

Background: Host complement-mediated opsonophagocytosis is an innate immune response to GAS.
Results: Inactivation of the covS component of the covRS system allows GAS to avoid opsonization.
Conclusion: Bacterial genes that subvert opsonization of GAS are regulated by the covRS system.
Significance: The covRS system regulates complement inhibitor binding to GAS via the products of the fba and enn genes.
Keywords: Bacteria, Bacterial Genetics, Bacterial Transcription, Gene Regulation, Gene Transcription, Streptococcus pyogenes, Transcription Regulation, Transcription Target Genes
Abstract
Group A Streptococcus pyogenes (GAS) strain AP53 is a primary isolate from a patient with necrotizing fasciitis. These AP53 cells contain an inactivating mutation in the sensor component of the cluster of virulence (cov) responder (R)/sensor (S) two-component gene regulatory system (covRS), which enhances the virulence of the primary strain, AP53/covR+S−. However, specific mechanisms by which the covRS system regulates the survival of GAS in humans are incomplete. Here, we show a key role for covRS in the regulation of opsonophagocytosis of AP53 by human neutrophils. AP53/covR+S− cells displayed potent binding of host complement inhibitors of C3 convertase, viz. Factor H (FH) and C4-binding protein (C4BP), which concomitantly led to minimal C3b deposition on AP53 cells, further showing that these plasma protein inhibitors are active on GAS cells. This resulted in weak killing of the bacteria by human neutrophils and a corresponding high death rate of mice after injection of these cells. After targeted allelic alteration of covS− to wild-type covS (covS+), a dramatic loss of FH and C4BP binding to the AP53/covR+S+ cells was observed. This resulted in elevated C3b deposition on AP53/covR+S+ cells, a high level of opsonophagocytosis by human neutrophils, and a very low death rate of mice infected with AP53/covR+S+. We show that covRS is a critical transcriptional regulator of genes directing AP53 killing by neutrophils and regulates the levels of the receptors for FH and C4BP, which we identify as the products of the fba and enn genes, respectively.
Introduction
Group A Streptococcus pyogenes (GAS)2 is a Gram-positive bacterial strain that is a causative agent for a variety of human infections ranging from simple pharyngeal and skin infections to more serious and often fatal pathologies, such as streptococcal toxic shock syndrome and necrotizing fasciitis. The pathogenesis of invasive GAS infections involves several stages among which are adhesion to epithelial surfaces, colonization, transmigration of the bacteria through the epithelium and subepithelium, survival in blood, penetration through the endothelium, and invasion into deep tissue. To accomplish these steps, invasive bacteria possess numerous genes encoding virulence factors, many of which need to be transcribed and/or repressed at specific stages of infection. To accomplish this, bacteria possess both monocistronic and polycistronic gene regulatory systems that activate and inactivate genes rapidly (1).
Among the best studied single component regulator of GAS virulence gene cluster expression under changing environmental conditions is the multiple gene activator (mga) system. This single transcription factor regulates expression of a variety of genes involved in virulence of the bacteria, such as the ubiquitous M-protein (emm gene), IgG and IgA receptors (fcR and enn genes, respectively), C5a peptidase (scpA gene), and a fibronectin-binding protein (fba gene) (2), which in many strains are arranged sequentially in a single virulence cluster (3). Two-component sensor/responder gene regulatory systems also exist in bacteria (4). In GAS, ∼13 such regulators are known (1). The most well studied of these is the cluster of virulence (cov) intracellular responder (covR)/extracellular sensor (covS) system (covRS) (5) in which environmental changes are “sensed” by the gene product (CovS) of covS and transmitted to its cognate protein product of covR (CovR) via phosphorylation-dephosphorylation (6). CovR then regulates repression or derepression of ∼15% of the GAS genome (7). Both of these genes are transcribed from a single promoter (8).
At several stages of dissemination of GAS, this microorganism must develop strategies to evade the host innate immune system, especially complement-mediated elimination of the microbe. The complement system is activated through antigen-antibody complexes (classical), host lectin binding to pathogen carbohydrate patterns (lectin pathway), and/or by small amounts of spontaneously generated complement Factor C3b, an opsonin, on microbial cell surfaces. Initially, C3b is generated by spontaneous activation (proteolysis) of Factor C3 (“tick over”), which then amplifies this pathway (alternate pathway) and deposits additional C3b on cells via covalent thioester binding (9). This is the triggering event for complement-mediated opsonization (10), which allows for phagocytotic cell recognition of the C3b on the pathogen via the receptor (C1Rg) (11). However, because uncontrolled activation of complement would be unfavorable to the host (e.g. anaphylactic response), natural inhibitors of complement activation exist, e.g. Factor H (FH) (12) and C4-binding protein (C4BP) (13). It has been shown that many types of pathogenic cells utilize FH to limit propagation of C3b deposition on their cell surfaces, thus protecting cells from opsonophagocytosis (14). With regard to GAS, it is widely assumed that the ubiquitous antiphagocytic bacterial M-protein is a major binding locus for sequestering host FH and C4BP to limit C3b deposition on bacterial surfaces (15). However, it has also been shown that not all M-proteins can bind FH and C4BP (16), and thus, M-proteins cannot function globally in antiphagocytosis by this mechanism. Therefore, other receptors for these complement inhibitors must exist in strains that can bind to these inhibitors.
Important concepts then emerge as to whether bacteria can control phagocytosis susceptibility by their natural regulatory systems, especially covRS, and whether FH and C4BP uptake by GAS is an important mechanism for control of complement-mediated opsonization in strains wherein the M-protein cannot function in this manner. In this investigation, we studied the role of covRS in opsonin deposition on an invasive skin-tropic GAS strain, AP53, and demonstrated a critical role for this regulatory system in opsonophagocytosis of this bacterium by neutrophils that is independent of complement inhibitor binding to M-protein.
EXPERIMENTAL PROCEDURES
Bacterial Strains
emm pattern D GAS strains AP53 (covR+S−pam+) (17) and NS931 (covR+S+pam−) (18) are primary patient isolates that have been used previously (19, 20).
Mice
C57BL/6 mice containing the human plasminogen (hPg) transgene (C57BL/6(hPg(Tg))) (21) were provided by Prof. D. Ginsburg (Ann Arbor, MI) and bred in our facilities.
Gene Sequencing and Identification
Sanger sequencing was accomplished on an ABI 3730xl 96-capillary sequencer (Gene Sequencing Facility, University of Notre Dame) using custom designed primers.
Isogenic GAS Strains
To construct isogenic mutants of strain AP53 with targeted deletions of Mga (AP53/mga−), SpeB (AP53/speB−), hPg-binding Group A streptococcal M-like protein (PAM) (AP53/pam−), the IgA-binding Enn protein (AP53/enn−), and fibronectin-binding protein (Fba) (AP53/fba−), targeting vectors containing the chloramphenicol acetyltransferase gene 5′-flanked by 300–400 bp of the AP53-specific DNA upstream of the ATG for the mga, speB, pam, enn, or fba genes and 3′-flanked by 300–400 bp of the AP53-specific DNA downstream of the TAA/TAG stop codon for these genes were constructed by routine methodology. The specific AP53 flanking sequences for each gene were obtained from the 13.4-kb sequence of the mga regulon that we published previously (20) and for speB from sequences surrounding the speB region of the AP53 genome. Restriction sites (typically 5′-NotI and 3′-SalI or -XhoI) were engineered into the two ends of the DNA segments and were used for insertion into the same sites of the temperature-sensitive plasmid pHY304, which also contained the erythromycin resistance (emr) gene. The resulting plasmids were then transformed into either or both of AP53/covR+S− and AP53/covR+S+ by electroporation. Chromosomal replacements of mga, speB, pam, enn, and fba by chloramphenicol acetyltransferase gene via allelic targeting were achieved by single crossover and double crossover strategies (20). In no case was mRNA for the specific gene that was replaced observed by RT-PCR, showing that total deletion was accomplished for each gene.
