

Background: Multiple protein amyloids can underlie both functional and pathological processes in various organisms.
Results: The common properties of different amyloids enable their isolation and identification.
Conclusion: A non-targeted proteomic approach can identify amyloid-forming and amyloid-associated proteins extracted directly from cells.
Significance: Novel amyloid-associated proteins can be identified in less tractable organisms and model systems, requiring no special genetic tools.
Keywords: Aggregation, Amyloid, Mass Spectrometry (MS), Polyglutamine, Prions, Stress Granule, Yeast, FUS, rnq1, sup35
Abstract
The formation of amyloid aggregates is implicated both as a primary cause of cellular degeneration in multiple human diseases and as a functional mechanism for providing extraordinary strength to large protein assemblies. The recent identification and characterization of several amyloid proteins from diverse organisms argues that the amyloid phenomenon is widespread in nature. Yet identifying new amyloid-forming proteins usually requires a priori knowledge of specific candidates. Amyloid fibers can resist heat, pressure, proteolysis, and denaturation by reagents such as urea or sodium dodecyl sulfate. Here we show that these properties can be exploited to identify naturally occurring amyloid-forming proteins directly from cell lysates. This proteomic-based approach utilizes a novel purification of amyloid aggregates followed by identification by mass spectrometry without the requirement for special genetic tools. We have validated this technique by blind identification of three amyloid-based yeast prions from laboratory and wild strains and disease-related polyglutamine proteins expressed in both yeast and mammalian cells. Furthermore, we found that polyglutamine aggregates specifically recruit some stress granule components, revealing a possible mechanism of toxicity. Therefore, core amyloid-forming proteins as well as strongly associated proteins can be identified directly from cells of diverse origin.
Introduction
Pathological aggregation of specific proteins is a hallmark of numerous neurodegenerative disorders, including Alzheimer, Parkinson, and Huntington diseases. For each disease a particular protein (or peptide) is considered the primary aggregating species. In many cases these proteins assemble into amyloid fibers that accumulate into larger deposits that resist normal protein quality control mechanisms. Amyloid is a highly ordered and stable type of protein aggregate that can form in intra- and extracellular spaces and can recruit multiple associated proteins (e.g. components of protein quality control machinery) in addition to the core protein(s). The identification of associated proteins suggested a toxicity mechanism by which functional cellular components are titrated out of solution. For example, sequestration of transcription factors and soluble motor proteins by polyglutamine aggregates can contribute to disease pathology (1, 2).
Beyond links to human disease, amyloid has been found in organisms from bacteria to humans, frequently serving beneficial functions (for review, see Refs. 3 and 4). The proposed roles of amyloid-forming proteins include biofilm formation, cellular adhesion, protein scaffolding, signaling cascades, and epigenetic inheritance. Yeast Saccharomyces cerevisiae naturally possesses a variety of proteins, called yeast prions, that form intracellular amyloid that can propagate within yeast culture for many generations (5, 6). Yeast prion infection occurs by transfer of amyloid aggregates between cells during mating or cell division. Prion formation is reversible and can create inherited phenotypes associated with modified functional activity of the prion protein. The accumulation of prion amyloid in yeast can be disadvantageous under standard growth conditions (7), but it is also proposed to be beneficial under certain stress conditions (8, 9).
The capacity to adopt an amyloid conformation is inherent to many different polypeptide sequences, suggesting that the amyloid configuration is a generic, widely accessible structure of polypeptides (10). Amyloids share an unbranched filamentous morphology with a common cross-β-core structure composed of stacked β strands that lie perpendicular to the fiber axis, forming a continuous sheet over the length of the fiber (for review, see Ref. 11). Abundant interlocking hydrogen bonds between β-sheets within fibers, known as “steric zippers” (12), results in protein assemblies of extraordinary thermal and chemical stability (13). Proteins of very different origin and sequence, once they adopt an amyloid conformation, share these common properties, including resistance to proteolysis, chaotropic agents, mechanical breakage, and strong detergents (13–16).
Several high throughput genetic screens were previously utilized to uncover >20 amyloid-based prion or prion-like proteins in yeast (for review, see Refs. 5 and 17). Genetic screening is powerful but is frequently limited to model systems or genetic reporters. Given the widespread existence of the prion phenomenon in S. cerevisiae and disease-associated aggregates in humans, there is a clear need for a multisystem approach to identify amyloid-forming proteins that does not rely on genetic manipulations. We found previously that SDS-resistant prion aggregates can be separated electrophoretically in agarose gels (in lieu of standard acrylamide gels) and then detected by immunostaining in a technique called SDD-AGE3 (14), which is conceptually similar to Western blotting. This method works successfully with many amyloid and prion protein substrates (18–20) and has been adapted for high throughput detection of known prions in wild yeast strains (21, 22). However, one major drawback of SDD-AGE is it requires prior knowledge of the aggregating species for immunodetection. In this work we present a complimentary approach where the extreme detergent resistance and high molecular weight of amyloid aggregates are utilized to enrich them from cellular lysates. We also developed a new filtration scheme that enables the subsequent identification of these enriched proteins by mass spectrometry. We show this unbiased proteomic strategy can be used to identify core amyloid-forming proteins both in yeast and in mammalian cells as well as proteins that are strongly associated with amyloid.
EXPERIMENTAL PROCEDURES
Yeast Plasmids, Strains, Growth Conditions, and Cell Imaging
Plasmids used in this study are: HttQ103-GFP and HttQ25-GFP, both GAL1-inducible expressers of exon 1 of human Huntingtin protein with 103 or 25 glutamines fused in-frame with green fluorescent protein (GFP) (23); HttQ103-RFP; Sup35NM-GFP, a CUP1 promoter-controlled derivative of Sup35NM-GFP plasmid (24); pRP1662 Pub1-CherryFP and pRP1362 Pab1-GFP (25); the GAL1-controlled RNQ1-GFP (26); Yep181-Sis1, a high copy with SIS1 under native promoter and the collection of HA-tagged plasmids for yeast expression (Yeast ORF library, Open Biosystems). Laboratory yeast strains: BY4741 (MATa his3 leu2 met15 ura3), BY4741 Pbp1-GFP (Invitrogen), 74D-694 (Mat a, ade1-14, ura3, leu2, trp1, his3) (27) [PSI+][RNQ+], [psi-][RNQ+], and [psi-][rnq-] (prion negative versions were confirmed by genetic tests and by fluorescent microscopy); BY241 (Mat a, ura3, leu2, trp1, Pdal5-ADE2, kar1, [URE3] or [ure-o]) (28); BY241 Δhsp104 (genomic deletion of HSP104); DK365 (Mat a, ade1-14, ura3, leu2, trp1, his3, [RNQ+]); W303 (MATa can1-100 his3-11,15 leu2-3,112 trp1-1 ura3–1 ade2–1) were used. We also collected (kind gifts of Drs. Fink and Kruglyak, also purchased from Davis Phaff Collection, University of California, Davis, SA) and analyzed the following wild yeast strains referenced by Halfmann et al. (22): ucd587, ucd939, ucd824, ucd2534, 5672, ncyc3467, ucd978, ucd521, ucd885, and ucd229 (all 10 contain [PSI+]) as well as ATCC26249, YJM428, YJM653, I14, WE372, F1634, F1535, F1640, F1545, and F1548. Yeast cells were grown at 30 °C in standard rich yeast extract/peptone/dextrose or synthetic media containing 2% glucose as a carbon source. For overexpression of genes under the copper promoter, cells were cultured in synthetic media supplemented with 50 μm CuSO4. For prion curing, cells were grown on yeast extract/peptone/dextrose supplemented with 4 mm GuHCl for at least 60 generations.
Imaging of live yeast cells expressing the appropriate GFP fusion proteins was performed on a Zeiss Pascal laser scanning confocal microscope equipped with a Plan-Apochromat 100×/1.4 oil objective (Carl Zeiss Inc.) and appropriate filters. The pinhole size was set at 0.6 μm. Image acquisition and analysis was carried out with LSM 5 software. 2% poly-l-lysine and 2% concanavalin A (Sigma) were used for coating glass slides (5 min at room temperature) to immobilize yeast cells.
Amyloid Enrichment and Identification from Yeast Cells
Yeast cells were grown in yeast extract/peptone/dextrose medium to A600 = 1 or 2. About 0.4 g of yeast pellet was used for the regular sample preparation. Cells were washed with cold TBS and disrupted with glass beads in 0.7 ml of buffer A (30 mm Tris-HCl, pH 7.5, 5 mm DTT, 40 mm NaCl, 3 mm CaCl2, 3 mm MgCl2, 5% glycerol, Halt protease inhibitors mixture (Thermo Scientific), 20 mm N-ethylmaleimide (Sigma), 0.5 μl of Benzonase nuclease (250 units/ul)) for 3 min by vortexing. The lysates were spun for 10 min at 800 × g at 4 °C to remove unbroken cells and cell debris. The supernatant was placed in a separate tube and treated with 100 μg/ml RNase A for 30 min on ice followed by 1% Triton X-100 for 10 min on ice. The lysate was then spun at 10,000 × g for 15 min at 4 °C and loaded onto the top of 1 ml of a 40% sucrose pad in the centrifuge tube. The lysate was spun for 1 h at 200,000 × g to concentrate the amyloid material. The pellet was rigorously suspended in 200 μl of buffer B (TBS, pH 7.5, 2% SDS, 5 mm DTT, Halt protease inhibitors with EDTA) and incubated at 37 °C for 15–30 min. During incubation the solution was mixed by pipetting multiple times to resuspend partially insoluble proteins, resulting in a clear solution with few visible particles. The solution was spun at 16,000 g for 12 min; the supernatant was mixed with 0.05% bromphenol blue and loaded on the top of the acrylamide gel (Any kDaTM, Bio-Rad) for SDS-PAGE. After electrophoresis, small pieces of the acrylamide gel (2–3 mm) around the base of the wells were removed with a razor blade, combined in one tube, and frozen at −20 °C for 30 min or overnight. For elution of aggregates, the gel pieces were resuspended in buffer C (10 mm Tris, pH 7.5, 0.3% SDS, 5 mm DTT) by pipetting and incubated at 37 °C for 10 min followed by a short centrifugation and collection of the supernatant. This procedure was repeated three times to ensure good elution. The aggregates were pelleted for 1 h at 200,000 × g and resuspended in a minimal volume of water before electron microscopy and immunogold labeling. For identification of the trapped proteins, the same elution procedure was performed at 95 °C instead of 37 °C, which provided conditions for partial disruption of aggregates. Alternatively, more complete elution of proteins from gel pieces can be done by electroelution (Eluter 422, Bio-Rad) according to the instructions. However, the efficiency of the electroelution was often comparable with only the heating (95 °C) and simple resuspension procedure described above.
