

Background: Twist1 is a transcriptional repressor that inhibits the development of Th1 cells.
Results: Twist1 impairs Th17 and Tfh cell development by decreasing IL-6-induced STAT3.
Conclusion: Twist1 represses the development of autoimmunity and germinal center B cell expansion and antibody production following immunization.
Significance: Twist1 is a common repressor of cell-mediated and humoral adaptive immunity.
Keywords: Autoimmunity, Differentiation, Inflammation, Interleukin, STAT Transcription Factor, STAT3, T Cell Biology, Transcription Factors, T Helper Cell
Abstract
Cytokine responsiveness is a critical component of the ability of cells to respond to the extracellular milieu. Transcription factor-mediated regulation of cytokine receptor expression is a common mode of altering responses to the external environment. We identify the transcription factor Twist1 as a component of a STAT3-induced feedback loop that controls IL-6 signals by directly repressing Il6ra. Human and mouse T cells lacking Twist1 have an increased ability to differentiate into Th17 cells. Mice with a T cell-specific deletion of Twist1 demonstrate increased Th17 and T follicular helper cell development, early onset experimental autoimmune encephalomyelitis, and increased antigen-specific antibody responses. Thus, Twist1 has a critical role in limiting both cell-mediated and humoral immunity.
Introduction
CD4 T helper cells control immunity to pathogens and the development of inflammatory disease by acquiring the ability to secrete effector cytokines. The differentiation of T helper subsets follows exposure to a specific cytokine environment. IL-12 promotes development of Th1 cells, IL-4 promotes Th2 differentiation, and there are partially redundant roles for IL-6 and IL-21 in T follicular helper (Tfh)3 cell development (1, 2). Th17 cells develop in response to several cytokines, including IL-6, TGF-β, IL-1β, and IL-23 (3–7). Restricted cytokine expression in Th17 cells result from coordinated expression of RORγt, BATF, IRF4, and other factors (8–10). Some of the factors in this network are required for the development of additional Th subsets and cooperate with specialized factors to promote acquisition of distinct phenotypes. BATF and IRF4, for example, function with BCL6 to promote development of Tfh cells (11). Cytokine signals that regulate T helper cell differentiation rely upon STAT proteins.
Responsiveness to the extracellular milieu is a core component of the adaptability of the immune system. Cytokines mediate intracellular communication and can promote the differentiation and proliferation of responsive cells. Regulating cytokine responsiveness is a recurring theme during the development of effector T cell subsets. Cytokine signaling can reinforce responsiveness by modulating receptor expression. The signal transducer and activator of transcription factor STAT5 promotes Il4ra and Il12rb2 expression, genes that are critical, respectively, for IL-4 and IL-12 signaling to stimulate Th2 and Th1 differentiation (12, 13). STAT3 promotes Il23r expression that is required for the development of inflammatory Th17 cells (14). Conversely, decreased receptor expression interferes with the ability of a cell to respond to the cytokine environment. STAT5 inhibits expression of Il6ra and Il6st, limiting Th17 differentiation (12). Similarly, the transcription factor GATA3 diminishes expression of Il12rb2 and Stat4 that mediate IL-12 responses and prevents Th2 cells from responding to a Th1-promoting environment (15, 16). Thus, regulation of cytokine signaling provides a very proximal point to control the differentiation of Th effector phenotypes.
STAT3 is required for multiple T helper cell lineages, including Th2, Th17, and Tfh (17–21). As part of its function, STAT3 activates genes that are common among these lineages (Maf, Batf, Irf4) and genes that are lineage-specific, such as Rorc for Th17 and Bcl6 for Tfh (22–27). However, a balance between positive and negative regulatory factors controls the differentiation of each of these subsets. The IL-2-STAT5 signaling pathway limits IL-17 production, and the balance between STAT3 and STAT5 activation determines the ability of cells to produce inflammatory cytokines (26, 28). STAT5 signaling similarly decreases the development of Tfh cells (29, 30). Whether additional transcription factors regulate the responsiveness of differentiating T cells to STAT3-activating cytokines has not been completely explored.
Twist1 is a basic helix-loop-helix protein important for developmental programs, including craniofacial, heart, and limb development during embryogenesis, and is induced by IL-12-STAT4 signaling in Th1 cells (31, 32). Twist1 displays preferential expression in Th1 cells and limits the expression of inflammatory cytokines, including IFN-γ and TNF-α in Th1 cells (31). Twist1 negatively regulates Th1 gene expression and cytokine production through several mechanisms, including decreasing the expression of Il12rb2, resulting in diminished STAT4 activation (33). Because Twist1 controls inflammatory cytokine production in Th1 cells, we speculated that Twist1 might play important roles in other T helper cell subsets. In this report, we show that Twist1 expression is induced following stimulation with STAT3-inducing cytokines and that it reduces IL-17 production in Th17 cells in vitro and in vivo. Moreover, Twist1 represses Tfh cell development in vivo. Twist1 represses Th17 and Tfh differentiation by directly binding to, and repressing expression of, the Il6ra locus, subsequently reducing STAT3 activation. Thus, Twist1 is a STAT3-induced negative regulator of Th17 and Tfh differentiation, limiting the development of cell-mediated and humoral immunity.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6 mice were purchased from Harlan Sprague-Dawley (Indianapolis, IN). Twist1fl/flCD4-Cre and Stat3fl/flCD4-Cre mice were described previously (17, 33). Twist1fl/flCD4-Cre mice were backcrossed to C57BL/6 mice for six generations with Cre-negative littermates as wild type mice for in vivo experiments. Mice were maintained under specific pathogen-free conditions. All experiments were performed with the approval of the Indiana University Institutional Animal Care and Use Committee.
In Vitro T Cell Differentiation
Naïve CD4+CD62L+ T cells were isolated from spleen and lymph nodes using MACS beads and columns (Miltenyi Biotec). CD4+ T cells were activated with plate-bound anti-CD3 (2 μg/ml 145–2C11) and soluble anti-CD28 (0.5 μg/ml BD Pharmingen) with additional cytokines (all from PeproTech) and antibodies (Bio X cell) to generate Th1 (5 ng/ml IL-12; and 10 μg/ml anti-IL-4, 11B11), Th2 (10 ng/ml IL-4; and 10 μg/ml anti-IFN-γ XMG), Th9 (20 ng/ml IL-4; 2 ng/ml TGF-β; and 10 μg/ml anti-IFN-γ, XMG), Th17 (100 ng/ml IL-6; 10 ng/ml IL-23; 10 ng/ml IL-1β; 2 ng/ml TGF-β;10 μg/ml anti-IL-4, 11B11; and 10 μg/ml anti-IFN-γ, XMG) or regulatory T (Treg; 2 ng/ml TGF-β, and 10 μg/ml anti-IL-4, 11B11) culture conditions. Cells were expanded after 3 days with half-concentration of the original cytokines in fresh medium. Cells were harvested on day 5 for analysis. To inhibit STAT3 activation, doses of cucurbitacin I (JSI-124, Sigma Aldrich) were added into WT and Twist1-deficient Th17 cell cultures. Phosphorylated STAT3 and cytokine production were measured using intracellular staining and ELISA, respectively. For receptor-blocking experiments, Th17 cells were cultured as above in the presence of control antibody or blocking antibody to IL-6R (15A7, Bio X cell). Cytokine production was measured using ELISA.
