

Background: Endophilin B2 is a BAR protein of previously unidentified function.
Results: Endophilin B2 interacts directly with plectin 1 and controls the perinuclear vimentin network.
Conclusion: The endophilin B2-plectin 1 complex functions as an anchoring device via BAR-mediated membrane binding activity.
Significance: Our data unravel a novel activity of a BAR protein that does not involve membrane deforming activity supporting membrane trafficking.
Keywords: Cell Biology, Cytoskeleton, Intermediate Filaments, Membrane, Protein Assembly, SH3 Domains, BAR Domain, Endophilin, Plectin
Abstract
Proteins of the Bin/amphiphysin/Rvs (BAR) domain superfamily are essential in controlling the shape and dynamics of intracellular membranes. Here, we present evidence for the unconventional function of a member of the endophilin family of BAR and Src homology 3 domain-containing proteins, namely endophilin B2, in the perinuclear organization of intermediate filaments. Using mass spectrometry analysis based on capturing endophilin B2 partners in in situ pre-established complexes in cells, we unravel the interaction of endophilin B2 with plectin 1, a variant of the cytoskeleton linker protein plectin as well as with vimentin. Endophilin B2 directly binds the N-terminal region of plectin 1 via Src homology 3-mediated interaction and vimentin indirectly via plectin-mediated interaction. The relevance of these interactions is strengthened by the selective and drastic reorganization of vimentin around nuclei upon overexpression of endophilin B2 and by the extensive colocalization of both proteins in a meshwork of perinuclear filamentous structures. By generating mutants of the endophilin B2 BAR domain, we show that this phenotype requires the BAR-mediated membrane binding activity of endophilin B2. Plectin 1 or endophilin B2 knockdown using RNA interference disturbed the perinuclear organization of vimentin. Altogether, these data suggest that the endophilin B2-plectin 1 complex functions as a membrane-anchoring device organizing and stabilizing the perinuclear network of vimentin filaments. Finally, we present evidence for the involvement of endophilin B2 and plectin 1 in nuclear positioning in individual cells. This points to the potential importance of the endophilin B2-plectin complex in the biological functions depending on nuclear migration and positioning.
Introduction
Cell motility and exchange of materials between intracellular compartments in eukaryotes involve dynamic remodeling of membrane curvature. Scaffolding, a process reversibly involving peripheral membrane proteins at the membrane-cytosol interface, can impose and/or stabilize curvature and is accomplished by several classes of proteins (1–3). The members of the superfamily of BAR3 domain proteins, initially associated with synaptic endocytic recycling, are key actors in scaffold-based membrane remodeling (2, 4).
Structure-function relationships of some BAR domain-containing proteins have benefited from the determination of tertiary and quaternary structures as well as development of in vitro assays of membrane reshaping activity (2, 5). Three unique families are distinguished, harboring either the initially characterized classical BAR (6, 7), the related F-BAR (Fes/CIP4 homology BAR), or the finally identified I-BAR (Inverse-BAR) (8, 9). BAR domains are dimers of a three-helix coiled coil bundle displaying a unique degree of curvature that is correlated to their preferential binding to tubules or vesicles of particular size (7, 10–13). The scaffolding action of the protein is favored by a high density of positively charged residues, often on the concave surface, interacting with negatively charged lipid of the cytosolic membrane leaflets (14). As described for the F-BAR domain, self-assembled helical coats propagate curvature necessary for membrane tubulation (8).
Members of the N-BAR family, such as amphiphysins, endophilins, and nadrins, possess an N-terminal sequence folded into an amphipathic α-helix in the membrane environment. This provides an additional membrane binding domain (15), reinforced in endophilins by a similar insert in the first helix, provoking increased constraint on membrane curvature (16–18). These helices confer N-BAR proteins the ability of either detecting and binding curved areas with lipid defects (19–21) or imposing membrane bending by pushing apart lipids in the monolayer (3, 16, 17, 20), thus contributing to curvature sensing or induction. The N-terminal α-helix was also proposed to favor the membrane scission activity of N-BAR domains (22). Structural studies of reconstructed membrane-bound N-BAR and F-BAR proteins revealed different principles for formation of helical lattices, relying on interactions between N-terminal α-helices and on extensive lateral interactions, respectively (8, 23).
BAR domain proteins participate in several cellular functions as most of them possess binding modules, such as the phosphoinositide binding pleckstrin and phox homology domains, or the protein recognition SH3 domain. These modules confer the ability to couple local membrane deformation and signaling functions (2).
Endophilins, consisting of an N-terminal N-BAR domain and a C-terminal SH3 domain flanking a variable intermediate region, are encoded by five genes in mammals and are distinguished in endophilins A1, A2, and A3 and endophilins B1 and B2, with highly similar structures (24, 25). The crystal structures of the endophilin A1 N-BAR and the endophilin A2 (endoA2) SH3 domains have been solved (26, 27), and the whole endoA2 molecule has only been modeled by small angle x-ray scattering reconstruction (28).
Mammalian endophilin A proteins are all highly enriched in the brain, with endoA2 being ubiquitously expressed. Numerous studies have investigated the functions of endophilins A in constitutive and regulated endocytosis, with emphasis on synaptic vesicle recycling (29–33). The two major binding partners of the SH3 domain of endophilins A, dynamin and synaptojanin, implicate the protein in the clathrin-mediated endocytic process, in particular during fission and uncoating (33, 34).
Endophilins B are found in most tissues and, initially identified based on sequence homology (24), were cloned following two-hybrid screens using Bax (B-cell lymphoma-2-associated X protein) as bait (35, 36). Bax binds the N-terminal sequence of endophilin B1 (endoB1) and not endophilin B2 (endoB2) (36). Importantly, endoB2 and endoB1 could interact in a yeast two-hybrid assay, showing their ability to form heterodimers (36). The idea of a pro-apoptotic role of endoB1, due to its transient interaction with Bax (hence its name Bif-1, Bax-interacting factor-1), has been largely substantiated (35, 37–40). However, the contribution of endoB1 in Bax-mediated permeabilization of the mitochondrial outer membrane, depending on Bax conformational changes, is not fully understood. Whether and how these events rely on self-assembly and membrane deforming activities of the BAR domain remain open questions. Consistently with its ability to reshape membranes (41), endoB1 was shown to control the morphology of the mitochondrial network and to participate in mitochondrial division (37). In agreement with its presence on Golgi membranes, endoB1 has also been involved in COPI-vesicle formation (41, 42). Similarly with the role of endophilins A in regulated endocytosis, endoB1 has been specifically implicated in the regulation of nerve growth factor/TrkA trafficking (43). Autophagy is a last process involving endoB1 because, in starved cells, it is translocated to early stage autophagosomes and recruits the ultraviolet irradiation resistant-associated gene (UVRAG), via its SH3 domain (44). Although the contribution of its membrane deforming activity, if any, needs to be determined, endoB1 could thus be part of a complex driving membrane biogenesis for the formation of autophagosomes. Relevant to this function, a key role of endoB1 in the fission of Golgi tubules during nutrient starvation was reported (44, 45).
EndoB1 is increasingly involved in tumor suppression (with autophagy and apoptosis being negative regulators of tumorigenesis (46)); strikingly, its close relative endoB2 has not been assigned any function. As lower organisms such as Caenorhabditis elegans and Drosophila have only one gene encoding for endophilin B, endoB1 and -B2 could exert redundant functions in mammals. However, because both can assemble as heterodimers, more subtle functions might exist in higher organisms, and at least some of the endoB1 described functions that might require endoB2 via heterodimerization. In this study, we present evidence for a specific function of endoB2 in mammalian cells that has never been described before and that involves interaction with plectin 1 (plec1), a plakin superfamily member controlling cytoskeleton architecture (47).
EXPERIMENTAL PROCEDURES
Cloning and Mutagenesis
For mammalian expression, the cDNA encoding for human endoB2 (short isoform or isoform1, Swiss-Prot accession Q9NR46-1) was cloned into pCDNA3.1/myc-His using BamHI/EcoRI, and the one encoding for N-BAR/endoB2 (encompassing amino-acids 1–280) was cloned into pEXPR-IBA3 using XbaI/BamHI. The plasmid encoding for vim-GFP was a kind gift from S. Etienne-Manneville (Institut Pasteur, France).
For bacterial expression, the cDNA encoding for endoB2 was cloned into pAsk-IBA3 using SacII/BamHI. Part of the cDNA sequence encoding for the N-terminal region of human plectin 1 (plectin isoform 1, Swiss-Prot accession Q15149-1), starting from glycine 121 and ending at threonine 174, was reconstituted by fill-in and PCR amplification using six overlapping sense and antisense primers with 5-′ and 3-′ primers containing BamHI and EcoRI restriction sites, respectively. The 3-′ primer also contained a stop codon. The resulting cDNA was subsequently cloned into pGEX-6P-1. For the mutant cDNA in which proline 153 was mutagenized into alanine, the same strategy as for the wild type sequence was used except that the relevant primer contained an alternative codon encoding for alanine. Recombinant proteins were designed by fusing to GST either a fragment of the wild type N-terminal sequence (residues 121–174) or a mutated form with the 150PETA153 sequence, giving rise to GST-plec1PETP and GST-plec1PETA, respectively (Fig. 3).
FIGURE 3.

Plec1 is a molecular partner of endoB2 SH3 domain. A, HeLa/endoB2-St, a stable cell line aimed at isolating in situ pre-formed complexes with endoB2 binding partners. Top two panels, Western blot (WB) analysis of lysates (L), supernatants (S) of precipitations, or eluates (E) from either immunoprecipitations (IP) on protein A-Sepharose (upper panel) or StrepTactin/Sepharose precipitations (lower panel) from HeLa or HeLa/endoB2-St cell lines as indicated (note that no background from beads was detected, neither for the protein A-Sepharose immunoprecipitations nor for the StrepTactin-Sepharose precipitations). Western blots were treated with anti-Bif-1 antibody recognizing both endoB2 and endoB1. Calibration curves using pure recombinant bacterial endoB1 and endoB2 showed that this antibody recognizes endoB1 with ∼10 times more affinity/avidity than endoB2. Detection of endoB2 in the lysates from either HeLa or HeLa/endoB2-St cells is stochastic, and in most experiments, immunoreactivity is undetectable unless HeLa/endoB2-St cells are treated with 5 mm sodium butyrate (compare in lower panel, −NaBut and +NaBut). From the immunoprecipitations (upper panel), it was estimated that the expression level of endoB2-St in HeLa/endoB2-St cells compares with endoB2 level in HeLa cells. From the StrepTactin-Sepharose precipitations (lower panel) and taking into account the 10-fold difference in anti-Bif-1 immunoreactivities mentioned above, it was calculated that a maximum of 40 or 2% of endoB2 is in a complex with endoB1 in the absence (− NaBut) or the presence (+ NaBut) of sodium butyrate, respectively. Bottom panel, immunodetection of endoB2-St in HeLa/endoB2-St cells treated overnight with 5 mm sodium butyrate and using an anti-StrepTag antibody. Arrowheads point at discrete immunoreactivity around nuclei (see also G). Note in addition the homogeneous staining that appears mainly cytosolic. Scale bar, 20 μm. B, alignment of mouse and human plec1 sequences (human amino acid residues 145–174) showing the putative PETP consensus motif for SH3 domain binding. The mutation (at position 153) is indicated to illustrate the difference between GST-plec1PETP and GST-plec1PETA. C, in vitro binding assay of endophilin B species to immobilized GST, GST-plec1PETP, and GST-plec1PETA (see under “Experimental Procedures”). Three molar ratios of GST chimera to endophilin were tested as indicated. The immunoblots of unbound fractions obtained from separate acrylamide gels are shown, with endophilin revealed by anti Bif-antibody. GST/endophilin 0 refers to control sample performed in the absence of GST or chimera. D, depletion curves of the unbound fraction as determined from the experiment illustrated in C. Chemiluminescence signals are plotted as a percentage of the value determined for GST/endophilin 0 unbound fraction (equivalent to the input material). E, immunoblot analysis of material bound on beads and corresponding to samples analyzed in C, with GST/endophilin molar ratio of 103. F, specific coimmunoprecipitation of plectin and endoB2 from HeLa cells. Cells transfected with the indicated two endoB2 species (or mock-transfected) were processed as described under “Experimental Procedures.” Immunoblots of fractions P2 and S2 are shown (similar results were obtained in two independent experiments). In the upper panels, the arrow points at the faint band corresponding to endogenous endoB1 visible in fractions S2 of the control and comigrating with an endoB2 degradation product visible in the two endoB2 lanes, full-length (*) and truncated (**) endoB2 species. In lower panels showing material with molecular mass above 130 kilodaltons, intact plectin (°) and two of its major proteolytic degradation products (<) are distinguished. G, z-section obtained from HeLa/endoB2-St cells treated overnight with 5 mm sodium butyrate and processed for double immunofluorescence analysis using anti-StrepTag and anti-plectin antibodies. Arrowheads point to discrete colocalization of endoB2 and plectin around nuclei. Scale bar, 20 μm.