After elution, the material was concentrated with a Spin-vac concentrator (Savant DNA 120, Thermo Scientific). For detection of the eluted proteins by immunostaining or Coomassie staining, the eluted material was subjected to SDS-PAGE. For mass spectrometry identification of the eluted proteins, the mixture was subsequently applied to the HiPPR Detergent Removal column and Zeba Desalting column (both from Thermo Scientific) to remove SDS and salts. The buffer was exchanged to 50 mm ammonium bicarbonate, pH 8.0. 0.1% RapiGest surfactant (Waters) was added to the purified mixture to help the following protease digestion. The sample was treated with 10 mm DTT for 5 min at 95 °C followed by 50 mm iodoacetamide for 30 min at room temperature. The sample was digested with Lys-C protease (Promega) for 3 h at 37 °C followed by trypsin (Promega) digestion for 16 h at 37 °C. The digestion reaction was stopped by the addition of 0.5% formic acid, incubated for 30 min at 37 °C to hydrolyze Rapigest, and spun 16,000 × g for 15 min. The supernatant was dried with the Spin-vac concentrator and sent for mass spectrometry identification.
We used in-house one-dimensional reverse phase ultra performance liquid chromatography (Waters) coupled in-line with a Xevo G2 QT of mass spectrometer (Waters) and ProteinLynx Global Server (Waters) for protein identification or two different commercial services, Mass Spectrometry and Proteomics Facility at Johns Hopkins School of Medicine and the Keck Proteomics Center at Yale University. The Johns Hopkins services were performed with a Thermo LTQ ion trap MS interfaced with a two-dimensional nanoLC system (Eksigent), and the Keck services utilized a Thermo Scientific LTQ-Orbitrap XL mass spectrometer. Protein identification was performed with the Mascot algorithm (Version 2.4.0) with the following search parameters: peptide mass tolerance, 15 ppm; fragment mass tolerance, 0.6 Da; maximum number of missed cleavages, 1; variable modifications carbamidomethyl (C) and oxidation (M). The in-house MS was used for the initial method development, and the commercial services were used for the majority of samples to obtain consistent and reproducible results and for sample comparisons.
Mammalian Cells Analysis and Amyloid Identification
The PC12 cells that model huntingtin (htt) aggregation expressing 74Q were previously described (29). The cells were maintained in high glucose Dulbecco's modified Eagle's medium with 100 units/ml penicillin/streptomycin, 2 mm l-glutamine, 10% horse serum, 5% Tet-free fetal bovine serum, and 100 μg/ml G418 at 37 °C and 10% CO2. Induction of htt expression was done by incubating the cells in media containing 1 μg/ml doxycycline for 48 h.
To prepare cell lysates for TAPI, five T75 flasks of PC12 cells with the induced expression of HttQ74-GFP were collected, washed with cold PBS, resuspended in 1 ml of radioimmune precipitation assay buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 1% deoxycholate, 0.1% SDS, 2 mm EDTA, Complete protease inhibitor mixture (Roche Applied Science), 5% glycerol), and lysed by pipetting on ice for 15 min. Lysates were clarified by 300 × g spin for 5 min and incubated with 0.2 mg/ml RNase I, 60 units/ml DNase I, 10 mm MgCl2 for 30 min on ice. After the second spin (300 × g, 13 min) lysates were loaded onto the top of 1 ml of 40% sucrose pad in the centrifuge tube followed by the standard TAPI procedure described above.
Analysis of Aggregates
Agarose Gel Electrophoresis (SDD-AGE) of prion aggregates was performed as previously described (14). Briefly, cell lysates were sedimented through a 30% sucrose pad (1 ml) in an SW55 rotor (Beckman) for 1 h at 45,000 rpm to enhance resolution of aggregates on the final picture. Recovered pellets were treated with 2% SDS for 10 min at 37 °C and separated by horizontal 1.8% agarose gel electrophoresis in Tris borate-EDTA buffer with 0.1% SDS. After electrophoresis, proteins were transferred from gels to Immobilon-P PVDF sheets (Millipore) overnight with a vacuum transfer unit (GE Healthcare) followed by immunostaining with anti-Sup35 antibodies.
Filter retardation assay were performed as described previously (30). Briefly, lysates of cells expressing HA-tagged yeast proteins for 5 h in SDGal were equalized, treated with 1% SDS for 10 min at room temperature, vacuum-blotted to cellulose acetate or PVDF membranes, washed with blotting buffer, and stained with an anti-HA antibody. As a modification of the method, SDS-treated cell lysates were loaded on acrylamide gel without boiling and subjected to SDS-PAGE under standard conditions. Resistant aggregates can be detected below loading wells at the start of the gel.
Transmission Electron Microscopy, Immuno-EM, and Antibodies Used in the Study
Extracted protein samples were dispensed as 10-μl aliquots on carbon/Formvar-coated copper grids (Ted Pella Inc.) and incubated for 15–20 min. Samples were blotted off with absorbent paper and washed by adding 10 μl of water quickly to the grid surface. This was repeated with a second round of water. Next, the water was blotted off, and 10 μl of 2% uranyl acetate stain was added to the grid surface. After about 2 min the stain was blotted off, and the grid was air-dried. Images were collected using a Jeol JEM-1011 operating at 80 kV.
The immuno-EM methodology was derived from Wischik et al. (31, 32). Briefly, samples were dispensed to grids as described above. Next, the grids were treated with 10 μl of blocking solution (2.5% bovine serum albumin, 0.1% cold fish gelatin) for 1 h. The blocking solution was blotted away, and then 10 μl of primary antibody in the same solution was added to the grid. After about 1 h, the primary antibody was removed, and the grid surface was washed 5 times with 10-μl aliquots of blocking solution. Secondary antibody in the same solution was dispensed to the grid and incubated for about 20 min. Finally, the grid surface was washed several times with blocking solution and then negative-stained with uranyl acetate as described above.
Antibodies used in the study were as follows: polyclonal anti-Sup35 and anti-Rnq1 (in house); anti-GFP and anti-HA (3F10) (Roche Applied Science); polyclonal anti-Sis1 (a gift of Dr. Cyr, University of North Carolina); anti-TDP-43 (Proteintech) and anti-FUS (Bethyl).
RESULTS
Proteomic Technique for Amyloid Identification
Strong anionic detergents like sodium dodecyl sulfate (SDS) and sarkosyl are known for their ability to solubilize and destroy most protein-protein complexes, ultimately leading to protein denaturation. In contrast, our previous work established that interactions within yeast prion aggregates are strong enough to survive the treatment of 2% SDS at 37 °C (14). We hypothesized that the biophysical properties of protein amyloid, such as its SDS resistance and large molecular weight would enable sufficient purification and enrichment from biological source material for identification by mass spectrometry.
The details of our approach to isolate amyloid aggregates are illustrated in Fig. 1 and are explicitly outlined under “Experimental Procedures.” The essential steps are summarized here. After cell disruption, we reduced the complexity of the cellular lysates by extensive nuclease treatment and differential centrifugation (Fig. 1, Step 2). The centrifugation isolated prion aggregates, ribosomes, and other high molecular weight assemblies. To disrupt most of these complexes but not prion aggregates, the mixtures were thoroughly treated with 2% SDS at 37 °C (Fig. 1, Step 3). To separate resistant prion aggregates from the solubilized material, the suspensions were loaded on an acrylamide gel and subjected to electrophoresis. Although solubilized proteins migrated into the gels, the resistant prion aggregates were trapped at the start of the gels due to their large size (Fig. 1, Step 4).
FIGURE 1.
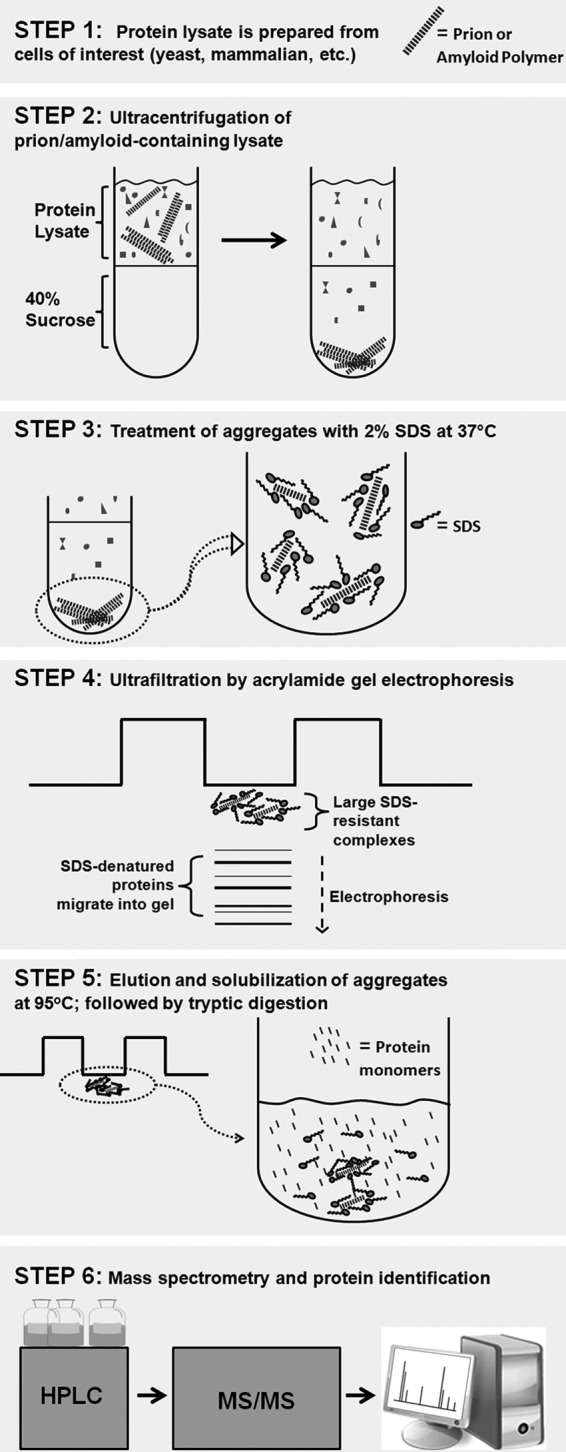
The workflow of the TAPI. See a detailed description under “Results” and “Experimental Procedures.”