Induction of EAE and ex Vivo Analyses
Induction and scoring of experimental autoimmune encephalomyelitis (EAE) disease has been described previously (34). In brief, a cohort of 8–12-week-old female WT and Twist1-deficient mice (7 mice/group) were immunized subcutaneously with 100 μg of myelin oligodendrocyte glycoprotein (MOGp35-55) peptide antigen (Genemed Synthesis) in a 150-μl emulsion of complete Freund's adjuvant (Sigma Aldrich) on days 0 and 7. The mice were injected (intraperitoneal) with 100 ng of pertussis toxin (Sigma Aldrich) on days 0 and 2. The clinical signs were scored daily for 30 days. On day 12 following induction of EAE, splenocytes were isolated and stimulated with MOG peptide for 48 h, and cytokine production was measured by ELISA. Mononuclear cells were isolated from brain using a 30%/70% Percoll gradient and stimulated with PMA and ionomycin for 2 h followed by monensin for a total of 6 h before staining for intracellular cytokine production.
Sheep Red Blood Cell (SRBC) Immunization and Antibody Titer Measurement
SRBC (VWR Intl.) were washed three times with PBS. Wild type and Twist1 mutant mice were injected with 1 × 109 cells (intraperitoneal). Mice were sacrificed after 9 days for the analysis. Serum was collected by cardiac puncture, and SRBC-specific antibodies were measured by ELISA as described previously (35). For in vivo receptor-blocking experiments, SRBC-immunized mice were injected (intraperitoneal) with 50 μg/ml of control antibody or blocking antibody to IL-6R (15A7, Bio X cell) on days 4, 6, and 8. Mice were sacrificed after 9 days for the analysis.
Retroviral Expression Vectors and Retroviral Transduction
Bicistronic retrovirus expressing enhanced GFP only (MIEG) or Twist1 and enhanced GFP (Twist1) and the preparation of retroviral stocks were described previously (33). CD4+ T cells were transduced on day 2 with control or retrovirus vector expressing gene of interest by centrifugation at 2000 rpm at 25 °C for 1 h in the presence of 8 μg/ml polybrene. Viral supernatant was replaced with the former culture supernatant supplemented with 50 units/ml human IL-2. After spin infection, cells were expanded on day 3 and analyzed on day 5.
Human Helper T Cell Differentiation
The use of human cells was approved by the Institutional Review Board of Indiana University. Naïve CD4+ T cells were isolated from PBMCs using magnetic beads (Miltenyi Biotec). For Th17 cell differentiation, naïve CD4+ cells were activated with anti-CD3 (2 μg/ml; HIT3a; BD Pharmingen) and soluble anti-CD28 (0.5 μg/ml; CD28.2; Biolegend) with additional cytokines and antibodies 10 ng/ml human IL-1β, 25 ng/ml human IL-6, 25 ng/ml human IL-23, 5 ng/ml human TGF-β, 10 μg/ml anti-IFN-γ, and 10 μg/ml anti-IL-4 (all from R&D Systems) and 25 ng/ml human IL-21 (Cell Sciences). On day 3, cells were expanded with additional medium and half-concentration of cytokines. Cells were harvested for analysis on day 5.
Transfection of siRNA
siRNAs targeting Twist1 or TWIST1 were purchased from Santa Cruz Biotechnology. For mouse Th17 cell transfection, CD4+ T cells were transfected with siRNA on day 2 using Amaxa Nucleofector kit (Lonza), rested overnight with hIL-2, and restimulated with anti-CD3 for 24 h for gene expression and cytokine production analyses. For human Th17 cell transfection, day 5-differentiated Th17 cells were transfected with siRNA using a human T cell nucleofector kit (Lonza), rested overnight with hIL-2, and restimulated with anti-CD3 for 24 h for gene expression analyses.
Luciferase Reporter Assay
The IL6RA promoter reporter was purchased from SwitchGear Genomics. For analyzing the effect of Twist1 on IL6RA promoter activity, Jurkat T cells were grown in RPMI 1640 with 10% FBS and transfected with 2–5 μg of the IL6RA luciferase reporter plasmid and control or increasing concentration of plasmid expressing Twist1 via FuGENE reagent (Roche Diagnostics). After 24 h, transfected cells were stimulated with PMA and ionomycin for 6 h before analyzing with the Dual-Luciferase system (Promega).
Analysis of Gene Expression, ELISA, and Flow Cytometry
Quantitative RT-PCR and ELISA were performed as described previously (36). For surface staining, resting T cells were stained for IL-2Rα-FITC and IL-6Rα-phycoerythrin (BD Pharmingen) and fixed with 2% paraformaldehyde for 10 min before analysis. For cytokine staining, CD4+ T cells were stimulated with PMA and ionomycin for 2 h followed by monesin for a total 5 h, fixed, permeabilized with 0.2% saponin, and stained for IL-17A-PE, IL-17F-Alexa Fluor 647, and IFNγ-phycoerythrin-Cy7 (BD Pharmingen). CD4-Alexa Fluor 700, ICOS-FITC, PD-1-PerCPCy5.5 (Biolegend), and biotinylated CXCR5 (eBioscience) were used to stain for Tfh cells. PNA-FITC (Vector labs), B220-phycoerythrin, GL-7-Alexa Fluor 647, biotinylated Fas (BD Pharmingen), and CD19-AF700 (Biolegend) were used to stain for germinal center B cells. A Foxp3 staining buffer set (eBioscience) was used for Bcl-6-phycoerythrin (BD Pharmingen) and Twist1-Alexa Fluor 647 (R&D Systems) intracellular staining. For phospho-STAT3 and phospho-STAT5 analyses, cells were fixed, permeabilized using 100% ice-cold methanol, and stained for phospho-STAT3-Alexa Fluor 647 and phospho-STAT5-phycoerythrin (BD Pharmingen) before analysis. For immunoblot analysis, whole-cell protein lysates were extracted from T cells and immunoblotted with Twist1 (Twist2C1a) or β-actin (C4) (Santa Cruz Biotechnology) as a control.
ChIP
ChIP assay was performed as described (37). In brief, resting Th17 cells were cross-linked for 10 min with 1% formaldehyde and lysed by sonication. After preclearing with salmon sperm DNA, bovine serum albumin, and protein agarose bead slurry (50%), cell extracts were incubated with either rabbit polyclonal STAT3 (C-20), Twist1 (H-81) (Santa Cruz Biotechnology), or normal rabbit IgG (Millipore) overnight at 4 °C. The immunocomplexes were precipitated with protein agarose beads at 4 °C for 2 h, washed, eluted, and cross-links were reversed at 65 °C overnight. DNA was purified, resuspended in H2O, and analyzed by quantitative PCR with Taqman or SYBR primers as described previously (17). Additional primers were as follows: Twist1 distal, 5′-AGCATGCAGGGCTTAATTTG-3′ (forward) and 5′-ACTGTGCTTCCAAAGGTGCT-3′ (reverse); Twist1 proximal, 5′-GCCAGGTCGGTTTTGAATGG-3′ (forward) and 5′-CGTGCGGGCGGAAAGTTTGG-3′ (reverse); Il6ra, 5′-CGTGGCTCAGATCGGTGT-3′ (forward) and 5′-GCCATCCTACTGGGCTTTC-3′ (reverse); Bcl6, 5′-CCCAACATAATTGTCCCAAA-3′ (forward) and 5′-GCGAGAGAGTTGAGCCGTTA-3′ (reverse); and Icos, 5′-ACACCA CATCAACCTCCACA-3′ (forward) and 5′-GAAGACAAAGACACGGCAGA-3′ (reverse).