In addition to the entire gene-deleted mutants, GAS strains with targeted mutations in specific genes, e.g. AP53/covR+S+ and NS931/covR+S−, were also generated. In these cases, specific mutations of interest were first constructed by mutagenesis of the genes in the above targeting vectors along with a new translationally silent restriction site near the designated mutation for purposes of screening. The single crossover/double crossover strategy was then used to recombine the variant genes as described above.
All mutated genes were fully sequenced for confirmation of the intended gene product. None of the mutant GAS strains showed differences in their growth rates in culture.
C3b Deposition on GAS
This assay was performed as described previously (22) with modifications. GAS strains were grown to log phase (LP; A600 nm ∼ 0.6), washed twice with PBS, and resuspended in gelatin-veronal buffer (Sigma-Aldrich). Bacteria (1 × 108) were incubated with 50% normal human serum (NHS; Atlanta Biologicals, Lawrenceville, GA) or gelatin-veronal buffer for 60 min at 37 °C. The GAS cells were washed three times with PBS, 10 mm EDTA, pH 7.4. The bound proteins were eluted in 60 μl of 4 mm Na2CO3, 46 mm NaHCO3, pH 9.2 for 2 h at 37 °C (22). The eluted proteins were separated by SDS-PAGE under non-reducing conditions, and bound C3-related proteins were detected by Western blotting. Mouse anti-human C3 monoclonal antibody (Abbiotec, San Diego, CA) was used as the primary antibody, and HRP-conjugated anti-mouse IgG (Cell Signaling Technology, Danvers, MA) was used as the secondary antibody. Purified human C3b protein (CompTech, Tyler, TX) was included as a positive control. The protein bands were visualized with aid of a Clarity Western ECL kit (Bio-Rad) in a ChemiDoc MP system (Bio-Rad). The band intensities were quantified using Image Lab Software (Bio-Rad).
Human Neutrophil Isolation and Bactericidal Assays
To obtain fresh human neutrophils, blood was drawn from normal volunteers by licensed phlebotomists consistent with the United States Code of Federal Regulations and International Conference on Harmonisation Guidelines on Good Clinical Practices. Institutional Review Board approval was obtained from the University of Notre Dame, and informed consent forms were signed by all patients in accord with the Declaration of Helsinki. Neutrophils were isolated from these blood samples and purified using the Lympholyte-poly system (Cedarlane Laboratories, Burlington, NC). The cells were resuspended in RPMI 1640 medium (Cellgro, Manassas, VA) containing 20% (v/v) plasma from nonimmune individuals.
GAS survival assays were performed as described previously with some modifications (23). The neutrophil suspension was placed into 96-well plates at 2 × 105 cells/well. GAS strains were grown to LP (A600 nm ∼ 0.6) in Todd-Hewitt + 1% yeast medium and diluted to the desired concentration in RPMI 1640 medium containing 20% (v/v) plasma. The suspension was then added to human neutrophils at a ratio of 1:1 (2 × 105 cfu GAS/2 × 105 neutrophils). The 96-well plate was centrifuged at 500 × g for 10 min and incubated at 37 °C in 5% CO2 for 60 min. The contents of the wells were diluted 1000× in sterile water for neutrophil lysis, plated on a Todd-Hewitt agar plate, and incubated overnight at 37 °C. Wells without neutrophils were used as internal controls to determine base-line bacterial counts at the assay end points. Percent survival of GAS was calculated using the following equation: ((cfu/ml experimental well)/(cfu/ml control well)) × 100. Percent GAS survival was plotted using GraphPad PRISM 6 software. The data were expressed as the mean ± S.E. from five independent experiments. Statistical analyses were performed using the Student's t test, and p < 0.05 was considered statistically significant.
Mouse Survival Studies
C57Bl/6 male mice (6–10 weeks of age) containing the hPg transgene (21) were used for survival studies with the desired GAS strains as described earlier (20). The mice were injected subcutaneously in the right flank with 4–5 × 108 cells and observed twice daily for survival for up to 10 days. Kaplan-Meier survival curves were compared by the paired log-rank test using GraphPad PRISM 6 software. p values <0.05 were considered statistically significant. These procedures were approved by the Institutional Animal Care and Use Committee of the University of Notre Dame.
Binding of C4BP and FH to GAS Cells
These assays were performed as described previously (24) with operational modifications. GAS strains were grown and incubated with or without NHS as described under “C3b Deposition on GAS.” After incubation, the cells were washed three times with PBS, and bound proteins were eluted with 50 μl of 0.1 m glycine, pH 2.0 by incubating the samples for 30 min at 37 °C. The eluted samples were neutralized with 50 μl of 1 m Tris-HCl, pH 9.5 and then analyzed by Western blotting under non-reducing conditions. Mouse monoclonal anti-human C4BP or anti-human FH (Thermo Scientific, Rockford, IL) was used as the primary antibody, and HRP-conjugated anti-mouse IgG (Cell Signaling Technology) was used as the secondary antibody. Purified FH and C4BP (CompTech) were the positive controls.
The percentage of C4BP or FH binding was calculated relative to AP53/covR+S−, which was set at 100%, and plotted using GraphPad PRISM 6 software. The data were calculated as the mean ± S.E. from four independent experiments. Statistical analyses were performed using the Student's t test. A p value of <0.05 was considered statistically significant.
mSpeB Activity Assay
mSpeB assays were measured on LP (A600 nm ∼ 0.6) and early SP (A600 nm ∼ 1.0) cell-free GAS culture supernatants (20) as modified from a published method (25).
Binding of hPg, FH, and C4BP to GAS Cells Using Flow Cytometric Analysis (FCA)
GAS cells were grown to an A600 nm of ∼0.6 at 37 °C, washed twice with PBS, and resuspended in PBS, 1% BSA for blocking. Bacterial cells (2 × 108 cfu) were then incubated with 20 μg/ml hPg (Enzyme Research Laboratories, South Bend, IN) in PBS, 1% BSA for 1 h at room temperature or with 10 μg/ml FH or C4BP for 1 h at 37 °C. The cells were next washed with PBS. The bound hPg, FH, and C4BP were detected using the primary antibodies mouse anti-hPg (Enzyme Research Laboratories) and mouse anti-human FH/C4BP (Thermo Scientific), respectively. A secondary antibody, Alexa Fluor 488-donkey anti-mouse IgG (Invitrogen), was used for detection. Finally, the cells were washed and fixed in 1% paraformaldehyde for FCA.
A FACSAria III (BD Biosciences) was used for FCA of the labeled bacteria using the 488 nm laser. Data acquisition and analysis were performed by gating on fluorescence (FITC-A) and side scatter with the scales set to logarithmic amplification. Cells in suspension were analyzed at a flow rate of 10 μl/min, and 10,000 events were recorded for analysis.
Histograms were analyzed using FCS Express Version 4 software (De Novo Software, Los Angeles, CA), which provided the statistical tool, viz. the median fluorescence intensity. This value for each histogram was used to calculate the events accepted by the gating formula that fall within the specified marker. This information was used to calculate the percentage of binding of hPg, FH, and C4BP relative to AP53/covR+S− cells, which was set at 100%. The data were plotted using GraphPad PRISM 6 software.
Activation of hPg by Washed GAS Cell Culture Supernatants
Filtered cell supernatants (35 ml) were concentrated to 1.5 ml using 10,000 molecular weight limit membrane centrifugation filters, rediluted with PBS, and reconcentrated to 1.5 ml. This procedure was repeated twice. The activation rates of hPg (0.2 μm) in the absence or presence of PAM (1.0 μm) were measured at 37 °C with 0.25 mm S2251 (Diapharma, West Chester, OH) as the chromogenic substrate. The A405 nm was measured at 1-min intervals for 2 h after addition of 100 μl of cell supernatants. To obtain initial rates of activation under these conditions, the mA405 nm was plotted against t2. The slopes of the lines and linear regressions of the data were obtained using GraphPad PRISM 6.
Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Three independent extractions of total RNA were obtained from AP53/covR+S− and AP53/covR+S+. Real time RT-PCRs were performed essentially as described (26) with the scpA, enn, and fba (or fbp) primers reported previously (20). For the bicistronic scpA-fba gene, the primers spanned regions within the scpA and fba genes (forward primer, residues 10517–10537 and reverse primer, residues 10897–10915) of the 13.3-kb clone of this entire mga regulon region from AP53 published earlier (20), leading to a 399-bp amplicon. The relative gene expression levels of scpA, enn, fba, and scpA-fba were analyzed by the 2−ΔΔCT method (27) in which CT represents the threshold cycle number of RT-PCR at which the amplified product was first detected. The statistical means of triplicate CT values were calculated for the target and reference genes (in this case gapdh) from both WT and mutant strains. The ΔΔCT and relative expression levels of the genes were calculated as we have described (20).
Recombinant (r) Proteins
r-PAM and r-SK2b were cloned from AP53 cells and expressed in Escherichia coli as detailed previously (20, 28).
RESULTS
This study was centered on examining the role of the covRS system in regulating complement-mediated opsonization of a skin-tropic invasive strain of GAS, AP53. We focused on a critical stage of this process, viz. deposition of the opsonin C3b on GAS cells through the functioning of C3 convertase. The AP53 strain contains a natural inactivating mutation in covS (AP53/covR+S−) due to a single base deletion (ΔT1404), resulting in a frameshift mutation that leads to early termination of covS(ΔT1404) translation (20). This defined mutation allowed the opportunity to reinsert this base in a targeted fashion to generate the strain AP53/covR+S+, thereby reestablishing full covRS function (20) and allowing direct comparison of isogenic strains to attempt to realize the goal of the investigation.
Regulation of C3b Deposition on AP53 Cells by CovRS
Fig. 1A shows a Western blot of C3b deposition at LP growth of AP53/covR+S− and AP53/covR+S+ cells. The primary antibody used reacts with both C3 and C3b, but C3b is known to be the form of C3 that is bound to GAS cells (29). The presence of C3b on GAS cells is a determinant for their opsonization (10). It is clearly demonstrated in the current work that AP53/covR+S+ cells, but not AP53/covR+S−cells, displayed C3b deposition (Fig. 1A, lane 6) in the presence of NHS, which is the source of complement factors. These data suggest that C3 convertase formation and/or activity is repressed by CovR, and this repression is reversed by CovS+. This lack of C3b deposition on AP53/covR+S− cells as compared with AP53/covR+S+ cells translated into a significantly higher level of survival of AP53/covR+S− cells in the presence of neutrophils (Fig. 1B) and a consequent substantially lower level of survival of mice injected with GAS AP53/covR+S− as compared with AP53/covR+S+ (Fig. 1C).
FIGURE 1.
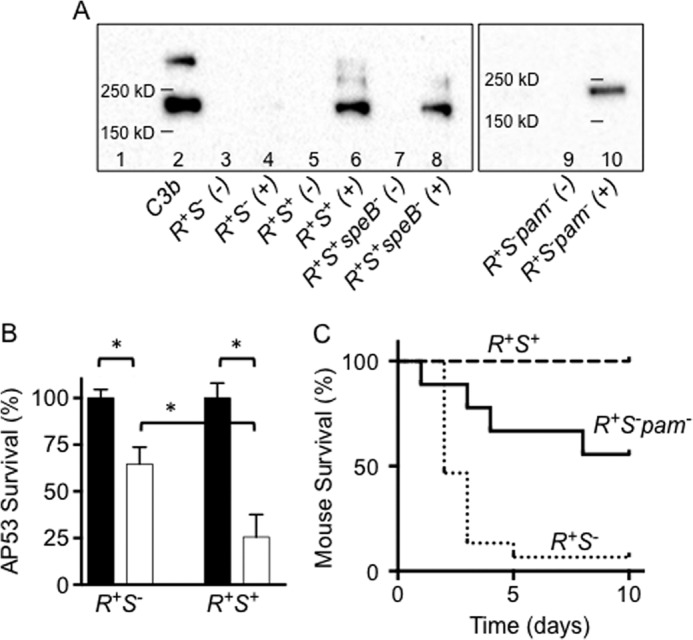
Relationship between C3b deposition and virulence of AP53 cells. A, Western blot analysis of C3b deposition on the indicated LP AP53 cells in the absence (−) and presence (+) of NHS. Molecular masses are indicated on the gel (lane 1) as determined from a standard molecular mass marker set. Purified C3b (1 μg; lane 2) is shown as a positive control. The gel containing lanes 9 and 10 was performed on a different occasion and is shown separately with molecular mass markers for that particular gel. B, survival of LP AP53/covR+S− (R+S−) and AP53/covR+S+ (R+S+) cells in human neutrophils. AP53 cells (2 × 105 cfu/well) and freshly isolated human neutrophils (2 × 105 cells/well) were incubated for 60 min at 37 °C. The neutrophils were then lysed, and the percentage of surviving AP53 cells was determined. Black-filled bars refer to AP53 survival of the indicated strains in the absence of neutrophils. White-filled bars refer to survival of the AP53 cells of the indicated strains in the presence of neutrophils. *, p < 0.05 between the sets indicated. n = 5 for each cell line. C, survival of C57BL/6(hPg(Tg)) mice after subcutaneous injection of AP53 cells. AP53 cells (4–5 × 108 cfu for each isogenic AP53 strain, viz. covR+S−, covR+S+, and covR+S−pam−, were injected into C57BL/6(hPg(Tg)) mice, and survival was monitored for a maximum of 10 days. p = 0.0001 when comparing covR+S+ (n = 7) with covR+S− (n = 15); p = 0.006 when comparing covR+S−pam− (n = 9) with covR+S− (n = 15). Error bars represent S.E.
Regulation of C3b Deposition on AP53 Cells by C3 Convertase Inhibitors
The activity of C3 convertase in both the classical/lectin and alternate complement pathways is naturally controlled by complement inhibitors C4BP (30, 31) and FH (32), respectively. The data of Fig. 2 illustrate the binding of FH (Fig. 2, A and B) and C4BP (Fig. 2, C and D) to various isogenic strains of AP53. Lanes 3 provide the binding data for serum FH (Fig. 2, A and B) and serum C4BP (Fig. 2, C and D) to AP53/covR+S− cells in the presence (+) of NHS. This was the reference point for both inhibitors for comparing other strains and was thus set at a value of 100% for relative quantitations in Fig. 2, B and D, respectively. AP53/covR+S+ cells display C3b on their cell surfaces (Fig. 1A, lane 6) and show greatly diminished cellular FH (Fig. 2A, lane 7) and C4BP (Fig. 2C, lane 7) binding, thus demonstrating an inverse relationship between C3 convertase inhibitor binding to cells and C3b deposition (Fig. 2, B and D). These results suggest for the first time that these relationships are part of the covRS regulatory system.
FIGURE 2.

Regulation of binding of FH and C4BP to AP53 cells and its isogenic strains at LP growth of cells. A, Western blot analysis of FH binding to the indicated LP AP53 cells in the absence (−) and presence (+) of NHS. Purified FH (1 μg; lane 1) is shown as a positive control. B, quantitation of the Western blot bands in A of the binding of FH to various AP53 lines. The percentage of relative binding to each strain is based on the binding of FH to AP53/covR+S− (R+S−), which was arbitrarily set to 100%. *, p < 0.05 when comparing the indicated strains with covR+S−. C, Western blot analysis of C4BP binding to the indicated cells in the absence (−) and presence (+) of NHS. Purified C4BP (1 μg; lane 1) is shown as a positive control. D, quantitation of the Western blot bands in C of the binding of C4BP to various AP53 lines. The percentage of relative binding to each strain is based on the binding of C4BP to covR+S−, which was arbitrarily set to 100%. *, p < 0.05 when comparing these strains with covR+S−. Error bars represent S.E.
