

Background: Hsp90 and co-chaperones are critical for the folding and activation of client proteins.
Results: Hsp90 and Cpr6, but not other tetratricopeptide repeat (TPR)-containing co-chaperones, interact with Ura2.
Conclusion: The TPR domain of Cpr6 has unique functions, including interaction with Ura2, an enzyme required for pyrimidine biosynthesis.
Significance: Identification of co-chaperone-specific interactions is crucial to understanding how to selectively target Hsp90 functions.
Keywords: Hsp90, Molecular Chaperone, Protein-Protein Interactions, Pyrimidine, Yeast Genetics
Abstract
The molecular chaperone heat shock protein 90 (Hsp90) is an essential protein required for the activity and stability of multiple proteins termed clients. Hsp90 cooperates with a set of co-chaperone proteins that modulate Hsp90 activity and/or target clients to Hsp90 for folding. Many of the Hsp90 co-chaperones, including Cpr6 and Cpr7, contain tetratricopeptide repeat (TPR) domains that bind a common acceptor site at the carboxyl terminus of Hsp90. We found that Cpr6 and Hsp90 interacted with Ura2, a protein critical for pyrimidine biosynthesis. Mutation or inhibition of Hsp90 resulted in decreased accumulation of Ura2, indicating it is an Hsp90 client. Cpr6 interacted with Ura2 in the absence of stable Cpr6-Hsp90 interaction, suggesting a direct interaction. However, loss of Cpr6 did not alter the Ura2-Hsp90 interaction or Ura2 accumulation. The TPR domain of Cpr6 was required for Ura2 interaction, but other TPR containing co-chaperones, including Cpr7, failed to interact with Ura2 or rescue CPR6-dependent growth defects. Further analysis suggests that the carboxyl-terminal 100 amino acids of Cpr6 and Cpr7 are critical for specifying their unique functions, providing new information about this important class of Hsp90 co-chaperones.
Introduction
Hsp90 is a highly conserved and abundant protein that is essential in eukaryotes and is required for the stability and activity of diverse clients involved in cell signaling and growth (1). Hsp90 exists as a dimer and contains three conserved domains: an amino-terminal ATP-binding domain, a middle domain, and a carboxyl-terminal dimerization domain. Although Hsp90 conformations are well characterized, the mechanism through which it is able to recognize and bind specific clients is unclear, and all three domains of Hsp90 have been implicated in client interaction (2–5).
Co-chaperone proteins regulate Hsp90 function throughout its ATPase cycle. The functions of these proteins include client loading, assisting Hsp90 conformational changes, and modulating ATPase activity. Hsp90 begins in an open, nucleotide-free conformation characterized by dimerization at its carboxyl-terminal domains. In this conformation the co-chaperones Cdc37 or Sti1 interact with Hsp90 to inhibit its ATPase cycle for client loading. Next, Hsp90 binds ATP and continues into a closed conformation characterized by dimerization of the amino-terminal domains as well as Cpr6 and Cpr7 interaction. Aha1 and p23 also bind the ATP bound form of Hsp90; Aha1 enhances the rate of ATP hydrolysis to allow Hsp90 to go back to its open conformation. Many of these co-chaperones (Sti1, Cpr6, Cpr7, Cns1, Ppt1, and Tah1) bind Hsp90 at its carboxyl-terminal MEEVD sequence through the use of tetratricopeptide repeat (TPR)2 domains (6–8). Although TPR containing co-chaperones share a common binding site on Hsp90, individual TPR domains have differing functions. Sti1 binds Hsp90 and Hsp70 through separate TPR domains, whereas the TPR domain of Sgt1 is used for client binding instead of Hsp90 interaction (9–11).
Cpr6 and Cpr7 of yeast and Cyp40 of mammals are homologous co-chaperones that contain an amino-terminal peptidyl-prolyl isomerase (PPIase) and carboxyl-terminal TPR domain. Each of these co-chaperones was able to suppress thermal aggregation of proteins (12, 13). Cpr7 has been shown to have higher chaperoning activity, whereas Cpr6 has higher PPIase activity, but the correlation between these activities and Hsp90 function is unknown. Cpr7 is required for growth and for the function of Hsp90 clients such as the glucocorticoid receptor, v-src, and heat shock factor 1. However, only minor Hsp90 client defects have been reported in cpr6 cells, and loss of CPR6 does not cause obvious growth defects (14–16). Prior to this study it was shown that Cpr6 has minor affects on Hsp90 ATPase activity (17), however, Cpr6-specific phenotypes or client interactions had not been identified.
We identified an interaction between Cpr6, Hsp90, and Ura2, a protein required for pyrimidine biosynthesis (18). Prior genome-wide studies suggested that Ura2 had genetic or physical interactions with Cpr6 and Hsp90 (19, 20), but no direct evidence had been shown. We show that Ura2 interacts with Hsp90, Hsp70, and Cpr6 and demonstrate that the Hsp90 mutation or inhibition results in reduced Ura2 accumulation, indicating it is an Hsp90 client. We further show that Cpr6, but not other TPR-containing co-chaperones, stably interacts with Ura2 even in the absence of the Cpr6-Hsp90 interaction, suggesting direct contacts between Ura2 and Cpr6. Other TPR-containing co-chaperones were also unable to rescue growth defects caused by loss of CPR6. Further evidence suggests that sequences near the end of the TPR domain through the carboxyl terminus are critical for the specialized functions of Cpr6 and Cpr7.
EXPERIMENTAL PROCEDURES
Media, Chemicals, Antibodies, and Plasmids
Standard yeast genetics were utilized. Yeast cells were grown in either YPD (yeast extract peptone dextrose) or defined synthetic complete media supplemented with 2% dextrose. Growth was examined by spotting 10-fold serial dilutions of yeast cultures onto the appropriate media, followed by incubation for 2 days at the indicated temperature. Radicicol was obtained from Sigma and dissolved in DMSO (dimethyl sulfoxide) to make a 10 mg/ml stock. 5-Fluoroorotic acid was obtained from Toronto Research Chemicals. The α-Xpress monoclonal antibody was obtained from Invitrogen. Polyclonal antibodies raised against Hsp90 as well as Sti1 and Cpr6 have been previously described (21, 22). The Ssa polyclonal antisera, which recognizes the last 56 amino acids of Ssa1, and the Tim44 antisera were gifts from Elizabeth Craig (University of Wisconsin, Madison, WI). Anti-Aha1 peptide antisera was raised against amino acids 1–16 of Aha1 conjugated to keyhole limpet hemocyanin. The Aha1 antibody was validated by establishing that it recognizes a protein of the correct size that is missing in cells lacking AHA1. Sti1 or Cpr6 was used as a loading control in most cases because their expression was unchanged under standard growth conditions. Tim44 was used as the loading control for Fig. 3B because the presence of radicicol may result in up-regulation of genes regulated by heat shock factor 1 (HSF1) in mammalian cells and fungi (15, 23).
FIGURE 3.
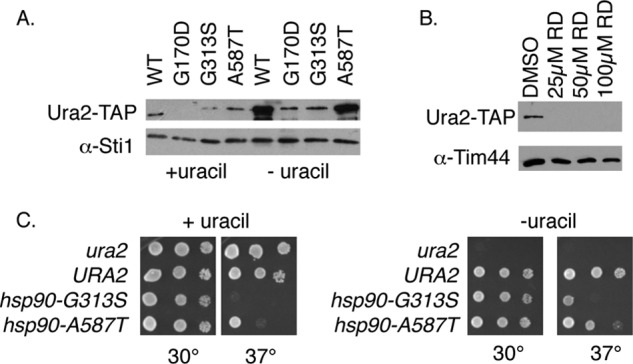
Hsp90 mutation or inhibition results in reduced Ura2-TAP accumulation. A, lysates of hsc82hsp82 URA2-TAP cells expressing WT or mutant Hsp90 grown in the presence or absence of uracil were probed for Ura2-TAP or Sti1 (as a loading control). B, WT cells expressing Ura2-TAP (JJ1069) were grown overnight in the absence of uracil prior to addition of DMSO (as a control) or radicicol (RD) at the indicated final concentration. After overnight growth, cell lysates were probed for Ura2-TAP or Tim44. C, growth of isogenic ura2, WT, hsc82hsp82/hsp90-G313S, or hsc82hsp82/hsp90-A587T strains on complete minimal media plates (left) or plates lacking uracil (right). All strains are URA3.