Statistical Analysis
Student's t test (two-tailed) was used to generate p values for all data.
RESULTS
STAT3-activating Cytokines Induce Twist1 Expression
Twist1 negatively regulates cytokine production in Th1 cells, although effects in other T helper subsets have not been defined (33). To test this, we compared cytokine production from in vitro polarized cultures of naïve CD4+ T cells from mice carrying a conditional mutant allele of Twist1 crossed to CD4-Cre mice (Twist1fl/flCD4-Cre+) and Twist1fl/flCD4-Cre− littermate controls (referred to as wild type). As shown previously, Th1 cells display increased production of IFN-γ (Fig. 1A). Cytokine production by Th2 and Th9 cells and percentages of Foxp3+ in vitro-derived Treg cells were similar between wild type and Twist1-deficient cultures (Fig. 1, A and B). In contrast, there was a marked increase in IL-17 production from Th17 cultures (Fig. 1A).
FIGURE 1.

Twist1 is regulated by STAT3-activating cytokines in Th17 cells. A, naive wild type and Twist1-deficient CD4+ T cells were cultured under Th1, Th2, Th9, Th17, and Treg cell polarizing conditions. Th1, Th2, Th9, and Th17 cells were restimulated with anti-CD3 for 24 h to access cytokine production by ELISA. B, percentage of Foxp3 expression in Treg cells following in vitro differentiation. C and D, on day 5, differentiated wild type Th17 cells generated as described in A were rested or stimulated with IL-6, IL-23, or IL-12 for 2 h before gene expression analysis by qRT-PCR (C) and Twist1 expression by immunoblot (IB) with densitometry normalized against β-actin (D). E, naïve wild type and Stat3-deficient CD4+ T cells were activated with anti-CD3 and anti-CD28 in the presence or absence of IL-6, TGF-β, or IL-12 and gene expression was analyzed by qRT-PCR after 3 days. F, schematic of Twist1 promoter containing STAT3 binding sites. G, cells prepared as described in C were used for ChIP analysis using STAT3 antibody. Data are mean of four independent experiments ± S.E. (A and B), or are mean of replicate samples ± S.D. and representative of three independent experiments with similar results (C–G).**, p < 0.01. unstim, unstimulated.
To begin to define a mechanism for Twist1 regulating Th17 development, we first examined the regulation of Twist1 in Th17 cells. Because STAT3 directly binds to the Twist1 promoter in breast cancer cells (38), we speculated that STAT3 might induce Twist1 expression in Th17 cultures. Stimulation of wild type Th17 cells with IL-6 or IL-23 to activate STAT3, or IL-12 to activate STAT4, led to increased Twist1 mRNA and protein expression compared with unstimulated cells (Fig. 1, C and D). Because Twist1 expression in Th17 cells is lower than Th1 cells (33), we hypothesized that an inhibitory signal represses Twist1 expression in developing Th17 cells. Indeed, IL-6 or IL-12 induced Twist1 expression in activated CD4+ T cells, and this was decreased when TGF-β was added to the culture (Fig. 1E). To confirm that Twist1 is a STAT3 target gene in Th17 cells, gene expression was compared in activated wild type and Stat3-deficient CD4+ T cells. In the absence of STAT3, IL-6 was unable to induce Twist1 expression, although expression was equally induced in IL-12-stimluated wild type and Stat3-deficient CD4+ T cells (Fig. 1E).
Given that the Twist1 promoter contains STAT3 binding sites (Fig. 1F) (38), we wanted to determine whether STAT3 could directly bind to the regulatory regions of Twist1. When ChIP assay was performed using Th17 cells, STAT3-activating cytokines, but not IL-12, resulted in STAT3 binding to the Twist1 promoter, with the greatest amounts in the proximal promoter segment (Fig. 1G). These results suggested that STAT3-activating cytokines and TGF-β play opposing roles in regulating Twist1 expression in Th17 cultures.
Twist1 Represses Cytokine Production in Th17 Cells
To define the scope of Twist1-dependent repression of the Th17 phenotype, we ectopically expressed Twist1 in Th17 cells and examined cytokine production. Ectopic Twist1 expression in Th17 cells resulted in decreased IL-17A and IL-17F production compared with control cells (Fig. 2A). Twist1-deficient Th17 cells produced more IL-17A, IL-17F, and GM-CSF than wild type cells, although IL-10 production was similar (Fig. 2, B and D, and data not shown).
FIGURE 2.

Twist1 suppresses cytokine production in Th17 cells. A, naïve CD4+ T cells were isolated from wild type mice and differentiated under Th17 culture conditions. On day 2, cells were transduced with either control or Twist1-GFP (Twist1)-expressing retrovirus. On day 5, cells were stimulated with PMA and ionomycin for 6 h before intracellular staining (ICS) for cytokine production. Data are gated on GFP+ cells. B, differentiated wild type and Twist1-deficient Th17 cells were stimulated with PMA and ionomycin for 6 h before ICS analysis. C and D, naïve wild type and Twist1-deficient CD4+ T cells were cultured under Th17 polarizing conditions with or without TGF-β. On day 5, cells were left unstimulated for gene expression analysis by qRT-PCR (C) or reactivated with anti-CD3 for 24 h to assess cytokine production by ELISA (D). E, naïve CD4+ T cells were isolated from PBMCs and differentiated under Th17 culture conditions. On day 5, cells were transfected with control or siRNA targeting TWIST1, rested overnight, and stimulated with anti-CD3 to assess gene expression by qRT-PCR. F and G, differentiated wild type and Twist1-deficient Th17 cells were used for gene expression analysis by qRT-PCR before (Rorc, Batf, and Maf) or after (Il17a) 6 h anti-CD3 stimulation (F) and ChIP analysis using STAT3 antibody (G). Data are mean of four to five independent experiments ± S.D (A–D) or are mean of replicate samples ± S.D. and representative of three independent experiments with similar results (E–G). *, p < 0.05; **, p < 0.01. ND, not detectable.