The sequence KARLKK (residues 173–178) in endoB2 was modified by site-directed mutagenesis (QuikChange II system, Stratagene, Agilent Technologies) into two distinct sequences as follows: AAALAA, using the primers 5′-GGATGCCTGCGCAGCGGCGCTGGCGGCGGCCAAGGCTGCAGAAGCC-3′ (forward) and 5′-GGCTTCTGCAGCCTTGGCCGCCGCCAGCGCCGCTGCGCAGGCATCC-3′ (reverse); and KARLAA, using the primers 5′-GGATGCCTGCAAAGCGAGGCTGGCGGCGGCCAAGGCTGCAGAAGCC-3′ (forward) and 5′-GGCTTCTGCAGCCTTGGCCGCCGCCAGCCTCGCTTTGCAGGCATCC-3′ (reverse), respectively. Both mutations were generated in parallel in the bacterial and mammalian recombinant expression vectors pASK3-endoB2-St and pcDNA3-endoB2-myc described earlier, respectively.
Recombinant Protein Expression and Purification
For protein purification, all proteins were expressed in transformed Escherichia coli BL21pLys (Novagen). Synthesis of endoB2-St, endoB1-St, their BAR-truncated species, and mutagenized forms (pASK-IBA3C vector) or GST, GST-plec1PETP, or GST-plec1PETA (pGEX-6P vector) was induced with 0.2 μg/ml anhydrotetracycline or 1 mm isopropyl β-thiogalactoside, respectively, for 3 h at 37 °C. Purification under native conditions at 4 °C was performed from cleared lysates by affinity chromatography on StrepTactin-Sepharose or glutathione-Sepharose according to the manufacturer's instructions (IBA, GmbH, Germany and Amersham Biosciences, respectively), yielding >95% pure proteins at 0.75–1.2 mg/ml. Unless freshly prepared material was required, proteins could be stored at −80 °C without loss in solubility or membrane binding ability. Protein concentration was determined using the Bio-Rad DC protein assay.
Cell Culture, Plasmid, and siRNA Transfections, Establishment of HeLa/endoB2-St Stable Cell Line
HeLa cells were grown in DMEM (PAA) supplemented with 10% fetal calf serum, 2 mm l-glutamine, and penicillin/streptomycin (PAA) at 37 °C in 5% CO2. For transient expression, cells grown at 60% confluency in 6-well plates (Nunc) were transfected with Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions, using 1 μg/ml plasmid DNA for 2 μl of reagent. Cells were used between 16 and 24 h post-transfection, except for vimentin-GFP short time expression (10 h), as mentioned in the text and the legends of figures.
For the establishment of the HeLa/endoB2-St and HeLa/vimentin-GFP stable cell lines, cells were transfected as for transient transfections and grown for 48 h in culture medium without G418. After trypsinization and appropriate dilutions, cells were grown for 3 weeks in culture medium supplemented with 0.8 mg/ml G418, and single colonies were isolated. Screening for endoB2-St-expressing clones was performed by immunofluorescence, using anti-StrepTag antibodies. Importantly, screening did not allow identification of positive clones unless cells were incubated overnight in the presence of 5 mm sodium butyrate. Hence, addition of sodium butyrate was always necessary to detect endoB2-St by immunofluorescence (see also legend of Fig. 3A for detailed analysis of endoB2 expression by HeLa/endoB2-St cells). Routine culture of HeLa/endoB2-St cells was performed in HeLa culture medium supplemented with 0.4 mg/ml G418.
siRNAs used were obtained from Dharmacon. siRNAs targeting endoB2 mRNAs that were named si endoB2-1 and si endoB2-2 were ON-TARGET plus siRNAs (human SH3GLB2 J-015810-05 and J-015810-06, respectively). Both worked with approximately the same efficiency (see Fig. 9). siRNAs targeting plec1 mRNAs that were named si plec1-1 and si plec1-2 were designed using the Dharmacon custom siRNA design software with the 5-′-specific sequence of plec1 (NCBI reference sequence NM_201380.2). Their sense sequences are GGAAGGAGCUUGAGGAGGUUU and GCGUGAUGGUGGCCAAGAAUU, respectively (the first one with ON-TARGET plus modifications). Control siRNA was ON-TARGET plus nontargeting siRNA-2 (reference D-001810-02). All siRNAs were resuspended according to manufacturer's instructions, in RNase-free water to reach 10 μm stock solution, aliquoted, and kept at −80 °C. For treatments with plec1 and endoB2 siRNAs, cells in 6-well plates were transfected with 100 nm final siRNAs using 2 μl of Lipofectamine 2000. For both plec1 and endoB2 siRNAs, 72-h treatments were applied because they allowed a satisfactory decrease of protein expression and observation of significant phenotypes in comparison with control siRNAs (with 48 h exhibiting virtually no reduction in protein expression for plectin). After 48-h siRNA treatments, cells were trypsinized and re-plated either in new 6-well plates (for cell lysis and subsequent biochemical analysis) or on glass coverslips (for immunofluorescence) and grown until reaching 72 h post-transfection. When endoB2 transient expression was coupled to plec1 siRNAs interference (see Fig. 7), cells re-plated after 48-h siRNA treatments were transfected and grown for 24 h.
FIGURE 9.

Decrease of endoB2 expression by siRNA interference and incidence on the vimentin network. HeLa/endoB2-St cells were transfected with control or endoB2 siRNAs (A, si endoB2-1 and si endoB2-2; B and C, si endoB2-2) for 72 h. A, Western blot analysis of endoB2 in cell lysates (top panel) or isolated after StrepTactin-Sepharose affinity chromatography (SA/seph eluate, bottom panel). In all experiments, endoB2 siRNAs allowed decreasing endoB2 expression by 75% at least in comparison with control. B, top and bottom panels correspond to two different fields. Images result from z-projections of five z-sections, each separated by a distance of 0.260 μm and obtained from cells processed for immunofluorescence using anti-vimentin antibodies. Note the mild bundling of vimentin filaments at the surface of nuclei accompanied by cytoplasmic extensions (arrowheads). Similar results were obtained with endoB2-1 siRNAs. Scale bar, 20 μm. C, top panel is a magnification of the region delimited in the top right image in B and shows the nuclear deformation imposed by the bundling of vimentin filaments. Increase in the indentation of nuclei was scored by the solidity index that measures the ratio between the surface of deformed nuclei divided by the theoretical surface whose contour is defined by the tips of indentations (see schematics). Right panel: n = 160 cells for si control and n = 153 cells for si endoB2-2.
FIGURE 7.

Decrease of plec1 expression by siRNA interference impairs the tight association of endoB2 filamentous structures with nuclei. HeLa cells were treated with control (si control) or plec1 siRNAs (si plec1-1 and si plec1-2) for 72 h. A, Western blot analysis of plectin and tubulin expression from cell lysates (equal protein amounts loaded) migrated in duplicates. Intact plectin (°) and its two major proteolytic degradation products (<) are indicated as in Fig. 3. Right panel shows quantifications of immunoblots for plectin obtained from three independent experiments (each color corresponding to a given experiment). B, quantification of cells that contain endoB2 filaments (values are means ± S.D. calculated from three independent experiments with n = 257 (si control), 213 (si plec1-1), and 226 (si plec1-2) cells analyzed). C, z-sections obtained from cells treated with siRNAs, transfected after 48 h to express endoB2 fused to a C-terminal Myc tag and processed for Myc and plectin immunostaining. Images were taken with the same optical settings. Note, for both plec1 siRNAs and in comparison with control, the modification of the localization of endoB2 filamentous structures that do not remain exclusively perinuclear and expand in the cytoplasm is shown. Scale bar, 16 μm.
Quantitative analysis of the two plec1 siRNAs efficiencies by Western blotting unraveled that knockdown of plectin expression was more or less efficient depending on cell lines; for HeLa/endoB2-St cells, plectin was expressed at levels corresponding to 73.5 ± 0.5 and 65.0 ± 11% of the control value (means of two independent determinations) for plec1-1 and plec1-2 siRNAs respectively; for HeLa/vim-GFP cells, plectin was expressed at levels corresponding to 50.0 ± 1.0 and 35.0 ± 8.5% of the control value (means of two independent determinations) for plec1-1 and plec1-2 siRNAs, respectively (which is in the range of what was measured for regular HeLa cells, see Fig. 7A).
Finally, it should be noticed that plec1-1 and plec1-2 siRNAs treatments, after 72 h, triggered diminished growth rate of cells (20–30% decrease of cell number in comparison with control siRNA due to cell death linked to aberrant mitosis; this has been reported for another plectin isoform (48)).
In the particular case of the short time window of vim-GFP expression, cells were transfected to express vim-GFP early on the next day of re-plating to allow 10-h transient expression. This short timing was determined after a kinetic of 4, 6, 8, 10, and 14 h of expression in which very few cells started to express vim-GFP in detectable amounts at 4 and 8–10 h being the most appropriate in terms of percentage of positive cells and level of expression (thus allowing a satisfactory dynamics between lower and highest signals, in a given cell).
Quantitative Analysis of plec1 siRNA Phenotypes
For the scoring of cells exhibiting bundling of vimentin filaments (Fig. 8A, top right panel), images were taken at the ×20 objective (see examples in supplemental Fig. S3). Significant bundling was considered when the length of a single vimentin bundle in a given cell exceeded the longest nucleus axis (nuclei were stained with DAPI to visualize their surface), typically ranging from 15 to 80 μm.
FIGURE 8.

Decrease of plec1 expression by siRNA interference and incidence on the vimentin network. HeLa/endoB2-St cells were transfected with control or plec1-1 siRNAs for 72 h, as indicated. A, top left panel, z-sections obtained from cells processed for plectin immunostaining to attest for the presence of cells with significant decrease in plec1 expression (top left (si plec1-1), note the presence of cells that express plectin at a level approaching the detection limit (*)). Top right panel, quantification of cells treated with si plec1-1 that express plectin at the detection limit and exhibit bundling of vimentin filaments (n = 646 and 780 cells analyzed, respectively) and of cells treated with si control that exhibit bundling of vimentin filaments (n = 430 cells analyzed). Values are means ± S.D. calculated from three independent experiments. Bottom panel, images are from z-projections of five z-sections, each separated by a distance of 0.260 μm and obtained from cells processed for immunofluorescence using anti-vimentin antibodies. Note the perinuclear bundling of IF and the decrease in the density of vimentin filaments encaging nuclei clearly visible in cells treated with control siRNAs. Similar results were obtained with si plec1-2 siRNAs. Scale bars, 20 μm. B, z-sections obtained from HeLa cells stably expressing vim-GFP, treated with control or plec1-1 siRNAs as indicated, and processed for immunofluorescence using anti-plectin antibodies. Images were taken with the same optical settings. Note that cells treated with plec1-1 siRNAs and expressing plectin at low level are characterized by the bundling of vimentin filaments in the close vicinity of nuclei and across the cytoplasm. Results were similar with si plec1-2 siRNAs. Scale bar, 20 μm.