It is thought that FH and C4BP bind to M-proteins expressed by GAS, particularly in strains expressing the M-proteins M5, M6, and M18 (16). This allowed a generally accepted assumption that GAS can accumulate FH and/or C4BP via M-protein and thereby inhibit C3b deposition on GAS. However, this cannot serve as a general mechanism because many GAS-derived M-proteins, e.g. M1, do not bind FH or C4BP (15, 16). The manner in which these considerations are relevant to important GAS strains that directly bind hPg have not been investigated. Thus, we studied whether such a mechanism could apply to strains containing a direct hPg-binding M-protein, viz. PAM, from GAS strain AP53. Our data dramatically show that deletion of PAM (AP53/covR+S−pam−) (Fig. 2, A and C, gel lanes 5) does not diminish binding of FH and C4BP, suggesting that PAM does not regulate binding of FH and C4BP. Surface plasmon resonance analysis using r-PAM and purified FH confirmed that PAM does not interact with FH (not shown).
Regulation of C3b Deposition on AP53 Cells by hPg/Human Plasmin (hPm) Binding
Despite the equivalent binding of C3 convertase inhibitors C4BP and FH to AP53/covR+S− and AP53/covR+S−pam− (Fig. 2), this latter cell line displays markedly higher deposition of C3b than AP53/covR+S− cells (Fig. 1A, lane 10), likely one important reason for the protection to lethality conferred on AP53/covR+S−pam− compared with AP53/covR+S− (Ref. 20 and Fig. 1C).
A well known property of PAM lies in its ability to interact very strongly with host serum hPg. In turn, hPg binding to PAM facilitates its activation to hPm by GAS-secreted streptokinase (SK), and as shown by FCA (Fig. 3A), hPg strongly interacts with both AP53/covR+S− and AP53/covR+S+ cells (Fig. 3B). However, hPg is not bound to AP53/covR+S+pam− (<10% relative to AP53/covR+S−), showing that PAM, when present, is the major receptor for hPg/hPm on GAS. The mechanism proposed thus suggests that C3b deposition relates also to hPg/hPm accumulation. It is likely that both FH and C4BP covR+S−-regulated receptor(s) and hPg/hPm accumulation by PAM are important factors in C3b deposition on this class of invasive GAS.
FIGURE 3.
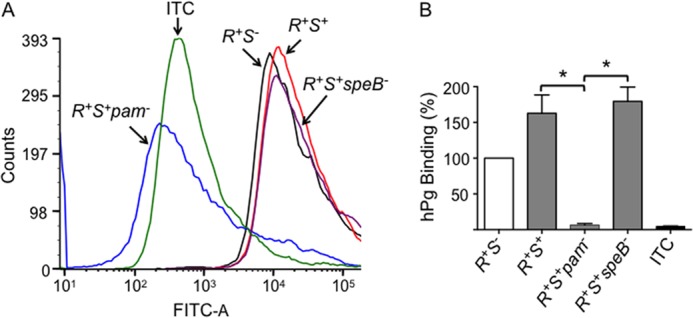
Influence of CovRS on the binding of hPg to PAM displayed on AP53 cells. A, typical FCA histograms of the binding of hPg to isogenic cell lines. Cells of each of the indicated cell lines (∼2 × 108 cfu) were incubated with hPg (20 μg/ml) at 25 °C. Mouse anti-human Pg was then added followed by Alexa Fluor 488-donkey anti-mouse IgG. The histograms of each strain were monitored at 488 nm. The data were collected with side scatter and fluorescence (FITC-A) gating using logarithmic amplification. Analyses of 10,000 events were obtained at a flow rate of 10 μl/min. B, the percentage of binding of hPg to AP53/covR+S− (R+S−) was arbitrarily taken as 100%, and the binding by other strains, viz. AP53/covR+S+ (R+S+), covR+S+pam− (R+S+pam−), and covR+S+speB− (R+S+speB−), was calculated using the median fluorescence intensity statistical value provided from analysis of each FCA histogram by FCS Express version 4 software. ITC (black-filled bar) refers to the isotype antibody control. *, p < 0.05 in comparisons of the indicated strains. n = 3. Error bars represent S.E.
Role of SpeB in C3b Deposition on AP53 Cells
In any comparison of the effects of covR+S+ and covR+S− strains, it is important to consider that covR+S+ positively regulates secretion and maturation of a cysteine protease, SpeB (mSpeB) (20). This protease affects virulence at different stages of infection through its cleavage of both secreted and membrane-bound proteins. The expression of mSpeB is suppressed, but not absent, in the media of LP AP53/covR+S+ cells as compared with early SP culture medium of this strain (Fig. 4A) and is severely down-regulated in culture media of AP53/covR+S− cells at all growth stages (20). We did not use late SP growth assays because it is known that mSpeB is greatly increased under late SP conditions and that many virulence proteins are affected by the presence of this protease. We used conditions (early SP) where mSpeB was moderately increased because these earlier growth stages (i.e. LP and early SP) are likely the conditions that C3b fixation would encounter. That these assays are specific for mSpeB was shown previously by the complete lack of activity seen when the mSpeB inhibitor E64 was added prior to substrate (Ref. 20 and this work (not shown)). Thus, mSpeB expression is unlikely to be a factor in the results obtained for AP53/covR+S− because very little mSpeB is produced but may influence the interpretation of the data for AP53/covR+S+ cells. To address this concern, we deleted the speB gene from AP53/covR+S+ cells to obtain strain AP53/covR+S+speB− and showed that C3b was deposited to a level equivalent to that on AP53/covR+S+ cells upon incubation with NHS (Fig. 1A, lanes 6 and 8). Concomitantly, FH binding of AP53/covR+S+speB− was very similar to that of AP53/covR+S+ cells (Fig. 4B), and hPg binding to AP53/covR+S+/speB− was equivalent to that of its parent strain (Fig. 3).
FIGURE 4.

Influence of speB expression on functional PAM display on AP53 cells and isogenic mutants. A, mSpeB assays in AP53 cell supernatants demonstrate that mSpeB is less strongly produced in LP (A600 nm ∼ 0.6) cultures than in the cultures at early SP (A600 nm ∼ 1.0). *, p < 0.05 for the comparisons indicated. n = 4. B, quantitation of Western blot bands of the binding of FH to various LP AP53 cell lines in the absence (−) and presence (+) of NHS where the percentage of relative binding of FH to AP53/covR+S+ (R+S+) and AP53/covR+S+speB− (R+S+speB−) is based on FH binding to AP53/covR+S− (R+S−) cells arbitrarily set to 100%. *, p < 0.05 when comparing the indicated strains with covR+S−. n = 4. C and D, activation of hPg by SK present in conditioned culture supernatants of various isogenic GAS cell lines in the absence of PAM (C) and in the presence of 1 μm PAM (D). Initial rates of activation are shown for each cell supernatant at LP and early SP growth of cells. r-SK (5 nm) is shown as a standard. *, p < 0.05 for the comparisons indicated. n = 4 for supernatants of each strain. Error bars represent S.E. mAbs, milliabsorbance units.
Effect of mSpeB on Functional PAM Expression on Cells
mSpeB is believed to catalyze degradation of M-protein and SK in vitro with purified proteins in cell cultures of various GAS strains as well as in mouse passaged isolates (33), and this may influence the levels of hPg bound to these cells (and consequently hPm production), especially at early SP where mSpeB production is approximately doubled from that produced mid-LP. The level of hPg binding to AP53/covR+S+/speB− is very similar to that of AP53/covR+S+ (Fig. 3, A and B) at early SP, thus minimizing the potential complication of mSpeB cleaving PAM in such a manner as to influence hPg binding.