Yeast Strains
All strains except URA2-TAP:HIS3 strain (Open Biosystems) (24) are isogenic to W303 (Table 1). JJ762 (WT), JJ816 (hsc82hsp82/YEp24HSP82), and JJ110 (cpr6 hsc82 hsp82/YEp24HSP82) have been described previously (7, 21). JJ1115 (cpr7) was derived from JJ547 (7). The Ura2-TAP:HIS3 strain was crossed to JJ762 five times to obtain the indicated strains. A ura2::HIS3 strain (a gift from Elizabeth Craig) was crossed to JJ1058 (a URA3 version of JJ762) to obtain the ura2 URA3 strain (JJ1093). JJ1055 and additional strains were derived from JJ110 or subsequent crosses.
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source or Ref. |
|---|---|---|
| JJ762 | MATa ade2-1 ura3–1 leu2–3,112 trp1–1 met2-Δ1 his3–11,15 lys2-Δ2 | E. A. Craig |
| JJ1055 | MATa cpr6::kanR | This study |
| JJ816 | MATa hsp82::LEU2 hsc82::LEU2/Yep24-HSP82 | 21 |
| JJ110 | MATα cpr6::kanR hsp82::LEU2 hsc82::LEU2/Yep24-HSP82 | 7 |
| JJ1069 | MATα URA2-TAP:HIS3 | This study |
| JJ1067 | MATα URA2-TAP:HIS3 cpr6::kanR MET2 | This study |
| JJ1097 | MATα URA2-TAP:HIS3 hsp82::LEU2 hsc82::LEU2/YEp24-HSP82 | This study |
| JJ1100 | MATα URA2-TAP cpr6 hsc82 hsp82/YEp24-HSP82 | This study |
| JJ1056 | MATα URA3 hsp82::LEU2 hsc82::LEU2/pTGPDhsp82-G313S | This study |
| JJ1057 | MATα URA3 hsp82::LEU2 hsc82::LEU2 pTGPDhsp82-A587T | This study |
| JJ1058 | MATα URA3 | This study |
| JJ1093 | MATα URA3 ura2::HIS3 | This study |
| JJ1115 | MATa cpr7::kanR | This study |
| MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 URA2-TAP::HIS3 | Open Biosystems |
Hsp82 and Co-chaperone Plasmids
YEp24-CNS1, pRS416GPDHis-Cpr6, pRS415GPDHis-Sti1, pRS414GPDHAProteinA, and pRS416GPDHis-Hsc82 plasmid were previously described (7, 25, 26). pRS416TEF-Ssa1 was a gift from Dr. Elizabeth Craig (University of Wisconsin). Genomic CPR7 or CNS1 sequences were amplified from yeast genomic DNA and cloned into the pRS416GPD vector containing an amino-terminal His6 sequence and the Xpress epitope using engineered BamHI and XhoI or BamHI and EcoRI sites, respectively. pRS416ADHHis-Cpr6 and pRS414GPDHis-Cpr6 were constructed by cloning the His-Cpr6 sequences into pRS416ADH or pRS414GPD (27). pRS316CPR6 was constructed by introduction of SacI and BamHI sites into genomic DNA and cloning into pRS316. pRS316His-CPR6, which lacks the Xpress epitope, was constructed by engineering 6 consecutive histidine residues into the amino terminus. pRS414GPDHis-Cpr6-HSP82 was created by cloning a genomic HSP82 ClaI fragment into pRS414GPDHis-Cpr6. Amino acid mutations were constructed using site-directed mutagenesis. Mutagenic oligonucleotide sequences are available upon request. His-Cpr6 truncation constructs were constructed using pRS416GPDHis-Cpr6. His-tagged Cpr6/Cpr7 chimeras were derived from FLAG-tagged chimeras (7). The Cpr6–6PPIase 6A/7B chimera was constructed using an engineered MfeI site at amino acid position 289 of Cpr7 that matched an endogenous MfeI site at amino acid position 267 in Cpr6. The resultant sequence at the junction is KINQLKMKIY.
Expression of either HSC82 or HSP82 is able to fully rescue the growth defects of an hsc82hsp82 strain. The Hsp82 isoform was used in all experiments except for Fig. 2A, which used Hsc82. Plasmids expressing WT or mutant Hsp90 were transformed into the indicated hsc82hsp82 strain harboring the YEp24-HSP82 plasmid. Transformants were grown in the presence of 5-fluoroorotic acid to lose the YEp24-HSP82 plasmid. Untagged or His-tagged Hsp82 was expressed under the GPD promoter (pTGPDHSP82 and pRS314GPDHis-Hsp82 plasmids, respectively) (7). The ΔMEEVD amino acid deletion was constructed in pRS314GPDHis-Hsp82. Other mutations were introduced into pTGPDHSP82. All mutations were confirmed using automated DNA sequencing.
FIGURE 2.
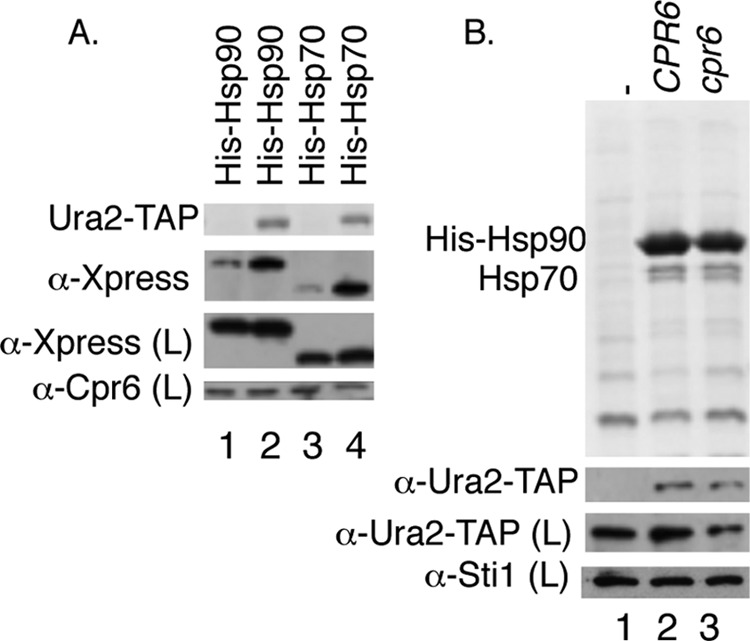
Hsp90 and Hsp70 also bind Ura2, and Hsp90 interacts with Ura2 in the absence of Cpr6. A, the WT strain (JJ762) expressing a control TAP-tag (odd numbered lanes) or a Ura2-TAP strain (Open Biosystems) (even numbered lanes) was transformed with plasmids expressing His- and Xpress-tagged Hsp90 or Hsp70. Ura2-TAP complexes were isolated using IgG-Sepharose. B, His-Hsp90 complexes were isolated from hsc82hsp82 URA2-TAP cells (lane 2) or cpr6 hsc82hsp82 URA2-TAP cells (lane 3). Lane 1 expressed untagged Hsp90.
Isolation of His-Cpr6 and Ura2-TAP Complexes
His-Cpr6 complexes were isolated as described for His-Hsp82 complexes (7). Briefly, cells were grown overnight and harvested at an A600 of 1.2–2.0. Cell pellets were resuspended in lysis buffer (20 mm Tris, pH 7.5, 100 mm KCl, 5 mm MgCl2, 5 mm imidazole containing a protease inhibitor tablet (Roche Applied Science)) and were disrupted in the presence of glass beads with 8 × 30-s pulses. Cell lysates were incubated with nickel resin (1 h with rocking, 4 °C) followed by washes with lysis buffer plus 35 mm imidazole and 0.1% or 0.5% Tween 20 (if cells expressed Ura2-TAP). Proteins were eluted from nickel resin by boiling in SDS-PAGE sample buffer and protein complexes were separated by gel electrophoresis (10% acrylamide unless otherwise indicated) followed by Coomassie Blue staining. Alternatively, proteins were transferred to nitrocellulose and chemiluminescence immunoblots were performed according to the manufacturer's suggestions (Pierce). A similar procedure was used to isolate Ura2-TAP using IgG-Sepharose (GE Healthcare).