Cell Lysis and Preparation of Protein Samples from Cultured Cells
For mass spectrometry analysis, a nonstringent and rapid protocol was used to ensure minimal loss of interacting proteins and reduce contamination. All steps were performed at 4 °C. HeLa/endoB2-St or HeLa/EB1-St stable cells were grown in two 10-cm dishes and treated with 5 mm sodium butyrate for 18–20 h, starting 1 day before reaching confluence. They were scraped in 1 ml of ice-cold culture medium and centrifuged at 800 × g for 10 min at 4 °C. The pellet was resuspended up to 0.8 ml in lysis buffer (1% (w/v) CHAPS, 50 mm Tris-Cl, pH 7.4, 100 mm NaCl, supplemented with 1 mm MgCl2, 20 μg/ml DNase, and the complete EDTA-free mixture of protease inhibitors (Roche Applied Science)), incubated for 30 min on ice, and then supplemented with 2 mm EDTA. CHAPS was used following our observation that vimentin is more efficiently extracted than with Triton X-100. We anticipated that these conditions would favor capturing complexes containing endoB2 and vimentin as suggested by colocalization of both proteins. Detergent-insoluble material was removed by centrifugation, twice for 10 min each at ∼22,000 × g at 4 °C. The clarified lysate (∼0.75 ml, 3.8 mg of protein) was chromatographed three times onto 55–60 μl of StrepTactin/Sepharose beads (IBA GmbH, Germany) in a 0.8-ml Mobicol column (MoBiTec GmbH) over 40–45 min. Beads were washed twice with 0.6 ml of lysis buffer and TBS (50 mm Tris-Cl, pH 7.4, 100 mm NaCl). They were then washed with 25 mm NH4HCO3 and drained for subsequent direct trypsin digestion. In some cases, material eluted with 10 mm desthiobiotin in 25 mm NH4HCO3 was directly trypsin-digested.
For coimmunoprecipitation experiments, transfected HeLa cells from 80 to 85% confluent 35-mm dishes were used. They were washed three times with ice-cold PBS and solubilized in the dish for 25–30 min on ice in 0.3 ml of HN buffer (25 mm Hepes-NaOH, pH 7.4, 100 mm NaCl) containing 0.5% (w/v) CHAPS and supplemented as above with MgCl2, DNase, and protease inhibitors. After addition of 2 mm EDTA, the scraped material was centrifuged for 8 min at 4000 × g at 4 °C. The supernatant was mixed with 80 μl of a 1:1 slurry of protein A-Sepharose equilibrated in the same buffer and incubated for preclearing for 30 min at 4 °C with end-over-end agitation in 500-μl tubes. Beads were removed by centrifuging 4 min at 300 × g (pellet P1), and the supernatant (S1, ∼0.3 ml) was incubated with anti-plectin rabbit monoclonal antibody (3 μl) (Epitomics) at 4 °C. After 20 min, 150 μl of a 1:1 slurry of protein A-Sepharose was added, and binding of IgGs was allowed for a further 80 min of incubation. The samples were then centrifuged for 4 min at 300 × g, and beads were washed three times with 400 μl of HN. Beads (P2) and supernatant (S2) were boiled in reducing Laemmli sample buffer (75 and 240 μl final volume, respectively) and submitted to SDS-PAGE and immunoblotting.
For routine lysate preparation, cells were grown on 6-well plates and rinsed once with ice-cold PBS, and cell lysis was performed in 300 μl of solubilization buffer (SB: 1% (w/v) Triton X-100, 100 mm NaCl, 2 mm EDTA, 50 mm Tris-HCl, pH 7.4) complemented with protease inhibitor mixture (Roche Applied Science). After 10 min, cells were scraped from the dish, and the lysates were incubated for 15 min before centrifugation for 10 min at 900 × g. When StrepTactin-Sepharose affinity isolation was performed, 50 μl of packed beads (IBA GmbH, Germany) prewashed with SB were added to 250 μl of supernatants and incubated for 1 h on a rocking platform. 50 μl of the lysates were kept for determination of signals in the input (referred to as lysates (L) in figures). Samples were centrifuged, and supernatants were kept (referred to as supernatants (S) in figures). Beads were washed three times with 500 μl of SB and resuspended to reach the final volume of ∼35 μl. Samples were boiled for 5 min in Laemmli sample buffer, and the solubilized material (referred to as eluates (E) in figures) was subsequently submitted to SDS-PAGE and immunoblotting. In all cases, protein concentration of the clarified extracts was determined using the Bio-Rad DC protein assay.
Mass Spectrometry Analysis
Proteins on StrepTactin-Sepharose beads were digested overnight at 37 °C by sequencing grade trypsin (12.5 μg/ml; Promega, Madison, WI) in 20 μl of 25 mm NH4HCO3. Peptides mixtures were analyzed by an LTQ Velos Orbitrap coupled to an Easy nano-LC Proxeon system (Thermo Fisher Scientific, San Jose, CA). Chromatographic separation of peptides was performed with the following parameters: column Easy Column Proxeon C18 (10 cm, 75 μm inner diameter., 120 Å), 300 nl/min flow, gradient rising from 95% solvent A (water, 0,1% formic acid) to 25% B (100% acetonitrile, 0,1% formic acid) in 20 min, then to 45% B in 40 min and finally to 80% B in 10 min. Peptides were analyzed in the Orbitrap in full ion scan mode with a mass resolution of 30,000 (at m/z 400) over the range m/z 400–1800. Fragments were obtained with a collision-induced dissociation activation with a collisional energy of 40%, an activation Q of 0.250 for 10 ms, and analyzed in the LTQ. MS/MS data were acquired in a data-dependent mode in which 20 most intense precursor ions were isolated. The maximum allowed ion accumulation times of 100 ms for MS scans and 50 ms for MS/MS scans were set. Raw files obtained were analyzed using Proteome Discoverer 1.2 software (Thermo Fisher Scientific, San Jose, CA) coupled to an in-house Mascot search server (version 2.1.2, Matrix Science, Boston). The mass tolerance of fragment ions was set to 10 ppm for precursor ions and 0.6 Da for fragments. Oxidation (methionine) and phosphorylations (serine, threonine, and tyrosine) were chosen as variable modifications. The maximum number of missed cleavages was limited to one for trypsin digestion. Raw files were searched against SwissProt database with the Homo sapiens taxonomy. A reversed database approach was used for the false discovery rate estimation. The maximum false discovery rate was set to 5%.
In Vitro Binding Assays
Tagged endophilin species and GST chimeras in PBS were used immediately after purification. Glutathione-Sepharose 4B beads loaded with recombinant GST, GST-plec1PETP, or GST-plec1PETA, respectively, were prepared upon production using BL21pLysS bacteria. A washed Sepharose 4B matrix with a protein content averaging 1.1 nmol/μl beads was used as a serial dilution in a final volume of 60 μl (3.75 μl up to 22.5 μl of beads). A control was designed using 22.5 μl of unloaded glutathione-Sepharose 4B beads. Samples were incubated in binding buffer A (1 mm EDTA, 2.5% (w/v) glycerol in PBS) for 2 h at 4 °C with 0.4 μm tagged endophilin, as indicated in the figure legends, with gentle agitation. Beads were washed (300 × g, 2 min) with 450 μl each of buffer A (once) and PBS (twice). Unbound and bound fractions, corresponding to last supernatant and bead pellet, were analyzed by SDS-PAGE followed by Western blotting.
For membrane binding assay, a liposome suspension (1.25 mm lipids) was prepared in HNE buffer (25 mm Hepes, pH 7.4, 0.1 m NaCl, 0.5 mm EDTA) and extruded three times through 0.4-μm (pore size) polycarbonate filters (Avanti Polar Lipids) as described previously (49). Binding was carried out in centrifuge polycarbonate tubes by mixing liposomes, at concentrations indicated in the figure legend (up to 1.11 μm final concentration), with wild type or mutant endoB2 (0.5 μm final concentration) in 125 μl of HNE buffer and incubating for 15 min at 24–25 °C. The reaction mixtures were then centrifuged for 15 min at 250,000 × g at 18 °C with the Beckman Coulter TLA 100 rotor. The supernatant was carefully removed, and the pellet was resuspended in HNE buffer. The two fractions (unbound and bound fractions, respectively) were diluted with Laemmli sample buffer and analyzed by SDS-PAGE and Western blotting.
Immunoblotting
Immunoblotting was performed exactly as described previously (49). Peroxidase activity using either conjugated secondary antibodies or conjugated StrepTactin was detected by enhanced chemiluminescence using the ECLPlus detection kit (GE Healthcare). Images were obtained by direct capture of chemiluminescence using the Intelligent Dark Box LAS-3000 (Fuji Sciences) imaging system, and signals were quantified using either MultiGauge (Fuji Sciences) or ImageJ software.
Immunofluorescence and Microscopy
For immunostaining, HeLa and HeLa/endoB2-St cells were grown on glass coverslips, rinsed with PBS, and fixed with either 3% paraformaldehyde for 20 min or on ice with ice-cold methanol for 5 min (the latter was used for anti-tubulin and anti-plectin immunostainings). After fixation, all subsequent steps were performed at room temperature. Coverslips were rinsed with PBS, and when fixed with paraformaldehyde, material was quenched for 20 min using 50 mm NH4Cl in PBS. After washing with PBS, cells fixed with paraformaldehyde were permeabilized for 10 min in PBS containing protease inhibitor mixture (Roche Applied Science) and 0.3% (w/v) Triton X-100. Further steps were performed using PBS, 0.2% (w/v) gelatin. Cells were rinsed three times for 30 min and incubated with primary antibodies for 60 min at room temperature. After washing, cells were incubated for 45 min with secondary antibodies. After washing, the coverslips were incubated for 10 min in PBS containing DAPI (0.5 μg/ml), rinsed in PBS, and rapidly soaked into distilled water before mounting in Dako medium (Dako). Coverslips were then kept at 4 °C until observation.
Fluorescence images were acquired under oil immersion ×63/1.4 or ×100/1.4 objective lenses with ApoTome system composed of an inverted microscope Zeiss Axiovert 200, a camera Axiocam MRm (pixel size 6.45 × 6.45 μm), the Zeiss ApoTome system, and acquisition software Axiovision 4.5. Images were treated with ImageJ and Photoshop software.
Unless specified in figure legends (see Figs. 8 and 9), images shown were obtained from a single z-section, and all consecutive z-sections were distant from 0.260 μm.
Nucleus Positioning Analysis
The nucleus position of siRNA-transfected HeLa cells was determined after normalization of cell morphology on adhesive micropatterns arrays. To this end, cells were plated 3 days after transfection at 25,000 cells/cm2 in 22-mm wells onto CYTOO ChipsTM Mini FN650 (Cytoo S.A., Grenoble, France) supporting H-shaped, fibronectin-coated patterns. They were then allowed to attach and spread for 5 h at 37 °C under a 5% CO2 atmosphere. After washing, cells were fixed with ice-cold methanol and processed as described above for immunofluorescent staining of tubulin and DAPI labeling. On the basis of preliminary experiments, small (30 μm) and medium (38 μm) size patterns were chosen as they allowed efficient cell attachment and morphological homogeneity, as ascertained by distribution of tubulin over printed fibronectin. With both pattern sizes, the nucleus centroid (as determined by Fiji Software) was localized on the long axis of the pattern. This allowed an accurate determination of the respective distances d1 and d2 (with d1 < d2) between the nucleus centroid and the two poles of the pattern (Fig. 11). Nucleus position was determined by the ratio d1/d2, with any deviation from 1 indicating an asymmetric positioning (off-centering).