Effect of mSpeB on Functional SK Expression in Cell Supernatants
The SK produced by AP53, classified as SK2b, shows maximal activity toward hPg activation when hPg is bound to PAM (34). Functional SK2b protein is optimally secreted at LP growth of GAS AP53 (Fig. 4C, compare LP AP53/covR+S− with early SP AP53/covR+S−).
Although absolute activities of r-SK2b are higher as expected in the presence of PAM, assays of culture supernatants show that slightly higher SK activities are seen in CovR+S− cells than in CovR+S+ cells. A significant feature of these data is that inactivation of the speB gene results in protection of SK. Specifically, 6–8-fold higher activities of cell supernatants toward hPg activation without (Fig. 4C, AP53/covR+S+speB−) or with (Fig. 4D, AP53/covR+S+speB−) r-PAM are noted, thus correlating well with the data of Fig. 4A. From these data, another interesting feature of these results can be explained. Whereas it has been shown previously that CovR+S− slightly represses sk transcription compared with CovR+S+ (20), a slightly higher SK protein activity is nonetheless seen in LP CovR+S− supernatants. Clearly, this dichotomy is due to the lower expression levels of mSpeB in CovR+S− cells that stabilizes SK levels. This effect is more dramatically seen in the higher SK activities found in AP53/CovR+S− cells compared with AP53/CovR+S+ cells, the latter of which produce higher levels of mSpeB. Although these effects are small, this feature nonetheless bears mention. These considerations influence the activation of hPg bound to PAM in that less proteolytic activity is provided to GAS cells with intact AP53/covR+S+, thus diminishing virulence of this strain as is seen from the data of Fig. 1.
Identification of the FH and C4BP Receptors on AP53 Cells
It is very unlikely that antigen-antibody complexes are present in these acute opsonophagocytosis experiments, thus minimizing the role of the classical complement pathway in C3b deposition. It is probable that the alternative and/or lectin complement pathways are operational in C3b deposition on GAS cells at early times of dissemination. These pathways utilize FH and C4BP (as does the classical pathway), respectively, as major inhibitors of C3b deposition on cells. Therefore, we sought to identify the relevant receptors for these inhibitors on AP53 cells, a topic that had not been addressed previously for pam+-expressing streptococcal cells.
It has been proposed that a wide variety of microbial human pathogens bind FH and/or C4BP to evade complement-mediated opsonophagocytosis. A number of streptococcal receptors for these inhibitors have been proposed, such as M5, M6, M18, M19, M24, and M28 proteins (15, 35, 36), enolase (37), Enn18 (35), Fba (38), and surface proteins from other microbes, e.g. surface proteins A1 and A2 from Moraxella catarrhalis (39), Lsa30 from Leptospira interrogans (40), OspE and CRASP-1/Bba68 proteins from various Borrelia strains (41), and many others. Among this group, if Fba and/or Enn is an important receptor for these complement inhibitors on AP53 cells, then their level of expression should be regulated by the mga gene product, Mga (20). Thus, a minimal requirement for these candidate receptors is that FH and C4BP binding would diminish in the absence of Mga expression. Thus, we examined the binding of these inhibitors to LP AP53/covR+S−mga− cells. The data obtained (Fig. 5A) demonstrate that large decreases (>75%) in binding of both FH and C4BP occur in strain AP53/covR+S−mga− as compared with strain AP53/covR+S−, showing that FH and C4BP are affected by the loss of mga expression. Although also diminished in AP53/mga− cells (20), other proximal gene products within the mga proximal virulence regulon, viz. ScpA and PAM, are not candidate receptors for FH or C4BP because elimination of PAM (Fig. 2) or ScpA (Fig. 5A) did not lead to significant loss of inhibitor binding. Overall, these experiments show that the receptor(s) for FH and C4BP on AP53 cells is also regulated by Mga and would be consistent with Fba and/or Enn as the receptor, respectively.
FIGURE 5.

A, binding of FH and C4BP to AP53 cells and isogenic mutants. Western blot gels were quantitated as in Fig. 2 to provide the binding data of FH and C4BP to individual proximal genes of the mga regulon. These data for pam− cells have been provided in Fig. 2. The percentage of relative binding for each inhibitor was calculated from Western blot band intensities relative to the binding of that inhibitor to covR+S−. *, p < 0.05 comparing the binding of the individual strains with covR+S−. B, relative expression of AP53 genes of the mga regulon, viz. scpA, fba, bicistronic scpA-fba, and enn, in the isolated total RNA from covR+S− (R+S−) and covR+S+ (R+S+) cells. The reference gene was gapdh. C and D, typical FCA histograms of the binding of FH (C) and C4BP (D) to isogenic cell lines. Cells of each of the indicated lines (∼2 × 108 cfu) were incubated at 37 °C with FH (10 μg/ml) (C) or C4BP (10 μg/ml) (D). Mouse anti-human FH or mouse anti-human C4BP was then added followed by Alexa Fluor 488-donkey anti-mouse IgG. Acquisition and analysis of the histograms were performed as in Fig. 3. E, the percentage of binding of FH and C4BP was calculated from histograms of C and D obtained from multiple runs (n = 4 for each cell line). The FH or C4BP binding to AP53/covR+S− was arbitrarily taken as 100%, and the relative binding of these inhibitors, each normalized to 100% binding to AP53/covR+S−, to other strains was calculated using the median fluorescence intensity statistical value provided from analysis of each FCA histogram by FCS Express version 4 software. ITC refers to the isotype antibody control. *, p < 0.05 in comparisons of FH binding of the indicated strains with AP53/covR+S−. ∧, p < 0.05 in comparisons of C4BP binding of the indicated strains with AP53/covR+S−. n = 4 for each strain. Error bars represent S.E.
Another requirement of the putative FH and C4BP receptors is that its expression must be regulated by CovRS as we demonstrate in the current study. Therefore, we performed quantitative RT-PCR on the fba and enn mRNAs in AP53/covR+S− and AP53/covR+S+. The data (Fig. 5B) show that a dramatic decrease in transcription of the fba and enn genes is found in AP53/covR+S+ as compared with AP53/covR+S− in full agreement with the comparative decrease in FH and C4BP binding to AP53/covR+S+. Because the sequential scpA-fba genes in the mga regulon are bicistronic and are transcriptionally driven by the proximal 5′ region of scpA, we also measured the transcription of scpA-fba in these two strains using the appropriate primer-probe set. The data obtained (Fig. 5B) for scpA-fba transcription are nearly identical to the fba and scpA genes alone in that scpA-fba is severely down-regulated in AP53/covR+S+ compared with AP53/covR+S−. This evidence shows that the products of the enn and fba genes remain major candidate receptors for FH and C4BP in this pam+ strain (AP53).
Further and more definitive proof of the hypothesis that the enn and fba gene products are the receptors for either or both FH and C4BP is obtained from the data of Fig. 5, C–E. Here, FCA analyses demonstrate that FH (Fig. 5C) interacts very weakly (∼5%) with AP53/covR+S−/fba− cells, which do not express fba mRNA, as compared with AP53/covR+S− cells, which display high expression of fba mRNA. However, binding of C4BP is not reduced in AP53/covR+S−fba− cells (Fig. 5, C and E). These data clearly show that Fba is a major receptor for FH on GAS AP53 cells.
With regard to the C4BP receptor, the data of Fig. 5, D and E, clearly show that Fba does not serve this role, but loss of the enn gene severely reduces C4BP binding when compared with the parent strain. FH binding to AP53/R+S−enn− is also reduced compared with AP53/R+S− but not to the same extent as C4BP binding. The data suggest that a major receptor for C4BP in these cells is the product of the enn gene, but FH may show some small level of cross-reactivity toward Enn.
