclusively toward proteasomal degradation. Fusion of the CL1 or MODC degron to the N terminus instead of the C terminus of mHtt-exon1 also did not change the degradation pattern compared with mHtt-exon1 without an additional degron signal (data not shown).
FIGURE 3.

Generation of Htt-exon1-97Q proteins with specific degron signals. A, schemata of expressed Htt-exon1-97Q proteins either without or with a CL1 or MODC degron signal. B, Western blot analysis of Neuro-2a cells after transient transfection of different Htt-exon1-97Q-degron constructs or an empty vector as control. Six hours after transfection, cells were treated for 16 h with DMSO, epoxomicin, or 3-MA and harvested. Soluble and insoluble Htt protein was detected on Western blot or on filter trap (doublets) by the 1C2 antibody. β-Actin was used as a loading control. As a control for the inhibitor treatment, the level of polyubiquitinated proteins was detected by an ubiquitin antibody. C, schemata of expressed Htt-exon1-97Q proteins either without or with an N-end rule degron signal. D, Western blot analysis of Neuro-2a cells after transient transfection of Htt-exon1-97Q constructs with or without an N-end rule degron or an empty vector as control. Six hours after transfection cells were treated for 16 h with DMSO, epoxomicin, or 3-MA and harvested. Soluble and insoluble mHtt was detected on Western blot or filter trap (doublets) by the polyQ and 1C2 antibody, respectively. β-Actin was used as a loading control, and for the inhibitor treatment, the level of polyubiquitinated proteins was detected by an ubiquitin antibody. E, quantification of Htt aggregates after expression of Htt-exon1-97Q-C4 with or without the N-end rule degron signal. Six hours after transfection, Neuro-2a cells were treated for 16 h with DMSO, epoxomicin, or 3-MA and fixed on coverslips for staining and aggregate scoring; **, p < 0.01 (n = 3). F, in vitro transcription/translation of mutant Htt constructs with a CL1 or N-end rule degron signal. Proteasomal degradation was inhibited by MG132 (100 μm). Htt was detected on Western blot by the 1C2 antibody. β-Actin was used as a loading control. Staining of endogenous β-actin, p62, and polyubiquitin was used as a control. Error bars, S.D.
Next, we generated a mHtt-exon1 fusion protein flanked on its N terminus by an “N-end rule” degradation signal, termed N-degron, composed of a ubiquitin moiety that is removed upon translation by deubiquitinases. This exposes the destabilizing amino acid arginine, followed by a 40-amino acid region consisting of two lysines for polyubiquitination as a target signal for the proteasomal degradation (40) (Fig. 3C). To test the effect of the specific N-terminal degron on mHtt-exon1 degradation and protein aggregation, Neuro-2a cells were transfected with the construct Htt-exon1-97Q or Ub-R-KK-Htt-exon1-97Q. Upon expression for 24 h, cells were treated with the inhibitors epoxomicin and 3-MA. In contrast to Htt-exon1-97Q, the N-end rule Htt protein Ub-R-KK-Htt-exon1-97Q has a low abundance of soluble protein level upon Western blot and no aggregated mHtt-exon1 detected on the filter trap. Due to the N-degron signal, the soluble monomeric Htt-exon1-97Q protein is efficiently targeted to the UPS, because treatment with the proteasomal inhibitor epoxomicin but not the autophagy inhibitor 3-MA resulted in an accumulation of Ub-R-KK-Htt-exon1-97Q in the soluble and SDS-insoluble fraction (Fig. 3D). This indicates that the specific N-degron signal transforms mHtt-exon1 into a short lived protein that is targeted via the proteasome before it can accumulate and aggregate within the cell. Smaller partial digest products of the Ub-R-KK-Htt-exon1-97Q protein by the proteasome consisting of putative intact polyQ tracts missing the flanking sequences were not detected upon Western blot with the specific polyQ antibody 3B5H10, suggesting that Ub-R-KK-Htt-exon1-97Q is fully degraded by the proteasome. Simultaneous inhibition of both the proteasome and autophagy by epoxomicin and 3-MA does not reveal a partial digest product of the Ub-R-KK-Htt-exon1-97Q protein by the proteasome that would be subsequently targeted by macroautophagy (data not shown). Next, the number of Neuro-2a cells containing mHtt-exon1 aggregates upon expression of Ub-R-KK-Htt-exon1-97Q or Htt-exon1-97Q with a C4 tag was determined. The amount of mHtt-exon1 aggregates in transfected cells confirms the filter trap data from Fig. 3D, because almost no aggregates of Ub-R-KK-Htt-exon1-97Q-C4 are detectable compared with Htt-exon1-97Q-C4 (Fig. 3E). The addition of the inhibitor epoxomicin or 3-MA for the last 16 h of expression shows an increase of mHtt-exon1 aggregation, which was mainly due to inhibition of autophagy, whereas mutant Ub-R-KK-Htt-exon1 aggregation only increases upon inhibition of the proteasomal function. Similarly, in vitro transcription and translation of mHtt-exon1 with the N-degron signal and the CL1 degron in rabbit reticulocyte lysate reveals a rapid degradation of the short lived Ub-R-KK-Htt-exon1-97Q protein by the proteasome compared with Htt-exon-97Q-CL1 protein after treatment with MG132, showing that the disappearance of the Ub-R-KK-Htt-exon1-97Q protein and its polyubiquitinated species is dependent on proteasomal function (Fig. 3F). As in Neuro-2a cells, Htt-exon1-97Q-CL1 expressed in rabbit reticulocyte lysate is not efficiently targeted by the proteasome.
These data show that the N-degron signal in Ub-R-KK-Htt-exon1 targets soluble mHtt-exon1 exclusively to the proteasomal pathway independent of macroautophagy and represents an adequate substrate suitable for studying proteasomal degradation of polyQ-expanded mHtt-exon1 within the cell.
Efficient Proteasomal Degradation of Short Lived mHtt
Expression of the Ub-R-KK-Htt-exon1-97Q protein reveals a direct clearance of the full mHtt-exon1 protein by the proteasome, because no expanded polyQ stretches as partial proteolytic products were detectable on Western blot by the polyQ antibody 3B5H10 (Fig. 3D). To prove that this particular antibody is able to detect pure polyQ tracts independent of the flanking Htt sequences by Western blot analysis, we expressed the constructs GFP-Ub-Q112 and, as a control, GFP-Ub in Neuro-2a cells and analyzed the polyQ peptides generated after N-terminal GFP-Ub hydrolysis by cellular deubiquitinases (23). The specific polyQ antibody 3B5H10 is able to detect polyQ peptides with a size of ∼30 kDa (arrow) and higher molecular species, which may represent polyQ oligomers (asterisks) (Fig. 4A). To examine whether a possible proteasomal polyQ product derived from mHtt-exon1 is more highly prone to aggregate than the mutant Ub-R-KK-Htt-exon1 protein and therefore not detectable on the soluble level by Western blotting, a soluble and insoluble fractionation of transfected Neuro-2a cell lysate treated with epoxomicin was performed. When proteins from the soluble and insoluble fractions were stained on a Western blot with the polyQ antibody 3B5H10, no additional polyQ fragments generated by the cellular proteasomes were detectable besides the low level of the proteasomal substrate Ub-R-KK-Htt-exon1-97Q, which increases in both fractions as monomeric and polyubiquitinated forms after epoxomicin treatment (asterisk) (Fig. 4B). To exclude the possibility that polyQ peptides generated by the proteasome accumulate within the cell and form formic acid-insensitive aggregates, we co-expressed wtHtt-exon1-25Q protein as an aggregation reporter for filter trap analysis and fluorescence microscopy. Untagged Htt-exon1-25Q co-expressed with GFP-Ub-Q112 in Neuro-2a cells co-aggregated with Q112 peptides detectable on a filter trap assay by the Htt-specific antibody N18 (Fig. 4C). Similarly, confocal microscopy of fixed Neuro-2a cells co-expressing non-fluorescent Ub-Q112 and the reporter Htt-exon1-25Q-GFP show nuclear and cytoplasmic inclusion bodies formed by polyQ peptides that co-sequester wtHtt-exon1 proteins (Fig. 4D). In contrast, co-expression of the short lived mHtt-exon1 variant Ub-R-KK-Htt-exon1-97Q and the reporter Htt-exon1-25Q-GFP reveals no detectable aggregation on filter trap by the antibody GFP except for epoxomicin-treated cells (Fig. 4E). In addition, co-expression of the aggregation reporter Htt-exon1-25Q-GFP with Htt-exon1-97Q-C4 or its short lived variant Ub-R-KK-Htt-exon1-97Q-C4 only shows ReAsH and GFP co-stained aggregates in cells expressing Htt-exon1-97Q-C4, whereas the reporter's GFP signal in cells co-expressing Ub-R-KK-Htt-exon1-97Q-C4 remains diffusely distributed in the cytoplasm (Fig. 4F). These results indicate that short lived mHtt-exon1 neither aggregates nor is proteolytically degraded into pure polyQ peptides that would co-sequester the reporter protein Htt-exon1-25Q-GFP.