FIGURE 11.
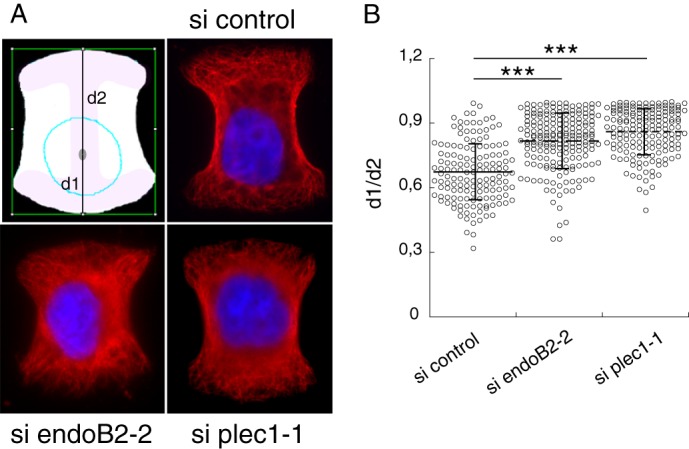
EndoB2 and plec1 control nuclear positioning. HeLa cells were transfected with control, plec1-1, or endoB2-2 siRNAs for 72 h, plated on micropatterns, and processed for tubulin immunostaining and DAPI staining. A, z-sections of cells from indicated siRNA treatments, immobilized on medium patterns, and showing the respective positions of nuclei. The schematic drawing shows distances d1 and d2 measured along the y axis of the pattern for scoring nuclei positioning (see “Experimental Procedures”). B, quantification and statistical analysis of nuclear positioning. n = 146, 163, and 192 cells analyzed for control, plec1-1, and endoB2-2 siRNAs, respectively, and pooled from two independent experiments. Horizontal lines and vertical bars indicate means ± S.D. p values are from Student's t test. *** indicates p values < 0.0001.
Electron Microscopy
For classical morphological analysis, cells were fixed in 2% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, for 1 h, postfixed for 1 h with 2% buffered osmium tetroxide, dehydrated in a graded series of ethanol solution, and then embedded in epoxy resin.
For immunolabeling, cells were prepared following Tokuyasu, and immunogold labeling was carried out as described previously (50). The following primary antibodies were used: anti-vimentin (GeneTex) and anti-Myc (9E10, Chemicon International). Single or double immunogold labeling on ultrathin cryosections was performed using protein A-gold conjugates (Utrecht, Netherlands). Images were acquired with a digital camera Keen View (SIS) mounted on a Tecnai 12 transmission electron microscope (FEI Co.) operated at 80 kV.
Antibodies
For immunoblots and immunofluorescence, the primary antibodies used were as follows: mouse monoclonal anti-Myc (Cell Signaling); mouse monoclonal anti-StrepTag II (IBA); rabbit polyclonal anti-vimentin (GeneTex); rabbit monoclonal anti-plectin (Epitomics); mouse monoclonal anti-Bif1 (Imgenex); mouse monoclonal anti-α-tubulin (Sigma); and mouse monoclonal anti-emerin (Novocastra).
Secondary antibodies that were used are as follows: HRP-conjugated AffiniPure goat anti-rabbit (Jackson ImmunoResearch); goat anti-rabbit IgG coupled to Alexa Fluor 568; chicken anti-mouse IgG coupled to Alexa Fluor 488; and donkey anti-mouse coupled to Alexa Fluor 594 (Molecular Probes). Proteins fused to the StrepTag epitope were detected using HRP-conjugated StrepTactin (IBA).
RESULTS
Overexpression of Endophilin B2 Triggers Extensive and Specific Reorganization of the Vimentin Network
To address the cellular functions of endoB2, we first studied its localization in cells. Upon transient expression in HeLa cells, endoB2 expressed at low detection levels was found to be mainly cytosolic, with discrete enhanced staining in the perinuclear region and in close contact with the plasma membrane (Fig. 1A). At higher expression levels, the majority of cells was characterized by immunoreactivity localized on filamentous structures surrounding the nucleus (Fig. 1, A and D). Analysis of Epon-embedded cells revealed, in many cells, the accumulation of a meshwork of more or less densely packed filamentous structures often contacting the outer nuclear membrane (Fig. 1B and see also supplemental Fig. S1 for comparison of a control nontransfected cell with a pCDNA3.1/endoB2-transfected cell exhibiting perinuclear accumulation of filamentous structures at low magnification). Bunches of filaments also appeared contacting nucleopores (Fig. 1B, panel e). In addition, filamentous assemblies exerted mechanical tension on nuclear membranes that appeared compressed laterally with deep invaginations (Fig. 1B, panel f, and see also the immunofluorescence D). Interestingly, as could be estimated when they do not strongly intermingle (Fig. 1B, panel b), the filaments looked heterogeneous in size and contrast, with diameters ranging from ∼5 to 10 nm for the smallest to 20 to 25 nm for the widest (that also appeared more dense to the electrons). The size and shape of these filaments indicated that, at least in part, they might belong to the class of intermediate filaments. Immunoelectron microscopy revealed immunoreactivities of both vimentin and endoB2 on the same bunches of filaments, confirming that these are partly made of intermediate filament proteins (Fig. 1C). Also, endoB2 immunoreactivity was detected at the rim of nuclei, suggesting that the protein is recruited at the outer nuclear membrane (Fig. 1C). Deduced from their apparent diameter and structure, the filaments may represent single and bundled units with the thinnest most probably made of vimentin (compatible with the size of protofibrils (51, 52)) and the widest of either endoB2 or of both endoB2 and vimentin, after bundling.
FIGURE 1.

Overexpression of endoB2 triggers the accumulation of filamentous structures around nuclei and is partly composed of intermediate filaments. HeLa cells were transfected to express endoB2 fused to a C-terminal Myc tag. A, anti-Myc immunofluorescence of z-sections with low expression level of endoB2 (left panel) showing cytosolic and discrete perinuclear as well as plasma membrane localization (arrowheads). At high expression level (right), endoB2 accumulates mainly as filamentous structures surrounding nuclei. Note that the two images were taken with different settings of the camera. z-sectioning through nuclei attested that cells are not binucleated. Scale bar, 20 μm. B, electron microscopy of Epon-embedded cells. Panels b and d correspond to magnifications of the areas delimited in (panels a and c) and show the presence of filaments of 5–10 nm diameter (arrowheads in panel b) and of 20–25 nm diameter (arrow in panel b). Panels e and f, note the proximity of bunches of filaments to nuclear membranes and nucleopore complexes (NPC (arrowheads in panel e)). ER, endoplasmic reticulum; Nuc, nucleus. Scale bars, panels a and c–f, 1 μm; panel b, 0.5 μm. C, immunoelectron microscopy using anti-Myc or anti-vimentin antibodies. Magnified areas at top right and bottom panels show the very close proximity of anti-Myc immunoreactivity to the nucleus. Nuc, nucleus; 5 nm gold, anti-vimentin; 15 nm gold, anti-Myc. Scale bar, 0.5 μm. D, anti-Myc and anti-emerin (a protein of the inner nuclear membrane) immunofluorescence of two z-sections at planes between the nucleus and the membrane contacting the coverslip (top images) or crossing the top of the nucleus (bottom images). Insets show invagination of the nuclear membrane in close contact with endoB2-myc staining. Scale bar, 20 μm.
Immunofluorescence microscopy showed that in cells expressing endoB2 transiently, vimentin was significantly re-organized and colocalized extensively with endoB2 in the perinuclear region (Fig. 2). Neither actin nor tubulin was found enriched around nuclei where endoB2 filaments accumulate, indicating that these cytoskeletal elements are not significantly re-organized in this region. Altogether, these data suggested the existence of physical and functional links between endoB2 and vimentin.
FIGURE 2.
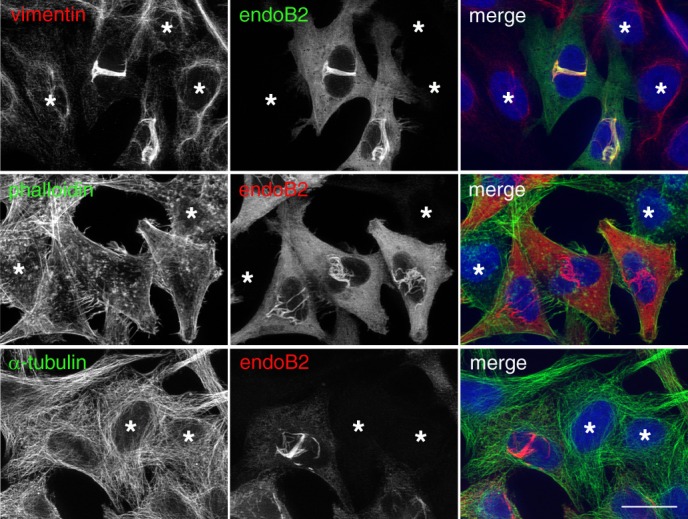
EndoB2 triggers the selective re-organization of the vimentin cytoskeleton. HeLa cells were transfected to express endoB2 fused to a C-terminal Myc tag and processed for double immunofluorescence analysis using anti-Myc and either anti-vimentin (top panels) and anti-tubulin (bottom panels) antibodies or phalloidin-Alexa Fluor 488 (middle panels). Compare cells that express endoB2 with the ones that do not (asterisks). Scale bar, 20 μm.
Plectin 1 Interacts with the SH3 Domain of Endophilin B2
We performed a proteomic analysis aimed at finding specific partners of endoB2 SH3 domain. To this end, a HeLa/endoB2-St cell line expressing endoB2 fused to a StrepTag was obtained. HeLa/endoB2-St cells express endoB2-St at a level that is barely detectable, both upon Western blot analysis in cell lysates and immunofluorescence staining. Importantly, signal intensity of immunoprecipitated proteins revealed that in this cell line endoB2-St is expressed at a level that is comparable with the endogenous protein (Fig. 3A). To enhance endoB2-St cellular content, cells were treated with sodium butyrate, which provided the advantage of increasing sensitivity of detection of endoB2-St in cell lysates (Fig. 3A, bottom, Western immunoblot panel) and immunodetection in cells (Fig. 3A, bottom panel). Concentration of signals by StrepTactin precipitation allowed accurate determination of endoB2 increase of expression after sodium butyrate treatment as well as the proportion of endoB2 interacting with endoB1 via heterodimerization (see legend of Fig. 3 for details). These experimental conditions, in addition to increasing the sensitivity of detection of endoB2 in complex with its interacting partners, allowed lowering quite significantly the proportion of endoB2/endoB1 heterodimers, thus increasing the chances of identifying molecular interactants of homodimeric and/or monomeric endoB2.
Endophilin B2 affinity-purified proteins were analyzed by LC-ESI-MS/MS using sodium butyrate-treated HeLa/endoB2-St cells as described under “Experimental Procedures.” The pulldown with the endoB2-St bait was designed to recover cytoplasmic proteins exhibiting a high affinity constant or low dissociation kinetics, or both, after synthesis and in situ assembly of complexes. The supplemental Fig. S2A shows proteins which, in addition to endoB2, were detected with high numbers of matching peptides as well as sequence coverage ranging from 20 to more than 50%; these proteins are ataxin-2 like protein, endoB1, plectin 1, and vimentin.