To evaluate how efficiently the prion aggregates could be trapped, we compared the isogenic yeast strain pair, where one strain has two prions ([PSI+] and [RNQ+], respectively, composed of amyloid forms of the proteins Sup35 and Rnq1), and the other has no prions ([psi-], [rnq-]; the proteins Sup35 and Rnq1 are in the soluble non-prion form). Both strains were grown and processed under identical conditions, treated with the procedure described above, and compared on the same gel after electrophoresis. For the strain with prions, substantially more material was visualized at the gel interface using Coomassie Blue stain, whereas the majority of the loaded proteins successfully migrated into the gel (Fig. 2A). Staining of the same gel with antibodies specific for the Sup35 and Rnq1 proteins clearly showed that these proteins were stuck at the start of the gel only when they are present in the prion form (Fig. 2B).
FIGURE 2.
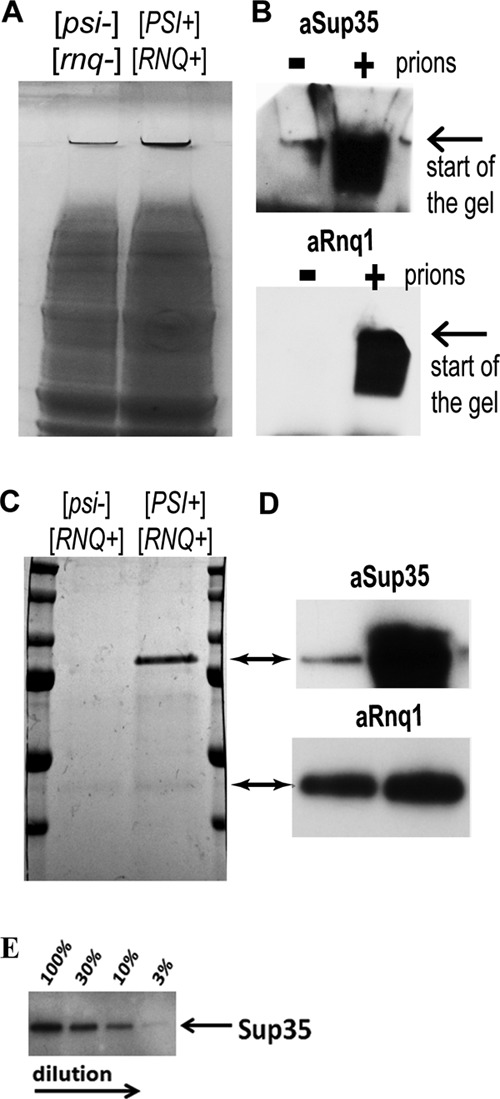
Purification of prion aggregates based on their SDS resistance. A, the isogenic yeast strain pair, 74D-694 [PSI+][RNQ+] and [psi-][rnq-], was lysed and treated with nucleases under identical conditions, as described under “Experimental Procedures.” After ultracentrifugation, the pellets were treated with 2% SDS at 37 °C, subjected to SDS-PAGE, and stained with Coomassie Blue. B, the same samples from A were subjected to SDS-PAGE and stained with anti-Sup35 and anti-Rnq1 antibodies. C, the isogenic yeast strain pair, differing only in its [psi] state (74D-694 [PSI+][RNQ+] and [psi-][RNQ+]), was lysed and treated as in A. Trapped proteins were eluted from the gel pieces by heating at 95 °C, concentrated, subjected to SDS-PAGE, and stained with Coomassie Blue. D, the same samples from C were subjected to SDS-PAGE and stained with anti-Sup35 and anti-Rnq1 antibodies. E, detection of Sup35 in a dilution series. 74D-694 [PSI+][RNQ+] and [psi-][rnq-] lysates were prepared and equalized by total protein concentration using a BCA assay. Four samples were prepared by diluting the prion lysate with the non-prion lysate in proportions where the final mixtures contained 100, 30, 10, and 3% of the prion lysate correspondingly. Samples were prepared by TAPI, and equal aliquots of the eluted and purified fractions (right before protease digestion) were subjected to SDS-PAGE and immunoblotting with anti-Sup35 antibodies.
We next attempted to elute the trapped amyloid aggregates from the acrylamide matrix. Thin gel pieces at the start of the gel were excised, placed in a small amount of the elution buffer, and heated to 95 °C with occasional mixing and pipetting (Fig. 1, Step 5). Because SDS works more efficiently at 95 °C, amyloid aggregates partially or completely fall apart. Eluted fractions were concentrated and subjected to SDS-PAGE. Remarkably, the Sup35 and Rnq1 proteins were detected by general protein staining (Coomassie Blue) specifically in the cases where these proteins were present in the amyloid form (Fig. 2C). The identities of the bands were confirmed by immunostaining with antibodies specific for the Sup35 and Rnq1 proteins (Fig. 2D). Very few other proteins were detected, showing that Sup35 and Rnq1 constitute a major proportion of the trapped aggregates. The yield for the Sup35 prion protein was estimated to be about 10 mg/liter of yeast culture grown on rich medium; the yield for Rnq1 was about 1–3 mg/liter of yeast culture (Rnq1 is less abundant in vivo).
We next determined if the isolated protein species from the aggregates could be identified by mass spectrometry. The heat-eluted fractions were depleted of SDS and salts and then digested with trypsin (Fig. 1, Step 6). Peptide mixtures were analyzed by tandem mass spectrometry (MS/MS), and the identities of the proteins were recovered by searching the S. cerevisiae protein database. Identified proteins were arranged by protein score, which reflects the number of unique peptides found for each protein and their relative abundance as well as the percentage of sequence coverage. In general, higher protein scores indicate a more abundant protein in the mixture. Sup35 and Rnq1 were consistently identified with the highest score in the [PSI+][RNQ+] strain, whereas they were absent in the isogenic [psi-] [rnq-] strain (Table 1). Strikingly, >70% of the possible tryptic peptides within a mass range of 500–3500 Da were identified by MS/MS for each protein, confirming their abundance in the mixture (supplemental Fig. S1, A and B, and S2). Other identified proteins had much lower scores and included mostly ribosomal proteins. We later found that the majority of ribosomal proteins can be removed by more thorough SDS treatment (>20-min incubation with occasional pipetting; see Table 1, right columns). Also, certain components of translation machinery (Tef1 and Tef3) were identified (see Tables 1 and 2); their presence may vary from sample to sample reflecting arbitrary distribution to the SDS-resistant fractions.
TABLE 1.
Identification of prion proteins by tandem mass spectrometry
The amyloid identification procedure (TAPI) was performed on the isogenic yeast strain pair, 74D-694 [PSI+][RNQ+] and [psi-][rnq-]. The trypsin-treated fractions were analyzed using the in-house mass spectrometer (two left columns) or by commercial service (Keck Biotechnology Resource Laboratory at Yale University; two right columns). Samples with asterisks (*) were treated more thoroughly with SDS to remove the majority of ribosomal proteins (marked by italics). The hits are arranged by protein score. Bold type indicates known prion proteins.
| 74D[PSI+][RNQ+] |
74D[psi-][rnq-] |
74D[PSI+][RNQ+]* |
74D[psi-][rnq-]* |
||||
|---|---|---|---|---|---|---|---|
| Protein name | Protein score | Protein name | Protein score | Protein name | Protein score | Protein name | Protein score |
| Sup35 | 3123 | RS11A | 247 | Sup35 | 1212 | RL30 | 55 |
| Rnq1 | 2042 | RS11B | 247 | Rnq1 | 328 | Tef3 | 46 |
| RL27A | 848 | RL30 | 211 | Nsr1 | 186 | Ecm33 | 42 |
| RL27B | 848 | RL27A | 198 | RS5 | 129 | Cdc28 | 37 |
| RL6A | 540 | RL27B | 198 | RL30 | 88 | Vps5 | 37 |
| RL6B | 505 | Pma1 | 194 | RL10 | 87 | Msc7 | 33 |
| RL2B | 373 | Pma2 | 183 | Cdc19 | 39 | Ptc2 | 29 |
| RL2A | 373 | RS3 | 140 | Msc7 | 30 | Ygr130c | 26 |
| RS9B | 342 | Tef1 | 115 | Cdc28 | 26 | Mnn9 | 21 |
| RS9A | 342 | RL24A | 102 | Sis1 | 24 | ||
| RL25 | 322 | RL24B | 102 | Mnn9 | 21 | ||
| RL4A | 315 | RS2 | 93 | Faa4 | 21 | ||
TABLE 2.