FIGURE 4.

Efficient proteasomal degradation of the short lived Htt-exon1-97Q protein. A, transient transfection of Neuro-2a cells with constructs encoding GFP-Ub-Q112 and GFP-Ub as a control. Within the cells, removal of N-terminal GFP-Ub by ubiquitinases releasing pure polyQ peptides (arrow) and its oligomeric species (asterisks) was detectable with the polyQ antibody on Western blot. β-Actin was used as a loading control. B, soluble and insoluble fractionation of Neuro-2a cells after transient transfection of Ub-R-KK-Htt-exon1-97Q. Six hours after transfection, cells were treated for 16 h with epoxomicin and DMSO as control and harvested for Western blot analysis. Expanded polyQ-containing Htt was detected on Western blot by the polyQ antibody (asterisk). β-Actin was used as a loading control, and for the inhibitor treatment, the level of polyubiquitinated proteins was detected by a ubiquitin antibody. C, filter trap analysis of Neuro-2a cells after transient co-transfection of GFP-Ub-Q112 and Htt-exon1-25Q. Aggregates of Q112 peptides co-sequestering the aggregation reporter protein Htt-exon1-25Q were detected by the 1C2 and N18 antibodies, whereas the N18 antibody recognized the N17 region of Htt-exon1-25Q. Filter trap analysis was performed in doublets. D, transient co-transfection of Neuro-2a cells with Ub-Q112 and Htt-exon1-25Q-GFP. Within the cells, ubiquitinases cleave off the N-terminal Ub, releasing polyQ peptides, which are prone to aggregate. After 24 h, cells were fixed, and aggregates of Q112 peptides co-sequestering the aggregation reporter Htt-exon1-25Q-GFP were detected by immunofluorescence. The nucleus was stained by DAPI. Scale bar, 10 μm. E, filter trap analysis of Neuro-2a cells after transient co-transfection of Ub-R-KK-Htt-exon1-97Q and Htt-exon1-25Q-GFP or the empty vector as control. Six hours after transfection, cells were treated for 16 h with epoxomicin and DMSO as a control and harvested for filter trap analysis (in doublets) with the GFP antibody. F, transient co-transfection of Neuro-2a cells with Htt-exon1-25Q-GFP and either Htt-exon1-97Q-C4 or Ub-R-KK-Htt-exon1-97Q-C4 for 48 h. Htt-exon1-97Q-C4 aggregates stained with ReAsH co-sequestered wtHtt, whereas Ub-R-KK-Htt-exon1-97Q-C4 neither aggregated nor was proteolytically degraded into pure polyQ peptides co-sequestering the reporter protein Htt-exon1-25Q-GFP. The nucleus was stained with DAPI. Scale bar, 10 μm. G, cellular uptake of propidium iodide (PI) in control cells transfected with an empty vector and cells expressing mutant Htt with or without the N-end rule degron signal; ***, p < 0.001 (n = 3). H, soluble and insoluble fractionation of TPPII−/− MEF cell lysate after transient transfection of Ub-R-KK-Htt-exon1-97Q or empty vector as control. Six hours after transfection, cells expressing Ub-R-KK-Htt-exon1-97Q were treated for 16 h with DMSO, PAQ22/bestatin, phenanthroline, or epoxomicin. Polyubiquitinated and monomeric (arrow) short lived mutant Htt was detected on Western blot by the polyQ antibody. β-Actin was used as a loading control. Error bars, S.D.
Next, we investigated whether targeting mHtt-exon1-97Q to the UPS not only reduces levels of soluble and insoluble mHtt-exon1 but would also reduce mHtt-exon1 induced toxicity, as measured by cellular uptake of PI. When Htt-exon1-97Q and its short lived variant Ub-R-KK-Htt-exon1-97Q were expressed in Neuro-2a cells for 48 h and stained with PI, mHtt-exon1-97Q increased PI-staining of the Neuro-2a cells, whereas fusion of the N-degron signal to mHtt-exon1 did not lead to mHtt-induced toxicity (Fig. 4G).
Previous in vitro studies suggested that in the case of expanded polyQ-containing proteins, eukaryotic proteasomes cannot cleave within the polyQ sequences releasing pure polyQ stretches without flanking sequences (25, 26). Subsequently, these polyQ fragments may be degraded by cytosolic peptidases before they start to aggregate. Peptides released by the proteasome are rapidly degraded into amino acids by various peptidases (42–46) like tripeptidyl peptidase II (TPPII), which can also target peptides longer than 15 amino acids (47, 48). The proposed cellular function of TPPII is an exo- and endopeptidase activity downstream of the proteasome. However, the proteolytic processing of proteasomal products has TPPII in common with other peptidases like puromycin-sensitive aminopeptidase (PSA). PSA may target released polyQ peptides because it was found to be capable of degrading short polyQ peptides (49). Recently, Menzies et al. (50) showed that PSA promotes autophagy independent of its cytosolic peptidase function, leading to enhanced clearance of aggregation-prone proteins.
To examine whether TPPII and PSA may target potential expanded polyQ peptides released by the proteasome upon degradation of Ub-R-KK-Htt-exon1-97Q, we expressed the short lived mHtt-exon1 in TPPII−/− MEF cells treated with a combination of the PSA inhibitors PAQ22 and bestatin or phenanthroline (Fig. 4H). As a control, transfected MEFs were treated with epoxomicin. Western blot analysis of the soluble and insoluble fraction of lysates showed the detection of the Ub-R-KK-Htt-exon1-97Q protein only after proteasomal inhibition (arrow), whereas no additional smaller polyQ-containing fragment was detectable by the polyQ antibody 3B5H10 in TPPII−/− MEF cells treated with inhibitors for PSA. This suggests that TPPII and PSA do not have an impact on the degradation of putative expanded polyQ stretches downstream of the proteasome and indicates that short lived mHtt-exon1 is efficiently targeted for proteasomal degradation, whereas no release of toxic, aggregation-prone polyQ peptides by the proteasome occurs.