Generality of CovRS in Regulating C3b Deposition on GAS
To demonstrate that CovRS regulation of complement-mediated opsonization of GAS is a more general effect, we chose to study the GAS cell line NS931, which contains an M-protein that does not directly interact with hPg (20). Consequently, NS931 secretes a form of SK (SK1) that does not require hPg to be bound to PAM for its optimal activation to hPm (34). Because NS931 contains the WT active forms of CovR and CovS (NS931/covR+S+), we generated an isogenic variant of NS931 in which T1404 was deleted from its covS gene, thus creating NS931/covR+S−, which is identical to the covS mutation in the AP53 patient isolate. We previously showed that NS931/covR+S− was substantially more lethal in mice than NS931/covR+S+ (20).
In accord with these lethality findings, we demonstrate herein that, like the AP53 system, C3b is more highly deposited on LP NS931/covR+S+ than on similarly grown LP NS931/covR+S− cells (Fig. 6A). Concomitantly enhanced accumulation of C4BP was observed on NS931/covR+S− as compared with NS931/covR+S+ cells (Fig. 6, B and C). Similarly, higher binding of FH was seen on the NS931/covR+S− strain compared with the NS931/covR+S+ strain (Fig. 6, D and E). Thus, the CovRS regulation of complement-mediated opsonization is a general feature of GAS mechanistically related to CovRS regulation of receptor levels for FH and C4BP and supported by hPg binding PAM. The down-regulation of SpeB, which occurs in both AP53/covR+S− and NS931/covR+S− (20), also assists survival of secreted SK, which can stimulate hPg activation and further reduce levels of C3b on these cells.
FIGURE 6.

Regulation of C3b deposition on LP NS931 cells by CovRS. A, Western blot of C3b deposition on the indicated NS931 cells in the absence (−) and presence (+) of NHS. The molecular masses indicated on the gel were determined from a standard molecular mass marker set. Purified C3b (1 μg; lane 1) is shown as a positive control. B, Western blot analysis of C4BP binding to NS931 cells in the absence (−) and presence (+) of NHS. Purified C4BP (1 μg; lane 1) is shown as a positive control. C, quantitation of the Western blot bands of the binding of C4BP to various AP53 lines (n = 4). The percentage of relative binding to each strain is based on NS931/covR+S−, which was arbitrarily set at 100%. *, p < 0.05 when comparing NS931/covR+S+ (R+S+) (+NHS) cells with NS931/covR+S− (R+S−) (+NHS) cells. n = 4. D and E, typical FCA of the binding of FH to various cell lines. D, cells of each of the indicated GAS lines (∼2 × 108 cfu) were incubated with FH (10 μg/ml) at 37 °C. Mouse anti-human FH was added as the primary antibody followed by Alexa Fluor 488-donkey anti-mouse IgG as the secondary antibody. Acquisition and analysis of the histograms were performed as in Fig. 3. E, the percentage of binding of FH calculated from multiple runs (n = 4). The FH binding to AP53/covR+S− was arbitrarily taken as 100%, and the binding by other strains was calculated using the median fluorescence intensity statistical value provided from analysis of each FCA histogram by FCS Express version 4 software. ITC refers to the isotype antibody control. *, p < 0.05 when comparing AP53/covR+S− with AP53/covR+S+. **, p < 0.05 when comparing NS931/covR+S− with NS931/covR+S+. n = 4 for each genotype. Error bars represent S.E.
DISCUSSION
Two-component gene regulatory systems are critical components of the virulence determinants of pathogenic bacteria. These systems allow “sensing” of the different environments that the bacteria encounter during host invasion and concomitantly permit a rapid transcriptional “response” to enhance or inhibit gene expression. These strategies are used by GAS for survival in different host defense mechanisms that attempt to eliminate these microorganisms. One of the best studied two-component regulatory gene systems is CovRS wherein the membrane-bound extracellular sensor protein (CovS) can function as an autophosphorylase, a kinase, or a phosphatase (1), ultimately phosphorylating or dephosphorylating CovR protein to modulate CovR-based transcription of genes that GAS use to survive upon pressure from the host (e.g. high salt levels in perspiration, low O2 tension in tissues, iron starvation, elevated temperature, etc.). CovR represses (e.g. speB) or stimulates (e.g. the hyaluronic acid capsule) genes needed for GAS dissemination, and these properties are modulated by CovS (20, 42, 43). Thus, it is pertinent to assess the role of CovRS in complement-mediated opsonophagocytosis of GAS to examine the importance of this step in the overall virulence of GAS.
C3b deposition is not observed on AP53 cells that contain intact CovR and a naturally inactivated CovS (AP53/covR+S−) but is strongly present on an isogenic strain in which the CovRS system is fully active (AP53/covR+S+). A similar observation has been made in comparing NS931/covR+S+ and isogenic NS931/covR+S− cells, showing that the effect is general. Correspondingly, AP53/covR+S+ is more efficiently phagocytosed by neutrophils as compared with AP53/covR+S− and is much less lethal in mice (20). Thus, a linear relationship among C3b deposition, neutrophil killing, and diminished lethality is established in GAS with an intact covRS system. This provides a rationale for observations that in some invasive lines GAS undergo inactivating mutations in covRS at stages of infection where certain genes may assist bacteria in their attempt to survive challenges presented by the host. For example, it has been found that inactivating mutations occurred in the covR or covS genes in clinical isolates from approximately one-half of patients with GAS-induced streptococcal toxic shock syndrome but not in isolates from mild infection (44). Such changes account for decreased production of proteins, such as mSpeB, which is required for local invasion but must be down-regulated for systemic dissemination (45). Furthermore, in another example, increased expression of other genes controlled by CovRS is required, e.g. streptolysin O (slo) necrosis of neutrophils (46).
Factor H and C4BP are inhibitors of the alternate and classical/lectin pathways, respectively, at the level of C3 convertase, and therefore it is believed that binding of these inhibitors to GAS, primarily via M-proteins, would protect this microorganism from opsonization. However, although the present study supports this in principle, this conclusion as an overall mechanism of complement-mediated opsonization is incomplete.
FH and C4BP do not substantially bind to PAM in this highly invasive primary isolate that contains an inactivating mutation in covS, which is likely the result of a mutation in the patient sample that encouraged its dissemination. This mutation did not affect PAM levels as measured through its most well recognized property, viz. the binding and activation of hPg. However, after reversion of covS− to covS+ via targeted replacement of the single base mutation in this particular covS− variant, FH and C4BP binding to AP53/covR+S+ was greatly diminished, and C3b was deposited on cells. PAM levels remained intact or were even increased on AP53/covR+S+ cells. Thus, whereas FH and C4BP binding to cells appears indispensable, the receptor(s) for these inhibitors is not PAM. These receptors are strongly regulated, directly or indirectly, by CovRS, a novel finding of this study, and by Mga. Very strong evidence has emerged from this work that the product of the fba gene, a major fibronectin-binding protein (Fba), is the CovRS-mediated receptor for FH, and similarly, the product of the enn gene, an IgA-binding protein, is also a major receptor for C4BP. In such highly invasive GAS strains, cellular receptors for complement inhibitors are needed to prevent early opsonophagocytosis and bacterial killing. M-proteins can serve this role in some cases, but when these highly variable M-proteins do not contain FH and C4BP binding properties, perhaps Fba and Enn, the genes of which are not present in all GAS strains, serve this purpose. It is interesting to speculate a possible coinheritance in GAS strains containing fba and/or enn genes with strains that contain M-proteins that do not interact with FH and C4BP. Published data consistent with this hypothesis show that, as with AP53, other pam+ strains, e.g. NS88.2 and Alab49, contain the fba and enn genes, and their PAM proteins do not interact with FH. In natural pam− strains, it has been shown that GAS strains serotyped as M1, M2, M3, M4, and M22 contain the fba gene, and their M-proteins concomitantly do not interact with FH. On the other hand, GAS serotypes M5, M6, and M18 do not contain the fba gene, but their M-proteins interact with FH (16, 47, 48). Thus, GAS has likely evolved to a point that assures that these microorganisms display receptors for complement inhibitors FH and/or C4BP, utilizing the M-protein or other surface-regulated genes in this role.