The isolation of endoB1 can be accounted for by a minor endoB2 pool forming bona fide endoB1/endoB2 heterodimers (see Fig. 3A) (36). Ataxin-2-like protein is a member of the spinocerebellar ataxia protein family homologous with ataxin-2, a known partner of endophilins A1 and A3 (53). The present data suggest that homologous proteins of the spinocerebellar ataxia family exert functions via associations with either endophilins A or B, a topic beyond the scope of this work.
The discovery of vimentin among components of the endoB2-St affinity-purified complexes is consistent with the hypothesis of a functional link between endoB2 and vimentin (Figs. 1 and 2). This, along with the additional identification by mass spectrometry of a plectin variant among putative binding partners (supplemental Fig. S2A), prompted further investigation of this partnership.
The identification of plectin 1 (also known as plectin isoform 1) was established by two trypsin digestion products matching its exclusive alternative N-terminal sequence (residues 1–174) (supplemental Fig. S2B). Plectin-derived peptides were not detected when experiments were performed using a cell line expressing endoB1-St, although endoB2 peptides could be identified. This is in agreement with the ability of endoB1 and -B2 to form heterodimers. These data also indicate that plectin is part of a complex built up on endoB2 but not on endoB1, and would suggest that in both cell types plectin interacts with a pool of endoB2 homodimers.
The endoB1/endoB2-plectin association was further characterized using another in vitro approach. Plec1 harbors a PXXP consensus motif for the SH3 domain binding, present in its N-terminal sequence (150PETP153), shared with the mouse protein, and absent from other isoforms (Fig. 3B and supplemental Fig. S2B). Immobilized recombinant GST-plec1PETP and GST-plec1PETA (designed as described under “Experimental Procedures”) were incubated with purified endoB1, endoB2, and N-BAR/endoB2 (a truncated form of endoB2 lacking the SH3 domain) to characterize a possible differential binding of plectin-derived peptides to the SH3 domain of endophilins B (see “Experimental Procedures”) (Fig. 3, C–E). As shown by the immunoblots and curves of Fig. 3, C and D, N-BAR/endoB2 and endoB1 had no binding ability whatever the chimera tested. In contrast, full-length endoB2 could bind the wild type but not the mutated N-terminal sequence of plectin, as shown by its decrease in supernatants of binding assays (Fig. 3, C and D) and its recovery on beads (Fig. 3E). These results are consistent with significantly distinct sequences of endophilin B SH3 domains and confirm the direct interaction of plectin with the endoB2 SH3 domain, mediated by the consensus PETP motif.
The endoB2/plectin interaction also occurred upon transient expression of exogenous endophilin. A coimmunoprecipitation experiment using HeLa cells transfected with endoB2-St or N-BAR-St/endoB2 (Fig. 3F) showed that, in contrast with N-BAR-St/endoB2, endoB2-St could be recovered with plectin. This confirmed the specificity of the SH3 domain-mediated interaction and showed that exogenous endoB2 is incorporated into plectin-containing complexes. Altogether, these data demonstrate that endoB2 and plectin are molecular interactants in mammalian cells.
Finally, the cellular distribution of endoB2 and plectin was investigated. Using both HeLa/endoB2-St cells and HeLa cells expressing endoB2 transiently, we found that both proteins indeed colocalize partly, with small but significant overlap in the perinuclear region (Figs. 3G and 4, respectively).
FIGURE 4.
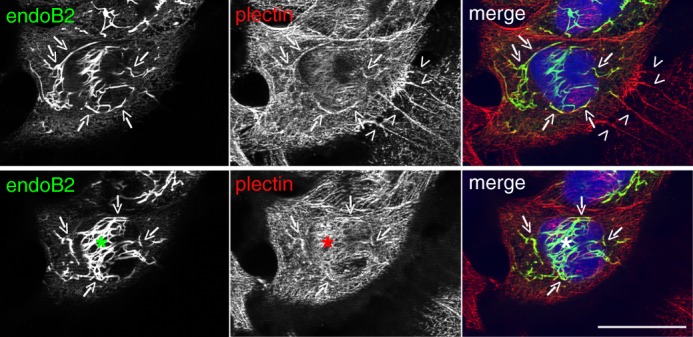
EndoB2 and plectin colocalize in the perinuclear region. HeLa cells were transfected to express endoB2 fused to a C-terminal Myc tag. Images are from two different z-sections obtained from cells processed for double immunofluorescence analysis using anti-Myc and anti-plectin antibodies. Plectin is localized all over the cell, with increased concentration at cell-to-cell contacts (top panels, arrowheads) and in filamentous structures surrounding the nucleus that partly colocalizes with endoB2 (arrows). The asterisks in the bottom panels are centered on a region above the nucleus that is immunoreactive for plectin and exhibits significant colocalization with endoB2. Scale bar, 30 μm.
The demonstration of a direct physical interaction between plec1 and endoB2, along with the pulldown of vimentin with endoB2-St, suggests the existence of a tripartite endoB2-plec1-vimentin complex, which we did not dissect further. The occupancy of the endoB2 SH3 domain-binding site by plectin implies that in this complex plectin 1 would bind the endoB2 SH3 domain via its N-terminal end and vimentin via its C-terminal end (54). Altogether with the drastic alteration of the vimentin network upon transient expression of endoB2, such evidence for an endoB2-plec1-vimentin complex strongly suggests that it is functionally involved in the spatial organization of intermediate filaments (IFs).
SH3 Domain of endoB2 Is Required for Colocalization with Vimentin
Because plectin interacts with endoB2 via the SH3 domain, deleting this domain should significantly reduce the colocalization of endoB2 and vimentin as well as the spatial reorganization of IFs. When N-BAR/endoB2 was transiently expressed, immunofluorescence analysis revealed that indeed the SH3 deletion mutant colocalized less extensively with vimentin than the full-length protein (Fig. 5A). This correlated to a lesser extent with vimentin reorganization, and N-BAR/endoB2 did not accumulate as filamentous structures encaging nuclei, albeit thick bundles in their close vicinity were observed (Fig. 5A). These were found to overlap only partly with vimentin. Instead, N-BAR/endoB2 filaments extended into the entire cytoplasm. The more distant from nuclei, the less these filaments were found colocalizing with vimentin. Electron microscopy performed on N-BAR/endoB2-expressing cells unraveled, in particular in regions in close proximity to the nucleus (Fig. 5B, left panel), the presence of small microfilaments with a diameter of ∼5–10 nm (as in the case of full-length endoB2 expression and that most probably correspond to vimentin protofibrils). Wider filaments were also visible, with a diameter around 10–15 nm (which is significantly smaller than the wider 20–25 nm and electron dense filaments observed upon expression of full-length endoB2). This meshwork of filaments appeared less electron dense in comparison with cells expressing full-length endoB2, which may be consistent with the decrease (or absence) of recruitment of proteinaceous material such as plectin large molecules. Interestingly, we also found large assemblies of filaments with a diameter around 10–15 nm that organize more or less linearly and as bundles as large as 1 μm (Fig. 5B, right panel). Consistently with immunofluorescence staining showing enrichment of endoB2 versus vimentin as distance from the nucleus increases, these filamentous structures are most probably largely, if not exclusively, composed of N-BAR/endoB2. These data suggest that, upon overexpression in cells, full-length endoB2 as well as its N-BAR domain have the propensity to organize as protein filaments. Altogether, these data show that the SH3 domain of endoB2 is required for assembling filamentous structures enriched in both endoB2 and vimentin around the nucleus.
FIGURE 5.
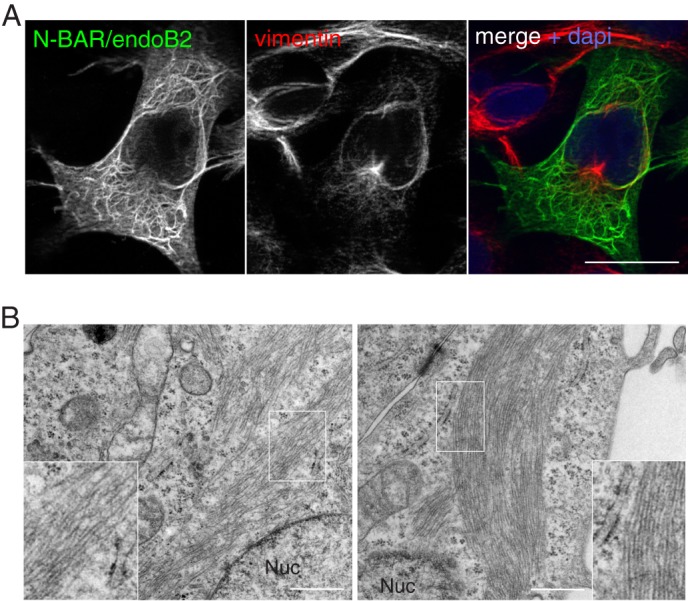
Colocalization with vimentin requires the SH3 domain of endoB2. HeLa cells were transfected to express N-BAR/endoB2 fused to a C-terminal StrepTag. A, z-section obtained from cells processed for double immunofluorescence analysis using anti-StrepTag and anti-vimentin antibodies. Scale bar, 20 μm. B, electron microscopy of Epon-embedded cells. Left, mixture of filaments of 5–10 and 10–15 nm in diameter in the perinuclear region (inset, magnified regions delimited by the white box, note the homogeneity in electron density). Right, large bunches of filaments homogeneous in diameter (10–15 nm) and distant from the nucleus (inset, magnified region delimited by the white box illustrating homogeneity in diameter and electron density). Nuc, nucleus. Scale bars, 0.5 μm.
BAR-mediated Membrane Binding Property of endoB2 Is Essential for Its Assembly into Perinuclear Filaments
The functions of BAR domain proteins rely on their membrane association via positively charged residues of the concave side and at the tips of the domain (7, 26). Hence, we addressed whether the propensity of endoB2 to assemble into filamentous structures around nuclei requires, at some point, the membrane binding activity of its BAR domain. Based on endophilin A structure (26, 49, 55), positively charged residues in the distal part of the second α-helix that should theoretically locate at the tips of the dimer were mutagenized to give rise to endoB2-KARLAA and endoB2-AAALAA as depicted in Fig. 6A. The consequences of the mutations, tested in transfected HeLa cells, were impressively drastic as the two mutants lost entirely the ability to trigger the formation of any accumulated filamentous structures and remained as a prominently diffuse cytosolic pool (Fig. 6B). In addition, expression of neither mutant triggered significant modification of vimentin organization (data not shown).
FIGURE 6.

Mutagenesis of critical residues in the BAR sequence involved in membrane binding abrogate the assembly of endoB2 into perinuclear filamentous structures. A, rod structure of endoB2 showing the domain organization and the two mutations used in this study of the 173–178 amino acid loop (KARLKK) between α-helices 2 and 3. Based on the crystal structure of human endophilin BAR domain (Protein Data Bank code 1X03 A), this sequence is located at both tips of the dimer. B, HeLa cells were transfected to express, as indicated, wild type endoB2 or the mutants fused to a C-terminal Myc tag. Images show z-sections obtained from cells processed for immunofluorescence analysis using anti-Myc antibodies. For both mutants, not a single cell on the entire coverslips showed the typical formation of perinuclear filamentous structures as in the case of wild type endoB2. Scale bar, 20 μm. C, membrane binding of wild type (wt) and doubly mutated (KARLAA) species was examined using phospholipid to endophilin molar ratio ranging from 0 (input) to 2222 as indicated. 11.1% of the supernatant (unbound) and 33% of the pellet (bound) (see “Experimental Procedures”) were immunoblotted using separate polyacrylamide gels and anti-Bif antibody. No precipitation of endophilins was observed in the absence of membranes. D, chemiluminescence signals obtained from the blot shown in C and representative of two experiments. Plotted is the bound (B) to free (F, unbound) ratio as a function of the phospholipid to endophilin ratio.