Mammalian 20 S Proteasomes Are Able to Degrade Expanded polyQ Sequences in Vitro
The previous results with the efficient cellular mHtt degradation, independent of peptidases like TPPII or PSA, predict that purified mammalian 20 S proteasomes are capable of degrading purified mutant Htt protein completely. To test this, both mHtt-exon1-97Q and wtHtt-exon1-25Q proteins were expressed in Neuro-2a cells and subsequently purified by immunoprecipitation via HA-agarose followed by dialysis. Purified open-gated mammalian 20 S proteasomes were incubated with purified mHtt-exon1 or wtHtt-exon1 for 12 or 24 h before the reaction was stopped by adding sample loading buffer. The digests were analyzed on a Western blot with the Htt-specific antibodies N18 against the Htt N terminus, polyQ 3B5H10 against the polyQ tract, and HA antibody against the C terminus. Within 24 h, both mHtt-exon1 (Fig. 5A) and wtHtt-exon1 (Fig. 5B) were almost fully degraded, with no remaining polyQ fragment as a partial proteolytic proteasomal product lacking the flanking N-terminal and C-terminal sequences. The addition of MG132 prevents the degradation of Htt-exon1 by the 20 S proteasome completely. Because the mHtt-exon1 protein is almost completely degraded within a 12-h incubation time, we followed the in vitro degradation kinetics for the time points 0, 3, 6, and 16 h to analyze earlier events (Fig. 5C). Although mHtt-exon1 diminishes in time, as detected by Western blot analysis using the polyQ antibody 3B5H10, marginal smaller proteasomal cleavage fragments appear within the first 3 h of digest as putative proteolytic intermediates. After 16 h of digest, only ∼10% of the purified mHtt-exon1 protein staining remains.
FIGURE 5.

In vitro proteasomal degradation of the Htt-exon1 protein. Purified Htt-exon1-97Q-H4 (A) and Htt-exon1-25Q-H4 proteins (B) were incubated with purified mammalian 20 S proteasomes at 37 °C for the times indicated. For inhibition of the 20 S proteasome, MG132 was added to the reaction. The proteins were subjected to SDS-PAGE and Western blot analysis using anti-Htt antibodies (N18 and polyQ) and anti-HA antibody. The amount of added 20 S proteasome was shown with an anti-α2 antibody used as a loading control. C, purified Htt-exon1-97Q-H4 proteins were incubated with purified mammalian 20 S proteasomes at 37 °C for the times indicated. For inhibition of the 20 S proteasome, MG132 was added to the reaction. The proteins were subjected to SDS-PAGE and Western blot analysis using anti-Htt antibody polyQ. The amount of added 20 S proteasome was shown with an anti-α2 antibody used as a loading control. Shown is a quantification of the remaining full-length purified Htt-exon1-97Q-H4 protein stained by the polyQ antibody within a 16-h digest by the 20 S proteasome. D, after incubation of purified mHtt with 20 S proteasome, aggregated proteins were separated from soluble protein material by centrifugation and solubilized at time point 16 h with formic acid (insoluble fraction) and subjected to SDS-PAGE and Western blot analysis using anti-Htt antibody polyQ. E, in vitro degradation of the mutant Htt protein with proteinase K. Purified mHtt was incubated with proteinase K at 37 °C for the times indicated. After digest, aggregates were captured by high speed centrifugation and solubilized by formic acid (insoluble fraction). The proteins were subjected to SDS-PAGE and Western blot analysis using anti-Htt antibody polyQ. The pure polyQ fragment (arrow) and a soluble oligomeric intermediate (asterisk) generated by the proteinase K digest were detected by the polyQ antibody. F, chymotrypsin-like activity of the purified mammalian 20 S proteasome monitored by Suc-LLVY-AMC digestion 16 h after incubation of proteasomes with purified wtHtt or mHtt treated with or without MG132 at 37 °C (n = 3). Error bars, S.D.
Next, we examined whether any partial processing products may aggregate within the 16 h of digest and would therefore not be detectable as soluble fragments on Western blot. However, analysis of the formic acid-treated insoluble fraction of the in vitro digest reveals no additional aggregation-prone polyQ fragments (Fig. 5D). To exclude the possibility that glutamine residues in the polyQ stretch are deamidated to glutamate prior to the in vitro digest, resulting in unintended protein degradation by the caspase-like activity of the proteasome, purified mHtt-exon1 was incubated with proteinase K, an enzyme that is capable of cleaving after glutamate but not glutamine. Incubation of mHtt-exon1 with proteinase K generates a proteolytic product within 2 h of incubation with a size of ∼27 kDa, which is the expected size of a pure 97Q peptide without the flanking Htt N-terminal and C-terminal sequences due to removal by proteinase K (Fig. 5E). Because glutamine deamidation in the mHtt-exon1 protein would result in various smaller fragments of different sizes after proteinase K digest, the appearance of a specific monomeric protein band (Fig. 5E, arrow) and a formic acid-soluble oligomeric species (asterisk) detectable by the polyQ antibody 3B5H10 upon proteinase K digest, but not proteasomal degradation, suggests that the mHtt-exon1 protein is completely degraded by the 20 S proteasome.
Although mHtt-exon1 and wtHtt-exon1 are efficiently degraded by the proteasome, we analyzed whether the expanded polyQ stretch affected in vitro proteasome activity, because impairment of proteasomes by polyQ proteins has been suggested before (25, 26). To test this, we monitored the chymotrypsin-like activity of purified 20 S proteasome after incubation with mHtt-exon1 and wtHtt-exon1. The expansion of the polyQ tract in the Htt-exon1 protein had no effect on the proteasomal activity, as measured with degradation of the proteasomal substrate Suc-LLVY-AMC in time (Fig. 5F). This supports the data indicating that mammalian proteasomes are capable of degrading expanded polyQ sequences, whereas the main proteasomal activity is not affected by the presence of mHtt-exon1.
Detection of Proteasome-mediated Cleavage Products from Htt-exon1 by Mass Spectrometry
As an additional step to evaluate cleavage of mHtt-exon1 by the mammalian 20 S proteasome, in vitro digests were analyzed by mass spectrometry (MS). Peptides generated by the proteasome within a 16-h digest of mHtt-exon1 and wtHtt-exon1 were separated and identified by LC-MS (Table 1). As a result of the analysis, we specified a number of N-terminal and C-terminal Htt peptides as proteasomal cleavage products that are represented in a schemata where the identified Htt peptides are summarized and marked in green (N-terminal peptides) and red (C-terminal peptides) (Fig. 6). The pattern of the Htt peptides suggests that there are one or more initial cleavage events in the purified Htt-exon1 protein, thereby generating longer peptides that may represent intermediate products that are sequentially trimmed at the C terminus to smaller peptides detected by MS as 8–23-amino acid-long peptides. Undetected proteasomal cleavage products exclusively consisting of only glutamine residues would be underestimated by MS due to their poor ionization (27).
TABLE 1.
Mass spectrometric identification of 20 S mammalian proteasome-mediated degradation products from Htt-exon1 97Q-H4 and Htt-exon1 25Q-H4
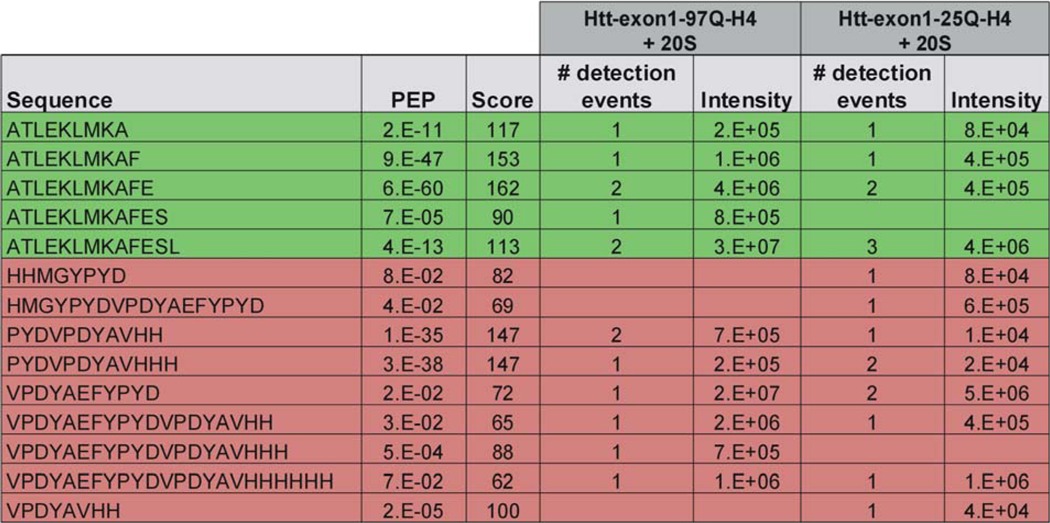
FIGURE 6.