Analysis of laboratory and wild yeast strains by TAPI
The amyloid identification procedure (TAPI) was performed on the listed yeast strains. The trypsin-treated fractions were analyzed blindly by commercial service (Keck Proteomics). Strains having GuHCl in their names were grown on YPD supplemented with 4 mm GuHCl for at least 60 generations to eliminate known amyloid-based prions. “Dilution” indicates the prion dilution experiment described under “Results” and in Fig. 2E. Yeast core ribosomal proteins as well as Tef1 and Tef3 were excluded from this table (they appear non-specifically in many samples).
| Strain name | Proteins identified by mass spectrometry |
|---|---|
| BY241[URE3] | Ure2, Sup35, Rnq1, Nop58, Sod2, Mrs3, Qri7, Nsr1, Cdc28, Pma1, Mnn9, Msc7 |
| BY241 GuHCl | Bmh1, Scg1, Mdh1, Yfr016c, Yor220w, Rad50, Cdc28, Coq5, UBI4, Ygr131w |
| BY4741 HttQ103-GFP | Q103-GFP, Rnq1, Pub1, Ynl208w, Bmh1, Glg2, Glg1, Snz2, Ptc2, Msc7, Sov1, Ctf18, Prm9, Ypr091c |
| 74D694[PSI+][RNQ+] 30% total (dilution) | Sup35, Rnq1, Nop58, Vps5, Msc7, Sis1, Cdc28, Mnn9, Ygr130c |
| 74D694[PSI+][RNQ+] 10% total (dilution) | Sup35, Nop58, Vps5, Ymr087w, Mrs3, Yir004w, Npp2, Ptc2, Sam4, Cdc28, Msc7, Tao3, Lys2, Opi3 |
| 74D694[PSI+][RNQ+] 3% total (dilution) | Vps5, Mrs3, Yir004w, Cdc28, Tao3, Mnn9 |
| DK365 [RNQ+] | Rnq1, Nop58, Nsr1, Pma1, Deg1, Ygr122w |
| ucd939[PSI+][RNQ+] | Rnq1, Sis1, Msc7, Nop58, Ynd1, Cdc28, Sam4, Ams1, Mnn9 |
| ucd939 GuHCl | Nop58, Ecm33, Cdc28, Yrf1, Yir004w, Ymr087w, Sdh1, Myo1, Lys2, Cue2, Ygr130c, Ysp1, Rtn1, Msl5 |
| ucd978 | Sup35, Rnq1, Msc7, Ykr015c, Mnn9 |
| ATCC26249 | Msc7, Myo1, Mnn9, Fhn1 |
| ATCC26249 GuHCl | Pma1, Sir1, Mrs3, Mnn9, Cdc28, Msc7 |
| I14 | Ssk1, Mnn9, Myo1, Cdc28, Pma1, Msc7 |
| I14 GuHCl | Med8, Sir1, Yfr016c, Rsc30, Rad50, Cdc28, Sam4, Hrd3 |
| YJM653 | Cdc19, Glg2, Pma1, Nop58, Cdc28, Sam4, Glg1, Msc7, Hrd3, Gir2 |
| YJM653 GuHCl | Vps5, Cdc28, Ptc2, Sov1, Msc7, Ylr290c, Mdn1 |
| F1535 | Pma1, Yil169, Nop58, Nsr1, Cdc28, Ygr131w |
| F1545 | Rsc3, Cdc28, Myo1, Mnn9, Msc7, Zip1, Ynd1 |
| F1545 GuHCl | Uba3, Ecm22, Coq5, Cdc28 |
| YJM428 | Cdc19, Yjr129c (SAM), Nop58, Deg1, Ptc2p, Has1, Msc7 |
| YJM428 GuHCl | YFR016C, Snz2, Ktr7, Gfd1, Mnn9 |
| BY4741 | Glg1, Glg2, Hst2, Msc7, Pma1, Def1, Rgd2, Mnn9, Myo1 |
| BY4741 Δhsp104 | Glg1, Glg2, Act1, Msc7, Myo1, Cdc28 |
| W303 | Med8, Bmh1, Mnn9, Ygr130c, Htb2, Cdc28, Yir004w, Msc7 |
We performed the same amyloid identification for the strain BY241 (28) with another well characterized yeast prion, [URE3], which is formed by the Ure2 protein. Proteomic data analysis after mass spectrometry resulted in Ure2 at the top of the protein-score list (along with the other prion proteins), confirming the method suitability for the detection of [URE3] (Table 2). We will subsequently refer to this method for identification of amyloid-based prions as “TAPI” (technique for amyloid purification and identification).
Detection of Prions in Wild Strains
The major advantage of TAPI is that it requires little genetic information of the tested organism and no specific detection tools, such as antibodies. Therefore, it could be applied to wild yeast strains and a variety of other organisms, including fungal pathogens, mammalian cell lines, etc. We tested if TAPI could be used to detect prions in wild S. cerevisiae strains. Prions in wild yeast strains occur quite rarely but have been documented (22, 24, 33, 34). For example, among 770 wild strains tested, 10 were reported to contain the [PSI+] prion (the same strains also contain [RNQ+], as [RNQ+] facilitates [PSI+] formation). We analyzed these wild S. cerevisiae strains by TAPI and unexpectedly identified the presence of the [PSI+] prion only in 3 of 10 reported strains. We performed SDD-AGE analysis on the set of reported [PSI+] wild strains and found strikingly that only the three strains that we confirmed by TAPI indeed contain large SDS-resistant aggregates composed of Sup35, whereas the remaining strains contain mostly low molecular weight oligomeric species (Fig. 3A). To confirm this finding, we transformed the reported [PSI+] wild strains with a low copy plasmid that encodes the reporter Sup35NM-GFP, which is widely used for detection of [PSI+] variants based on the formation of fluorescent foci (35). Fluorescent microscopy imaging strongly corroborated our SDD-AGE analysis; large visible foci were detected only in three strains with large SDS-resistant aggregates but not in strains with oligomeric Sup35 aggregates (Fig. 3B). In all strains a substantial fraction of cells (>50%) did not show formation of large foci. Therefore, these wild yeast contain [PSI+] with unusually small and/or SDS-sensitive Sup35 aggregates, which are distinct from laboratory variants and have not been previously reported. At the same time, Rnq1 was detected by TAPI in the same wild strain containing small Sup35 aggregates, indicating the presence of the typical [RNQ+] prion (Table 2, UCD939) and confirming that there is nothing particular to the strain preventing TAPI from working.
FIGURE 3.
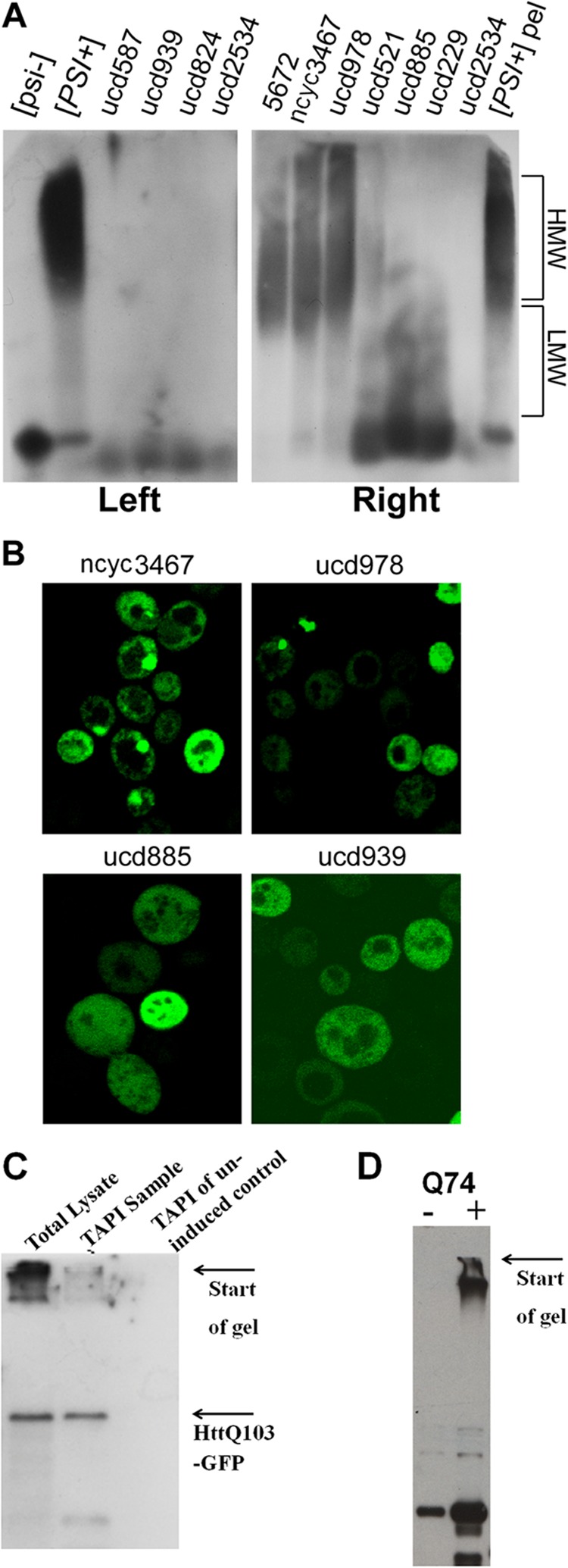
Verification of protein aggregation. A, 10 [PSI+] wild strains were analyzed by SDD-AGE (14); Sup35 was detected by immunostaining. The isogenic pair 74D-694 [PSI+][RNQ+] and [psi-][rnq-] was used as controls. HMW,- high molecular weight aggregates; LMW, low molecular weight aggregates. In the left panel, cell lysates were directly applied to SDD-AGE; in the right panel they were first pelleted (200,000 × g for 1 h), and pellets were applied to SDD-AGE (pelleting improves resolution of low molecular weight aggregates). B, the same wild strains were transformed with Sup35NM-GFP and analyzed by fluorescent microscopy. C and D, detection of polyglutamine-GFP by immunoblotting. C, yeast cells expressing HttQ103-GFP from the inducible GAL1 promoter for 5 h were lysed and subjected to TAPI. HttQ103-GFP was detected by immunoblotting with anti-GFP antibodies from crude cell lysate (lane 1) and after elution and purification before protease digestion (lane 2). No signal was detected from the uninduced control cells. D, Western analysis of the PC12 huntingtin cell model (29) with stably transfected exon 1 of the HD gene with 74 CAGs under the control of a tetracycline promoter.