Mass Spectrometry Fingerprint Analysis
Proteins were identified at the Environmental Biotechnology Institute at the University of Idaho. The analysis of peptides was done using reverse phase liquid chromatography on a Waters nanoAcquity Ultra Performance Liquid Chromatograph. This was followed by tandem mass spectrometry using a Waters Micromass Q-Tof Premier quadrupole-time of flight mass spectrometer using a nanospray electrospray ionization inlet. The protein of interest was excised from the Coomassie-stained gel and dried using acetonitrile. Sequence grade trypsin (Sigma) was used to digest the band at 37 °C for 20 h. Peptides were extracted three times using 100 mm ammonium bicarbonate for one extraction and 50% acetonitrile containing 5% formic acid for the other two extractions. The extracts were evaporated to complete dryness and concentrated in 20 μl of redissolvation solution (5% ACN and 0.1% formic acid). Peptides were identified using the MASCOT program with a peptide and MS/MS tolerance of 0.2 Da. Twenty-seven peptides corresponding to Ura2 were identified, representing coverage of 14.36% and a score of 341. Thirteen peptides matched Ssa1/2, with 36% coverage and a score of 706.
RESULTS
Cpr6 Interacts with Ura2
A plasmid expressing His-Cpr6 under the strong GPD promoter was transformed into cpr6 cells. Whole cell extracts were incubated with nickel resin followed by SDS-PAGE and immunoblot analysis. As shown in Fig. 1A, Hsp90 (encoded by HSC82 and HSP82) bound nickel resin in the presence, but not absence of His-Cpr6. Two additional proteins bound only in the presence of His-Cpr6 were identified by protein mass spectrometry fingerprint analysis. The 250-kDa protein was Ura2, which has a predicted molecular mass of 245 kDa (18). We confirmed that the 250-kDa band was lost when His-Cpr6 complexes were isolated from ura2 cells (not shown). The 70-kDa protein was Ssa1 or Ssa2, nearly identical members of an essential cytosolic Hsp70 gene family (28, 29). Immunoblot analysis confirmed that Ssa1/2, hereafter referred to as Hsp70, co-purifies with His-Cpr6.
FIGURE 1.
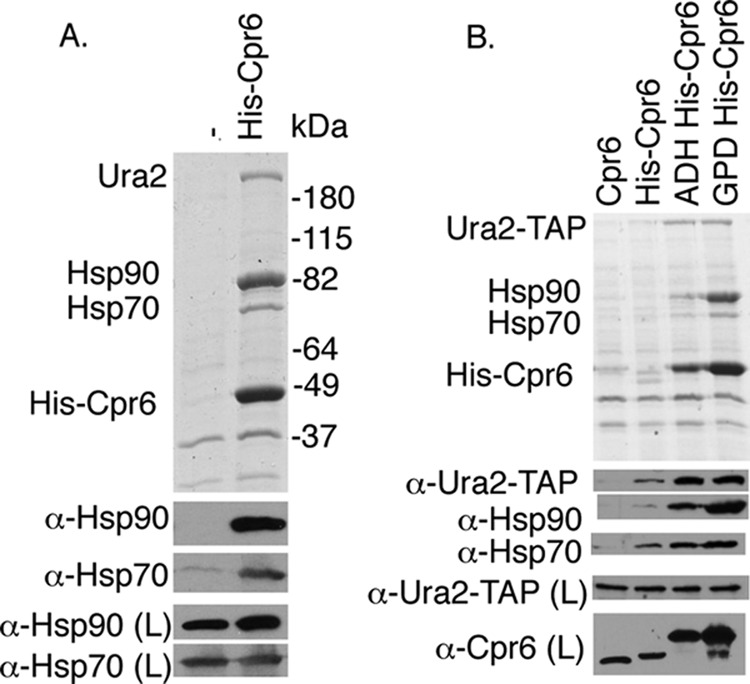
Cpr6 binds Hsp90, Hsp70, and Ura2. A, Δcpr6 cells were transformed with a plasmid expressing untagged CPR6 or pRS416GPDHIS-CPR6. Extracts from cells grown in media lacking uracil were incubated with nickel resin and complexes were analyzed using SDS-PAGE and immunoblot analysis. The upper panel displays the Coomassie Blue-stained gel of the resin samples, whereas the lower panels show immunoblot analysis. Immunoblots of the lysate (L) are provided as a loading control. B, Δcpr6 cells expressing URA2-TAP at the normal chromosomal location were transformed with pRS316CPR6 or the indicated plasmid. His-Cpr6 complexes were analyzed as described.
Next we examined the ability of His-Cpr6 expressed from its endogenous promoter or the ADH promoter (27) to interact with Ura2 using a cpr6 strain that expresses TAP-tagged Ura2 from the normal chromosomal location (24). As shown in Fig. 1B, the ADH and GPD promoters result in dramatically enhanced recovery of His-Cpr6, but all three constructs interacted with Ura2-TAP. His-Cpr6 expressed under its own promoter migrated faster than the other tagged version because it lacks the Xpress epitope. This indicates that the Cpr6-Ura2 interaction is not a consequence of Cpr6 overexpression. Because the level of associated Ura2-TAP increased upon His-Cpr6 expression, this suggests that only a portion of Ura2 is normally associated with Cpr6.
Changes in Cpr6 expression had some effect on Hsp90 interaction, but did not dramatically affect Hsp70 interaction. In prior studies, Cpr6 co-purified with His-Hsp90 only in the presence of the non-hydrolyzable ATP analog, AMP-PNP (7, 22). This result indicates that overexpression of Cpr6 results in Hsp90 interaction in the presence or absence of nucleotide.
Hsp90 and Hsp70 Also Interact with Ura2
We next examined whether Hsp90 and Hsp70 interact with Ura2 (Fig. 2A). Ura2-TAP complexes were isolated from strains expressing His- and Xpress-tagged Hsp90 (Hsc82 isoform) or Hsp70 (Ssa1 isoform). Although some background binding of Hsp90 and Hsp70 was observed in control samples that lacked Ura2-TAP, elevated levels of Hsp90 and Hsp70 were recovered in the presence of Ura2-TAP.
Cdc37 and Sgt1 have been shown to be able to target clients to Hsp90 for folding (9, 30). However, mammalian Cyp40 interacts with Hsp90 after Hsp90 has bound receptor (31). As shown in Fig. 2B, His-Hsp90 interacted with Ura2-TAP even in the absence of CPR6, indicating Cpr6 is not required for the Hsp90-Ura2 interaction.
Hsp90 Mutation or Inhibition Results in Reduced Steady State Levels of Ura2
Genetic or pharmacological inhibition of Hsp90 disrupts client activity and/or accumulation (32–34). We examined the impact of temperature-sensitive hsp90 mutations that are known to disrupt client activity (35) on Ura2-TAP accumulation and Ura2 function. Because the expression of URA2 is regulated by the availability of uracil (36, 37), Ura2-TAP levels were examined when cells were grown either the presence or absence of uracil (Fig. 3A). As expected, the accumulation of Ura2-TAP was increased in WT cells grown in the absence of uracil. Under both growth conditions, the level of Ura2-TAP was dramatically reduced in the presence of the G170D and G313S mutations. However, the A587T mutation had little or no effect on Ura2-TAP accumulation. The Hsp90 inhibitor radicicol also resulted in greatly reduced Ura2-TAP levels (Fig. 3B), demonstrating the importance of Hsp90 function for Ura2 stability or folding.
Because the Hsp90 mutation resulted in reduced accumulation of Ura2-TAP, and URA2 is essential for growth in the absence of uracil (18), we examined whether cells expressing Hsp90 mutants would also display growth defects when grown on media lacking uracil. As shown in Fig. 3C, ura2 cells are able to grow on complete minimal media, but are unable to grow in the absence of uracil. However, cells expressing Hsp90-G313S or Hsp90-A587T did not display enhanced growth defects in the absence of uracil. The dramatic increase of Ura2 levels when cells are grown in the absence of uracil (Fig. 3A) may mitigate the effect of reduced Ura2 protein accumulation, explaining why uracil auxotrophy is not observed in Hsp90 mutant cells. Loss of CPR6 also had no effect on the ability of cells to grow in the absence of uracil (not shown).
Further Analysis of the Role of Hsp90 and Cpr6 on Ura2 Function
URA2 expression and Ura2 enzymatic activities are highest when cells are grown in the absence of uracil (18, 37, 38). Because some Hsp90 clients, such as steroid hormone receptors, only interact with Hsp90 under certain conditions (39), we determined whether growth conditions affect the Cpr6-Ura2 interaction. Our prior analyses of Ura2 interaction with Cpr6 were conducted with cells grown in the absence of uracil. To monitor Cpr6-Ura2 interaction and Ura2-TAP levels under a variety of growth conditions, we constructed a plasmid that expressed both WT Hsp90 and His-Cpr6. This plasmid is essential for viability in an hsc82hsp82 Ura2-TAP strain.