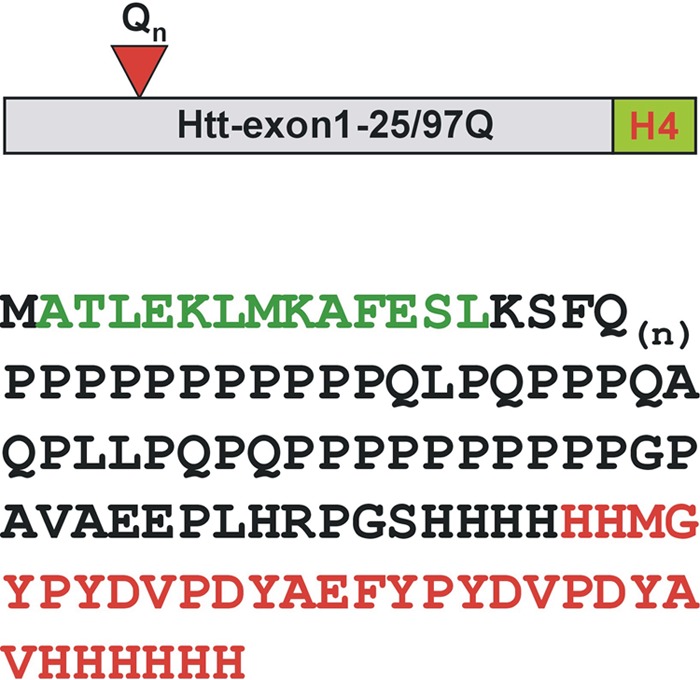
Peptide coverage of identified 20 S proteasomal Htt cleavage products. Purified Htt-exon1-25/97Q-H4 proteins were incubated with purified mammalian 20 S proteasomes for 16 h at 37 °C. Fractionation of the <3-kDa peptide pool with a flow-through 3-kDa microconcentrator was performed, and separated Htt peptides generated by the 20 S proteasome were used for subsequent mass spectrometry analysis. Htt-exon1-25/97Q-H4 sequences of peptides identified as proteasome-mediated degradation products are shown in green (N-terminal) and red (C-terminal).
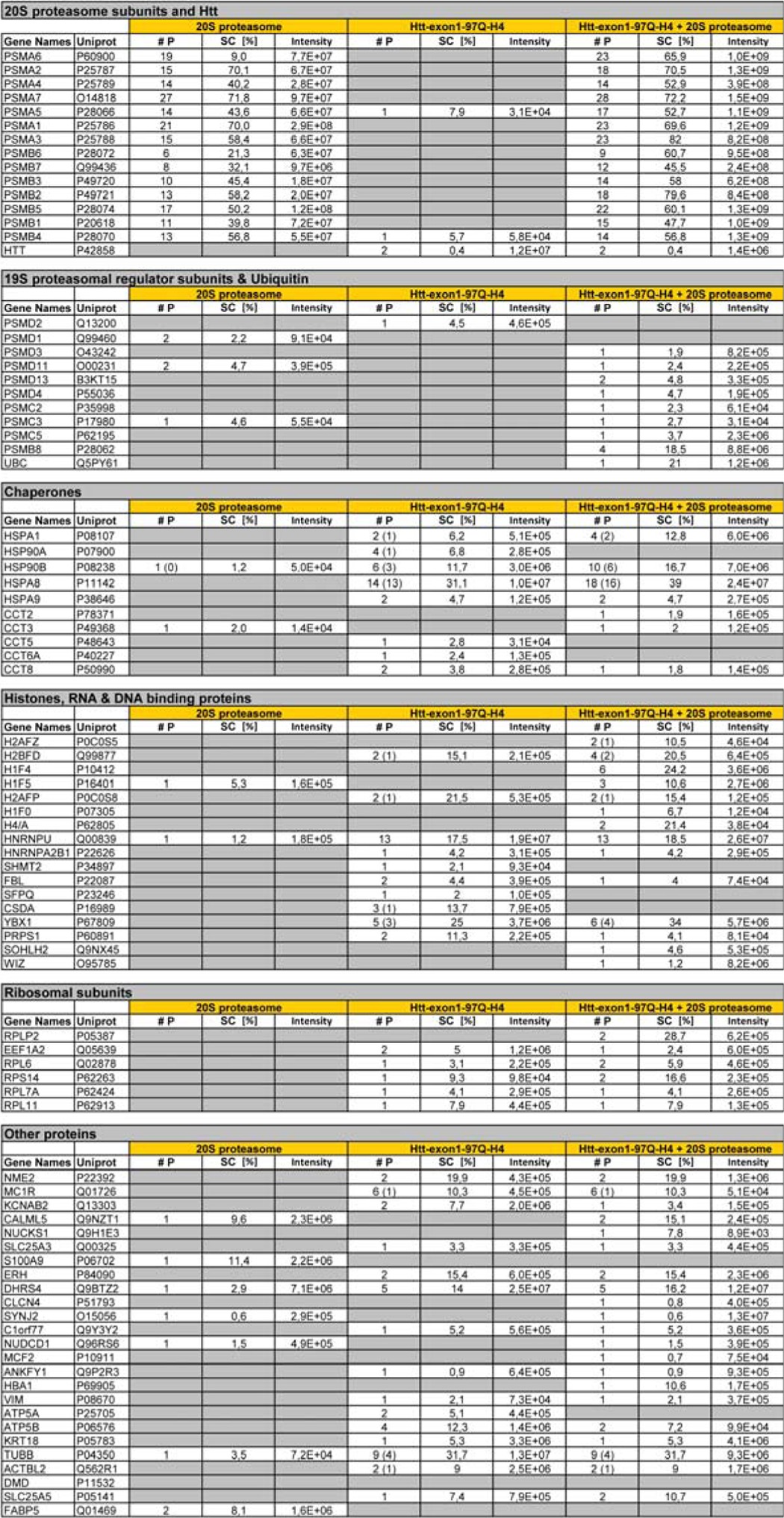
To exclude the possibility that mHtt-exon1 degradation is attributed to a contaminating protease potentially co-purified with the 20 S proteasome or the mHtt-exon1 protein, we identified co-purified proteins by MS analysis of peptides generated by trypsin cleavage (Table 2). However, no co-purified contaminating protease could be detected, indicating that Htt-exon1 was exclusively degraded by the proteasome. Furthermore, MS analysis of the Htt-exon1 in vitro digest, including the proteasomal inhibitor MG132 or merely without the 20 S proteasome, shows no Htt peptide generation (data not shown). Interestingly, some chaperones were identified by MS analysis that have been co-immunoprecipitated with mHtt-exon1 and could facilitate unfolding and degradation both in living cells and in vitro.