Identification of Polyglutamine Amyloid and Associated Proteins from Yeast and Mammalian Cells
Next, we tested whether TAPI is suitable for identification of the intracellular amyloid formed by a disease-related human protein during yeast expression. For this purpose we expressed in yeast HttQ103-GFP, a model protein that consists of the exon 1 of human Huntingtin protein with the expanded polyglutamine track fused in-frame with GFP. The glutamine expansion promotes aggregation of the protein and is linked with the development of clinical symptoms in Huntington disease (for review, see Ref. 36). Exon 1 models have been powerful tools for studying aggregate formation in relation to cell death in various systems from yeast to mice, mimicking truncated versions of Huntingtin found in intraneuronal aggregates (37, 38). This protein readily forms amyloid during yeast expression with properties that resemble yeast prion aggregates, such as SDS resistance (20, 23).
We applied TAPI to yeast cells expressing HttQ103-GFP from the inducible GAL1 promoter for 5 h, which yielded HttQ103-GFP at the top of the identification list (see Table 2; 7 unique peptides were detected), implying that TAPI is suitable for identification of a non-prion intracellular amyloid when such amyloid is sufficiently abundant. The presence of protein in the purified fraction was also confirmed by immunoblotting (Fig. 3C). Interestingly, together with HttQ103-GFP, we also identified two glutamine-rich proteins: Pub1 and Ynl208w (Table 2). The stress-granule protein Pub1 was recently predicted to have high intrinsic potential for prion formation (21, 39), and both proteins were previously shown to co-aggregate with polyglutamine (40, 41). These proteins were specific to the expression of HttQ103-GFP, as we could not detect them in the wild type strain (BY4741) nor in other strains tested. Their identification by TAPI argues that they physically co-polymerize with polyglutamine during co-expression.
Finally, we examined if we could identify similar amyloid-forming protein directly from mammalian cells, thus greatly expanding the applicability of TAPI. We utilized the stable, inducible PC12 cell line (29), where expression of human Huntingtin protein exon 1 with 74 glutamines fused in-frame with GFP (HttQ74-GFP) is driven by a cytomegalovirus promoter under the control of a tetracycline-responsive element. The expression of HttQ74-GFP results in progressive accumulation of insoluble ubiquitin-positive intracellular aggregates leading to cell death. Overall, this inducible cell line recapitulates many of the features of Huntington disease (29). PC12 cells were grown and induced for 48 h by the addition of doxycycline, which results in accumulation of GFP-positive aggregates within cells (29). Cells were collected and lysed, and protein expression was confirmed by Western blot (Fig. 3D). TAPI analysis returned HttQ74-GFP as the top identified protein, with excellent peptide coverage (eight unique peptides, supplemental Fig. S1C). Polyubiquitin was also identified together with HttQ74-GFP (5 unique peptides, 50% coverage, supplemental Fig. SD), confirming the abundant ubiquitination of polyglutamine aggregates as reported previously (29). Other proteins were also detected, but proving their significance will require further studies. Yet at least two detected proteins, FUS and TDP-43, are of high interest, as they form intracellular aggregates in patients with amyotrophic lateral sclerosis (42).
Recruitment of Stress Granule Proteins to Polyglutamine Aggregates
Interestingly, Pub1, FUS, and TDP-43 are components of stress granules; that, is structures forming under stress conditions for protection and processing of cellular RNAs. Their efficient recruitment to polyglutamine aggregates could disturb cellular RNA homeostasis and contribute to disease conditions. To confirm the mass spectrometry results, we tested if Pub1 along with other stress granule proteins would co-localize with polyglutamine in cellular inclusions. Pub1-CherryFP (a fusion of Pub1 with mCherry fluorescent protein) was co-expressed with HttQ103-GFP or HttQ25-GFP (a non-aggregating version containing 25 glutamines). Although Pub1 had mostly diffuse cytoplasmic staining during non-pathological HttQ25-GFP induction, it was very efficiently recruited to aggregates formed by HttQ103-GFP (Fig. 4, A and B). We then tested if other stress granule markers, Pbp1 and Pab1, would be recruited to polyglutamine aggregates. Pbp1 showed significant level of recruitment during HttQ103-GFP induction, whereas Pab1 localization remained diffuse (Fig. 4, C and D). Furthermore, we confirmed by immunostaining the specific co-aggregation of FUS and TDP-43 with polyglutamine from the PC12 cells expressing HttQ74-GFP (Fig. 5A). Further work is required to determine if other stress granule components may also be trapped.
FIGURE 4.
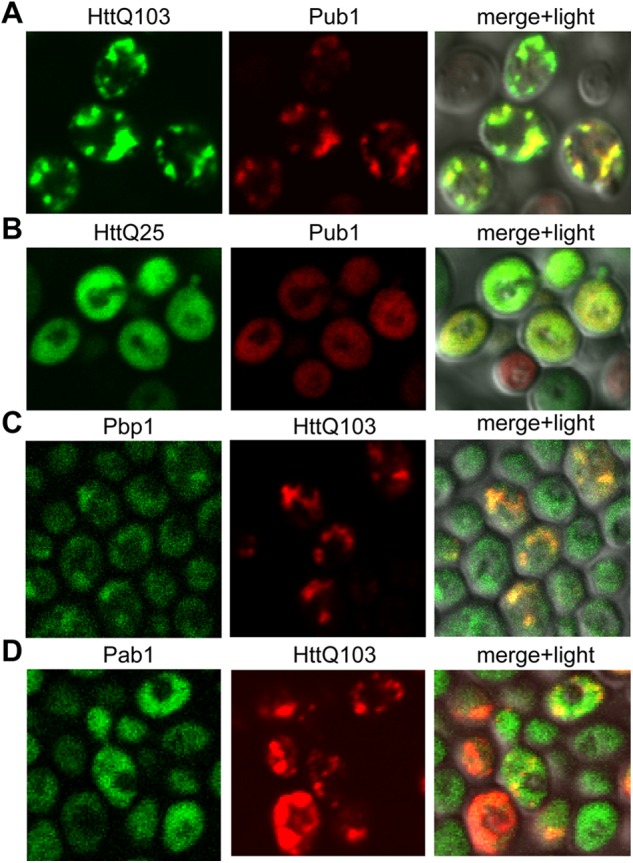
Co-localization of polyglutamine aggregates with stress granule proteins in yeast. A and B, co-expression of the GAL1-controlled HttQ103-GFP (A) or HttQ25-GFP (B) and Pub1-CherryFP in the W303. C and D, co-expression of Pbp1-GFP (C) or Pab1-GFP (D) and the GAL1-controlled HttQ103-RFP in the BY4741.
FIGURE 5.
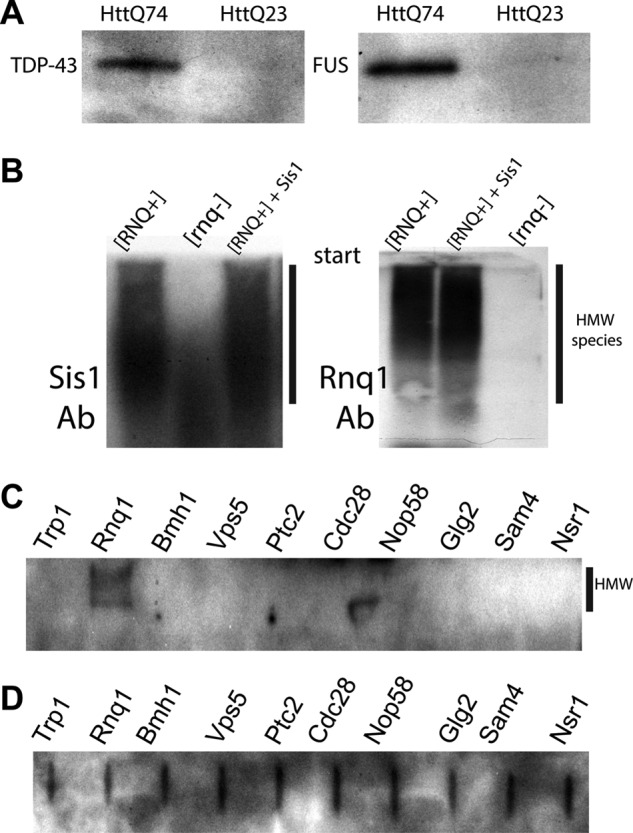
Follow-up analysis of the identified proteins. A, immunostaining of elution fractions (before protease digestion) prepared from PC12 cells expressing either HttQ74-GFP or HttQ23-GFP. Anti-TDP-43 or anti-FUS antibodies were used. B, SDD-AGE analysis of 74D-694 [RNQ+] and [rnq-] strains that expressed the GAL1-controlled RNQ1-GFP for 5 h. As a positive control, 74D-694 [RNQ+] with overproduction of Sis1 was used. Immunostaining was performed with anti-Sis1 antibodies (Ab, left) or anti-Rnq1 antibodies (right). HMW, high molecular weight aggregates. C and D, analysis of amyloid-forming potential of the identified proteins. Eight common proteins (HA-tagged, from the yeast ORF library) were expressed in BY4741. Lysates were treated with 1% SDS and loaded on an acrylamide gel for SDS-PAGE (C) or directly on PVDF membrane (D, to estimate the expression level) followed by immunostaining with the anti-HA antibodies. In C, only a portion of the gel just below the loading wells is shown.
Search for New Prions and Identification of Proteins with Amyloid-like SDS-resistant Properties
Three prions ([RNQ+], [MOT3+], and rarely, [PSI+]) were identified in wild yeast strains (22, 24), and additional unknown prions have been proposed to also exist in wild strains based on the growth comparisons of strains under specific conditions before and after GuHCl treatment (22). GuHCl is a chemical inhibitor of chaperone Hsp104 that is required for propagation of the vast majority of amyloid-based prions (for review, see Ref. 43). Assuming the GuHCl treatment of yeast cells specifically eliminates prions, the growth differences before and after treatment may indicate the presence of prions responsible for this phenotype. We have obtained several strains that do not contain [RNQ+], [MOT3+], or [PSI+] prions and were reported to show growth differences under several independent conditions (22). TAPI applied to these strains identified a set of specific proteins that were consistently present in samples, yet their identification scores were lower than for traditional prions, and most of them can be detected after GuHCl treatment in the same or different strain (Table 2). Therefore, our TAPI results suggest that any putative prions in these strains are likely distinct from the better-characterized prions.