Because TGF-β inhibits Twist1 expression and Th17 differentiation in the presence of IL-23 and absence of TGF-β results in highly encephalitogenic Th17 cells (39), we compared the differentiation of wild type and Twist1-deficient CD4+ T cells in the presence or absence of TGF-β in Th17 cell culture conditions. Th17 cells derived in the absence of TGF-β had increased Twist1 gene expression, compared with those derived under conventional Th17 conditions (Fig. 2C). Moreover, Twist1-deficient Th17 cells derived in the absence of TGF-β had increased secretion of IL-17A and GM-CSF (Fig. 2D). Although TGF-β represses Twist1 expression and has differential effects on IL-17 and GM-CSF production (Fig. 2, C and D) (4, 5), IL-6 was able to induce Twist1 expression, resulting in altered cytokine production in the presence or absence of TGF-β. Thus, Twist1 repressed IL-17 and GM-CSF even when TGF-β is present in Th17 culture conditions to limit Twist1 expression.
To demonstrate that Twist1 function is conserved in human Th17 cells, naïve CD4+ T cells isolated from the peripheral blood of healthy individuals were differentiated into Th17 cells, transfected with siRNA encoding TWIST1, and assessed for gene expression. Knockdown of TWIST1 in human Th17 cells resulted in increased IL17A and IL17F gene expression (Fig. 2E). TWIST1 knockdown in human Th17 cells also resulted in increased expression of the Th17-inducing genes RORC, BATF, and MAF, compared with control cells (Fig. 2E). Messenger RNA for Il17a, Rorc, Batf, and Maf were similarly increased in Twist1-deficient Th17 cells compared with wild type cells (Fig. 2F). Because each of these genes is a direct target of STAT3 (22, 23, 25–27), we tested whether binding of STAT3 to the promoters of these genes was altered. We observed increased STAT3 binding to gene promoters in Twist1-deficient Th17 cells compared with wild type cells (Fig. 2G). Together, these data demonstrate that Twist1 impairs differentiation of mouse and human IL-17-secreting T cells.
Twist1 Impairs IL-6-STAT3 Signaling by Repressing Il6ra Expression
Twist1-deficiency resulted in increased binding of STAT3 to Th17 target genes, and the balance between STAT3 and STAT5 signaling is crucial in regulating Th17 cell differentiation (28). We hypothesized that Twist1 was altering cytokine signaling and investigated the kinetics of phospho-STAT3 and phospho-STAT5 during Th17 differentiation using wild type and Twist1-deficient naïve CD4+ T cells. The frequency of phospho-STAT3 was higher in Twist1-deficient Th17 cells on day 2 and day 3 compared with wild type cells, although phospho-STAT5 was comparable between the two cell types (Fig. 3A). The increase in phospho-STAT3 but not phospho-STAT5 in Twist1-deficient Th17 cells correlates with higher IL-6Rα expression but similar IL-2Rα expression on days 2 and 3 compared with wild type cells (Fig. 3, B and C). Il6st, the gp130 chain of IL-6 receptor, and Stat3 expression were similar between wild type and Twist1-deficient Th17 cells, although Il6ra mRNA reflected the same pattern as protein expression (Fig. 3C). Given that IL-21 and IL-23 induce phospho-STAT3, we wanted to determine whether Twist1 also has a negative effect on Il23r and Il21r expression. Twist1-deficient Th17 cells had similar levels of Il23r and Il21r expression compared with wild type cells (Fig. 3C). Because IL-6Rα expression was increased at early time points, we examined cytokine production from Th17 cells during differentiation and observed similar increases of cytokine production from T cells that lack expression of Twist1 (Fig. 3D).
FIGURE 3.

Twist1 impairs IL-6-STAT3 signaling in Th17 cells. A–D, naïve CD4+ T cells were isolated from WT and Twist1fl/flCD4-Cre mice and differentiated under Th17 polarizing conditions. The levels of phospho-STAT3 (pSTAT3) and phospho-STAT5 (pSTAT5) were measured by ICS each day (A). T cells cultured under Th17 conditions for 2 or 3 days were used for surface marker analysis (B), gene expression analysis by qRT-PCR (C), or analysis of cytokine production after anti-CD3 stimulation (D). E and F, naïve CD4+ T cells were isolated from WT and Twist1fl/flCD4-Cre mice and differentiated under Th17 polarizing conditions with increased doses of STAT3 inhibitor (JSI-124). Cells were harvested on days 3 (D3) and 5 and used to measure the level of pSTAT3 by ICS (E) or restimulated with anti-CD3 to assess cytokine production by ELISA (F). G, T cells were cultured as above in the presence of control antibody or blocking antibody to IL-6R, harvested on days 3 and 5, and restimulated with anti-CD3 to assess cytokine production using ELISA. H, schematic of Il6ra promoter containing Twist1 binding sites. I and J, T cells cultured under Th17 conditions for 2 or 3 days were used for gene expression analysis by qRT-PCR (I) or used for ChIP analysis using Twist1 antibody (J). K, luciferase activity in Jurkat T cells transfected with various concentrations of plasmid encoding Twist1 along with IL6RA or NFAT luciferase reporter and then activated for 6 h with PMA and ionomycin. Data are mean of four independent experiments ± S.D. (A, B, and D) or are mean of replicate samples ± S.D. and representative of three independent experiments with similar results (C and E–K). *, p < 0.05; **, p < 0.01. ND, not detectable, RU, relative units.
To test the requirement for STAT3 in this process, we treated wild type and Twist1-deficient Th17 cultures with an inhibitor of STAT3 activation during differentiation. Addition of the inhibitor decreased STAT3 phosphorylation at days 3 and 5 of cultured wild type and Twist1-deficient T cells (Fig. 3E). There was a corresponding dose-dependent decrease in IL-17 production at all time points (Fig. 3F), with lower doses of the inhibitor resulting in production of IL-17 production from Twist1-deficient Th17 cells similar to that in untreated wild type cells (Fig. 3F). Similarly, blocking IL-6R in Twist1-deficient Th17 cultures resulted in IL-17 production comparable with untreated wild type cells (Fig. 3G). These results suggested that Twist1 specifically targets IL-6-STAT3 signaling in Th17 cells.
We next wanted to determine whether Twist1 represses Il6ra expression by directly binding to the E-box sites in the Il6ra promoter that is conserved in mouse and human genes (Fig. 3H). When ChIP was performed using wild type and Twist1-deficient Th17 cells, the binding of Twist1 to the promoter of Il6ra was observed by days 2 and 3 in wild type cell cultures, with the peak of binding following the peak of Twist1 expression (Fig. 3, I and J). To further demonstrate the direct consequences of Twist1 binding to the Il6ra promoter, Jurkat T cells were transfected with an IL6RA luciferase reporter and a plasmid encoding Twist1. Notably, Twist1 repressed the transcriptional activity of the IL6RA promoter, but not an NFAT reporter, in a dose-dependent manner (Fig. 3K).