TABLE 2.
Mass spectrometric identification of 20 S proteasome and Htt-exon1 97Q-H4 co-purified proteins
#P, number of detected peptides (unique peptides if different); SC [%], sequence coverage in percent; 20 S proteasome, ≥3-kDa fraction of purified mammalian 20 S proteasome; Htt-exon1 97Q-H4, ≥3-kDa fraction of purified mutant Htt; Htt-exon1 97Q-H4 + 20 S proteasome, ≥3-kDa fraction of 20 S proteasomal digest of Htt (16 h, 37 °C).
DISCUSSION
Our data show that mHtt-exon1 is a substrate for both macroautophagy and the proteasome. Although treatment of cells with the proteasome inhibitor did not show an increase in monomeric and aggregated mHtt-exon1 levels, this may be explained by the subsequent induction of macroautophagy upon UPS inhibition (35). A clear contribution of Htt-exon1 proteasomal degradation could be visualized when macroautophagy was impaired, such as in Atg5−/− MEF cells. To analyze whether the cellular proteasome is capable of cleaving within an expanded polyQ stretch, considering the native Htt-exon1 protein context, we targeted the mHtt-exon1 protein to the proteasomal pathway in ubiquitin-dependent and -independent manners by the fusion of mHtt-exon1 to various degron signals. We show that the mHtt-exon1 short lived variant with the N-degron signal (Ub-R-KK) is efficiently cleared by the proteasome before aggregation can occur, independent of macroautophagy. This finding clearly indicates that the eukaryotic UPS can degrade mHtt-exon1 completely, including the expanded polyQ sequence. Fusion of pure polyQ sequences to GFP with an N-terminal degradation signal also decreased soluble and insoluble GFP-polyQ protein levels in transfected cells (51, 52). However, expression of the GFP-polyQ protein carrying the Ub-R degron signal still led to the formation of aggregates within cells, suggesting that this degron signal is not as efficient as the Ub-R-KK degron signal used in our study, where no aggregation of short lived mHtt-exon1 was detectable. In contrast to previous studies (25, 51), we analyzed the proteasomal degradation of mHtt-exon1 without a fusion to large fluorescent proteins, like GFP, and in addition we determined the role of polyQ clearance by macroautophagy and putative cytoplasmic peptidases next to the proteasomal degradation.
Intriguingly, C-terminal degrons, such as the CL1 degron and the C-terminal PEST sequence from MODC, are known to reduce the half-life of GFP (24, 38, 39), and a previous study showed that the degron signal of ODC converts mHtt with a length of 163 amino acids to an unstable protein (53). However, our data using mHtt-exon1-CL1 and mHtt-exon1-MODC show that these proteins are not proper proteasomal substrates, suggesting that the protein context next to the specific degron signal plays an important role in substrate recognition and its subsequent proteasomal degradation. It also underscores the importance of targeting mHtt fragments to the proteasome, which is most likely dependent on an appropriate ubiquitination of the specific substrate similar to the N-end rule. Improving the targeting of monomeric mutant Htt fragments to the nuclear and cytoplasmic proteasomal degradation pathway before aggregation occurs is certainly a very important issue that should be addressed in follow-up studies. Enhancing the processes of efficient ubiquitination to target nuclear and cytoplasmic mutant Htt to the proteasomal pathway might be a potential therapeutic approach, but this requires the identification of involved E2/E3 ligases or deubiquitinating enzymes and determination of whether their activity can be specifically triggered.
Rapid degradation of mHtt-exon1 with an N-degron by cellular proteasomes might occur through several sequential rounds in ubiquitin-dependent and -independent manners. First, polyubiquitinated mHtt-exon1 is recognized, deubiquitinated, and unfolded by the 19 S regulatory particle, followed by entry of the unfolded mHtt-exon1 into the hollow cavity of the 20 S core particle. Here, Htt fragments of different lengths are generated as proteolytic products containing the full or shortened polyQ tracts. Second, some of these generated Htt fragments may either again become ubiquitinated and processed into smaller peptides by the 26 S proteasome, or third, a ubiquitin-independent proteasomal way is responsible for the degradation of the released unfolded Htt fragments into smaller peptides that are subsequently recycled into amino acids by cytoplasmic peptidases (54). The ubiquitin-independent proteasomal processing of Htt-exon1 or subsequent Htt peptides might be performed by 20 S proteasomes associated with activating caps. Whereas the 26 S proteasome is composed of a 19 S cap and a 20 S catalytic core particle, the 19 S cap can be replaced by 11 S activators, termed PA28αβγ (55–58). PA28α and PA28β form a heteroheptameric ring, and PA28γ forms a homoheptameric ring (59–62). PA28αβ or PA28γ activators dock on the 20 S proteasome, recognize unfolded peptides and proteins in an ATP-independent manner, and stimulate the proteasomal catalytic activity (63–65). Alternatively, 11 S activators function in the context of hybrid proteasomes, where a 20 S proteasome is capped by one 11 S activator and on the opposite site by a 19 S cap (58). Interestingly, in vitro studies revealed that the mutant PA28γ (K188E) cap increases the activity of all three 20 S catalytic subunits, and proteasomal degradation of short polyQ peptides is 10-fold greater compared with WT PA28γ, suggesting a beneficial effect of activators toward polyQ stretch degradation (27, 66).
Importantly, the in vitro data in our study were obtained with mHtt-exon1 without the specific N-terminal degron, showing that also non-modified mHtt-exon1 can be degraded entirely by 20 S proteasomes. The in vitro digests confirm our observations in living cells that the proteasome efficiently degrades the expanded polyQ repeat. Our results are in contrast with a study reporting the release of pure polyQ sequences during in vitro proteasomal degradation of a polyQ-fusion protein due to inefficient digestion (26). This previous in vitro study shows that enhancing the proteasomal degradation of expanded-polyQ proteins is not valid as a therapeutic approach for polyQ diseases. In this study, we show for the first time that mammalian proteasomes can entirely degrade mutant Htt fragments in living cells and that both cellular and in vitro proteasomal destruction of mHtt-exon1 were devoid of long polyQ peptides as partial cleavage products. Subsequently, reducing the amount of monomeric mHtt by accelerating the mHtt proteasomal destruction obviates the accumulation of toxic Htt fragments and finally represents a therapeutic strategy for HD.
However, the discrepancy between the different in vitro results could be a consequence of distinct substrate or proteasome purification for the degradation assay. Nevertheless, Pratt and Rechsteiner (27) showed by MS that activation of the proteasome by the mutant proteasome activator PA28γ (K188E) appears to improve in vitro degradation of peptides containing 10 glutamines with cleavage after each of the 10 glutamines, indicating that the proteasome is capable of digesting short polyQ sequences. Our MS data together with the Western blot analysis of the in vitro degraded Htt-exon1 gives a first glimpse of how the proteasome may mechanistically degrade Htt fragments independent of the length of the polyQ stretch. We identified N- and C-terminal Htt peptides that appear to be cleaved off from the polyQ/polyP tract as initial cleavage products and, in addition, peptides resulting from C-terminal single amino acid trimming of these Htt cleavage products, suggesting a subsequent shortening of the intermediate Htt peptides (Table 1). Because proteasomal cleavage products consisting of only glutamine or proline residues would be underestimated by mass spectrometry due to their poor ionization, pure polyQ peptides cannot be identified by mass spectrometry. Therefore, additional Western blot analysis with the specific polyQ antibody clearly reveals a complete degradation of the polyQ tract by the proteasome but not by proteinase K, which cannot cleave after glutamine residues.