We then used endoB2-KARLAA and wild type endoB2 to compare their in vitro membrane binding properties. In contrast to wild type, the binding of endoB2-KARLAA to liposomes was severely diminished (Fig. 6, C and D). Hence, a loss of the endoB2 phenotype due to the mutagenesis of residues 177 and 178 can be directly correlated with a decrease in membrane binding activity. Altogether, these data strongly suggest that binding to a membrane is a prerequisite for the assembly of endoB2 into filamentous structures.
Interfering with plec1 Expression Impairs the Localization of endoB2 Filaments around the Nucleus
To address the need of plec1 in endoB2 perinuclear assembly, the cellular level of plec1 was lowered by siRNA treatment followed by transient expression of endoB2 using regular HeLa cells. Two siRNAs designed to target specifically the plec1 mRNA were obtained and allowed decreasing 40–70% of plectin detected by Western blotting after 72 h (see Fig. 7A for quantification of siRNAs efficiency). Importantly, immunofluorescence experiments unraveled a heterogeneous efficiency of plectin knockdown, with 26.15 ± 4.00% of cells (mean value ± S.D. obtained from three independent experiments) expressing plectin close to the detection limit. This strongly suggested that the targeted plectin isoform is the predominant isoform in our HeLa cells. After treatment with both plec1 siRNA and transfection to express endoB2 transiently, quantitative analysis showed that the percentage of cells containing endoB2 filaments decreased significantly in comparison with control (Fig. 7B). In addition, in cells in which endoB2 organized as filaments, their perinuclear organization was disturbed, and the majority of filamentous structures spread across the cytoplasm (Fig. 7C). These data reinforce the idea that the plec1/endoB2 interaction is required for the formation and maintenance of endoB2 filaments in the perinuclear region.
Interfering with plec1 and endoB2 Expression Triggers Alteration of Intermediate Filament Perinuclear Architecture
We then investigated if interfering with either endoB2 or plec1 expression would modify the spatial organization of the vimentin network.
To compare the effects of plec1 and endoB2 siRNAs interference, HeLa/endoB2-St cells were used because they allow rigorous quantification of endoB2 siRNA efficiency after StrepTactin precipitation (endoB2 cannot be determined accurately from cell lysates, see Fig. 3A). We first analyzed the organization of endogenous vimentin by immunofluorescence after treatment with plec1 siRNAs. As observed for regular HeLa cells, plec1 siRNA treatment of HeLa/endoB2-St cells was characterized by heterogeneous efficiency (Fig. 8A, top left panel), with 19.5% of cells expressing plectin at a level approaching the detection limit (mean value obtained from three independent experiments, see Fig. 8A, top right panel). This number is in good correlation with the 15.1% of cells exhibiting significant change in vimentin organization (Fig. 8A, top right panel and see supplemental Fig. S3 for a broad appreciation of the phenotype at low magnification. See also “Experimental Procedures” for the criteria applied for this quantification.). Change in vimentin organization was essentially characterized by the bundling of vimentin filaments around nuclei and across the cytoplasm, the latter observed occasionally in cells treated with control siRNAs (Fig. 8A, top right panel). At high magnification, bundling was accompanied by disappearance of vimentin filaments encaging nuclei (Fig. 8A, bottom panel). Parallel experiments performed in HeLa cells stably expressing vimentin-GFP (vim-GFP) demonstrated the direct correlation between the decrease of plectin immunoreactivity and abnormal vimentin phenotype (see Fig. 8B).
Treatment with endoB2 siRNAs had a milder effect on endogenous vimentin in comparison with plec1 siRNAs (Fig. 9). As in the case of plec1 siRNA treatment, bundling of vimentin filaments in the close vicinity of nuclei and across cytoplasm was visible (Fig. 9B). However, in comparison with plec1 siRNAs, bundled filaments also appeared to encage nuclei in the three dimensions of space, which correlated with a significant increase in nuclear indentation (Fig. 9C). This may result from mechanical constraints exerted by filaments strengthened by bundling and whose anchorage at the nuclear membrane remains significantly maintained by the activity of plec1.
We also investigated the process of vimentin filament assembly de novo using newly synthesized vim-GFP in the context of siRNA treatments because we noticed that this approach leads to more homogeneous patterns of vimentin organization in comparison with the steady state endogenous pool (thus allowing classification of vimentin patterns in categories to perform quantitative analysis of phenotypes). In addition, following the de novo incorporation of vimentin structural units into a filamentous network has previously been instrumental for underlining the involvement of another plectin isoform in the assembly of a central cage-like core structure stabilizing the perinuclear vimentin network (56). Superimposition of newly synthesized vimentin-GFP after a short period of transient expression (10 h, see “Experimental Procedures” for details) with endogenous plectin unraveled significant vimentin and plectin overlaps, in particular at dense plectin spots in the periphery of nuclei (Fig. 10A). Using these experimental conditions, we have been able to identify four main categories of cells in regard to vimentin organization (categories a, b, c, and d in Fig. 10B, see legend for details). These were characterized by specific organization patterns of the newly assembled vimentin network and whose relative abundance appeared to change depending on siRNAs treatments. Upon decrease of either plec1 or endoB2 expression, the milder effect (likely not significant because of small variations) concerned the population of cells belonging to “category a” in which vim-GFP may be localized, in pre-existing protofilaments, at sites of exchange of nonfluorescent elementary units with newly synthesized fluorescent ones (which is consistent with the described dynamics of filament subunits in protofibrils (57)). The main effects that were consistently obtained when decreasing the expression of either protein were as follows: (i) a significant reduction in the number of cells in which newly synthesized vimentin assembles as tight cages (category c), and (ii) an increase in the number of cells exhibiting cytoplasmic dispersion of the vimentin network (category d). Decrease of plec1 expression was also characterized by a reduced percentage of cells belonging to “category b,” which was not the case for endoB2. This suggests that plec1 may be more of a determinant for anchoring the newly synthesized pool of vimentin in the putative pericentriolar region. This is consistent with a previous report showing the localization of, and a role for, plectin at centrosomes (58). Finally and importantly, similar results were obtained with the same experimental conditions using regular HeLa instead of the HeLa/endoB2-St cells (see the legend of Fig. 10 for details). Altogether, these data reinforced the significance of our findings and are consistent with the notion that plec1 and endoB2 contribute to the organization/stabilization of IFs encaging nuclei.
FIGURE 10.

Establishment of a tight perinuclear vimentin network requires plec1 and endoB2. A, HeLa/endoB2-St cells were transfected to express vim-GFP (10 h of expression). Cells were processed for immunofluorescence using anti-plectin antibodies. Images show two z-sections of the same cell, with top images obtained at a plane between the nucleus and the membrane contacting the coverslip and the bottom images obtained at a plane partly crossing the nucleus. Middle panels correspond to magnification of the areas delimited in the top panels and show plectin-enriched spots in contact with vimentin filaments (arrowheads). B, HeLa/endoB2-St cells were transfected with control, plec1 (si plec1-1 and si plec1-2) or endoB2 (si endoB2-1 and si endoB2-2) siRNAs for 72 h. They were subsequently transfected to express vim-GFP for 10 h. Top panels are z-sections showing four main different vim-GFP organization patterns exhibited by cells and defining categories, respectively, characterized as follows: category a, dispersion of small vimentin filamentous units; category b, localization of the vimentin network both in the perinuclear region and across the cytoplasm, with a concentrated spot nearby the nucleus; category c, organization of vimentin filaments as dense, tight cages around the nucleus; category d, dispersion of the meshwork across the entire cellular volume. Images of categories –c were obtained from cells treated with control siRNAs and of category d from cells treated with plec1-1 siRNAs (similar extensive dispersion of the network was also obtained for plec1-2 and endoB2-1 and endoB2-2 siRNAs). Bottom panels were obtained from numeration of cells belonging to each category, for both plec1 and endoB2 siRNAs. In the bottom left panel, n = 449, 476, and 221 counted cells for si control, si plec1-1, and si plec1-2, respectively. In the bottom right panel, n = 423, 451, and 435 counted cells for si control, si endoB2-1, and si endoB2-2, respectively. Similar results were obtained in independent experiments performed with regular HeLa cells in which the most significant effect was the major decrease in the proportion of cells characterized by the presence of tight vimentin cages (category c) for both plec1 and endoB2 siRNAs treatments and in comparison with the respective controls (12.8 and 15.1% of cells belonging to category c for si plec1-1 (n = 251 cells) and si plec1-2 (n = 239 cells), respectively, in comparison with 39.4% for the control (n = 323 cells); 9.7 and 10.1% of cells belonging to category c for si endoB2-1 (n = 277 cells) and si endoB2-2 (n = 289 cells), respectively, in comparison with 30.2% for the control (n = 215 cells)). Scale bars, 20 μm.
Interfering with plec1 and endoB2 Expression Impairs Nuclear Positioning
The results shown above illustrate a function of endoB2 in IF perinuclear architecture. Nuclear positioning has recently emerged to be placed under the control of diverse components of the cytoskeleton and to be critical for cell polarity, cell differentiation, and cell orientation during migration (59). To evaluate the contribution of endoB2 in cellular features involving perinuclear cytoskeletal organization, we investigated the incidence of destabilizing IF perinuclear architecture on nuclear positioning upon a decrease in endoB2 expression. We performed a quantitative analysis of the nuclear position in HeLa cells treated with siRNAs prior to plating on micropatterns (Fig. 11). Interfering with plec1 expression was also analyzed for comparison. For both plec1 and endoB2 siRNA treatments and in comparison with control, significant decrease in nuclei off-centering was measured.
DISCUSSION
In this study, we unravel the specific activity of endophilin B2, a member of the endophilin family, in the control of IF spatial organization. We provide evidence for the existence of a complex between endoB2 and isoform 1 of plectin that appears to be involved in the establishment and maintenance of the vimentin network, in particular in the region surrounding the nuclei. This activity, involving endoB2 specifically (and not its close relative endoB1), is of quite a significant impact, as it is the first time that a BAR domain-containing protein is shown to function in the organization of cytoskeletal components other than actin. So far, many of these proteins were known to control actin branching and polymerization upon recruitment of actin nucleation promoting factors via SH3 domains (60). Several members of the BAR domain superfamily coordinate their BAR-mediated membrane shaping activity with pushing forces generated by actin polymerization. This allows the modification of the membrane curvature during endocytosis as well as during the formation of lamellipodia and filopodia (15, 60). Although these processes are, in general, highly dynamic, the spatial organization of IFs that is placed under the control of the endoB2-plectin complex is most probably more static. In this scheme, endoB2 would be part of a plectin-dependent membrane-anchoring device ensuring the cohesion and maintenance of IF organization.
The specific interaction of endoB2 and plec1, one of the nine plectin human isoforms, is also a significant finding. Very few proteins have been found to bind to the N-terminal part of this longest plectin variant. This is also the case for β-synemin, which putatively links IFs to the dystrophin-glycoprotein complex at the plasma membrane of striated muscle cells (61) and of Siah (Seven in absentia homolog), a RING domain protein that functions in E3 ubiquitin ligase complexes involved in the degradation of a wide range of proteins, including β-catenin (62). For the latter, a consensus peptide in plec1 required for binding has been identified and is located upstream of the PETP motif involved in the interaction with endoB2. Importantly, it has been proposed that the binding of proteins to the N-terminal region of plec1 regulates that of interactants of the actin-binding domain that is contiguous with this sequence (47, 63). The plectin actin-binding domain can bind to a series of proteins other than actin, among which are integrin β-4 (a component of hemidesmosomes at the plasma membrane (64, 65)) and nesprin-3α, a protein of the outer nuclear membrane (66)). If endoB2 were to modulate the binding of plec1 actin-binding domain-interacting proteins, an appealing possibility that would require further investigations in the future, this would add to the layers of complexity taking part in the regulation of the activity of proteins involved in plectin-mediated anchorage of IFs, potentially both at the plasma and nuclear membranes. This leads to an important aspect of our findings that concern the molecular mechanisms underlying the specificity of endoB2 recruitment on intracellular membranes.