Mice with Twist1-deficient T Cells Display more Severe Clinical Symptoms of MOG-induced EAE
Although Th1 and Th17 cells have been demonstrated to be crucial in mediating the development of EAE, the role of IFN-γ and IL-17 in EAE disease has been controversial (40, 41). Recently, GM-CSF, produced by Th1 and Th17 cells, has been identified as a contributor to the development of EAE (5, 42). As Twist1 negatively regulates IL-17 and GM-CSF in Th17 cells (Fig. 2) and IFN-γ in Th1 cells (33), we wanted to compare the development of MOG peptide-induced EAE in wild type and Twist1fl/flCD4-Cre+ mice. Twist1fl/flCD4-Cre+ mice manifested severe clinical symptom of MOG-induced EAE than wild type mice, although maximal severity and recovery were similar (Fig. 4A). Increased disease resulted in a 26% increase in the area under the mean clinical disease score curve of Twist1fl/flCD4-Cre+ mice, compared with control mice (area under the curve, WT (22.6 clincial score × time); Twist1-mutant mice (28.6)). The number of days with a mean clinical score greater than one was an average of 16.5 for control mice and 21 for Twist1fl/flCD4-Cre+ mice, an increase of 27%. Earlier disease development correlated with an increase in CD4+IL-17A+, CD4+IFN-γ+, and CD4+IL-17A+IFN-γ+ mononuclear cells isolated from the brain of Twist1-mutant mice compared with wild type mice at day 12 (Fig. 4B). In addition, MOG-stimulated Twist1-deficient splenocytes produced significantly more IL-17, GM-CSF, and IFN-γ compared with wild type cells (Fig. 4C). The earlier onset of MOG-induced EAE in Twist1 mutant mice is not likely due to a defect in regulatory T cells because Twist1 mutant mice have percentages of nTreg and in vitro development of iTreg that are comparable with wild type mice (data not shown and Fig. 1A). Together, these data suggest that Twist1 limits the development of inflammatory T cell subsets and autoimmune disease.
FIGURE 4.

Clinical symptoms of EAE in the absence of Twist1. A–C, wild type and Twist1fl/flCD4-Cre mice were immunized with MOGp(35–55) to induce EAE. Mean clinical score in MOG-induced EAE disease is shown in A. On day 12, mononuclear cells were isolated from brain and stimulated with PMA and ionomycin for 6 h to measure cytokine production by ICS (gated on CD4+ T cells) (B), or splenocytes were stimulated with MOG peptide for 48 h, and cytokine production was assessed by ELISA (C). Data are mean ± S.E. of seven mice per group (A) or four mice per group (B and C) and representative of two independent experiments with similar results. **, p < 0.01.
Twist1 Limits T Follicular Helper Cell Development
Because Twist1 impacts IL-6 signaling, and IL-6-induced STAT3 signaling is required for Tfh development, we wanted to determine if Twist1 deficiency in T cells affected Tfh generation. Twist1 is expressed at greater amounts in Tfh cells (CD4+CD44+CXCR5+PD-1+; mean fluoresence intensity, 4954) than in non-Tfh effector cells (CD4+CD44+CXCR5−PD-1−; mean fluorescence intensity, 3096) or naïve T cells (CD4+CD44−CD62L+; mean fluorescence intensity-1926) as determined by intracellular staining for Twist1. We initially examined Tfh development in mice with EAE. Following immunization with MOG peptide, splenocytes from Twist1fl/flCD4-Cre+ mice had significantly more Tfh cells (defined as CD4+CXCR5+PD-1hiICOS+) than wild type splenocytes (Fig. 5A). To further explore the ability of Twist1 to regulate Tfh development, we immunized wild type and Twist1fl/flCD4-Cre+ mice with SRBC. As observed following MOG peptide immunization, SRBC immunization resulted in increased Tfh cell development in Twist1fl/flCD4-Cre+ mice, compared with wild type mice (Fig. 5B). Percentages of Tfh cells in the absence of Twist1 were similarly increased defining cells with either ICOS or Bcl-6 expression (Fig. 5B). Moreover, in the absence of Twist1, there was an increase in the percentages of CD4+CXCR5+PD-1hi cells that were phospho-STAT3-positive and IL-6Rα-positive, and in the amount (mean fluorescence intensity) of IL-6Rα expression (Fig. 5B).
FIGURE 5.

Mice with Twist1-deficient T cells have more T follicular helper cells. A, WT and Twist1fl/flCD4-Cre mice were immunized with MOGp(35–55) as described in Fig. 4. Twenty days following immunization, splenocytes were stained for Tfh cells. B and C, WT and Twist1fl/flCD4-Cre mice were immunized with SRBC. On day 9, splenocytes were analyzed by flow cytometry with percentages of PD-1+ICOS+, PD-1+pSTAT3+, and PD-1+IL-6Rα+ cells indicated (B). Following immunization, cell populations were sorted for CD4+CXCR5+PD-1+ICOS+ (Tfh) or CD4+CXCR5−PD-1−ICOS− (non-Tfh), and gene expression was analyzed (C). D, SRBC-immunized WT and Twist1fl/flCD4-Cre mice were injected (intraperitoneal) with control antibody or blocking antibody to IL-6R on days 4, 6, and 8. On day 9, splenocytes were analyzed by flow cytometry with percentages of PD-1+ICOS+ and PD-1+pSTAT3+ cells indicated. (A, B, and D). Data are gated on CD4+CXCR5+. Percentages are mean ± S.E. of four to five mice per group and representative of two independent experiments with similar results (A and B), are mean ± S.E. of five mice per group (D), or are mean of replicate samples ± S.D. and representative of three independent experiments with similar results (C). *, p < 0.05. MFI, mean fluorescence intensity. ND, not detected.
We then sorted Tfh and non-Tfh cells from SRBC-immunized wild type and Twist1fl/flCD4-Cre+ mice to examine changes in gene expression following normalization for the increased Tfh cell number in the absence of Twist1. Consistent with flow cytometry, Twist1 was expressed in greater amounts in the Tfh population than in non-Tfh cells, and no Twist1 mRNA was detected in Cre+ cells (Fig. 5C). We observed little difference in gene expression of Batf, Bcl6 and Irf4 between wild type and Twist1fl/flCD4-Cre+ cells in the non-Tfh population. In the Tfh population, the absence of Twist1 resulted in modest increases of Batf and Bcl6 and a more dramatic increase of Irf4 (Fig. 5C). Similar to observations in Th17 cells, the gene most increased in Twist1-deficient Tfh cells was Il6ra (Fig. 5C). When we blocked IL-6 signaling using anti-IL-6R antibody, we observed a decrease in the percentages of CD4+CXCR5+PD-1hi cells that were phospho-STAT3-positive in wild type and Twist1fl/flCD4-Cre+ mice (Fig. 5D). In addition, the Tfh population in anti-IL-6R treated Twist1fl/flCD4-Cre+ mice was less than the percentage of Tfh cells in untreated wild type mice (Fig. 5D). This result identifies the IL-6-STAT3 signaling pathway as a critical Twist1 target during Tfh cell development.
We then tested whether T cells activated in the absence or presence of IL-6 (Tfh-like conditions) demonstrated Twist1-dependent regulation of Tfh genes. Addition of IL-6 to activated T cell cultures resulted in increased pSTAT3, increased STAT3 binding to the Twist1 promoter, and increased Twist1 expression over 48 h of culture (Fig. 6, A and B). Paralleling the induction of Twist1 expression, Twist1 binding to the Il6ra, Bcl6, and Icos promoters was also induced by IL-6 (Fig. 6C). Thus, as in Th17 cells, Twist1 is a component of a STAT3-inducible negative feedback loop in Tfh cells.
