

Background: SK1 is widely involved in promoting inflammatory diseases.
Results: Unexpectedly, loss of SK1 caused an increase in chemokines upon TNF stimulation. This occurred through regulation of p38 MAPK but independently of NF-κB.
Conclusion: SK1 negatively regulates RANTES expression through the p38 MAPK pathway.
Significance: Targeting SK1 in therapy may affect inflammatory conditions by up-regulating chemokines independently of NF-κB.
Keywords: Chemokines, Inflammation, NF-kappa B (NF-KB), p38 MAPK, Sphingosine-1-phosphate, Tumor Necrosis Factor (TNF), RANTES, Sphingosine Kinase
Abstract
Sphingosine kinase 1 (SK1) produces the pro-survival sphingolipid sphingosine 1-phosphate and has been implicated in inflammation, proliferation, and angiogenesis. Recent studies identified TRAF2 as a sphingosine 1-phosphate target, implicating SK1 in activation of the NF-κB pathway, but the functional consequences of this connection on gene expression are unknown. Here, we find that loss of SK1 potentiates induction of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted; also known as CCL5) in HeLa cells stimulated with TNF-α despite RANTES induction being highly dependent on the NF-κB pathway. Additionally, we find that SK1 is not required for TNF-induced IKK phosphorylation, IκB degradation, nuclear translocation of NF-κB subunits, and transcriptional NF-κB activity. In contrast, loss of SK1 prevented TNF-induced phosphorylation of p38 MAPK, and inhibition of p38 MAPK, like SK1 knockdown, also potentiates RANTES induction. Finally, in addition to RANTES, loss of SK1 also potentiated the induction of multiple chemokines and cytokines in the TNF response. Taken together, these data identify a potential and novel anti-inflammatory function of SK1 in which chemokine levels are suppressed through SK1-mediated activation of p38 MAPK. Furthermore, in this system, activation of NF-κB is dissociated from SK1, suggesting that the interaction between these pathways may be more complex than currently thought.
Introduction
Originally considered structural molecules, sphingolipids have emerged as biological mediators in a variety of cellular processes (1). Because multiple sphingolipids have bioactive functions, their levels are under tight control by a complex, interlinked network of enzymes. The sphingosine kinases (SK)2 serve as key points in the sphingolipid pathway by converting pro-apoptotic sphingosine to pro-survival sphingosine 1-phosphate (S1P), and considerable research has implicated both SKs and S1P in a variety of pathologies including cardiovascular diseases (2), ischemia and reperfusion injuries in the brain and kidneys (3, 4), inflammatory diseases, and cancer (5). Thus, SKs have emerged as potential therapeutic targets.
Currently, two isoforms of SK with distinct cellular localizations are known: SK1 and SK2. SK1 is primarily cytoplasmic and translocates to the plasma membrane upon activation (6), whereas SK2 can be found in the endoplasmic reticulum, nucleus, and mitochondria (7). The generation of S1P by SK1 can exert its signaling effects through two routes. In the first, S1P is exported to the outer leaflet via transporter proteins (8, 9), where it can act on any of five identified G-protein-coupled receptors (S1P receptors 1–5) (10). Alternatively, S1P is also thought to function as an intracellular signaling molecule acting directly on intracellular targets. One recently identified S1P target is TRAF2 (11), an important mediator of NF-κB activation in response to the cytokine TNF. However, the functional consequences of this proposed connection between SK1 and NF-κB in regulating TNF-mediated gene expression have yet to be fully explored.
TNF is a key player in inflammation and is a major target of therapy in diseases such as rheumatoid arthritis, inflammatory bowel disease, and asthma, among others. In the inflammatory response, TNF plays a role in the recruitment of circulating immune cells to sites of inflammation by up-regulating adhesion molecules (intracellular adhesion molecule and vascular cell adhesion molecule) and inducing the secretion of chemokines such as IL-8, monocyte chemotactic protein 1, and CCL5 (RANTES). Considerable research has shown that TNF mediates pro-survival and pro-inflammatory pathways through activation of NF-κB, c-Jun, Ras/ERK, and Akt. However, activation of sphingolipid metabolism is also well established as a central part of TNF signaling (12–14). Indeed, recent studies from our laboratory and others have implicated bioactive sphingolipids as important mediators of pro-inflammatory signaling by TNF (15, 16); however, the roles of specific enzymes of sphingolipid metabolism and the relevant bioactive lipid mediators are not clear. Defining the specific roles of individual enzymes and specific lipid mediators is not only critical for understanding these pathways but is also important for delineating candidate therapeutic targets and defining downstream mechanisms.