In comparison with other BAR domain-containing proteins, endophilins do not have a lipid-binding domain able to recognize specific membrane phospholipids. Thus, specific membrane targeting relies on SH3 domain-mediated interactions. This would theoretically target endoB2 to loci where plec1 is recruited. The results presented here are compatible with a plec1-driven recruitment of endoB2 at nuclear membranes, plec1 anchoring being most probably assigned by nesprin-3α on those membranes (66, 67). This possibility is consistent with the observation that knockdown of nesprin-3α triggers a reduction in vimentin networking around the nucleus (68, 69), a phenotype that is similar to the one described here upon decreasing expression of either plec1 or endoB2.
Our data strongly suggest that, upon overexpression in mammalian cells, endoB2 and its N-BAR domain self-assemble into filamentous structures apparently devoid of a membrane component. To our knowledge, this has never been reported for any protein of the BAR superfamily. Rather, many overexpressed BAR proteins have been shown, by electron microscopy, to trigger the appearance of unambiguous tubular membrane structures whose diameters correlate with the intrinsic curvature radius of the BAR domain (8). However, the in vitro formation of filaments was observed for recombinant proteins such as the F-BAR domain of the endocytic protein CIP4 (8) or the BAR proteins Pil1 and Lsp1, the core constituents of eisosomes in Saccharomyces cerevisiae (70–72). This indicates that several proteins of the BAR superfamily can assemble into helical structures that do not surround a membrane. Although endoB2 and its N-BAR domain appear to assemble into protein filaments upon overexpression in vivo, we think that endogenous concentration of endoB2 would not favor the formation of stable protein helical assemblies (in agreement with the apparent absence of such structures in cells expressing the protein at low levels under conditions used in this work). However, we cannot rule out totally that small oligomers of such nature, difficult to visualize because of scarcity, irrespectively of the methodology used, may form more or less transiently.
The fact that endoB2 does not trigger the formation of typical membrane tubular structures in cells although it polymerizes extensively is intriguing, in particular because the protein can assemble onto liposomes in vitro and deform them into tubules,4 as observed for other BAR domain proteins. However, our data on the endoB2-KARLAA mutant with no membrane binding ability and no filamentous assembly property indicate that binding to a membrane is most probably required for significant polymerization to occur. Hence, this suggests that the nature of membranes onto which endoB2 is recruited cannot undergo BAR-mediated tubulation, in particular not with the high curvature imposed by the helical assembly of an N-BAR domain (20–25 nm; our estimation for endoB2 decorated tubules obtained in vitro).4 Because our data are most compatible with a model in which endoB2 is engaged in a complex with plectin at specific domains of the nuclear membrane, these would not deform because of intrinsic rigidity. This may be due to the presence of trans-interacting proteins bridging the outer and inner membranes such as proteins containing a Klarsicht/ANC-1/SYNE homology (KASH) domain (among which the already mentioned nesprin-3α and proteins containing a SUN (Sad1 and UNC-84) domain (73)). Because we could visualize, by EM, bunches of filaments anchoring close to nucleopores, nearby membrane microdomains might be sites where plectin-endoB2 complexes are at work.
Presently, we believe that in cells at steady state, endoB2 would act in complex with plectin either as single BAR domains or as small oligomers that may organize in two dimensions. Two-dimensional linear arrays of side-lying BAR domains have been described, at least in vitro and with F-BAR domains bound to rigid membranes (8). By analogy, the N-BAR domain of endoB2 may be side-lying on the cytosolic face of the membrane, and the two residues Lys-177 and Lys-178 (mutagenized in endoB2-KARLAA) may be essential for membrane binding in these conditions. Alternatively, endoB2 may organize on the membrane as in the case of the yeast proteins Pil1 and Lsp1 whose assembly is limited to semi-ring structures (70).
Finally, although still limited in the sense that obtained in immortalized and poorly differentiated cells, our data suggest a potential role of endoB2 in the control of nucleus positioning, possibly in concert with its binding partner plec1. Because decreased expression of either protein impairs perinuclear vimentin organization, alteration of nucleus positioning may be a consequence of IF disorganization. It should be stressed that because plec1 is a predominant isoform in the cells that were used in this study, it may have been an advantage to link plec1/endoB2 activities to IF-mediated nuclear positioning which, obviously, requires further investigations in an appropriate differentiated system. To our knowledge, the main observations made so far concerning the phenotypes associated with the loss of plec1 expression are on impairment in migration potential, in particular of dermal fibroblasts and immune cells (74). Because links between oriented cell migration and rearward positioning of the nucleus have been described (59), our work establishes a basis for future investigations on plec1 N-terminal binding partners, including endoB2, on plec1/IFs-regulated motility of cells that prominently express that plectin isoform. Also, because plec1 is the major linker protein between IFs and the nuclear membrane in muscle cells (75, 76), it should be quite relevant to combine genetic tools to investigate the function of its binding partners, in particular endoB2, in nuclear positioning in this cell type.
Supplementary Material
Acknowledgments
We thank M. Chapelle and T. Leger (Structural and Functional Proteomics Platform, Institut Jacques Monod) for mass spectrometry analysis and V. Contremoulins for image analysis (ImagoSeine, Institut Jacques Monod).
This work was supported by CNRS and by a grant from La Ligue National Contre le Cancer, Comité de Paris.

This article contains supplemental Figs. S1–S3.
C. Vannier and A. A. Schmidt, unpublished data.
- BAR
- Bin/amphiphysin/Rvs
- endoA/B
- endophilin A/B
- IF
- intermediate filament
- SH3
- Src homology 3
- plec1
- plectin 1
- vim-GFP
- vimentin-GFP.
REFERENCES
- 1. McMahon H. T., Gallop J. L. (2005) Membrane curvature and mechanisms of dynamic cell membrane remodeling. Nature 438, 590–596 [DOI] [PubMed] [Google Scholar]
- 2. Itoh T., De Camilli P. (2006) BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim. Biophys. Acta 1761, 897–912 [DOI] [PubMed] [Google Scholar]
- 3. Zimmerberg J., Kozlov M. M. (2006) How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 7, 9–19 [DOI] [PubMed] [Google Scholar]
- 4. Mim C., Unger V. M. (2012) Membrane curvature and its generation by BAR proteins. Trends Biochem. Sci. 37, 526–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Masuda M., Mochizuki N. (2010) Structural characteristics of BAR domain superfamily to sculpt the membrane. Semin. Cell Dev. Biol. 21, 391–398 [DOI] [PubMed] [Google Scholar]
- 6. Tarricone C., Xiao B., Justin N., Walker P. A., Rittinger K., Gamblin S. J., Smerdon S. J. (2001) The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. Nature 411, 215–219 [DOI] [PubMed] [Google Scholar]
- 7. Peter B. J., Kent H. M., Mills I. G., Vallis Y., Butler P. J., Evans P. R., McMahon H. T. (2004) BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303, 495–499 [DOI] [PubMed] [Google Scholar]
- 8. Frost A., Perera R., Roux A., Spasov K., Destaing O., Egelman E. H., De Camilli P., Unger V. M. (2008) Structural basis of membrane invagination by F-BAR domains. Cell 132, 807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saarikangas J., Zhao H., Pykäläinen A., Laurinmäki P., Mattila P. K., Kinnunen P. K., Butcher S. J., Lappalainen P. (2009) Molecular mechanisms of membrane deformation by I-BAR domain proteins. Curr. Biol. 19, 95–107 [DOI] [PubMed] [Google Scholar]
- 10. Itoh T., Erdmann K. S., Roux A., Habermann B., Werner H., De Camilli P. (2005) Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev. Cell 9, 791–804 [DOI] [PubMed] [Google Scholar]
- 11. Henne W. M., Kent H. M., Ford M. G., Hegde B. G., Daumke O., Butler P. J., Mittal R., Langen R., Evans P. R., McMahon H. T. (2007) Structure and analysis of FCHo2 F-BAR domain: a dimerizing and membrane recruitment module that effects membrane curvature. Structure 15, 839–852 [DOI] [PubMed] [Google Scholar]
- 12. Mattila P. K., Pykäläinen A., Saarikangas J., Paavilainen V. O., Vihinen H., Jokitalo E., Lappalainen P. (2007) Missing-in-metastasis and IRSp53 deform PI(4,5)P2-rich membranes by an inverse BAR domain-like mechanism. J. Cell Biol. 176, 953–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimada A., Niwa H., Tsujita K., Suetsugu S., Nitta K., Hanawa-Suetsugu K., Akasaka R., Nishino Y., Toyama M., Chen L., Liu Z. J., Wang B. C., Yamamoto M., Terada T., Miyazawa A., Tanaka A., Sugano S., Shirouzu M., Nagayama K., Takenawa T., Yokoyama S. (2007) Curved EFC/F-BAR-domain dimers are joined end to end into a filament for membrane invagination in endocytosis. Cell 129, 761–772 [DOI] [PubMed] [Google Scholar]
- 14. Qualmann B., Koch D., Kessels M. M. (2011) Let's go bananas: revisiting the endocytic BAR code. EMBO J. 30, 3501–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frost A., Unger V. M., De Camilli P. (2009) The BAR domain superfamily: membrane-molding macromolecules. Cell 137, 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gallop J. L., Jao C. C., Kent H. M., Butler P. J., Evans P. R., Langen R., McMahon H. T. (2006) Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 25, 2898–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Masuda M., Takeda S., Sone M., Ohki T., Mori H., Kamioka Y., Mochizuki N. (2006) Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J. 25, 2889–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jao C. C., Hegde B. G., Gallop J. L., Hegde P. B., McMahon H. T., Haworth I. S., Langen R. (2010) Roles of amphipathic helices and the bin/amphiphysin/rvs (BAR) domain of endophilin in membrane curvature generation. J. Biol. Chem. 285, 20164–20170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhatia V. K., Madsen K. L., Bolinger P. Y., Kunding A., Hedegård P., Gether U., Stamou D. (2009) Amphipathic motifs in BAR domains are essential for membrane curvature sensing. EMBO J. 28, 3303–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Madsen K. L., Bhatia V. K., Gether U., Stamou D. (2010) BAR domains, amphipathic helices and membrane-anchored proteins use the same mechanism to sense membrane curvature. FEBS Lett. 584, 1848–1855 [DOI] [PubMed] [Google Scholar]
- 21. Cui H., Lyman E., Voth G. A. (2011) Mechanism of membrane curvature sensing by amphipathic helix containing proteins. Biophys. J. 100, 1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boucrot E., Pick A., Çamdere G., Liska N., Evergren E., McMahon H. T., Kozlov M. M. (2012) Membrane fission is promoted by insertion of amphipathic helices and is restricted by crescent BAR domains. Cell 149, 124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mim C., Cui H., Gawronski-Salerno J. A., Frost A., Lyman E., Voth G. A., Unger V. M. (2012) Structural basis of membrane bending by the N-BAR protein endophilin. Cell 149, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huttner W. B., Schmidt A. (2000) Lipids, lipid modification and lipid-protein interaction in membrane budding and fission–insights from the roles of endophilin A1 and synaptophysin in synaptic vesicle endocytosis. Curr. Opin. Neurobiol. 10, 543–551 [DOI] [PubMed] [Google Scholar]