Although cellular Htt-exon1-97Q is aggregation-prone and forms Htt-positive IBs in Neuro-2a cells, no additional mHtt-exon1 aggregation occurred upon purification and during the incubation time of several h at 37 °C, indicating that this purified protein mainly persists in a probably monomeric unfolded state suitable for processing by the open-gated 20 S proteasome. The maintenance of the mHtt native monomeric form might be explained by the mHtt-exon1 co-purified proteins identified by MS. Our data show that mHtt-exon1 is associated with a number of chaperones that may keep purified mHtt-exon1 in a monomeric state (Table 2). We identified HspA1 (Hsp70), HspA8 (Hsc70), HspA9 (mtHsp70), Hsp90, and subunits of the chaperonin TRiC (CCT) in the purified Htt-exon1 protein solution. Molecular chaperones are important modulators of polyQ-expanded protein aggregation in the cell, and several reports evaluated the effects of chaperone function on polyQ aggregation and toxicity in vivo and in vitro (67–71). Overexpression of Hsp70 suppresses polyQ-induced neuropathology in a spinocerebellar ataxia 1 (SCA1) mouse model and in an SCA3 fly model (72), and in vitro experiments demonstrated that Hsp70 and its cochaperone Hsp40 suppressed the assembly of mHtt into amyloid-like fibrils (68). Furthermore, a recent study reported that mHtt interacts with Hsp90, and cell treatment with a selective Hsp90 inhibitor enhanced mHtt clearance by the UPS (73). The role of TRiC in polyQ aggregation was addressed, showing that the ring-shaped, hetero-oligomeric chaperonin TRiC inhibits mHtt-exon1 aggregation in yeast and in cell culture (74, 75). In a follow up study, Tam et al. (76) reported that TRiC binds to the N17 domain of Htt, thereby stabilizing the monomeric conformation by acting as a “cap” preventing aggregation. The latter might be a reasonable explanation for why purified mHtt-exon1 from Neuro-2a cells is kept in a soluble monomeric form.
Although mHtt-exon1 is polyubiquitinated within the cell, this post-translational modification is not a sufficient signal for fast proteasomal destruction when compared with the mHtt-exon1 with an N-degron signal that is targeted for proteasomal clearance via the Lys-48-ubiquitin linkage (40). The fact that the level of polyubiquitinated mHtt-exon1 without a specific degron signal is not increased after proteasomal inhibition suggests either that the ubiquitin linkage pattern is different compared with short lived Htt-exon1 or that mHtt-exon1 mainly becomes polyubiquitinated upon aggregation, and this Htt species cannot be cleared by the proteasome.4 Previously, a selective degradation of phosphorylated Htt was proposed, which involved both the lysosomal and proteasomal pathway (15). Overexpression of the kinase IKK increased phosphorylation and reduced polyubiquitination of Htt-exon1 in transfected St14A cells, indicating that phosphorylation of the Htt N terminus influences the post-translational modifications of neighboring lysine residues, such as ubiquitination, and consequently the half-life of Htt.
To avoid undesired post-translational modifications independent of Htt-exon1, we expressed an Htt-exon1 construct with a stop codon after amino acid 90 with no additional tag for immunoblotting, and for imaging of IBs and quantification, we made use of a short tetracysteine tag (C4 tag) instead of a GFP fusion protein. This tag is lacking lysine residues for putative additional ubiquitination compared with GFP to exclude an impact of GFP post-translational modifications on Htt clearance because it was shown that Ub-R-GFP as a proteasomal reporter is ubiquitinated after the removal of the N-terminal ubiquitin moiety and rapidly degraded by the proteasome (39, 77).
In the aggregation process from soluble monomeric Htt to large IBs, a variety of intermediate species have been described (78, 79). The role of the different Htt species in the HD pathology is controversial, because a protective role for IBs has been suggested in striatal cells transfected with Htt by sequestration of toxic soluble mutant Htt species, thereby reducing neuronal death (80). Whether monomeric or oligomeric forms represent toxic species is currently still under debate, although evidence supports a more toxic role for soluble polyQ oligomers than polyQ monomers (81). However, there is a strong correlation between detection of mutant Htt monomers and small oligomers by the polyQ antibody 3B5H10 and prediction of neuronal toxicity (82). Consequently, decreasing the level of monomeric mHtt by accelerating the mHtt proteasomal degradation obviates the accumulation of toxic species within the cell and represents a beneficial therapeutic strategy for HD.
Acknowledgments
We thank J. Steffan (University of California), M.D. Kaytor (Emory University), R. Kopito (Stanford University), N. Dantuma (Karolinska Institute), N. Mizushima (Tokyo Medical and Dental University), K. Rock (University of Massachusetts Medical School) and H. Overkleeft (Leiden University) for generously sharing reagents and plasmids. We thank N. de Wilde for technical assistance and B. Florea (Leiden University) for the mass spectrometric analysis.
This work was supported by Dutch Organization for Scientific Research VIDI Grant NWO-Zon-MW, 91796315; by Prinses Beatrix Fonds Grant W.OR10-25; and by Hersenstichting Grant KS2010(1)-06.
K. Juenemann, S. Schipper-Krom, A. Wiemhoefer, A. Kloss, A. Sanz Sanz, and E. A. J. Reits, manuscript in preparation.
- HD
- Huntington disease
- Htt
- huntingtin
- mHtt
- mutant Htt
- 3-MA
- 3-methyladenine
- MEF
- mouse embryonic fibroblast
- MODC
- mouse ornithine decarboxylase
- polyQ
- polyglutamine
- PSA
- puromycin-sensitive aminopeptidase
- Ub
- ubiquitin
- UPS
- ubiquitin-proteasome system
- TPPII
- tripeptidyl peptidase II
- IB
- inclusion body
- AMC
- 7-amido-4-methylcoumarin
- Suc
- succinyl.