FIGURE 6.
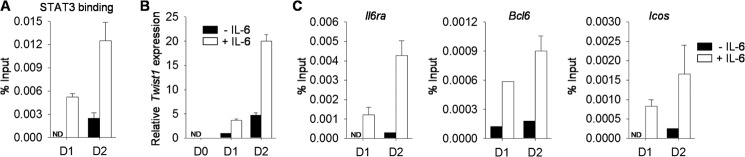
Twist1 binds to Tfh cell-associated genes. A–C, naïve WT CD4+ T cells were activated with or without IL-6 for 2 days. Cells were harvested daily to analyze STAT3 binding to the Twist1 promoter (A) or Twist1 binding to the indicated promoters (C) by ChIP assay or to assess gene expression by qRT-PCR (B). A, percentages are mean ± S.E. of four to five mice per group. Data are mean of replicate samples ± S.D. and representative of three independent experiments with similar results. ND, not detectable; D1, day 1; D2, day 2.
To determine the functional consequences of the increased Tfh cells that develop in mice with Twist1-deficient T cells, we examined the development of germinal center B cells and antibody production following SRBC immunization. We observed a 3-fold increase in the percentages of germinal center B cells (defined as B220+CD19+Fas+ GL-7+PNA+) (Fig. 7, A and B). Analysis of SRBC-specific antibody production demonstrated increased serum IgG antibody titers in Twist1fl/flCD4-Cre+ mice, compared with wild type mice (Fig. 7C). Isotype-specific analysis demonstrated greater IgG1 and IgG2a/c serum antibody titers in mice that lack Twist1 expression in T cells than in wild type cells (Fig. 7C). Thus, Twist1 limits Tfh development and humoral immunity.
FIGURE 7.
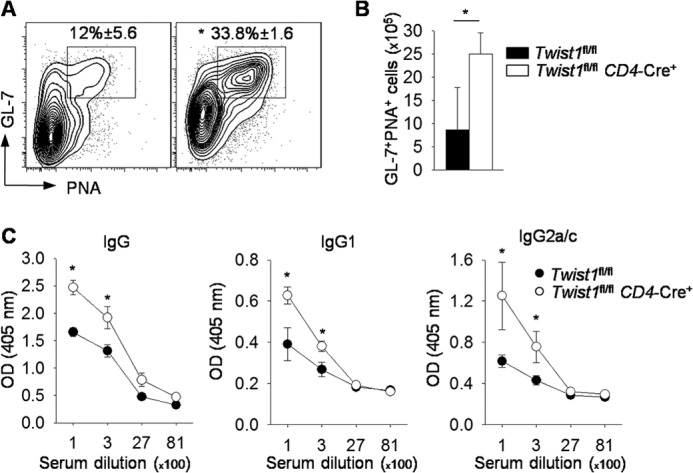
Twist1 represses germinal center B cells and antibody production in SRBC-immunized mice. A–C, WT and Twist1fl/flCD4-Cre mice were immunized with SRBC. On day 9, splenocytes were stained for germinal center B cells (A) with total cell count shown in B. Data are gated on B220+CD19+Fas+. Serum from WT and Twist1fl/flCD4-Cre mice was diluted and used to measure antibody titers by ELISA (C). Data are mean ± S.E. of four to five mice per group and representative of two independent experiments with similar results. *, p < 0.05. PNA, peanut agglutinin.
DISCUSSION
The ability of cells to respond to their environment is essential in immunity. Integrating the responses to the cytokine milieu is critical in cellular differentiation and can alter responses to subsequent cytokine exposure. In this report, we identify a cytokine signaled feedback loop that regulates T helper cell differentiation. Cytokines, including IL-6, induce the STAT3-dependent expression of Twist1, which then binds to the promoter of the Il6ra gene, repressing transcription and thus limiting IL-6 responsiveness and STAT3 activation. The ability of Twist1 to repress IL-6 signaling limits the development of Th17 cells and Tfh cells in vivo, thereby controlling cell-mediated and humoral components of the immune response. This observation is consistent with recent findings that Twist1 can also regulate the cell fate decisions of multipotential cardiac neural crest between neurons and smooth muscle via its direct transcriptional repression of Phox2b (43).
Twist1 functions as either a homodimer or heterodimer with other basic helix-loop-helix factors where the dimerization partners dictate the function (44). Altering the balance between Twist1 and Hand2 has a significant impact on limb and craniofacial defects in humans with Saethre-Chotzen syndrome (45). Twist1 has been shown to form a dimer with E47 protein, which is inhibited by the Id3 (44–46). Interestingly, Id3-deficient mice have a defect in regulatory T cell generation and an enhancement in Th17 differentiation linked to the ability of E47 to induce Rorc expression (47). Maruyama et al. (47) suggested that the ability of E47 to transactivate Rorc expression might require other factors downstream of IL-6. Consistent with this, we observed an increase in E47 binding at the Rorc promoter in Twist1-deficient Th17 cells compared with WT cells, although there was no change in either Tcfe2a (encoding E47) or Id3 expression (data not shown). E2A and Id3 also have opposing roles in the generation of Tfh-like cells, and E2A contributes to germinal center B cell development, suggesting a similar role in this subset (48, 49). Moreover, Twist1 can also function through non-canonical basic helix-loop-helix protein-protein interactions. We have previously shown that Twist1 inhibits IFN-γ production by forming a complex with Runx3 through its Runt DNA binding domain and preventing it from binding DNA (33). Because Runx1 transactivates Rorc expression, it is possible that Twist1 interacts with Runx1, thus repressing Rorc expression. Whether Runx1 or Runx3 contribute to Tfh development has not been defined. Further studies need to be performed to dissect the relationship between Twist1, E47, and the lineage determining factors for the development of each subset.
Although Twist1 may regulate T helper subset development through several mechanisms, one paradigm that emerges is Twist1 being an essential component of a cytokine-induced feedback loop. In Th1 cells, STAT4 induces Twist1, which subsequently decreases Il12rb2 expression and STAT4 activation (33). Similarly, in Th17 and Tfh cells, STAT3 induces Twist1, which represses Il6ra, resulting in decreased STAT3 activation. In Th17 cells, and likely in Tfh cells as well, this alters the balance of activation between STAT3 and STAT5 that have opposing roles in both of these subsets (19, 28–30). Thus, Twist1 functions as a balancing factor that regulates signal integration.
Many transcription factors inhibit the development of T helper cell lineages (1, 50, 51). However, the majority of factors promote one lineage at the expense of another. For example, GATA3 promotes Th2 differentiation as it inhibits the development of Th1 and Th17 cells (15, 52). In this respect, Twist1 is novel in that it represses the development of Th1, Th17, and Tfh cells, without any corresponding increase in cytokine secretion characteristic of other T helper subsets. In targeting these subsets, Twist1 regulates particular components of the inflammatory T cell-mediated immune response. The additional ability of Twist1 to limit B cell responses suggests that signaling pathways regulating Twist1 expression represent potential therapeutic targets for broad modulation of the immune response.