- 25. Kjaerulff O., Brodin L., Jung A. (2011) The structure and function of endophilin proteins. Cell Biochem. Biophys. 60, 137–154 [DOI] [PubMed] [Google Scholar]
- 26. Weissenhorn W. (2005) Crystal structure of the endophilin-A1 BAR domain. J. Mol. Biol. 351, 653–661 [DOI] [PubMed] [Google Scholar]
- 27. Loll P. J., Swain E., Chen Y., Turner B. T., Zhang J. F. (2008) Structure of the SH3 domain of rat endophilin A2. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 243–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Q., Kaan H. Y., Hooda R. N., Goh S. L., Sondermann H. (2008) Structure and plasticity of endophilin and Sorting Nexin 9. Structure 16, 1574–1587 [DOI] [PubMed] [Google Scholar]
- 29. de Heuvel E., Bell A. W., Ramjaun A. R., Wong K., Sossin W. S., McPherson P. S. (1997) Identification of the major synaptojanin-binding proteins in brain. J. Biol. Chem. 272, 8710–8716 [DOI] [PubMed] [Google Scholar]
- 30. Ringstad N., Gad H., Löw P., Di Paolo G., Brodin L., Shupliakov O., De Camilli P. (1999) Endophilin/SH3p4 is required for the transition from early to late stages in clathrin-mediated synaptic vesicle endocytosis. Neuron 24, 143–154 [DOI] [PubMed] [Google Scholar]
- 31. Guichet A., Wucherpfennig T., Dudu V., Etter S., Wilsch-Bräuniger M., Hellwig A., González-Gaitán M., Huttner W. B., Schmidt A. A. (2002) Essential role of endophilin A in synaptic vesicle budding at the Drosophila neuromuscular junction. EMBO J. 21, 1661–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verstreken P., Kjaerulff O., Lloyd T. E., Atkinson R., Zhou Y., Meinertzhagen I. A., Bellen H. J. (2002) Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell 109, 101–112 [DOI] [PubMed] [Google Scholar]
- 33. Milosevic I., Giovedi S., Lou X., Raimondi A., Collesi C., Shen H., Paradise S., O'Toole E., Ferguson S., Cremona O., De Camilli P. (2011) Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 72, 587–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meinecke M., Boucrot E., Camdere G., Hon W. C., Mittal R., McMahon H. T. (2013) Cooperative recruitment of Dynamin and BAR domain-containing proteins leads to GTP-dependent membrane scission. J. Biol. Chem. 288, 6651–6661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cuddeback S. M., Yamaguchi H., Komatsu K., Miyashita T., Yamada M., Wu C., Singh S., Wang H. G. (2001) Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J. Biol. Chem. 276, 20559–20565 [DOI] [PubMed] [Google Scholar]
- 36. Pierrat B., Simonen M., Cueto M., Mestan J., Ferrigno P., Heim J. (2001) SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics 71, 222–234 [DOI] [PubMed] [Google Scholar]
- 37. Karbowski M., Jeong S. Y., Youle R. J. (2004) Endophilin B1 is required for the maintenance of mitochondrial morphology. J. Cell Biol. 166, 1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takahashi Y., Karbowski M., Yamaguchi H., Kazi A., Wu J., Sebti S. M., Youle R. J., Wang H. G. (2005) Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol. Cell. Biol. 25, 9369–9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Etxebarria A., Terrones O., Yamaguchi H., Landajuela A., Landeta O., Antonsson B., Wang H. G., Basañez G. (2009) Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large scale membrane morphological rearrangements. J. Biol. Chem. 284, 4200–4212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rostovtseva T. K., Boukari H., Antignani A., Shiu B., Banerjee S., Neutzner A., Youle R. J. (2009) Bax activates endophilin B1 oligomerization and lipid membrane vesiculation. J. Biol. Chem. 284, 34390–34399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Farsad K., Ringstad N., Takei K., Floyd S. R., Rose K., De Camilli P. (2001) Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J. Cell Biol. 155, 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang J. S., Zhang L., Lee S. Y., Gad H., Luini A., Hsu V. W. (2006) Key components of the fission machinery are interchangeable. Nat. Cell Biol. 8, 1376–1382 [DOI] [PubMed] [Google Scholar]
- 43. Wan J., Cheung A. Y., Fu W. Y., Wu C., Zhang M., Mobley W. C., Cheung Z. H., Ip N. Y. (2008) Endophilin B1 as a novel regulator of nerve growth factor/TrkA trafficking and neurite outgrowth. J. Neurosci. 28, 9002–9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi Y., Coppola D., Matsushita N., Cualing H. D., Sun M., Sato Y., Liang C., Jung J. U., Cheng J. Q., Mulé J. J., Pledger W. J., Wang H. G. (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9, 1142–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takahashi Y., Meyerkord C. L., Hori T., Runkle K., Fox T. E., Kester M., Loughran T. P., Wang H. G. (2011) Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy 7, 61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kang R., Zeh H. J., Lotze M. T., Tang D. (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sonnenberg A., Liem R. K. (2007) Plakins in development and disease. Exp. Cell Res. 313, 2189–2203 [DOI] [PubMed] [Google Scholar]
- 48. Valencia R. G., Walko G., Janda L., Novacek J., Mihailovska E., Reipert S., Andra-Marobela K., Wiche G. (2013) Intermediate filament-associated cytolinker plectin 1c destabilizes microtubules in keratinocytes. Mol. Biol. Cell 24, 768–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gortat A., San-Roman M. J., Vannier C., Schmidt A. A. (2012) Single point mutation in Bin/Amphiphysin/Rvs (BAR) sequence of endophilin impairs dimerization, membrane shaping, and Src homology 3 domain-mediated partnership. J. Biol. Chem. 287, 4232–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Slot J. W., Geuze H. J. (2007) Cryosectioning and immunolabeling. Nat. Protoc. 2, 2480–2491 [DOI] [PubMed] [Google Scholar]
- 51. Kirmse R., Bouchet-Marquis C., Page C., Hoenger A. (2010) Three-dimensional cryo-electron microscopy on intermediate filaments. Methods Cell Biol. 96, 565–589 [DOI] [PubMed] [Google Scholar]
- 52. Herrmann H., Strelkov S. V., Burkhard P., Aebi U. (2009) Intermediate filaments: primary determinants of cell architecture and plasticity. J. Clin. Invest. 119, 1772–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ralser M., Nonhoff U., Albrecht M., Lengauer T., Wanker E. E., Lehrach H., Krobitsch S. (2005) Ataxin-2 and huntingtin interact with endophilin-A complexes to function in plastin-associated pathways. Hum. Mol. Genet. 14, 2893–2909 [DOI] [PubMed] [Google Scholar]
- 54. Rezniczek G. A., Janda L., Wiche G. (2004) Plectin. Methods Cell Biol. 78, 721–755 [PubMed] [Google Scholar]
- 55. Rao Y., Ma Q., Vahedi-Faridi A., Sundborger A., Pechstein A., Puchkov D., Luo L., Shupliakov O., Saenger W., Haucke V. (2010) Molecular basis for SH3 domain regulation of F-BAR-mediated membrane deformation. Proc. Natl. Acad. Sci. U.S.A. 107, 8213–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Burgstaller G., Gregor M., Winter L., Wiche G. (2010) Keeping the vimentin network under control: cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol. Biol. Cell 21, 3362–3375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vikstrom K. L., Lim S. S., Goldman R. D., Borisy G. G. (1992) Steady state dynamics of intermediate filament networks. J. Cell Biol. 118, 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Niwa T., Saito H., Imajoh-ohmi S., Kaminishi M., Seto Y., Miki Y., Nakanishi A. (2009) BRCA2 interacts with the cytoskeletal linker protein plectin to form a complex controlling centrosome localization. Cancer Sci. 100, 2115–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dupin I., Etienne-Manneville S. (2011) Nuclear positioning: mechanisms and functions. Int. J. Biochem. Cell Biol. 43, 1698–1707 [DOI] [PubMed] [Google Scholar]
- 60. Suetsugu S., Gautreau A. (2012) Synergistic BAR-NPF interactions in actin-driven membrane remodeling. Trends Cell Biol. 22, 141–150 [DOI] [PubMed] [Google Scholar]
- 61. Hijikata T., Nakamura A., Isokawa K., Imamura M., Yuasa K., Ishikawa R., Kohama K., Takeda S., Yorifuji H. (2008) Plectin 1 links intermediate filaments to costameric sarcolemma through β-synemin, α-dystrobrevin and actin. J. Cell Sci. 121, 2062–2074 [DOI] [PubMed] [Google Scholar]
- 62. House C. M., Frew I. J., Huang H. L., Wiche G., Traficante N., Nice E., Catimel B., Bowtell D. D. (2003) A binding motif for Siah ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 100, 3101–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Litjens S. H., Koster J., Kuikman I., van Wilpe S., de Pereda J. M., Sonnenberg A. (2003) Specificity of binding of the plectin actin-binding domain to β4 integrin. Mol. Biol. Cell 14, 4039–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rezniczek G. A., de Pereda J. M., Reipert S., Wiche G. (1998) Linking integrin α6β4-based cell adhesion to the intermediate filament cytoskeleton: direct interaction between the β4 subunit and plectin at multiple molecular sites. J. Cell Biol. 141, 209–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Geerts D., Fontao L., Nievers M. G., Schaapveld R. Q., Purkis P. E., Wheeler G. N., Lane E. B., Leigh I. M., Sonnenberg A. (1999) Binding of integrin α6β4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J. Cell Biol. 147, 417–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wilhelmsen K., Litjens S. H., Kuikman I., Tshimbalanga N., Janssen H., van den Bout I., Raymond K., Sonnenberg A. (2005) Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 171, 799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ketema M., Wilhelmsen K., Kuikman I., Janssen H., Hodzic D., Sonnenberg A. (2007) Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J. Cell Sci. 120, 3384–3394 [DOI] [PubMed] [Google Scholar]
- 68. Postel R., Ketema M., Kuikman I., de Pereda J. M., Sonnenberg A. (2011) Nesprin-3 augments peripheral nuclear localization of intermediate filaments in zebrafish. J. Cell Sci. 124, 755–764 [DOI] [PubMed] [Google Scholar]
- 69. Morgan J. T., Pfeiffer E. R., Thirkill T. L., Kumar P., Peng G., Fridolfsson H. N., Douglas G. C., Starr D. A., Barakat A. I. (2011) Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Mol. Biol. Cell 22, 4324–4334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Karotki L., Huiskonen J. T., Stefan C. J., Ziółkowska N. E., Roth R., Surma M. A., Krogan N. J., Emr S. D., Heuser J., Grünewald K., Walther T. C. (2011) Eisosome proteins assemble into a membrane scaffold. J. Cell Biol. 195, 889–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kabeche R., Baldissard S., Hammond J., Howard L., Moseley J. B. (2011) The filament-forming protein Pil1 assembles linear eisosomes in fission yeast. Mol. Biol. Cell 22, 4059–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Olivera-Couto A., Graña M., Harispe L., Aguilar P. S. (2011) The eisosome core is composed of BAR domain proteins. Mol. Biol. Cell 22, 2360–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Starr D. A., Fridolfsson H. N. (2010) Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu. Rev. Cell Dev. Biol. 26, 421–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abrahamsberg C., Fuchs P., Osmanagic-Myers S., Fischer I., Propst F., Elbe-Bürger A., Wiche G. (2005) Targeted ablation of plectin isoform 1 uncovers role of cytolinker proteins in leukocyte recruitment. Proc. Natl. Acad. Sci. U.S.A. 102, 18449–18454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Konieczny P., Fuchs P., Reipert S., Kunz W. S., Zeöld A., Fischer I., Paulin D., Schröder R., Wiche G. (2008) Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J. Cell Biol. 181, 667–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wiche G., Winter L. (2011) Plectin isoforms as organizers of intermediate filament cytoarchitecture. Bioarchitecture 1, 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.