First we examined the interaction of Ura2-TAP with His-Cpr6 when cells were grown under a variety of conditions, including rich media (YPD), complete minimal media, limiting uracil (1 mg/liter), or media lacking uracil (Ura D.O.) (Fig. 4A). Ura2-TAP co-purified with His-Cpr6 under all conditions. Thus, Ura2 appears to interact with Cpr6 under a wide range of growth conditions, it is just more evident when cells are grown in the absence of uracil because more Ura2 is present.
FIGURE 4.
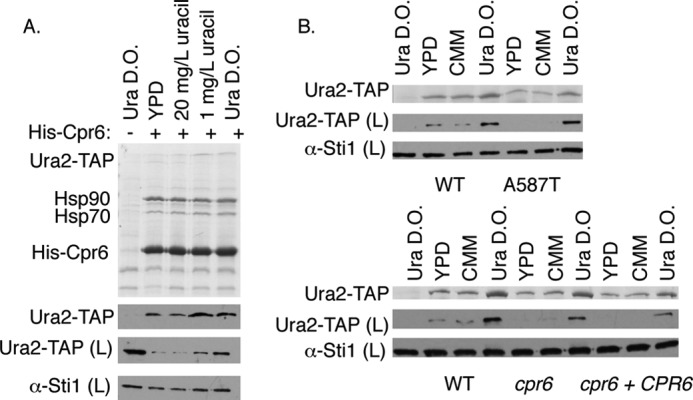
Effect of growth conditions on Ura2-Cpr6 interaction and Ura2 accumulation. A, hsc82hsp82 URA2-TAP cells expressed either untagged WT Hsp90 (lane 1) or a plasmid expressing both His-Cpr6 and WT Hsp90. Complete minimal media is 20 mg/liter of uracil. Ura D.O. media lacks uracil. B, upper, hsc82hsp82 URA2-TAP cells expressed WT or mutant Hsp90 along with the pRS316 vector. Lower, hsc82hsp82 URA2-TAP cells or cpr6hsc82hsp82 URA2-TAP cells expressed WT Hsp90 along with the pRS316 vector or a plasmid expressing both His-Cpr6 and WT Hsp90. In lane 1, upper and lower, hsc82hsp82 cells expressed untagged WT Hsp90 along with the pRS316 vector. His-Cpr6 complexes were isolated from cells grown overnight in the indicated media.
Other potential ways that Hsp90 or Cpr6 could affect Ura2 function is by regulating URA2 transcription or differentially stabilizing Ura2 in the presence or absence of uracil. To broadly assess these possibilities, we examined whether Hsp90 mutation or CPR6 deletion affected the steady state levels of Ura2-TAP when cells were grown under differing growth conditions. The accumulation of Ura2-TAP in cells expressing the Hsp90-A587T mutation was essentially the same as in cells expressing WT Hsp90 (Fig. 4B, upper panels). Deletion of CPR6 also had little effect on overall accumulation of Ura2-TAP (Fig. 2B and Fig. 4B, lower panels).
Cpr6 Interacts with Ura2 in an Hsp90-independent Manner
Although the functional relevance of the Cpr6 interaction with Ura2 remains unclear, little is known about Cpr6 function or interactions between TPR containing co-chaperones and Hsp90 clients. To determine whether Cpr6 interaction with Ura2 is Hsp90-dependent, we examined the Cpr6-Ura2 interaction in cells expressing Hsp90ΔMEEVD, which does not stably interact with Cpr6 due to loss of the TPR binding site (7, 11). We used the construct overexpressing His-Cpr6 under the GPD promoter because it allowed us to simultaneously monitor the effect of Cpr6 alteration on interaction with Hsp90, Hsp70, Ura2, and the co-chaperone Aha1, which was previously shown to co-purify with Cpr6 (28). As expected, His-Cpr6 interaction with Hsp90ΔMEEVD was disrupted (Fig. 5A). The Aha1 interaction was also disrupted. However, Ura2 and Hsp70 were still able to interact with Cpr6 in cells expressing Hsp90ΔMEEVD, indicating that Cpr6 is able to interact with those proteins in the absence of stable Hsp90 interaction.
FIGURE 5.
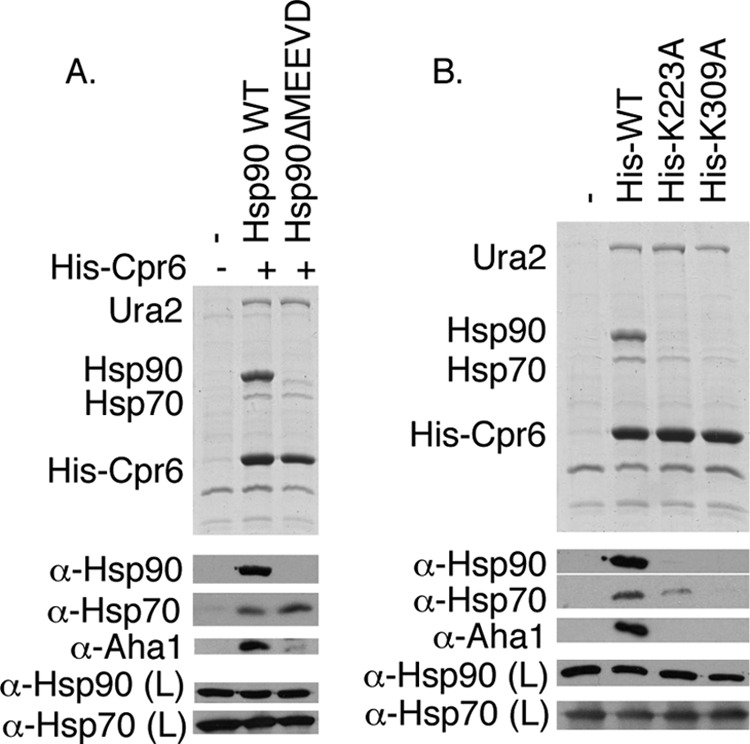
Cpr6 interacts with Ura2 in the absence of Hsp90-Cpr6 interaction. A, His-Cpr6 was isolated from cpr6hsc82hsp82 cells expressing WT Hsp90 or Hsp90ΔMEEVD. The top panel shows the Coomassie-stained gel of the resin samples, whereas the bottom panels display immunoblot analysis of resin or lysate (L) samples as indicated. B, His-Cpr6 complexes were isolated from Δcpr6 cells expressing His-tagged versions of Cpr6 WT, -K223A, or -K309A. Cells were grown in uracil dropout media.
The crystal structure of bovine Cyp40 is available, and a prior study identified amino acid alterations within the TPR domain of Cyp40 that disrupted the Hsp90 interaction (40, 41). Based on that information we predicted that His-Cpr6-K223A and His-Cpr6-K309A would not stably interact with Hsp90. We compared the interaction of WT and mutant Cpr6 with Hsp90, Hsp70, Aha1, and Ura2. As shown in Fig. 5B, the K223A and K309A alterations disrupted the His-Cpr6 interaction with Hsp90, Hsp70, and Aha1, but the Cpr6-Ura2 interaction was not affected, suggesting that the Cpr6-Ura2 interaction is direct. Hsp70 bound Cpr6 in the absence of the Hsp90 interaction (Fig. 5A), and the Cpr6-Hsp70 interaction was disrupted by K223A and K309A alterations. Thus, these residues in the TPR domain may directly interact with the terminal MEEVD of Hsp90 or the similarly conserved terminal EEVD sequence of Hsp70 (42). This is consistent with a prior study that showed that Cyp40 was able to directly interact with Hsp70 (43).
The TPR Domain of Cpr6 Interacts with Ura2
Cpr6 contains an amino-terminal PPIase domain and a carboxyl-terminal TPR domain separated by an acidic linker (40). Additionally, Cpr6 contains a short basic region following its TPR domain (Fig. 6A). We isolated truncation mutants of Cpr6 that expressed the isolated PPIase domain of Cpr6 including the linker region (1–212), the TPR domain including the linker and basic region (171–371), the TPR domain without the linker (212–371), or Cpr6 with a truncated basic carboxyl-terminal region (1–358) (Fig. 6B). In whole cell lysates, each of the mutants was expressed at near wild-type levels except for Cpr6-(212–371), which accumulated at lower levels. Cpr6-(171–371), but not Cpr6-(1–212), was able to interact with Hsp90, Hsp70, Aha1, and Ura2, indicating the PPIase is dispensable for those interactions. Mutations that trimmed charged regions adjacent to the TPR domains (212–371 and 1–358) resulted in reduced interaction with Hsp90 and Hsp70. However, Cpr6-(212–371) and Cpr6-(1–358) maintained interaction with Ura2, indicating that the TPR domain of Cpr6 mediates Ura2 interaction.