We extended the analysis of proteins that are present in the SDS-resistant fractions for a few laboratory strains and combined the results with the wild yeast strain data to confirm the presence of a specific pool of proteins that is consistently identified by TAPI (see Table 2). We intentionally excluded core ribosomal components and two abundant translational factors (Tef1, Tef3) that were frequent contaminants in our sample preparations. Importantly, we identified chaperone Sis1 specifically in samples with the [RNQ+] prion (Tables 1 and 2). Sis1 is known to interact with Rnq1 (44) and to be required for the propagation of the [RNQ+] prion (45, 46). We confirmed this association by showing that Sis1 is specifically present in the SDS-resistant Rnq1 aggregates (Fig. 5B). In this experiment we compared the high molecular weight aggregates by SDD-AGE from strains 74D-694 [RNQ+] and [rnq-] that expressed the GAL1-controlled RNQ1-GFP for 5 h. As a positive control, 74D-694 [RNQ+] with overproduction of both Rnq1-GFP and Sis1 was used. As was reported previously, Sis1 overexpression partially protects cells from Rnq1-associated toxicity (data not shown and Ref. 44); however, it did not reduce Rnq1 aggregation (Fig. 5B).
The remaining proteins that were present independently in more than two individual strains are Pma1, Msc7, Cdc28, Mrs3, Sam4, Mnn9, Glg2, Cdc19, Nsr1p, Nop58, Myo1, Ptc2, Bmh1, Vps5, and Ygr130c. Most of these proteins are not abundant in vivo (according to high throughput analysis of protein expression in yeast) (47), so they would unlikely appear by chance in our samples. Instead, to be consistently present in the SDS-resistant fractions these proteins must form stable and large complexes. Consistent with this conclusion, high throughput localization studies of the yeast proteome (48) showed that many of these proteins (10 from 15 total) form punctate structures inside yeast cells. To address their amyloid-forming potential, eight selected proteins were expressed in yeast with the C-terminal HA tag, and formation of intracellular amyloid was determined by detection of high molecular weight SDS-resistant species trapped on the start of an acrylamide gel. Rnq1 was included as a positive control and Trp1 as a negative control. Only Rnq1 was consistently detected at the start of the gel showing efficient amyloid formation (Fig. 5C). The other proteins failed to show the formation of large SDS-resistant species like Rnq1 (Fig. 5C). A filter retardation assay, where protein aggregates are trapped on low binding cellulose membrane for detection (30), was independently employed and showed similar results (data not shown). Therefore, tested proteins do not readily form amyloid during in vivo expression.
Unstable Prions May Escape Detection
The majority of prions in S. cerevisiae are intrinsically unstable and are lost in the absence of selection. It has been proposed that the dynamic nature of prion formation and loss creates an additional regulation mechanism for protein activity (49, 50). Prion instability results in a mixed population of cells with and without prion aggregates and may create challenges for prion identification when the percentage of cells with a prion is low. To determine the threshold for identification of the known prions by TAPI, we performed a dilution series of the prion lysate (both [PSI+] and [RNQ+] were present) with the lysate of the isogenic strain that did not contain prions. TAPI was performed on samples that were prepared to contain 100, 30, 10 and 3% of the prion lysate by total protein amount. Before tryptic digestion, aliquots of purified samples were subjected to Western and immunostaining with antibodies specific to Sup35. By immunostaining we were able to detect Sup35 in all dilution samples (Fig. 2E), but by mass spectrometry Sup35 was not identified in the 3% dilution sample (Table 2), indicating that mass spectrometry is a less sensitive identification method in our hands compared with immunostaining. Rnq1 was detected by mass spectrometry only in the 30% dilution sample; this is likely explained by the limited possible peptides that can be produced with trypsin digestion due to Rnq1 lack of sequence complexity (all possible peptides reside in the first third of the protein; supplemental Fig. S1B). A certain quantity of protein is likely necessary for faithful identification by mass spectrometry. Nonetheless, TAPI can identify amyloid-based prions even if they are only present in a large minority of the total cell population.
Recovery of SDS-resistant Prion Polymers for Morphological Characterization
During the elution step of TAPI the trapped aggregates are disrupted by heating at 95–100 °C. Interestingly, we found that we could extract, albeit less efficiently, some trapped amyloid aggregates by simply resuspending the gel pieces in elution buffer and pipetting intensively without heating. When this elution procedure was performed for prion-positive and isogenic negative controls, there was clearly more proteinaceous material recovered from the prion-containing samples as judged by the total amounts of material that adhered to the hydrophobic surface of carbon/Formvar electron microscopy grids (Fig. 6, A and B). This corroborates the greater recovery of protein from prion strains as viewed by Coomassie staining in Fig. 2A. Moreover, Western analysis indicated that Sup35 and Rnq1 were specifically recovered from the prion-containing samples (Fig. 6G). Negative-stain transmission electron microscopy of material extracted from the prion-containing samples revealed several morphologically distinct species (examples shown in Fig. 6, C–F), including short barrel-like structures (Fig. 6E) that somewhat resembled structures from earlier attempts to visualize natural prion aggregates (51, 52). When recovered protein was allowed to sit for several days at room temperature, some longer filamentous structures were observed including helical structures (Fig. 6F) morphologically similar to amyloid prepared in vitro with recombinant protein (53). Such sample evolution is reminiscent of how isolated amorphous bacterial inclusion bodies contain short protofibrils that can seed the growth of longer distinct filaments that form over time (54). We attempted immunogold transmission electron microscopy to specifically identify Sup35- or Rnq1-containing species. This technique worked for highly enriched recombinant protein samples (data not shown), but with the heterogeneous mixtures and small species extracted from acrylamide gel it was difficult to objectively and unambiguously assign specific structures.
FIGURE 6.
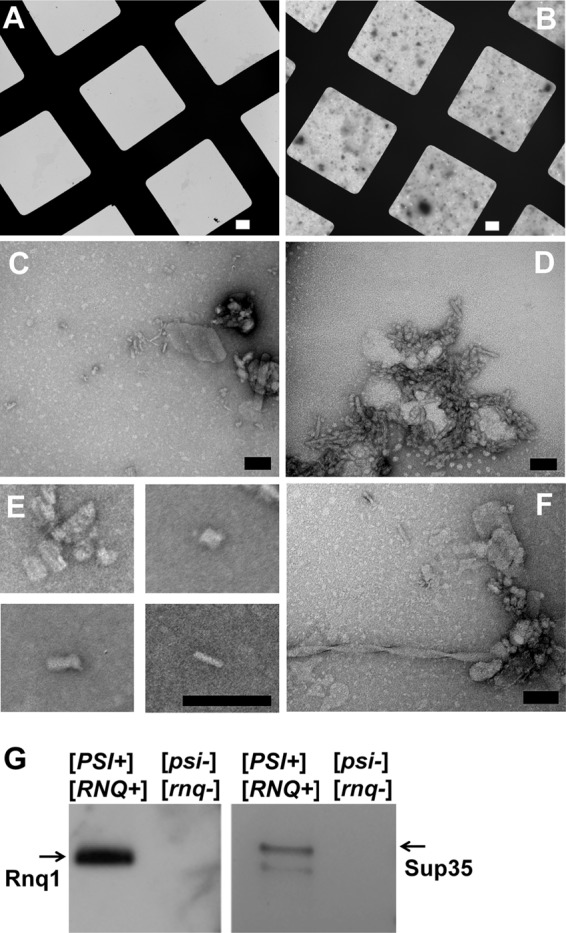
Electron micrographs of the recovered SDS-resistant protein aggregates from yeast cells. A, and B, the gel-recovered material from the isogenic strain pair 74D-694 [PSI+][RNQ+] and [psi-][rnq-] acquired at 800× direct magnification on carbon/Formvar-covered copper grids; A shows a field from the strain with no prions; B shows the field from the strain harboring both [PSI+] and [RNQ+]. C and D, representative images of the proteinaceous debris from the prion-containing strain at 80,000× and 150,000× direct magnification. E, small barrel-shaped structures observed from prion-containing samples. F, example of a fiber observed after recovered material was incubated several days at room temperature. White scale bars = 10 μm; black scale bars = 100 nm. G, Western blotting and immunostaining of the gel-recovered material from A and B above with anti-Sup35 and anti-Rnq1 antibodies.
DISCUSSION
The recent discoveries of functional amyloids in a variety of organisms including humans as well as multiple amyloid-based prion and prion-like proteins in S. cerevisiae have reshaped our traditional view of amyloid as only related to disease conditions. The pervasiveness of these mechanisms in nature, however, remains to be determined. The discoveries of prions in S. cerevisiae were often based on the genetic manipulation of the host, so it is not clear whether these proteins can efficiently acquire and maintain the prion status in wild type strains (only 3 of >12 reported prions were confirmed to exist in wild yeast thus far). In this work we found that the biophysical properties of amyloid aggregates enabled their isolation from the majority of cellular proteins. We developed these observations into a novel technique, TAPI, that we show is capable of identifying amyloid-like proteins directly from fungal and mammalian cells without prior knowledge of aggregating species.
Protein identification by TAPI is restricted to complexes with structural resistance to SDS treatment. The oligomeric intermediates or amyloid forms of many proteins that are linked to human diseases demonstrate SDS resistance, including Aβ peptide, Tau, mammalian prion protein PrP, and proteins with polyglutamine stretches (55–57). Other functional amyloid proteins and yeast prion proteins also share this property (16, 58–60). Therefore, SDS resistance appears to be a universal characteristic that distinguishes interactions within amyloid polymers. The molecular basis of this stability is attributed to the cross-β-structure with its abundance of interlocking hydrogen bonds within fibers. Interestingly, most proteins that are resistant to SDS-induced denaturation in their native states appear to be oligomeric β-sheet proteins (61), indicating that the β sheet-based structure can provide SDS resistance even without amyloid formation. Therefore, it is expected that TAPI will deliver identification of some complexes that are not amyloid. We argue that the identification of amyloid polymers will usually be superior based on their larger total molecular weight. Multiple copies of an amyloid-forming protein within the trapped aggregate provide an abundance of peptides, facilitating identification by mass spectrometry.