Although we show that PAM is not an FH or C4BP receptor and we demonstrate that functional pam expression as expressed by hPg binding is not regulated to any large extent by CovRS, we provide data that suggest that its presence on GAS is critical for inhibition of C3b deposition. This is likely due to the ability of PAM to bind hPg/hPm. hPm bound to cells is thought to inhibit C3b deposition via degradation of this opsonin (49, 50). Thus, the increase in C3b deposition and partial rescue of lethality in AP53/covR+S+pam− can be attributed both to down-regulation of the FH and C4BP receptor on the cells and to the loss of hPg/hPm binding ability. That the hPm-PAM interaction on cells is pathophysiologically important in this regard is underlined by the data presented demonstrating that increased SK production via a targeted inactivation of the speB gene leads to increased cellular hPm display on LP AP53/covR+S+speB− cells as compared with LP AP53/covR+S+ cells.
In conclusion, using invasive strains of GAS, we have demonstrated that the CovRS system provides a dominant influence on complement-mediated innate immunity via regulation of C3b opsonin deposition on GAS surfaces. Thus, mechanistically, the gene switching to covR+S− that bacteria use to assist their survival and dissemination in the host allows for down-regulation of mSpeB, resistance to opsonophagocytosis, and an increase in SK production, all of which are selective advantages for GAS in avoiding their complement-mediated clearance. These mechanisms provide a rationale that explains the high invasive potential of skin-tropic PAM-containing S. pyogenes strains and considerably expand knowledge of the role of the covRS operon in GAS virulence.
This work was supported, in whole or in part, by National Institutes of Health Grant HL013423.
- GAS
- Group A S. pyogenes
- CovRS
- cluster of virulence responder/sensor
- FH
- Factor H
- C4BP
- C4-binding protein
- Mga
- multiple gene activator
- hPg
- human plasminogen
- PAM
- Group A streptococcal M-like protein
- LP
- log phase
- NHS
- normal human serum
- SpeB
- streptococcal pyrogenic endotoxin B
- mSpeB
- mature SpeB
- SP
- stationary phase
- FCA
- flow cytometric analysis
- CT
- threshold cycle number
- r
- recombinant
- hPm
- human plasmin
- SK
- streptokinase.
REFERENCES
- 1. Churchward G. (2007) The two faces of Janus: virulence gene regulation by CovR/S in group A streptococci. Mol. Microbiol. 64, 34–41 [DOI] [PubMed] [Google Scholar]
- 2. McLandsborough L. A., Cleary P. P. (1995) Insertional inactivation of virR in Streptococcus pyogenes M49 demonstrates that VirR functions as a positive regulator of ScpA, FcRA, OF, and M protein. FEMS Microbiol. Lett. 128, 45–51 [DOI] [PubMed] [Google Scholar]
- 3. Podbielski A. (1993) Three different types of organization of the vir regulon in group A streptococci. Mol. Gen. Genet. 237, 287–300 [DOI] [PubMed] [Google Scholar]
- 4. Laub M. T., Goulian M. (2007) Specificity in two-component signal transduction pathways. Annu. Rev. Genet. 41, 121–145 [DOI] [PubMed] [Google Scholar]
- 5. Kondo H., Nakagawa A., Nishihira J., Nishimura Y., Mizuno T., Tanaka I. (1997) Escherichia coli positive regulator OmpR has a large loop structure at the putative RNA polymerase interaction site. Nat. Struct. Biol. 4, 28–31 [DOI] [PubMed] [Google Scholar]
- 6. Dalton T. L., Scott J. R. (2004) CovS inactivates CovR and is required for growth under conditions of general stress in Streptococcus pyogenes. J. Bacteriol. 186, 3928–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Graham M. R., Smoot L. M., Migliaccio C. A., Virtaneva K., Sturdevant D. E., Porcella S. F., Federle M. J., Adams G. J., Scott J. R., Musser J. M. (2002) Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc. Natl. Acad. Sci. U.S.A. 99, 13855–13860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gryllos I., Levin J. C., Wessels M. R. (2003) The CsrR/CsrS two-component system of group A Streptococcus responds to environmental Mg2+. Proc. Natl. Acad. Sci. U.S.A. 100, 4227–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pangburn M. K., Müller-Eberhard H. J. (1980) Relation of putative thioester bond in C3 to activation of the alternative pathway and the binding of C3b to biological targets of complement. J. Exp. Med. 152, 1102–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ross G. D., Medof M. E. (1985) Membrane complement receptors specific for bound fragments of C3. Adv. Immunol. 37, 217–267 [DOI] [PubMed] [Google Scholar]
- 11. Wiesmann C., Katschke K. J., Yin J., Helmy K. Y., Steffek M., Fairbrother W. J., McCallum S. A., Embuscado L., DeForge L., Hass P. E., van Lookeren Campagne M. (2006) Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature 444, 217–220 [DOI] [PubMed] [Google Scholar]
- 12. Pangburn M. K., Schreiber R. D., Müller-Eberhard H. J. (1977) Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein β1H for cleavage of C3b and C4b in solution. J. Exp. Med. 146, 257–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujita T., Gigli I., Nussenzweig V. (1978) Human C4-binding protein. II. Role in proteolysis of C4b by C3b-inactivator. J. Exp. Med. 148, 1044–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lindahl G., Sjöbring U., Johnsson E. (2000) Human complement regulators: a major target for pathogenic microorganisms. Curr. Opin. Immunol. 12, 44–51 [DOI] [PubMed] [Google Scholar]
- 15. Horstmann R. D., Sievertsen H. J., Knobloch J., Fischetti V. A. (1988) Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc. Natl. Acad. Sci. U.S.A. 85, 1657–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gustafsson M. C., Lannergård J., Nilsson O. R., Kristensen B. M., Olsen J. E., Harris C. L., Ufret-Vincenty R. L., Stålhammar-Carlemalm M., Lindahl G. (2013) Factor H binds to the hypervariable region of many Streptococcus pyogenes M proteins but does not promote phagocytosis resistance or acute virulence. PLoS Pathog. 9, e1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berge A., Sjöbring U. (1993) PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J. Biol. Chem. 268, 25417–25424 [PubMed] [Google Scholar]
- 18. McKay F. C., McArthur J. D., Sanderson-Smith M. L., Gardam S., Currie B. J., Sriprakash K. S., Fagan P. K., Towers R. J., Batzloff M. R., Chhatwal G. S., Ranson M., Walker M. J. (2004) Plasminogen binding by group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infect. Immun. 72, 364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McArthur J. D., McKay F. C., Ramachandran V., Shyam P., Cork A. J., Sanderson-Smith M. L., Cole J. N., Ringdahl U., Sjöbring U., Ranson M., Walker M. J. (2008) Allelic variants of streptokinase from Streptococcus pyogenes display functional differences in plasminogen activation. FASEB J. 22, 3146–3153 [DOI] [PubMed] [Google Scholar]
- 20. Liang Z., Zhang Y., Agrahari G., Chandrahas V., Glinton K., Donahue D. L., Balsara R. D., Ploplis V. A., Castellino F. J. (2013) A natural inactivating mutation in the CovS component of the CovRS regulatory operon in a pattern D Streptococcal pyogenes strain influences virulence-associated genes. J. Biol. Chem. 288, 6561–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun H., Ringdahl U., Homeister J. W., Fay W. P., Engleberg N. C., Yang A. Y., Rozek L. S., Wang X., Sjöbring U., Ginsburg D. (2004) Plasminogen is a critical host pathogenicity factor for Group A streptococcal infection. Science 305, 1283–1286 [DOI] [PubMed] [Google Scholar]