FIGURE 6.
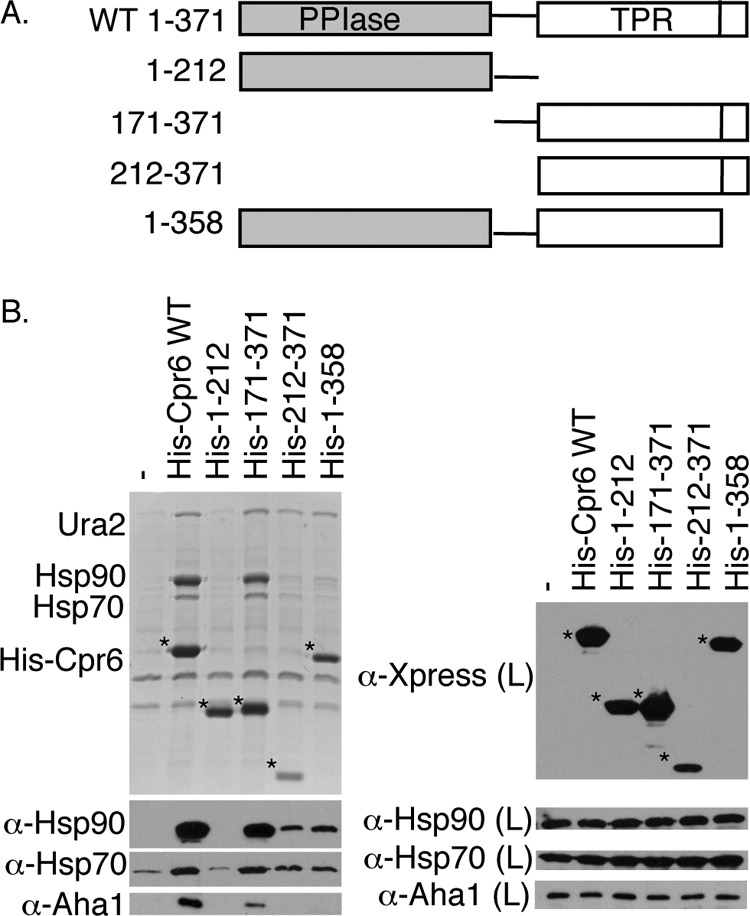
The TPR domain of Cpr6 is required for Ura2 interaction. A, schematic diagram of WT Cpr6 and truncation mutants. All constructs contain an amino-terminal His tag and Xpress epitope. B, His-Cpr6 complexes were isolated from Δcpr6 cells. His-Cpr6 constructs (denoted by an asterisk (*)) were detected with an α-Xpress monoclonal antibody.
Sequences that Flank the TPR Domain Are Critical for in Vivo Functions of Cpr6
Under normal laboratory growth conditions loss of CPR6 does not cause noticeable growth defects (16). However, previous studies showed that cells that lack non-essential co-chaperones exhibit growth defects in cells expressing mutant Hsp90 (21, 44). We compared the growth of a panel of Hsp90 temperature-sensitive mutants in WT (JJ816) or Δcpr6 (JJ110) cells that lacked chromosomal Hsp90 genes. Strains expressing Hsp90-T22I, -G170D, -Δ211–259, -Δ211–264, -W300A, -G313S, -F349A, or -ΔMEEVD (7, 35, 45) grew similarly in the presence or absence of CPR6 (not shown). However, a growth defect was observed upon combination of the hsp90-A587T mutation and Δcpr6. As shown in Fig. 7A, hsp90-A587T cells grow well at 25, 30, and 34 °C, but are dead at 37 °C. Δcpr6 hsp90-A587T cells exhibit slow growth at 25, 30, and 34 °C relative to cells expressing hsp90-A587T alone, but this growth defect was rescued by the presence of a plasmid expressing CPR6. Cpr6-(171–371) was the only truncation construct able to restore growth as well as WT Cpr6 (Fig. 7A). We also examined the ability of Cpr6-K223A and Cpr6-K309A, which disrupt the TPR-MEEVD interaction to rescue growth (Fig. 7B). Surprisingly, both of these mutants were able to support growth as well as WT Cpr6 despite their inability to stably interact with Hsp90, Hsp70, and Aha1 (Fig. 5B). Although the results in Fig. 7B were obtained when cells overexpressed Cpr6 constructs under the GPD promoter, similar results were obtained when we examined the Cpr6-K223A and Cpr6-K309A expressed under their own promoter (data not shown). Together these results suggest that Cpr6 interactions with Hsp90 are critical for function, because loss of the linker or amino acids within the basic carboxyl-terminal region disrupted Hsp90 interaction as well as Cpr6 function. However, Cpr6 and Hsp90 may have additional contacts outside the TPR-MEEVD interaction site.
FIGURE 7.
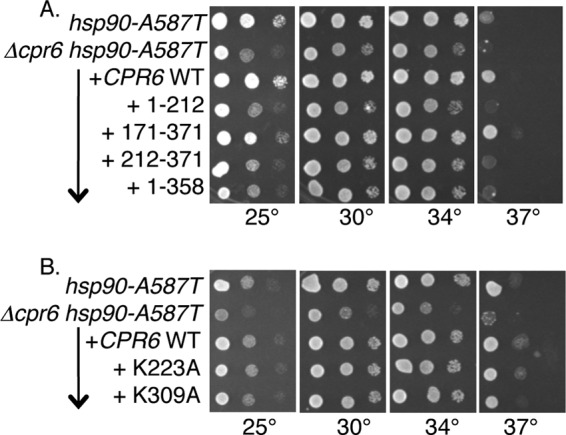
Cpr6 function requires sequences flanking the TPR domain. A and B, strains JJ816 (hsc82hsp82) and JJ110 (cpr6hsc82hsp82) expressed hsp90-A587T. Cells were transformed with pRS416 or a plasmid expressing WT or mutant Cpr6. Growth of 10-fold serial dilutions on selective media was monitored after 2 days at the indicated temperatures.
To determine whether other TPR-containing co-chaperones bind Ura2, His- and Xpress-tagged versions of each TPR-containing co-chaperone were expressed under the GPD promoter. Co-chaperone complexes were isolated from a strain expressing Ura2-TAP (Fig. 8A). Comparable levels of Hsp90 bound His-Cpr6 and His-Sti1. Consistent with a prior analysis (46), little Hsp90 binding to Cpr7 or Cns1 was observed under these conditions. Ura2-TAP was only isolated in complex with His-Cpr6, indicating the Ura2 interaction is not a function shared with other TPR containing co-chaperones.
FIGURE 8.
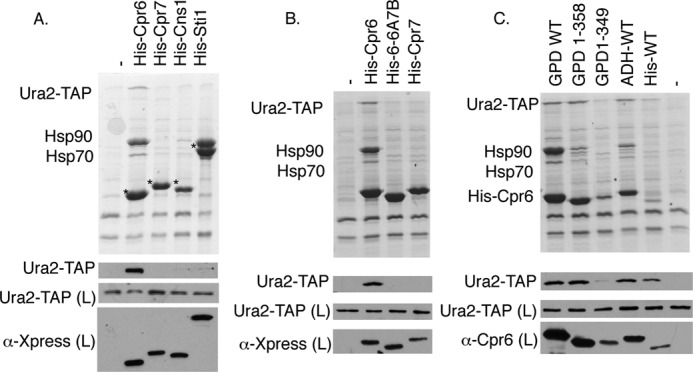
Specificity of the Ura2-Cpr6 interaction. A, URA2-TAP strain JJ1069 was transformed with empty vector (pRS316) or plasmids expressing His- and Xpress-tagged Cpr6, Cpr7, Cns1, or Sti1 under the GPD promoter. Cell extracts were incubated with nickel resin and analyzed as described. Migration of various co-chaperones is denoted with an asterisk (*). B and C, His-tagged WT or mutant Cpr6, Cpr7, or Cpr6-PPI-6A/7B complexes were isolated from the cpr6 URA2-TAP strain (JJ1067). All complexes were isolated from cells grown in media lacking uracil. Ura2-TAP was detected using α-TAP antisera.