Our application of TAPI provided important biological information beyond amyloid identification. We successfully identified proteins that are strongly associated with aggregates. Although other mass spectrometry-based approaches were employed previously to characterize proteins associated with amyloid (Ref. 62 and references therein), our method is unique as it includes both stringent SDS treatment and a novel separation of aggregates. As a consequence, only the strongest interactors survived, reducing the burden of analyzing hundreds of hits. Although some important interactors may be missing, this method may be used in combination with other proteomic approaches.
As an example, the chaperone Sis1 was identified by TAPI together with Rnq1 specifically in [RNQ+] strains. Sis1, along with other chaperones, is known to be associated with Rnq1 prion aggregates (51), and mutations in SIS1 can affect Rnq1 aggregation (45, 46). Yet, no other chaperones were identified by TAPI, implying that among chaperones Sis1 has the strongest binding or a distinct mode of binding to prion aggregates.
In a more intriguing example, we identified two glutamine-rich proteins specifically with amyloid-forming HttQ103-GFP (Pub1 and Ynl208w; Table 2). This suggests that these proteins may co-polymerize with polyglutamine aggregates during co-expression. Pub1 is required for stability of mRNAs and is involved in stress granule assembly. Other proteins related to RNA post-transcriptional modifications have been previously identified as interactors of Huntingtin with expanded polyglutamine track (63); some of them may contribute to Huntington disease pathology similar to recruitment of glutamine-rich transcriptional factors (2). Accordingly, we identified RNA-binding proteins FUS and TDP-43 from PC12 cells expressing HttQ74. These proteins are involved in stress granule formation and have low complexity domains that are similar to yeast prion-forming domains as well as domains found in many stress granule components (64). We extended our finding by showing that another stress granule marker, Pbp1, is also recruited to polyglutamine aggregates. Therefore, efficient recruitment of stress granule components to polyglutamine aggregates may disturb RNA homeostasis and contribute to disease pathology.
From the strains that do not harbor known amyloids or prions, we have identified a specific set of 15 yeast proteins that are consistently present in SDS-resistant complexes but do not form conventional prion aggregates, as they were not affected by the specific prion inhibitor, GuHCl. This set does not overlap with the others reported from proteome-wide screens for proteins that form filaments (65) or large assemblies (66) in S. cerevisiae. The set is enriched in proteins involved in cell communication, cell cycle, developmental, homeostatic, and system processes (supplemental Fig. S3). Presumably, the identified proteins form complexes with special structures that are large and/or sufficiently stable to partially withstand the SDS treatment. It would be of interest to investigate whether such SDS-resistant structures have specific biological roles. Two of these proteins, Bmh1 and Vps5, are known to influence polyglutamine aggregation and toxicity (40, 67). Their identification by TAPI may be a consequence of their general association with intracellular protein aggregates. Sequence analysis of the identified proteins show that they are mostly distinct from each other and from the known yeast prion proteins (such as Sup35, Rnq1, and Ure2), which have characteristic unfolded domains that are rich in glutamines and asparagines. The identified proteins do not overlap with proposed prion-like proteins from previous bioinformatics searches (for examples Refs. 21 and 68), which means TAPI can provide a different data set without certain amino acid sequence biases that could result from bioinformatics algorithms. Only one of our candidate proteins, Glg2, has a region (∼90 amino acids) that scores highly in the prion-predicting algorithm (PAPA) developed in the laboratory of Eric Ross (69).
Although TAPI was reliable for identification of the well characterized stable prions in yeast strains, it shows limitations for the unstable or unusual prions or prion variants. Accuracy of detection may be compromised when the prion is present in less than 10% of the cells (Table 2). Prions with SDS-soluble or oligomeric aggregates may also escape the detection by TAPI (Fig. 3, Table 2). Therefore, the described approach works best for amyloids or amyloid-based prions when large aggregates are present in the majority of cells, making them more abundant in the sample preparation. To improve the sensitivity of the method, selection of cells with amyloid aggregates or enrichment of aggregates within lysates (for example, by FACS analysis (55)) may be performed. For known amyloids, the presence of amyloid protein in the sample can be detected with specific antibodies rather than mass spectrometry, which may lead to an increase in the detection limit (Fig. 2E, Table 2). The sensitivity of TAPI will improve with advances in mass spectrometry technology.
Finally, to our knowledge no comparison has been reported between prions that exist in the laboratory and those from wild yeast strains. Here, we observed that the properties of Sup35 aggregates present in many of these wild [PSI+] strains were quite different from aggregates present in common laboratory strains. SDD-AGE analysis revealed the absence of high molecular weight SDS-resistant Sup35 species in 7 of 10 analyzed wild strains, and this finding was corroborated by the absence of large fluorescent foci formed by Sup35NM-GFP. Presumably, although the [PSI+] prion is present in these strains (independently confirmed by Ref. 70), it has acquired a special form where Sup35 prion aggregates are smaller and/or more SDS-sensitive compared with the Sup35 aggregates from laboratory strains. Such properties could enable the [PSI+] prion to be more easily transmitted to daughter cells during cellular division (i.e. greater infectivity), increasing prion retention within a yeast population. Additional studies are required to determine whether other amyloid-based prions can similarly adapt for higher infectivity.
Supplementary Material
Acknowledgments
We thank Maggie Wear (Uniformed Services University of the Health Sciences) for critical reading of the manuscript. We also thank Greg Mueller, Mike Flora, and Sean Moran (all, Uniformed Services University of the Health Sciences) for help with initial sample preparations, Christopher Grunseich (NINDS, National Institutes of Health (NIH)) and Michail Alterman and Melkamu Getie-Kebtie (both, Food and Drug Administration and Center for Biologics Evaluation and Research) for helpful suggestions and discussion about proteomic techniques, Gerry Fink (Massachusetts Institute of Technology), Leonid Kruglyak (Princeton University), Reed Wickner, David Bateman, and Herman Edskes (all, NIH), Roy Parker (University of Colorado), David Rubinsztein (Medical Research Council, Cambridge, UK), and Michael Sherman (Boston University) for sharing plasmids and strains. We thank Dennis McDaniel (Uniformed Services University of the Health Sciences) for providing assistance with confocal and electron microscopy and Ulrich Baxa (NCI, National Institutes of Health) for help with immuno-EM and Keck Proteomics (Yale University) and the Johns Hopkins University Mass Spectrometry facility for the protein identification.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R03DA035194. This work was also supported by Uniformed Services University of the Health Sciences Grants RO75QF and CS75QF.

This article contains supplemental Figs. S1–S3.
- SDD-AGE
- semi-denaturing detergent agarose gel electrophoresis
- htt
- huntingtin
- TAPI
- technique for amyloid purification and identification
- GuHCl
- guanidine HCl.