REFERENCES
- 1. The Huntington's Disease Collaborative Research Group, T. H. s. D. C. R. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. Ross C. A., Becher M. W., Colomer V., Engelender S., Wood J. D., Sharp A. H. (1997) Huntington's disease and dentatorubral-pallidoluysian atrophy. Proteins, pathogenesis and pathology. Brain Pathol. 7, 1003–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang C. C., Faber P. W., Persichetti F., Mittal V., Vonsattel J. P., MacDonald M. E., Gusella J. F. (1998) Amyloid formation by mutant huntingtin. Threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 24, 217–233 [DOI] [PubMed] [Google Scholar]
- 4. Ordway J. M., Tallaksen-Greene S., Gutekunst C. A., Bernstein E. M., Cearley J. A., Wiener H. W., Dure L. S., 4th, Lindsey R., Hersch S. M., Jope R. S., Albin R. L., Detloff P. J. (1997) Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 91, 753–763 [DOI] [PubMed] [Google Scholar]
- 5. Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., Ross C. A., Scherzinger E., Wanker E. E., Mangiarini L., Bates G. P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548 [DOI] [PubMed] [Google Scholar]
- 6. La Spada A. R., Paulson H. L., Fischbeck K. H. (1994) Trinucleotide repeat expansion in neurological disease. Ann. Neurol. 36, 814–822 [DOI] [PubMed] [Google Scholar]
- 7. Ikeda H., Yamaguchi M., Sugai S., Aze Y., Narumiya S., Kakizuka A. (1996) Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 13, 196–202 [DOI] [PubMed] [Google Scholar]
- 8. DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 [DOI] [PubMed] [Google Scholar]
- 9. Gutekunst C. A., Li S. H., Yi H., Mulroy J. S., Kuemmerle S., Jones R., Rye D., Ferrante R. J., Hersch S. M., Li X. J. (1999) Nuclear and neuropil aggregates in Huntington's disease. Relationship to neuropathology. J. Neurosci. 19, 2522–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lunkes A., Lindenberg K. S., Ben-Haïem L., Weber C., Devys D., Landwehrmeyer G. B., Mandel J. L., Trottier Y. (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 10, 259–269 [DOI] [PubMed] [Google Scholar]
- 11. Schilling G., Klevytska A., Tebbenkamp A. T., Juenemann K., Cooper J., Gonzales V., Slunt H., Poirer M., Ross C. A., Borchelt D. R. (2007) Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J. Neuropathol. Exp. Neurol. 66, 313–320 [DOI] [PubMed] [Google Scholar]
- 12. Juenemann K., Weisse C., Reichmann D., Kaether C., Calkhoven C. F., Schilling G. (2011) Modulation of mutant huntingtin N-terminal cleavage and its effect on aggregation and cell death. Neurotox. Res. 20, 120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S. W., Bates G. P. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 [DOI] [PubMed] [Google Scholar]
- 14. Rubinsztein D. C. (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786 [DOI] [PubMed] [Google Scholar]
- 15. Thompson L. M., Aiken C. T., Kaltenbach L. S., Agrawal N., Illes K., Khoshnan A., Martinez-Vincente M., Arrasate M., O'Rourke J. G., Khashwji H., Lukacsovich T., Zhu Y. Z., Lau A. L., Massey A., Hayden M. R., Zeitlin S. O., Finkbeiner S., Green K. N., LaFerla F. M., Bates G., Huang L., Patterson P. H., Lo D. C., Cuervo A. M., Marsh J. L., Steffan J. S. (2009) IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 187, 1083–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Wang C. E., Huang S., Xu X., Li X. J., Li H., Li S. (2010) Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum. Mol. Genet. 19, 2445–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wyttenbach A., Carmichael J., Swartz J., Furlong R. A., Narain Y., Rankin J., Rubinsztein D. C. (2000) Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 97, 2898–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Waelter S., Boeddrich A., Lurz R., Scherzinger E., Lueder G., Lehrach H., Wanker E. E. (2001) Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 12, 1393–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ravikumar B., Duden R., Rubinsztein D. C. (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 20. Qin Z. H., Wang Y., Kegel K. B., Kazantsev A., Apostol B. L., Thompson L. M., Yoder J., Aronin N., DiFiglia M. (2003) Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 12, 3231–3244 [DOI] [PubMed] [Google Scholar]
- 21. Yamamoto A., Lucas J. J., Hen R. (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101, 57–66 [DOI] [PubMed] [Google Scholar]
- 22. Zhou H., Cao F., Wang Z., Yu Z. X., Nguyen H. P., Evans J., Li S. H., Li X. J. (2003) Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J. Cell Biol. 163, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raspe M., Gillis J., Krol H., Krom S., Bosch K., van Veen H., Reits E. (2009) Mimicking proteasomal release of polyglutamine peptides initiates aggregation and toxicity. J. Cell Sci. 122, 3262–3271 [DOI] [PubMed] [Google Scholar]
- 24. Bence N. F., Sampat R. M., Kopito R. R. (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555 [DOI] [PubMed] [Google Scholar]
- 25. Holmberg C. I., Staniszewski K. E., Mensah K. N., Matouschek A., Morimoto R. I. (2004) EMBO J. 23, 4307–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Venkatraman P., Wetzel R., Tanaka M., Nukina N., Goldberg A. L. (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95–104 [DOI] [PubMed] [Google Scholar]
- 27. Pratt G., Rechsteiner M. (2008) Proteasomes cleave at multiple sites within polyglutamine tracts. Activation by PA28γ(K188E). J. Biol. Chem. 283, 12919–12925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaytor M. D., Wilkinson K. D., Warren S. T. (2004) Modulating huntingtin half-life alters polyglutamine-dependent aggregate formation and cell toxicity. J. Neurochem. 89, 962–973 [DOI] [PubMed] [Google Scholar]
- 29. Bachmair A., Varshavsky A. (1989) The degradation signal in a short-lived protein. Cell 56, 1019–1032 [DOI] [PubMed] [Google Scholar]
- 30. Florea B. I., Verdoes M., Li N., van der Linden W. A., Geurink P. P., van den Elst H., Hofmann T., de Ru A., van Veelen P. A., Tanaka K., Sasaki K., Murata S., den Dulk H., Brouwer J., Ossendorp F. A., Kisselev A. F., Overkleeft H. S. (2010) Activity-based profiling reveals reactivity of the murine thymoproteasome-specific subunit β5t. Chem. Biol. 17, 795–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zoeger A., Blau M., Egerer K., Feist E., Dahlmann B. (2006) Circulating proteasomes are functional and have a subtype pattern distinct from 20S proteasomes in major blood cells. Clin. Chem. 52, 2079–2086 [DOI] [PubMed] [Google Scholar]
- 32. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation, and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 33. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 34. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda. A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 35. Korolchuk V. I., Menzies F. M., Rubinsztein D. C. (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398 [DOI] [PubMed] [Google Scholar]
- 36. Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., Yoshimori T. (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanada T., Noda N. N., Satomi Y., Ichimura Y., Fujioka Y., Takao T., Inagaki F., Ohsumi Y. (2007) The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282, 37298–37302 [DOI] [PubMed] [Google Scholar]
- 38. Corish P., Tyler-Smith C. (1999) Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 12, 1035–1040 [DOI] [PubMed] [Google Scholar]
- 39. Dantuma N. P., Lindsten K., Glas R., Jellne M., Masucci M. G. (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18, 538–543 [DOI] [PubMed] [Google Scholar]
- 40. Bachmair A., Finley D., Varshavsky A. (1986) In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186 [DOI] [PubMed] [Google Scholar]
- 41. Griffin B. A., Adams S. R., Tsien R. Y. (1998) Specific covalent labeling of recombinant protein molecules inside live cells. Science 281, 269–272 [DOI] [PubMed] [Google Scholar]
- 42. Beninga J., Rock K. L., Goldberg A. L. (1998) Interferon-γ can stimulate post-proteasomal trimming of the N terminus of an antigenic peptide by inducing leucine aminopeptidase. J. Biol. Chem. 273, 18734–18742 [DOI] [PubMed] [Google Scholar]
- 43. Mo X. Y., Cascio P., Lemerise K., Goldberg A. L., Rock K. (1999) Distinct proteolytic processes generate the C and N termini of MHC class I-binding peptides. J. Immunol. 163, 5851–5859 [PubMed] [Google Scholar]