The data in this report demonstrate that Twist1 is a STAT3 target gene that directly represses Il6ra, impairing IL-6-STAT3 signaling. This limits the expression of subset-associated transcription factors, including Rorc, Batf, Bcl6, and Maf, resulting in decreased cytokine production and effector function. These results reveal a negative regulatory feedback loop controlling the transcriptional network required for the development of multiple T helper subsets.
Acknowledgment
Support for the Herman B Wells Center was provided in part by the Riley Children's Foundation.
This work was supported by National Institutes of Health Grants R01AI045515 (to M. H. K.), R01 AR061392 (to A. B. F.), R21 AI099825 (to A. L. D.), P01 AI056097 (to M. H. K. and J. S. B.), R01 AI079065 (to J. S. B.), and P30 DK090948.
- Tfh
- T follicular helper
- SRBC
- sheep red blood cell(s)
- MOG
- myelin oligodendrocyte glycoprotein
- EAE
- experimental autoimmune encephalomyelitis
- nTreg
- natural regulatory T cells
- qRT-PCR
- quantitative real-time PCR
- Treg
- regulatory T cell
- ICS
- intracellular staining
- ROR
- retinoic acid-related orphan receptor
- BATF
- B cell activating transcription factor-like
- IRF4
- interferon regulatory factor 4
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Zhu J., Yamane H., Paul W. E. (2010) Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 28, 445–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vahedi G., Takahashi H., Nakayamada S., Sun H. W., Sartorelli V., Kanno Y., O'Shea J. J. (2012) STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stritesky G. L., Yeh N., Kaplan M. H. (2008) IL-23 promotes maintenance but not commitment to the Th17 lineage. J. Immunol. 181, 5948–5955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mangan P. R., Harrington L. E., O'Quinn D. B., Helms W. S., Bullard D. C., Elson C. O., Hatton R. D., Wahl S. M., Schoeb T. R., Weaver C. T. (2006) Transforming growth factor-β induces development of the T(H)17 lineage. Nature 441, 231–234 [DOI] [PubMed] [Google Scholar]
- 5. El-Behi M., Ciric B., Dai H., Yan Y., Cullimore M., Safavi F., Zhang G. X., Dittel B. N., Rostami A. (2011) The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 12, 568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Veldhoen M., Hocking R. J., Atkins C. J., Locksley R. M., Stockinger B. (2006) TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189 [DOI] [PubMed] [Google Scholar]
- 7. Harrington L. E., Hatton R. D., Mangan P. R., Turner H., Murphy T. L., Murphy K. M., Weaver C. T. (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132 [DOI] [PubMed] [Google Scholar]
- 8. Ciofani M., Madar A., Galan C., Sellars M., Mace K., Pauli F., Agarwal A., Huang W., Parkurst C. N., Muratet M., Newberry K. M., Meadows S., Greenfield A., Yang Y., Jain P., Kirigin F. K., Birchmeier C., Wagner E. F., Murphy K. M., Myers R. M., Bonneau R., Littman D. R. (2012) A validated regulatory network for Th17 cell specification. Cell 151, 289–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu C., Yosef N., Thalhamer T., Zhu C., Xiao S., Kishi Y., Regev A., Kuchroo V. K. (2013) Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yosef N., Shalek A. K., Gaublomme J. T., Jin H., Lee Y., Awasthi A., Wu C., Karwacz K., Xiao S., Jorgolli M., Gennert D., Satija R., Shakya A., Lu D. Y., Trombetta J. J., Pillai M. R., Ratcliffe P. J., Coleman M. L., Bix M., Tantin D., Park H., Kuchroo V. K., Regev A. (2013) Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X., Nurieva R. I., Dong C. (2013) Transcriptional regulation of follicular T-helper (Tfh) cells. Immunol. Rev. 252, 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liao W., Lin J. X., Wang L., Li P., Leonard W. J. (2011) Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat. Immunol. 12, 551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liao W., Schones D. E., Oh J., Cui Y., Cui K., Roh T. Y., Zhao K., Leonard W. J. (2008) Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor α-chain expression. Nat. Immunol. 9, 1288–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou L., Ivanov I. I., Spolski R., Min R., Shenderov K., Egawa T., Levy D. E., Leonard W. J., Littman D. R. (2007) IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 8, 967–974 [DOI] [PubMed] [Google Scholar]
- 15. Usui T., Nishikomori R., Kitani A., Strober W. (2003) GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL-12Rβ2 chain or T-bet. Immunity 18, 415–428 [DOI] [PubMed] [Google Scholar]
- 16. Szabo S. J., Dighe A. S., Gubler U., Murphy K. M. (1997) Regulation of the interleukin (IL)-12R β2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J. Exp. Med. 185, 817–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stritesky G. L., Muthukrishnan R., Sehra S., Goswami R., Pham D., Travers J., Nguyen E. T., Levy D. E., Kaplan M. H. (2011) The transcription factor STAT3 is required for T helper 2 cell development. Immunity 34, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang X. O., Pappu B. P., Nurieva R., Akimzhanov A., Kang H. S., Chung Y., Ma L., Shah B., Panopoulos A. D., Schluns K. S., Watowich S. S., Tian Q., Jetten A. M., Dong C. (2008) T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity 28, 29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nurieva R. I., Chung Y., Hwang D., Yang X. O., Kang H. S., Ma L., Wang Y. H., Watowich S. S., Jetten A. M., Tian Q., Dong C. (2008) Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 29, 138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma C. S., Avery D. T., Chan A., Batten M., Bustamante J., Boisson-Dupuis S., Arkwright P. D., Kreins A. Y., Averbuch D., Engelhard D., Magdorf K., Kilic S. S., Minegishi Y., Nonoyama S., French M. A., Choo S., Smart J. M., Peake J., Wong M., Gray P., Cook M. C., Fulcher D. A., Casanova J. L., Deenick E. K., Tangye S. G. (2012) Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 119, 3997–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mathur A. N., Chang H. C., Zisoulis D. G., Stritesky G. L., Yu Q., O'Malley J. T., Kapur R., Levy D. E., Kansas G. S., Kaplan M. H. (2007) Stat3 and Stat4 direct development of IL-17-secreting Th cells. J. Immunol. 178, 4901–4907 [DOI] [PubMed] [Google Scholar]
- 22. Bauquet A. T., Jin H., Paterson A. M., Mitsdoerffer M., Ho I. C., Sharpe A. H., Kuchroo V. K. (2009) The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 10, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ivanov I. I., McKenzie B. S., Zhou L., Tadokoro C. E., Lepelley A., Lafaille J. J., Cua D. J., Littman D. R. (2006) The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 [DOI] [PubMed] [Google Scholar]
- 24. Yu D., Rao S., Tsai L. M., Lee S. K., He Y., Sutcliffe E. L., Srivastava M., Linterman M., Zheng L., Simpson N., Ellyard J. I., Parish I. A., Ma C. S., Li Q. J., Parish C. R., Mackay C. R., Vinuesa C. G. (2009) The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31, 457–468 [DOI] [PubMed] [Google Scholar]