- 22. Li K., Zhou W., Hong Y., Sacks S. H., Sheerin N. S. (2009) Synergy between type 1 fimbriae expression and C3 opsonisation increases internalisation of E. coli by human tubular epithelial cells. BMC Microbiol. 9, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cole J. N., Pence M. A., von Köckritz-Blickwede M., Hollands A., Gallo R. L., Walker M. J., Nizet V. (2010) M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. MBio 1, e00191-e00210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dieudonné-Vatran A., Krentz S., Blom A. M., Meri S., Henriques-Normark B., Riesbeck K., Albiger B. (2009) Clinical isolates of Streptococcus pneumoniae bind the complement inhibitor C4b-binding protein in a PspC allele-dependent fashion. J. Immunol. 182, 7865–7877 [DOI] [PubMed] [Google Scholar]
- 25. Hytönen J., Haataja S., Gerlach D., Podbielski A., Finne J. (2001) The SpeB virulence factor of Streptococcus pyogenes, a multifunctional secreted and cell surface molecule with strepadhesin, laminin-binding and cysteine protease activity. Mol. Microbiol. 39, 512–519 [DOI] [PubMed] [Google Scholar]
- 26. Roberts S. A., Scott J. R. (2007) RivR and the small RNA RivX: the missing links between the CovR regulatory cascade and the Mga regulon. Mol. Microbiol. 66, 1506–1522 [DOI] [PubMed] [Google Scholar]
- 27. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 28. Zhang Y., Liang Z., Glinton K., Ploplis V. A., Castellino F. J. (2013) Functional differences between Streptococcus pyogenes cluster 1 and cluster 2b streptokinases are determined by their β-domains. FEBS Lett. 587, 1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weis J. J., Law S. K., Levine R. P., Cleary P. P. (1985) Resistance to phagocytosis by group A streptococci: failure of deposited complement opsonins to interact with cellular receptors. J. Immunol. 134, 500–505 [PubMed] [Google Scholar]
- 30. Blom A. M., Kask L., Dahlbäck B. (2001) Structural requirements for the complement regulatory activities of C4BP. J. Biol. Chem. 276, 27136–27144 [DOI] [PubMed] [Google Scholar]
- 31. Rawal N., Rajagopalan R., Salvi V. P. (2009) Stringent regulation of complement lectin pathway C3/C5 convertase by C4b-binding protein (C4BP). Mol. Immunol. 46, 2902–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wyatt R. J., Julian B. A., Weinstein A., Rothfield N. F., McLean R. H. (1982) Partial H (β1H) deficiency and glomerulonephritis in two families. J. Clin. Immunol. 2, 110–117 [DOI] [PubMed] [Google Scholar]
- 33. Rezcallah M. S., Boyle M. D., Sledjeski D. D. (2004) Mouse skin passage of Streptococcus pyogenes results in increased streptokinase expression and activity. Microbiology 150, 365–371 [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y., Liang Z., Hsueh H. T., Ploplis V. A., Castellino F. J. (2012) Characterization of streptokinases from Group A streptococci reveals a strong functional relationship that supports the coinheritance of plasminogen-binding M-protein and cluster 2b streptokinase. J. Biol. Chem. 287, 42093–42103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pérez-Caballero D., García-Laorden I., Cortés G., Wessels M. R., de Córdoba S. R., Albertí S. (2004) Interaction between complement regulators and Streptococcus pyogenes: binding of C4b-binding protein and factor H/factor H-like protein 1 to M18 strains involves two different cell surface molecules. J. Immunol. 173, 6899–6904 [DOI] [PubMed] [Google Scholar]
- 36. Jenkins H. T., Mark L., Ball G., Persson J., Lindahl G., Uhrin D., Blom A. M., Barlow P. N. (2006) Human C4b-binding protein, structural basis for interaction with streptococcal M protein, a major bacterial virulence factor. J. Biol. Chem. 281, 3690–3697 [DOI] [PubMed] [Google Scholar]
- 37. Agarwal V., Hammerschmidt S., Malm S., Bergmann S., Riesbeck K., Blom A. M. (2012) Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J. Immunol. 189, 3575–3584 [DOI] [PubMed] [Google Scholar]
- 38. Pandiripally V., Gregory E., Cue D. (2002) Acquisition of regulators of complement activation by Streptococcus pyogenes serotype M1. Infect. Immun. 70, 6206–6214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nordström T., Blom A. M., Forsgren A., Riesbeck K. (2004) The emerging pathogen Moraxella catarrhalis interacts with complement inhibitor C4b binding protein through ubiquitous surface proteins A1 and A2. J. Immunol. 173, 4598–4606 [DOI] [PubMed] [Google Scholar]
- 40. Souza N. M., Vieira M. L., Alves I. J., de Morais Z. M., Vasconcellos S. A., Nascimento A. L. (2012) Lsa30, a novel adhesin of Leptospira interrogans binds human plasminogen and the complement regulator C4bp. Microb. Pathog. 53, 125–134 [DOI] [PubMed] [Google Scholar]
- 41. Pietikäinen J., Meri T., Blom A. M., Meri S. (2010) Binding of the complement inhibitor C4b-binding protein to Lyme disease Borreliae. Mol. Immunol. 47, 1299–1305 [DOI] [PubMed] [Google Scholar]
- 42. Heath A., DiRita V. J., Barg N. L., Engleberg N. C. (1999) A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect. Immun. 67, 5298–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Treviño J., Perez N., Ramirez-Peña E., Liu Z., Shelburne S. A., 3rd, Musser J. M., Sumby P. (2009) CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect. Immun. 77, 3141–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ikebe T., Ato M., Matsumura T., Hasegawa H., Sata T., Kobayashi K., Watanabe H. (2010) Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog. 6, e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cole J. N., McArthur J. D., McKay F. C., Sanderson-Smith M. L., Cork A. J., Ranson M., Rohde M., Itzek A., Sun H., Ginsburg D., Kotb M., Nizet V., Chhatwal G. S., Walker M. J. (2006) Trigger for group A streptococcal M1T1 invasive disease. FASEB J. 20, 1745–1747 [DOI] [PubMed] [Google Scholar]
- 46. Ato M., Ikebe T., Kawabata H., Takemori T., Watanabe H. (2008) Incompetence of neutrophils to invasive group A Streptococcus is attributed to induction of plural virulence factors by dysfunction of a regulator. PLoS One 3, e3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blackmore T. K., Fischetti V. A., Sadlon T. A., Ward H. M., Gordon D. L. (1998) M protein of the group A Streptococcus binds to the seventh short consensus repeat of human complement factor H. Infect. Immun. 66, 1427–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Terao Y., Kawabata S., Kunitomo E., Murakami J., Nakagawa I., Hamada S. (2001) Fba, a novel fibronectin-binding protein from Streptococcus pyogenes, promotes bacterial entry into epithelial cells, and the fba gene is positively transcribed under the Mga regulator. Mol. Microbiol. 42, 75–86 [DOI] [PubMed] [Google Scholar]
- 49. Barthel D., Schindler S., Zipfel P. F. (2012) Plasminogen is a complement inhibitor. J. Biol. Chem. 287, 18831–18842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vieira M. L., Atzingen M. V., Oliveira R., Mendes R. S., Domingos R. F., Vasconcellos S. A., Nascimento A. L. (2012) Plasminogen binding proteins and plasmin generation on the surface of Leptospira spp.: the contribution to the bacteria-host interactions. J. Biomed. Biotechnol. 2012, 758513. [DOI] [PMC free article] [PubMed] [Google Scholar]