The TPR domains of Cpr6 and Cpr7, including the linker and carboxyl-terminal basic region, share ∼33% identity, and the TPR domain of Cpr6 contains three separate repeats. To further narrow down the region of Cpr6 that interacts with Ura2, we constructed a chimeric protein that combined the PPIase domain, acidic linker, and TPR1 of Cpr6 (amino acids 1–269 of Cpr6) with TPR2, TPR3, and the basic carboxyl terminus of Cpr7 (Cpr6-PPI-6A/7B) (amino acids 292–393). We compared the interaction of this construct, as well as Cpr6 and Cpr7 with Ura2-TAP (Fig. 8B). Cpr6-PPI-6A/7B failed to interact with Hsp90 or Ura2-TAP, indicating it was no longer able to function like Cpr6. We then constructed one additional truncation mutant, Cpr6-(1–349). That construct, which was expressed at levels similar to that of His-Cpr6 expressed from its own promoter (Fig. 8C), also failed to interact with Ura2-TAP. Together, these results indicate that amino acid residues 270–358 of Cpr6 are critical for Ura2 interaction.
The above results suggest that Cpr6-PPI-6A/7B functions like Cpr7, not Cpr6. To confirm this, we examined the ability of Cpr6-PPI-6A/7B to rescue in vivo functions of Cpr6 and Cpr7. Because overexpression of CNS1 had previously been shown to rescue defects of cpr7 cells (47, 48), we also examined the ability of CNS1 to rescue cpr6-dependent growth defects. As shown in Fig. 9A, overexpression of CNS1, CPR7, and CPR6-PPI-6A/7B failed to rescue the growth defect of the Δcpr6 hsp90-A587T strain. Overexpression of STI1 was also unable to rescue growth (not shown). However, CPR6-PPI-6A/7B was able rescue the defects of a cpr7 strain (Fig. 9B). This indicates that this construct is functional and that the sequences required for Cpr7 functions are dictated by the last 100 amino acids.
FIGURE 9.
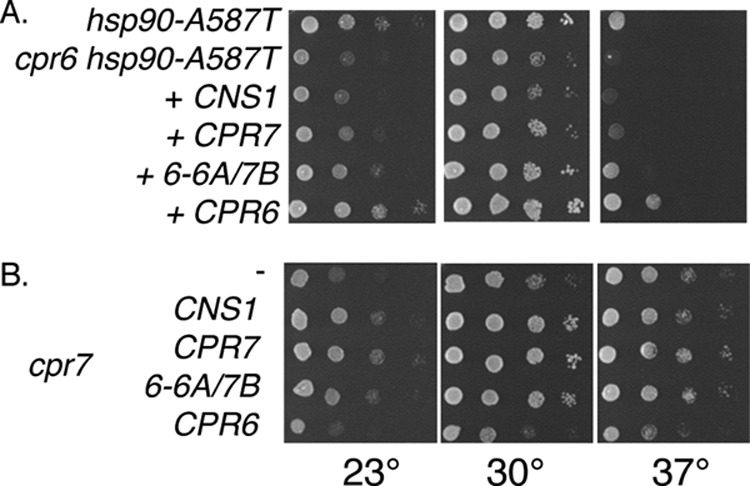
Cpr6 function cannot be rescued by overexpression of CPR7, CNS1, or CPR6-PPI-6A/7B. A, strains JJ816 (hsc82hsp82) and JJ110 (cpr6hsc82hsp82) expressed hsp90-A587T. JJ110 cells expressing empty vector or the indicated plasmid were grown for 2 days at the indicated temperatures. B, growth of cpr7 cells expressing the indicated plasmid after 2 days at the indicated temperatures.
DISCUSSION
We identified a physical interaction between Cpr6, Hsp90, and Ura2, a protein required for pyrimidine biosynthesis, and present strong evidence that Cpr6 directly binds Ura2. Although the human homolog of Cpr6, Cyp40, has been shown in complex with the progesterone and estrogen receptors (31, 49), a direct interaction between Cyp40 and an Hsp90 client has not been previously described. Our results indicate that the Ura2-Cpr6 interaction site localizes to the TPR domain, which is also a major site of interaction between Cpr6 and Hsp90, raising the intriguing possibility that the TPR domain of Cpr6 may be able to simultaneously bind Hsp90 and Ura2.
The charged linker, TPR domain, and basic region of Cpr7 was previously shown to be sufficient for in vivo functions (7, 14, 46), and our results suggest that the last 100 amino acids contain the critical determinants. Prior studies also suggest that the TPR domains of Cpr6, Cpr7, and Cyp40, including flanking sequences, directly contact other proteins but our studies are the first to show that Cpr6 has a specific interaction with an Hsp90 client that is native to yeast. Cyp40 was able to directly bind the DNA-binding domain of the c-Myb transcription factor and inhibit its DNA binding activity (50). Cpr6 and Cpr7 were both shown to interact with different parts of the p33 replication protein from tomato bushy stunt virus, although only Cpr7 inhibited virus replication in a cell-free assay (51). Analysis of Cpr6 mutants indicates that residues 270–358 are critical for Ura2 interaction. We have not yet identified specific amino acids within the TPR domain of Cpr6 that dictate Ura2 interaction, but surfaces distinct from the MEEVD-binding groove have previously been shown to be important for TPR-protein interactions (52).
Regions outside of the TPR domain of Cpr6 are critical for Hsp90 interaction and in vivo functions. Specifically, deletion of the linker region or truncation of the basic carboxyl-terminal residues reduced the ability of Cpr6 to stably interact with Hsp90. These same regions have previously been found to be important for the Cyp40-Hsp90 interaction and Cyp40 chaperone activity (53, 54). Surprisingly, alteration of residues that specifically disrupt the interaction of Cpr6 with the MEEVD residues of Hsp90 did not disrupt Cpr6 function. Similar results were obtained upon mutation of Sti1 (21). Additional sites of contact between Hsp90 and Sti1 have been identified (55), and it seems likely that Cpr6 also contacts multiple sites on Hsp90. The function of the PPIase domain of Cpr6 remains unknown.
Cpr6 appears to directly interact with Hsp70, but the in vivo significance of the Cpr6-Hsp70 interaction is unknown. Like Hsp90, Hsp70 contains a conserved carboxyl-terminal EEVD motif (42), and the K223A and K309A alterations that target the EEVD binding groove disrupted Cpr6 interaction with both Hsp90 and Hsp70. Cyp40 was previously shown to interact with Hsp70 in vitro, but Cyp40 did not affect the ATPase activity of Hsp70 (43).
Ura2 is an Hsp90 client that exhibits reduced accumulation upon Hsp90 inhibition, but we were unable to demonstrate a role for Cpr6 in Ura2 accumulation, regulation, or activity. The carbamoyl-phosphate synthetase domains of Ura2 have homology with Cpa1 and Cpa2 in yeast (38). It is possible that those proteins compensate for reduced Ura2 function in cells expressing Hsp90 or Cpr6 mutants. Alternatively, Cpr6 may have more modest effects on Ura2 function that are not detectable using our assays. Cyp40, along with FKBP51 and FKBP52 bind Hsp90 during the late stage of the steroid hormone chaperone cycle, once receptor is bound to Hsp90 in the ATP-bound, closed conformation (31). However, Cyp40, FKBP51, and FKBP52 were not required during in vitro folding of steroid hormone receptors using purified proteins (56, 57).
Hsp90 interacts with over 10 co-chaperones in yeast (6), and one question is whether co-chaperones have unique, overlapping, or redundant functions. Among the TPR co-chaperones, it has been shown that FKBP51/52 and Cyp40 exhibit different binding specificities for steroid hormone receptors and in some cases have differing effects on receptor activity. FKBP52 was able to bind the GR directly, but that interaction did not require the TPR domain (58, 59). We identified the first known functions unique to Cpr6. Cpr6, but not other TPR-containing co-chaperones, was specifically able to interact with Ura2, and CPR6-specific growth defects were not rescued by overexpression of CPR7, CNS1, or STI1. The TPR domains of Cpr6 and Cpr7 consist of a tandem array of three repeat units, each of which forms a pair of anti-parallel α helices. Our results with the Cpr6-PPI-6A/7B chimera indicate that the last 100 amino acids of Cpr7, encompassing the second and third TPR repeats, as well as additional sequences after the TPR domain, are sufficient to confer Cpr7 functions. Unfortunately the mirror chimera, Cpr7-PPI-7A/6B, was not stably expressed (not shown), so we were unable to determine whether those same regions are sufficient to dictate Cpr6-specific functions. Cpr7 may play a role in regulating Hsp90 conformational changes (7), whereas Cpr6 cooperates with Aha1 to promote the ATPase activity of Hsp90 (8). Further studies are necessary to determine whether distinct Cpr6 and Cpr7 functions are due to differential binding to Hsp90 clients or due to differing effects on Hsp90.