- 44. Brömme D., Rossi A. B., Smeekens S. P., Anderson D. C., Payan D. G. (1996) Human bleomycin hydrolase. Molecular cloning, sequencing, functional expression, and enzymatic characterization. Biochemistry 35, 6706–6714 [DOI] [PubMed] [Google Scholar]
- 45. Stoltze L., Schirle M., Schwarz G., Schröter C., Thompson M. W., Hersh L. B., Kalbacher H., Stevanovic S., Rammensee H. G., Schild H. (2000) Two new proteases in the MHC class I processing pathway. Nat. Immunol. 1, 413–418 [DOI] [PubMed] [Google Scholar]
- 46. Johnson G. D., Hersh L. B. (1990) Studies on the subsite specificity of the rat brain puromycin-sensitive aminopeptidase. Arch. Biochem. Biophys. 276, 305–309 [DOI] [PubMed] [Google Scholar]
- 47. Reits E., Neijssen J., Herberts C., Benckhuijsen W., Janssen L., Drijfhout J. W., Neefjes J. (2004) A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation. Immunity 20, 495–506 [DOI] [PubMed] [Google Scholar]
- 48. York I. A., Bhutani N., Zendzian S., Goldberg A. L., Rock K. L. (2006) Tripeptidyl peptidase II is the major peptidase needed to trim long antigenic precursors, but is not required for most MHC class I antigen presentation. J. Immunol. 177, 1434–1443 [DOI] [PubMed] [Google Scholar]
- 49. Bhutani N., Venkatraman P., Goldberg A. L. (2007) Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO J. 26, 1385–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Menzies F. M., Hourez R., Imarisio S., Raspe M., Sadiq O., Chandraratna D., O'Kane C., Rock K. L., Reits E., Goldberg A. L., Rubinsztein D. C. (2010) Puromycin-sensitive aminopeptidase protects against aggregation-prone proteins via autophagy. Hum. Mol. Genet. 19, 4573–4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Michalik A., Van Broeckhoven C. (2004) Proteasome degrades soluble expanded polyglutamine completely and efficiently. Neurobiol. Dis. 16, 202–211 [DOI] [PubMed] [Google Scholar]
- 52. Verhoef L. G., Lindsten K., Masucci M. G., Dantuma N. P. (2002) Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 11, 2689–2700 [DOI] [PubMed] [Google Scholar]
- 53. Rousseau E., Kojima R., Hoffner G., Djian P., Bertolotti A. (2009) Misfolding of proteins with a polyglutamine expansion is facilitated by proteasomal chaperones. J. Biol. Chem. 284, 1917–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rockel B., Kopec K. O., Lupas A. N., Baumeister W. (2012) Structure and function of tripeptidyl peptidase II, a giant cytosolic protease. Biochim. Biophys. Acta 1824, 237–245 [DOI] [PubMed] [Google Scholar]
- 55. Voges D., Zwickl P., Baumeister W. (1999) The 26S proteasome. A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068 [DOI] [PubMed] [Google Scholar]
- 56. Smith D. M., Kafri G., Cheng Y., Ng D., Walz T., Goldberg A. L. (2005) ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol. Cell 20, 687–698 [DOI] [PubMed] [Google Scholar]
- 57. Bar-Nun S., Glickman M. H. (2012) Proteasomal AAA-ATPases. Structure and function. Biochim. Biophys. Acta 1823, 67–82 [DOI] [PubMed] [Google Scholar]
- 58. Rechsteiner M., Hill C. P. (2005) Mobilizing the proteolytic machine. Cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 15, 27–33 [DOI] [PubMed] [Google Scholar]
- 59. Johnston S. C., Whitby F. G., Realini C., Rechsteiner M., Hill C. P. (1997) The proteasome 11S regulator subunit REG α (PA28 α) is a heptamer. Protein Sci. 6, 2469–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Knowlton J. R., Johnston S. C., Whitby F. G., Realini C., Zhang Z., Rechsteiner M., Hill C. P. (1997) Structure of the proteasome activator REGα (PA28α). Nature 390, 639–643 [DOI] [PubMed] [Google Scholar]
- 61. Zhang Z., Krutchinsky A., Endicott S., Realini C., Rechsteiner M., Standing K. G. (1999) Proteasome activator 11S REG or PA28. Recombinant REG α/REG β hetero-oligomers are heptamers. Biochemistry 38, 5651–5658 [DOI] [PubMed] [Google Scholar]
- 62. Masson P., Lundin D., Söderbom F., Young P. (2009) Characterization of a REG/PA28 proteasome activator homolog in Dictyostelium discoideum indicates that the ubiquitin- and ATP-independent REGγ proteasome is an ancient nuclear protease. Eukaryot. Cell 8, 844–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Förster A., Masters E. I., Whitby F. G., Robinson H., Hill C. P. (2005) The 1.9 Å structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions. Mol. Cell 18, 589–599 [DOI] [PubMed] [Google Scholar]
- 64. Chu-Ping M., Slaughter C. A., DeMartino G. N. (1992) Purification and characterization of a protein inhibitor of the 20S proteasome (macropain). Biochim. Biophys. Acta 1119, 303–311 [DOI] [PubMed] [Google Scholar]
- 65. Dubiel W., Pratt G., Ferrell K., Rechsteiner M. (1992) Purification of an 11 S regulator of the multicatalytic protease. J. Biol. Chem. 267, 22369–22377 [PubMed] [Google Scholar]
- 66. Li J., Gao X., Ortega J., Nazif T., Joss L., Bogyo M., Steven A. C., Rechsteiner M. (2001) Lysine 188 substitutions convert the pattern of proteasome activation by REGγ to that of REGs α and β. EMBO J. 20, 3359–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kobayashi Y., Kume A., Li M., Doyu M., Hata M., Ohtsuka K., Sobue G. (2000) Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J. Biol. Chem. 275, 8772–8778 [DOI] [PubMed] [Google Scholar]
- 68. Muchowski P. J., Schaffar G., Sittler A., Wanker E. E., Hayer-Hartl M. K., Hartl F. U. (2000) Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 97, 7841–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wacker J. L., Zareie M. H., Fong H., Sarikaya M., Muchowski P. J. (2004) Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 11, 1215–1222 [DOI] [PubMed] [Google Scholar]
- 70. Warrick J. M., Chan H. Y., Gray-Board G. L., Chai Y., Paulson H. L., Bonini N. M. (1999) Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 23, 425–428 [DOI] [PubMed] [Google Scholar]
- 71. Glover J. R., Lindquist S. (1998) Hsp104, Hsp70, and Hsp40. A novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82 [DOI] [PubMed] [Google Scholar]
- 72. Cummings C. J., Sun Y., Opal P., Antalffy B., Mestril R., Orr H. T., Dillmann W. H., Zoghbi H. Y. (2001) Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum. Mol. Genet. 10, 1511–1518 [DOI] [PubMed] [Google Scholar]
- 73. Baldo B., Weiss A., Parker C. N., Bibel M., Paganetti P., Kaupmann K. (2012) A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein. J. Biol. Chem. 287, 1406–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tam S., Geller R., Spiess C., Frydman J. (2006) The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 8, 1155–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kitamura A., Kubota H., Pack C. G., Matsumoto G., Hirayama S., Takahashi Y., Kimura H., Kinjo M., Morimoto R. I., Nagata K. (2006) Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 8, 1163–1170 [DOI] [PubMed] [Google Scholar]
- 76. Tam S., Spiess C., Auyeung W., Joachimiak L., Chen B., Poirier M. A., Frydman J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Menéndez-Benito V., Heessen S., Dantuma N. P. (2005) Monitoring of ubiquitin-dependent proteolysis with green fluorescent protein substrates. Methods Enzymol. 399, 490–511 [DOI] [PubMed] [Google Scholar]
- 78. Ross C. A., Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 [DOI] [PubMed] [Google Scholar]
- 79. Hands S. L., Wyttenbach A. (2010) Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta Neuropathol. 120, 419–437 [DOI] [PubMed] [Google Scholar]
- 80. Arrasate M., Mitra S., Schweitzer E. S., Segal M. R., Finkbeiner S. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810 [DOI] [PubMed] [Google Scholar]
- 81. Takahashi T., Kikuchi S., Katada S., Nagai Y., Nishizawa M., Onodera O. (2008) Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 17, 345–356 [DOI] [PubMed] [Google Scholar]
- 82. Miller J., Arrasate M., Brooks E., Libeu C. P., Legleiter J., Hatters D., Curtis J., Cheung K., Krishnan P., Mitra S., Widjaja K., Shaby B. A., Lotz G. P., Newhouse Y., Mitchell E. J., Osmand A., Gray M., Thulasiramin V., Saudou F., Segal M., Yang X. W., Masliah E., Thompson L. M., Muchowski P. J., Weisgraber K. H., Finkbeiner S. (2011) Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 7, 925–934 [DOI] [PMC free article] [PubMed] [Google Scholar]