- 25. Brüstle A., Heink S., Huber M., Rosenplänter C., Stadelmann C., Yu P., Arpaia E., Mak T. W., Kamradt T., Lohoff M. (2007) The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 8, 958–966 [DOI] [PubMed] [Google Scholar]
- 26. Durant L., Watford W. T., Ramos H. L., Laurence A., Vahedi G., Wei L., Takahashi H., Sun H. W., Kanno Y., Powrie F., O'Shea J. J. (2010) Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 32, 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schraml B. U., Hildner K., Ise W., Lee W. L., Smith W. A., Solomon B., Sahota G., Sim J., Mukasa R., Cemerski S., Hatton R. D., Stormo G. D., Weaver C. T., Russell J. H., Murphy T. L., Murphy K. M. (2009) The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460, 405–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang X. P., Ghoreschi K., Steward-Tharp S. M., Rodriguez-Canales J., Zhu J., Grainger J. R., Hirahara K., Sun H. W., Wei L., Vahedi G., Kanno Y., O'Shea J. J., Laurence A. (2011) Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 12, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnston R. J., Choi Y. S., Diamond J. A., Yang J. A., Crotty S. (2012) STAT5 is a potent negative regulator of TFH cell differentiation. J. Exp. Med. 209, 243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nurieva R. I., Podd A., Chen Y., Alekseev A. M., Yu M., Qi X., Huang H., Wen R., Wang J., Li H. S., Watowich S. S., Qi H., Dong C., Wang D. (2012) STAT5 protein negatively regulates T follicular helper (Tfh) cell generation and function. J. Biol. Chem. 287, 11234–11239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Niesner U., Albrecht I., Janke M., Doebis C., Loddenkemper C., Lexberg M. H., Eulenburg K., Kreher S., Koeck J., Baumgrass R., Bonhagen K., Kamradt T., Enghard P., Humrich J. Y., Rutz S., Schulze-Topphoff U., Aktas O., Bartfeld S., Radbruch H., Hegazy A. N., Löhning M., Baumgart D. C., Duchmann R., Rudwaleit M., Häupl T., Gitelman I., Krenn V., Gruen J., Sieper J., Zeitz M., Wiedenmann B., Zipp F., Hamann A., Janitz M., Scheffold A., Burmester G. R., Chang H. D., Radbruch A. (2008) Autoregulation of Th1-mediated inflammation by twist1. J. Exp. Med. 205, 1889–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barnes R. M., Firulli A. B. (2009) A twist of insight - the role of Twist-family bHLH factors in development. Int J Dev. Biol. 53, 909–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pham D., Vincentz J. W., Firulli A. B., Kaplan M. H. (2012) Twist1 regulates Ifng expression in Th1 cells by interfering with Runx3 function. J. Immunol. 189, 832–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mo C., Chearwae W., O'Malley J. T., Adams S. M., Kanakasabai S., Walline C. C., Stritesky G. L., Good S. R., Perumal N. B., Kaplan M. H., Bright J. J. (2008) Stat4 isoforms differentially regulate inflammation and demyelination in experimental allergic encephalomyelitis. J. Immunol. 181, 5681–5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Temple L., Kawabata T. T., Munson A. E., White K. L., Jr. (1993) Comparison of ELISA and plaque-forming assays for measuring the humoral immune response to SRBC in rats and mice treated with benzo[a]pyrene or cyclophosphamide. Fundam. Appl. Toxicol. 21, 412–419 [DOI] [PubMed] [Google Scholar]
- 36. Mathur A. N., Chang H. C., Zisoulis D. G., Kapur R., Belladonna M. L., Kansas G. S., Kaplan M. H. (2006) T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood 108, 1595–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thieu V. T., Yu Q., Chang H. C., Yeh N., Nguyen E. T., Sehra S., Kaplan M. H. (2008) Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng G. Z., Zhang W. Z., Sun M., Wang Q., Coppola D., Mansour M., Xu L. M., Costanzo C., Cheng J. Q., Wang L. H. (2008) Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J. Biol. Chem. 283, 14665–14673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghoreschi K., Laurence A., Yang X. P., Tato C. M., McGeachy M. J., Konkel J. E., Ramos H. L., Wei L., Davidson T. S., Bouladoux N., Grainger J. R., Chen Q., Kanno Y., Watford W. T., Sun H. W., Eberl G., Shevach E. M., Belkaid Y., Cua D. J., Chen W., O'Shea J. J. (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467, 967–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pierson E., Simmons S. B., Castelli L., Goverman J. M. (2012) Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol. Rev. 248, 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Becher B., Segal B. M. (2011) T(H)17 cytokines in autoimmune neuro-inflammation. Curr. Opin. Immunol. 23, 707–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Codarri L., Gyülvészi G., Tosevski V., Hesske L., Fontana A., Magnenat L., Suter T., Becher B. (2011) RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 12, 560–567 [DOI] [PubMed] [Google Scholar]
- 43. Vincentz J. W., Firulli B. A., Lin A., Spicer D. B., Howard M. J., Firulli A. B. (2013) Twist1 controls a cell-specification switch governing cell fate decisions within the cardiac neural crest. PLoS Genet. 9, e1003405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Castanon I., Von Stetina S., Kass J., Baylies M. K. (2001) Dimerization partners determine the activity of the Twist bHLH protein during Drosophila mesoderm development. Development 128, 3145–3159 [DOI] [PubMed] [Google Scholar]
- 45. Firulli B. A., Krawchuk D., Centonze V. E., Vargesson N., Virshup D. M., Conway S. J., Cserjesi P., Laufer E., Firulli A. B. (2005) Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat. Genet. 37, 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hayashi M., Nimura K., Kashiwagi K., Harada T., Takaoka K., Kato H., Tamai K., Kaneda Y. (2007) Comparative roles of Twist-1 and Id1 in transcriptional regulation by BMP signaling. J. Cell Sci. 120, 1350–1357 [DOI] [PubMed] [Google Scholar]
- 47. Maruyama T., Li J., Vaque J. P., Konkel J. E., Wang W., Zhang B., Zhang P., Zamarron B. F., Yu D., Wu Y., Zhuang Y., Gutkind J. S., Chen W. (2011) Control of the differentiation of regulatory T cells and T(H)17 cells by the DNA-binding inhibitor Id3. Nat. Immunol. 12, 86–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miyazaki M., Rivera R. R., Miyazaki K., Lin Y. C., Agata Y., Murre C. (2011) The opposing roles of the transcription factor E2A and its antagonist Id3 that orchestrate and enforce the naive fate of T cells. Nat. Immunol. 12, 992–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kwon K., Hutter C., Sun Q., Bilic I., Cobaleda C., Malin S., Busslinger M. (2008) Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 28, 751–762 [DOI] [PubMed] [Google Scholar]
- 50. Kanno Y., Vahedi G., Hirahara K., Singleton K., O'Shea J. J. (2012) Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Ann. Rev. Immunol. 30, 707–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou L., Chong M. M., Littman D. R. (2009) Plasticity of CD4+ T cell lineage differentiation. Immunity 30, 646–655 [DOI] [PubMed] [Google Scholar]
- 52. Yagi R., Junttila I. S., Wei G., Urban J. F., Jr., Zhao K., Paul W. E., Zhu J. (2010) The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-γ. Immunity 32, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]