Acknowledgments
We thank Elizabeth Craig (University of Wisconsin-Madison) for the anti-Ssa1/2 antisera and ura2 yeast strain. We also thank Lee Deobald (University of Idaho Environmental Biotechnology Institute) for assistance in preparing samples for mass spectrometry.
This work was supported by National Science Foundation Grant MCB-0744522 (to J. L. J.).
- TPR
- tetratricopeptide repeat
- PPIase
- peptidyl-prolyl isomerase
- DMSO
- dimethyl sulfoxide
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate.
REFERENCES
- 1. Wegele H., Müller L., Buchner J. (2004) Hsp70 and Hsp90. A relay team for protein folding. Rev. Physiol. Biochem. Pharmacol. 151, 1–44 [DOI] [PubMed] [Google Scholar]
- 2. Vaughan C. K., Gohlke U., Sobott F., Good V. M., Ali M. M., Prodromou C., Robinson C. V., Saibil H. R., Pearl L. H. (2006) Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 23, 697–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richter K., Buchner J. (2011) Closing in on the Hsp90 chaperone-client relationship. Structure 19, 445–446 [DOI] [PubMed] [Google Scholar]
- 4. Street T. O., Lavery L. A., Agard D. A. (2011) Substrate binding drives large-scale conformational changes in the Hsp90 molecular chaperone. Mol. Cell 42, 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Genest O., Reidy M., Street T. O., Hoskins J. R., Camberg J. L., Agard D. A., Masison D. C., Wickner S. (2013) Uncovering a region of heat shock protein 90 important for client binding in E. coli and chaperone function in yeast. Mol. Cell 49, 464–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li J., Soroka J., Buchner J. (2012) The Hsp90 chaperone machinery. Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 1823, 624–635 [DOI] [PubMed] [Google Scholar]
- 7. Zuehlke A. D., Johnson J. L. (2012) Chaperoning the chaperone. A role for the co-chaperone Cpr7 in modulating Hsp90 function in Saccharomyces cerevisiae. Genetics 191, 805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li J., Richter K., Reinstein J., Buchner J. (2013) Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nat. Struct. Mol. Biol. 20, 326–331 [DOI] [PubMed] [Google Scholar]
- 9. Catlett M. G., Kaplan K. B. (2006) Sgt1p is a unique co-chaperone that acts as a client-adaptor to link Hsp90 to Skp1p. J. Biol. Chem. 281, 33739–33748 [DOI] [PubMed] [Google Scholar]
- 10. Chen S., Smith D. F. (1998) Hop as an adaptor in the heat shock protein 70 (Hsp70) and hsp90 chaperone machinery. J. Biol. Chem. 273, 35194–35200 [DOI] [PubMed] [Google Scholar]
- 11. Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H., Hartl F. U., Moarefi I. (2000) Structure of TPR domain-peptide complexes. Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101, 199–210 [DOI] [PubMed] [Google Scholar]
- 12. Freeman B. C., Toft D. O., Morimoto R. I. (1996) Molecular chaperone machines. Chaperone activities of the cyclophilin Cyp-40 and the steroid aporeceptor-associated protein p23. Science 274, 1718–1720 [DOI] [PubMed] [Google Scholar]
- 13. Mayr C., Richter K., Lilie H., Buchner J. (2000) Cpr6 and Cpr7, two closely related Hsp90-associated immunophilins from Saccharomyces cerevisiae, differ in their functional properties. J. Biol. Chem. 275, 34140–34146 [DOI] [PubMed] [Google Scholar]
- 14. Duina A. A., Chang H. C., Marsh J. A., Lindquist S., Gaber R. F. (1996) A cyclophilin function in Hsp90-dependent signal transduction. Science 274, 1713–1715 [DOI] [PubMed] [Google Scholar]
- 15. Duina A. A., Kalton H. M., Gaber R. F. (1998) Requirement for Hsp90 and a CyP-40-type cyclophilin in negative regulation of the heat shock response. J. Biol. Chem. 273, 18974–18978 [DOI] [PubMed] [Google Scholar]
- 16. Duina A. A., Marsh J. A., Gaber R. F. (1996) Identification of two CyP-40-like cyclophilins in Saccharomyces cerevisiae, one of which is required for normal growth. Yeast 12, 943–952 [DOI] [PubMed] [Google Scholar]
- 17. Panaretou B., Siligardi G., Meyer P., Maloney A., Sullivan J. K., Singh S., Millson S. H., Clarke P. A., Naaby-Hansen S., Stein R., Cramer R., Mollapour M., Workman P., Piper P. W., Pearl L. H., Prodromou C. (2002) Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 10, 1307–1318 [DOI] [PubMed] [Google Scholar]
- 18. Denis-Duphil M. (1989) Pyrimidine biosynthesis in Saccharomyces cerevisiae. The ura2 cluster gene, its multifunctional enzyme product, and other structural or regulatory genes involved in de novo UMP synthesis. Biochem. Cell Biol. 67, 612–631 [DOI] [PubMed] [Google Scholar]
- 19. McClellan A. J., Xia Y., Deutschbauer A. M., Davis R. W., Gerstein M., Frydman J. (2007) Diverse cellular functions of the hsp90 molecular chaperone uncovered using systems approaches. Cell 131, 121–135 [DOI] [PubMed] [Google Scholar]
- 20. Krogan N. J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A. P., Punna T., Peregrín-Alvarez J. M., Shales M., Zhang X., Davey M., Robinson M. D., Paccanaro A., Bray J. E., Sheung A., Beattie B., Richards D. P., Canadien V., Lalev A., Mena F., Wong P., Starostine A., Canete M. M., Vlasblom J., Wu S., Orsi C., Collins S. R., Chandran S., Haw R., Rilstone J. J., Gandi K., Thompson N. J., Musso G., St Onge P., Ghanny S., Lam M. H., Butland G., Altaf-Ul A. M., Kanaya S., Shilatifard A., O'Shea E., Weissman J. S., Ingles C. J., Hughes T. R., Parkinson J., Gerstein M., Wodak S. J., Emili A., Greenblatt J. F. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 [DOI] [PubMed] [Google Scholar]
- 21. Flom G., Weekes J., Williams J. J., Johnson J. L. (2006) Effect of mutation of the tetratricopeptide repeat and aspartate-proline 2 domains of Sti1 on Hsp90 signaling and interaction in Saccharomyces cerevisiae. Genetics 172, 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson J. L., Halas A., Flom G. (2007) Nucleotide-dependent interaction of Saccharomyces cerevisiae Hsp90 with the cochaperone proteins Sti1, Cpr6, and Sba1. Mol. Cell. Biol. 27, 768–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leach M. D., Budge S., Walker L., Munro C., Cowen L. E., Brown A. J. (2012) Hsp90 orchestrates transcriptional regulation by Hsf1 and cell wall remodelling by MAPK signalling during thermal adaptation in a pathogenic yeast. PLoS Pathog. 8, e1003069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 25. Flom G., Behal R. H., Rosen L., Cole D. G., Johnson J. L. (2007) Definition of the minimal fragments of Sti1 required for dimerization, interaction with Hsp70 and Hsp90 and in vivo functions. Biochem. J. 404, 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flom G. A., Langner E., Johnson J. L. (2012) Identification of an Hsp90 mutation that selectively disrupts cAMP/PKA signaling in Saccharomyces cerevisiae. Curr. Genet. 58, 149–163 [DOI] [PubMed] [Google Scholar]
- 27. Mumberg D., Müller R., Funk M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 [DOI] [PubMed] [Google Scholar]
- 28. Li J., Richter K., Buchner J. (2011) Mixed Hsp90-cochaperone complexes are important for the progression of the reaction cycle. Nat. Struct. Mol. Biol. 18, 61–66 [DOI] [PubMed] [Google Scholar]
- 29. Werner-Washburne M., Stone D. E., Craig E. A. (1987) Complex interactions among members of an essential subfamily of hsp70 genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 7, 2568–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stepanova L., Leng X., Parker S. B., Harper J. W. (1996) Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 10, 1491–1502 [DOI] [PubMed] [Google Scholar]