REFERENCES
- 1. Gunawardena S., Her L. S., Brusch R. G., Laymon R. A., Niesman I. R., Gordesky-Gold B., Sintasath L., Bonini N. M., Goldstein L. S. (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40, 25–40 [DOI] [PubMed] [Google Scholar]
- 2. Nucifora F. C., Jr., Ellerby L. M., Wellington C. L., Wood J. D., Herring W. J., Sawa A., Hayden M. R., Dawson V. L., Dawson T. M., Ross C. A. (2003) Nuclear localization of a non-caspase truncation product of atrophin-1, with an expanded polyglutamine repeat, increases cellular toxicity. J. Biol. Chem. 278, 13047–13055 [DOI] [PubMed] [Google Scholar]
- 3. Blanco L. P., Evans M. L., Smith D. R., Badtke M. P., Chapman M. R. (2012) Diversity, biogenesis, and function of microbial amyloids. Trends Microbiol. 20, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shewmaker F., McGlinchey R. P., Wickner R. B. (2011) Structural insights into functional and pathological amyloid. J. Biol. Chem. 286, 16533–16540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liebman S. W., Chernoff Y. O. (2012) Prions in yeast. Genetics 191, 1041–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wickner R. B., Shewmaker F., Kryndushkin D., Edskes H. K. (2008) Protein inheritance (prions) based on parallel in-register β-sheet amyloid structures. Bioessays 30, 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McGlinchey R. P., Kryndushkin D., Wickner R. B. (2011) Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. U. S. A. 108, 5337–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. True H. L., Lindquist S. L. (2000) A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407, 477–483 [DOI] [PubMed] [Google Scholar]
- 9. Tyedmers J., Madariaga M. L., Lindquist S. (2008) Prion switching in response to environmental stress. PLoS Biol. 6, e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dobson C. M. (1999) Protein misfolding, evolution, and disease. Trends Biochem. Sci. 24, 329–332 [DOI] [PubMed] [Google Scholar]
- 11. Tycko R. (2006) Molecular structure of amyloid fibrils. Insights from solid-state NMR. Q. Rev. Biophys. 39, 1–55 [DOI] [PubMed] [Google Scholar]
- 12. Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., Eisenberg D. (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 13. Dong J., Castro C. E., Boyce M. C., Lang M. J., Lindquist S. (2010) Optical trapping with high forces reveals unexpected behaviors of prion fibrils. Nat. Struct. Mol. Biol. 17, 1422–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kryndushkin D. S., Alexandrov I. M., Ter-Avanesyan M. D., Kushnirov V. V. (2003) Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278, 49636–49643 [DOI] [PubMed] [Google Scholar]
- 15. MacPhee C. E., Dobson C. M. (2000) Chemical dissection and reassembly of amyloid fibrils formed by a peptide fragment of transthyretin. J. Mol. Biol. 297, 1203–1215 [DOI] [PubMed] [Google Scholar]
- 16. Serio T. R., Cashikar A. G., Kowal A. S., Sawicki G. J., Moslehi J. J., Serpell L., Arnsdorf M. F., Lindquist S. L. (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289, 1317–1321 [DOI] [PubMed] [Google Scholar]
- 17. MacLea K. S., Ross E. D. (2011) Strategies for identifying new prions in yeast. Prion 5, 263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bagriantsev S. N., Kushnirov V. V., Liebman S. W. (2006) Analysis of amyloid aggregates using agarose gel electrophoresis. Methods Enzymol. 412, 33–48 [DOI] [PubMed] [Google Scholar]
- 19. Guo W., Chen Y., Zhou X., Kar A., Ray P., Chen X., Rao E. J., Yang M., Ye H., Zhu L., Liu J., Xu M., Yang Y., Wang C., Zhang D., Bigio E. H., Mesulam M., Shen Y., Xu Q., Fushimi K., Wu J. Y. (2011) An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation, and neurotoxicity. Nat. Struct. Mol. Biol. 18, 822–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salnikova A. B., Kryndushkin D. S., Smirnov V. N., Kushnirov V. V., Ter-Avanesyan M. D. (2005) Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its non-heritable amyloids. J. Biol. Chem. 280, 8808–8812 [DOI] [PubMed] [Google Scholar]
- 21. Alberti S., Halfmann R., King O., Kapila A., Lindquist S. (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137, 146–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halfmann R., Jarosz D. F., Jones S. K., Chang A., Lancaster A. K., Lindquist S. (2012) Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 482, 363–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meriin A. B., Zhang X., He X., Newnam G. P., Chernoff Y. O., Sherman M. Y. (2002) Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakayashiki T., Kurtzman C. P., Edskes H. K., Wickner R. B. (2005) Yeast prions [URE3] and [PSI+] are diseases. Proc. Natl. Acad. Sci. U. S. A. 102, 10575–10580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buchan J. R., Muhlrad D., Parker R. (2008) P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 183, 441–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kryndushkin D., Ihrke G., Piermartiri T. C., Shewmaker F. (2012) A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol. Microbiol. 86, 1531–1547 [DOI] [PubMed] [Google Scholar]
- 27. Chernoff Y. O., Lindquist S. L., Ono B., Inge-Vechtomov S. G., Liebman S. W. (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [PSI+]. Science 268, 880–884 [DOI] [PubMed] [Google Scholar]
- 28. Kryndushkin D. S., Shewmaker F., Wickner R. B. (2008) Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 27, 2725–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wyttenbach A., Swartz J., Kita H., Thykjaer T., Carmichael J., Bradley J., Brown R., Maxwell M., Schapira A., Orntoft T. F., Kato K., Rubinsztein D. C. (2001) Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum. Mol. Genet. 10, 1829–1845 [DOI] [PubMed] [Google Scholar]
- 30. Muchowski P. J., Schaffar G., Sittler A., Wanker E. E., Hayer-Hartl M. K., Hartl F. U. (2000) Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U. S. A. 97, 7841–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wischik C. M., Novak M., Edwards P. C., Klug A., Tichelaar W., Crowther R. A. (1988) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 85, 4884–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wischik C. M., Novak M., Thøgersen H. C., Edwards P. C., Runswick M. J., Jakes R., Walker J. E., Milstein C., Roth M., Klug A. (1988) Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 85, 4506–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kelly A. C., Shewmaker F. P., Kryndushkin D., Wickner R. B. (2012) Sex, prions, and plasmids in yeast. Proc. Natl. Acad. Sci. U. S. A. 109, E2683--E2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Resende C. G., Outeiro T. F., Sands L., Lindquist S., Tuite M. F. (2003) Prion protein gene polymorphisms in Saccharomyces cerevisiae. Mol. Microbiol. 49, 1005–1017 [DOI] [PubMed] [Google Scholar]
- 35. Patino M. M., Liu J. J., Glover J. R., Lindquist S. (1996) Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 273, 622–626 [DOI] [PubMed] [Google Scholar]
- 36. Zuccato C., Valenza M., Cattaneo E. (2010) Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol. Rev. 90, 905–981 [DOI] [PubMed] [Google Scholar]
- 37. Cooper J. K., Schilling G., Peters M. F., Herring W. J., Sharp A. H., Kaminsky Z., Masone J., Khan F. A., Delanoy M., Borchelt D. R., Dawson V. L., Dawson T. M., Ross C. A. (1998) Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum. Mol. Genet. 7, 783–790 [DOI] [PubMed] [Google Scholar]
- 38. Martindale D., Hackam A., Wieczorek A., Ellerby L., Wellington C., McCutcheon K., Singaraja R., Kazemi-Esfarjani P., Devon R., Kim S. U., Bredesen D. E., Tufaro F., Hayden M. R. (1998) Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 18, 150–154 [DOI] [PubMed] [Google Scholar]
- 39. Toombs J. A., McCarty B. R., Ross E. D. (2010) Compositional determinants of prion formation in yeast. Mol. Cell. Biol. 30, 319–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y., Meriin A. B., Costello C. E., Sherman M. Y. (2007) Characterization of proteins associated with polyglutamine aggregates. A novel approach towards isolation of aggregates from protein conformation disorders. Prion 1, 128–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Urakov V. N., Vishnevskaya A. B., Alexandrov I. M., Kushnirov V. V., Smirnov V. N., Ter-Avanesyan M. D. (2010) Interdependence of amyloid formation in yeast. Implications for polyglutamine disorders and biological functions. Prion 4, 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lagier-Tourenne C., Polymenidou M., Cleveland D. W. (2010) TDP-43 and FUS/TLS. Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romanova N. V., Chernoff Y. O. (2009) Hsp104 and prion propagation. Protein Pept. Lett. 16, 598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Douglas P. M., Treusch S., Ren H. Y., Halfmann R., Duennwald M. L., Lindquist S., Cyr D. M. (2008) Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc. Natl. Acad. Sci. U. S. A. 105, 7206–7211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lopez N., Aron R., Craig E. A. (2003) Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+]. Mol. Biol. Cell 14, 1172–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sondheimer N., Lopez N., Craig E. A., Lindquist S. (2001) The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J. 20, 2435–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 48. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 49. Newby G. A., Lindquist S. (2013) Blessings in disguise. Biological benefits of prion-like mechanisms. Trends Cell Biol. 23, 251–259 [DOI] [PubMed] [Google Scholar]
- 50. Suzuki G., Shimazu N., Tanaka M. (2012) A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 336, 355–359 [DOI] [PubMed] [Google Scholar]
- 51. Bagriantsev S. N., Gracheva E. O., Richmond J. E., Liebman S. W. (2008) Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Mol. Biol. Cell 19, 2433–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. King C. Y., Diaz-Avalos R. (2004) Protein-only transmission of three yeast prion strains. Nature 428, 319–323 [DOI] [PubMed] [Google Scholar]
- 53. Glover J. R., Kowal A. S., Schirmer E. C., Patino M. M., Liu J. J., Lindquist S. (1997) Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89, 811–819 [DOI] [PubMed] [Google Scholar]
- 54. Wang L., Maji S. K., Sawaya M. R., Eisenberg D., Riek R. (2008) Bacterial inclusion bodies contain amyloid-like structure. PLoS Biol. 6, e195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Doi H., Mitsui K., Kurosawa M., Machida Y., Kuroiwa Y., Nukina N. (2004) Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS Lett. 571, 171–176 [DOI] [PubMed] [Google Scholar]
- 56. Nübling G., Bader B., Levin J., Hildebrandt J., Kretzschmar H., Giese A. (2012) Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with α-synuclein at the single molecule level. Mol. Neurodegener. 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walsh D. M., Klyubin I., Fadeeva J. V., Rowan M. J., Selkoe D. J. (2002) Amyloid-β oligomers. Their production, toxicity, and therapeutic inhibition. Biochem. Soc. Trans. 30, 552–557 [DOI] [PubMed] [Google Scholar]
- 58. Chapman M. R., Robinson L. S., Pinkner J. S., Roth R., Heuser J., Hammar M., Normark S., Hultgren S. J. (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Collinson S. K., Emödy L., Müller K. H., Trust T. J., Kay W. W. (1991) Purification and characterization of thin, aggregative fimbriae from Salmonella enteritidis. J. Bacteriol. 173, 4773–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schwartz K., Syed A. K., Stephenson R. E., Rickard A. H., Boles B. R. (2012) Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 8, e1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Manning M., Colón W. (2004) Structural basis of protein kinetic stability. Resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward β-sheet structure. Biochemistry 43, 11248–11254 [DOI] [PubMed] [Google Scholar]
- 62. Xu G., Stevens S. M., Jr., Moore B. D., McClung S., Borchelt D. R. (2013) Cytosolic proteins lose solubility as amyloid deposits in a transgenic mouse model of Alzheimer-type amyloidosis. Hum. Mol. Genet. 22, 2765–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ratovitski T., Chighladze E., Arbez N., Boronina T., Herbrich S., Cole R. N., Ross C. A. (2012) Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle 11, 2006–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li Y. R., King O. D., Shorter J., Gitler A. D. (2013) Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Narayanaswamy R., Levy M., Tsechansky M., Stovall G. M., O'Connell J. D., Mirrielees J., Ellington A. D., Marcotte E. M. (2009) Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. U. S. A. 106, 10147–10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Noree C., Sato B. K., Broyer R. M., Wilhelm J. E. (2010) Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J. Cell Biol. 190, 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Manogaran A. L., Hong J. Y., Hufana J., Tyedmers J., Lindquist S., Liebman S. W. (2011) Prion formation and polyglutamine aggregation are controlled by two classes of genes. PLoS Genet. 7, e1001386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Michelitsch M. D., Weissman J. S. (2000) A census of glutamine/asparagine-rich regions. Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. U. S. A. 97, 11910–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Toombs J. A., Petri M., Paul K. R., Kan G. Y., Ben-Hur A., Ross E. D. (2012) De novo design of synthetic prion domains. Proc. Natl. Acad. Sci. U. S. A. 109, 6519–6524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bateman D. A., Wickner R. B. (2013) The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genet. 9, e1003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.