- 31. Barent R. L., Nair S. C., Carr D. C., Ruan Y., Rimerman R. A., Fulton J., Zhang Y., Smith D. F. (1998) Analysis of FKBP51/FKBP52 chimeras and mutants for Hsp90 binding and association with progesterone receptor complexes. Mol. Endocrinol. 12, 342–354 [DOI] [PubMed] [Google Scholar]
- 32. Pearl L. H., Prodromou C., Workman P. (2008) The Hsp90 molecular chaperone. An open and shut case for treatment. Biochem. J. 410, 439–453 [DOI] [PubMed] [Google Scholar]
- 33. Mollapour M., Tsutsumi S., Donnelly A. C., Beebe K., Tokita M. J., Lee M. J., Lee S., Morra G., Bourboulia D., Scroggins B. T., Colombo G., Blagg B. S., Panaretou B., Stetler-Stevenson W. G., Trepel J. B., Piper P. W., Prodromou C., Pearl L. H., Neckers L. (2010) Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell 37, 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mandal A. K., Gibney P. A., Nillegoda N. B., Theodoraki M. A., Caplan A. J., Morano K. A. (2010) Hsp110 chaperones control client fate determination in the hsp70-hsp90 chaperone system. Mol. Biol. Cell 21, 1439–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nathan D. F., Lindquist S. (1995) Mutational analysis of Hsp90 function. Interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol. 15, 3917–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kwapisz M., Wery M., Després D., Ghavi-Helm Y., Soutourina J., Thuriaux P., Lacroute F. (2008) Mutations of RNA polymerase II activate key genes of the nucleoside triphosphate biosynthetic pathways. EMBO J. 27, 2411–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thiebaut M., Colin J., Neil H., Jacquier A., Séraphin B., Lacroute F., Libri D. (2008) Futile cycle of transcription initiation and termination modulates the response to nucleotide shortage in S. cerevisiae. Mol. Cell 31, 671–682 [DOI] [PubMed] [Google Scholar]
- 38. Antonelli R., Estevez L., Denis-Duphil M. (1998) Carbamyl-phosphate synthetase domain of the yeast multifunctional protein Ura2 is necessary for aspartate transcarbamylase inhibition by UTP. FEBS Lett. 422, 170–174 [DOI] [PubMed] [Google Scholar]
- 39. Pratt W. B., Toft D. O. (2003) Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. (Maywood) 228, 111–133 [DOI] [PubMed] [Google Scholar]
- 40. Ward B. K., Allan R. K., Mok D., Temple S. E., Taylor P., Dornan J., Mark P. J., Shaw D. J., Kumar P., Walkinshaw M. D., Ratajczak T. (2002) A structure-based mutational analysis of cyclophilin 40 identifies key residues in the core tetratricopeptide repeat domain that mediate binding to Hsp90. J. Biol. Chem. 277, 40799–40809 [DOI] [PubMed] [Google Scholar]
- 41. Taylor P., Dornan J., Carrello A., Minchin R. F., Ratajczak T., Walkinshaw M. D. (2001) Two structures of cyclophilin 40. Folding and fidelity in the TPR domains. Structure 9, 431–438 [DOI] [PubMed] [Google Scholar]
- 42. Freeman B. C., Myers M. P., Schumacher R., Morimoto R. I. (1995) Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J. 14, 2281–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carrello A., Allan R. K., Morgan S. L., Owen B. A., Mok D., Ward B. K., Minchin R. F., Toft D. O., Ratajczak T. (2004) Interaction of the Hsp90 cochaperone cyclophilin 40 with Hsc70. Cell Stress Chaperones 9, 167–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Armstrong H., Wolmarans A., Mercier R., Mai B., LaPointe P. (2012) The co-chaperone Hch1 regulates Hsp90 function differently than its homologue Aha1 and confers sensitivity to yeast to the Hsp90 inhibitor NVP-AUY922. Plos One 7, e49322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meyer P., Prodromou C., Hu B., Vaughan C., Roe S. M., Panaretou B., Piper P. W., Pearl L. H. (2003) Structural and functional analysis of the middle segment of hsp90. Implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell 11, 647–658 [DOI] [PubMed] [Google Scholar]
- 46. Tesic M., Marsh J. A., Cullinan S. B., Gaber R. F. (2003) Functional interactions between Hsp90 and the co-chaperones Cns1 and Cpr7 in Saccharomyces cerevisiae. J. Biol. Chem. 278, 32692–32701 [DOI] [PubMed] [Google Scholar]
- 47. Dolinski K. J., Cardenas M. E., Heitman J. (1998) CNS1 encodes an essential p60/Sti1 homolog in Saccharomyces cerevisiae that suppresses cyclophilin 40 mutations and interacts with Hsp90. Mol. Cell. Biol. 18, 7344–7352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marsh J. A., Kalton H. M., Gaber R. F. (1998) Cns1 is an essential protein associated with the hsp90 chaperone complex in Saccharomyces cerevisiae that can restore cyclophilin 40-dependent functions in cpr7[/Delta] cells. Mol. Cell. Biol. 18, 7353–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ratajczak T., Carrello A., Mark P. J., Warner B. J., Simpson R. J., Moritz R. L., House A. K. (1993) The cyclophilin component of the unactivated estrogen receptor contains a tetratricopeptide repeat domain and shares identity with p59 (FKBP59). J. Biol. Chem. 268, 13187–13192 [PubMed] [Google Scholar]
- 50. Leverson J. D., Ness S. A. (1998) Point mutations in v-Myb disrupt a cyclophilin-catalyzed negative regulatory mechanism. Mol. Cell 1, 203–211 [DOI] [PubMed] [Google Scholar]
- 51. Lin J. Y., Mendu V., Pogany J., Qin J., Nagy P. D. (2012) The TPR domain in the host Cyp40-like cyclophilin binds to the viral replication protein and inhibits the assembly of the tombusviral replicase. PLoS Pathog 8, e1002491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zeytuni N., Zarivach R. (2012) Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 20, 397–405 [DOI] [PubMed] [Google Scholar]
- 53. Ratajczak T., Carrello A. (1996) Cyclophilin 40 (CyP-40), mapping of its hsp90 binding domain and evidence that FKBP52 competes with CyP-40 for hsp90 binding. J. Biol. Chem. 271, 2961–2965 [DOI] [PubMed] [Google Scholar]
- 54. Mok D., Allan R. K., Carrello A., Wangoo K., Walkinshaw M. D., Ratajczak T. (2006) The chaperone function of cyclophilin 40 maps to a cleft between the prolyl isomerase and tetratricopeptide repeat domains. FEBS Lett. 580, 2761–2768 [DOI] [PubMed] [Google Scholar]
- 55. Schmid A. B., Lagleder S., Gräwert M. A., Röhl A., Hagn F., Wandinger S. K., Cox M. B., Demmer O., Richter K., Groll M., Kessler H., Buchner J. (2012) The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 31, 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kosano H., Stensgard B., Charlesworth M. C., McMahon N., Toft D. (1998) The assembly of progesterone receptor-hsp90 complexes using purified proteins. J. Biol. Chem. 273, 32973–32979 [DOI] [PubMed] [Google Scholar]
- 57. Morishima Y., Kanelakis K. C., Silverstein A. M., Dittmar K. D., Estrada L., Pratt W. B. (2000) The Hsp organizer protein hop enhances the rate of but is not essential for glucocorticoid receptor folding by the multiprotein Hsp90-based chaperone system. J. Biol. Chem. 275, 6894–6900 [DOI] [PubMed] [Google Scholar]
- 58. Silverstein A. M., Galigniana M. D., Kanelakis K. C., Radanyi C., Renoir J. M., Pratt W. B. (1999) Different regions of the immunophilin FKBP52 determine its association with the glucocorticoid receptor, hsp90, and cytoplasmic dynein. J. Biol. Chem. 274, 36980–36986 [DOI] [PubMed] [Google Scholar]
- 59. Riggs D. L., Cox M. B., Cheung-Flynn J., Prapapanich V., Carrigan P. E., Smith D. F. (2004) Functional specificity of co-chaperone interactions with Hsp90 client proteins. Crit. Rev. Biochem. Mol. Biol. 39, 279–295 [DOI] [PubMed] [Google Scholar]