RANTES induces the chemotaxis of multiple immune cells (17) and is secreted by many cell types including fibroblasts (18), epithelial cells (19), and vascular smooth muscle cells (20). High levels of RANTES are associated with several pathologies including atherosclerosis (21), various cancers (22, 23), diabetes (24), glomerulonephritis (25), and rheumatoid arthritis (26). In contrast, failure to secrete RANTES impairs many T cell functions such as the clearance of viral infections (27). Regulation of RANTES is thought to occur primarily at the transcriptional level, because its mRNA is reported to be extremely stable (28). However, transcriptional regulation of RANTES is both stimulus- and cell type-dependent. In the TNF response, many transcription factors have been implicated in RANTES induction including NF-κB and interferon regulatory factor 1 (29, 30), and activator protein 1 and NF-AT in airway epithelial and smooth muscle cells, respectively (31). Moreover, upstream of transcription factors, other signaling cascades have been implicated in RANTES regulation such as JNK (32), ERK (33), and p38 MAPK (34). More recently, our group identified sphingolipids as regulators of RANTES through the acid sphingomyelinase/acid ceramidase pathway (16). Strikingly, our data indicated that loss of SK1 significantly enhanced RANTES induction. However, the mechanism by which this was mediated was unclear.
Here, we have explored the mechanism by which SK1 regulates RANTES induction. Using HeLa cells as a model system, we find that TNF induction of RANTES is strongly NF-κB-dependent, yet loss of SK1 strongly up-regulates RANTES independently of effects on NF-κB activation. In contrast, SK1 is required for activation of p38 MAPK, and inhibiting p38 MAPK also leads to induction of RANTES, thus suggesting that p38 MAPK is the mediator of SK1 effects on suppression of RANTES. Finally, the effect of SK1 knockdown on RANTES is part of a broader response with several other chemokines responding in a similar manner. Taken together, we conclude that SK1 negatively regulates RANTES induction through activation of p38 MAPK in TNF-stimulated HeLa cells and suggests that it could play a modulatory effect on the immune system that may not necessarily be pro-inflammatory. Moreover, SK1 was dispensable for TNF-induced activation of the NF-κB pathway in this system.
EXPERIMENTAL PROCEDURES
Materials
High glucose DMEM, RPMI 1640 medium, FBS, penicillin-streptomycin, Oligofectamine, and Superscript III first strand synthesis kit were purchased from Invitrogen. Essentially fatty acid-free BSA, and monoclonal anti-β-actin antibody were from Sigma-Aldrich. Anti-phospho-IKKα/β, anti-total IKKα, anti-total IKKβ, anti-total IκBα, anti-p65, and anti-p50 antibodies were from Cell Signaling Technology (Danvers, MA). Anti-phospho-p38 was from Promega (Madison, WI). Anti-total p38 and HRP-labeled secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Chemiluminescence kit and BCA kit was from ThermoScientific (Suwanee, GA). Draq5 and 488-conjugated secondary antibody were purchased from Alexis Biochemicals (San Diego, CA). SYBR Green and iTAQ master mixes were purchased from Bio-Rad. Human and mouse TNF-α were purchased from PeproTech (Rocky Hill, NJ). SKi-II (4-[4-(4-chloro-phenyl)-thia-zol-2-ylamino]-phenol) was purchased from Cayman Chemical (Ann Arbor, MI).
Cell Culture and siRNA
HeLa and A549 cells were originally purchased from American Type Culture Collection (Manassas, VA). DMEM and RPMI were supplemented with 1% penicillin-streptomycin and 10% FBS, and cells were incubated in standard culture conditions: 37 °C and 5% CO2. When serum-free medium was used, 0.1% BSA, 1% penicillin-streptomycin, and 10 mm HEPES were supplemented to DMEM or RPMI. In all cases before treatment with TNF, the media were changed on cells to fresh new media. Gene silencing was carried out using siRNA directed against human SK1 (target sequence 5′-AAGGGCAAGGCCTTGCAGCTC-3) and human SK2: Hs_SPHK2_5 FlexiTube siRNA SI00288561 (Qiagen) and with Qiagen all-star siRNA as a negative control. The siRNA directed against p65 was a validated predesigned sequence from Invitrogen. Transfections were carried out using Oligofectamine (Invitrogen) according to the manufacturer's protocol. For siRNA experiments, HeLa cells were seeded into 60-mm plates at ∼200,000 cells/dish and treated with 20 nm for 48 h prior to stimulation.
Generation of Mouse Embryonic Fibroblasts (MEFs)
WT and SK1−/− fibroblasts were obtained as previously described (35). MEFs were generated from SK1± littermate matings in a C57BL6.129S background. MEFs were isolated from E13.5 embryos. The cells were maintained in high glucose DMEM supplemented with 10% FBS in standard culture conditions. For genotyping, genomic DNA was extracted using DNeasy blood and tissue kit (Qiagen), and 2 μl of DNA was combined with 1 μl each of either SK1 primer 1 and SK1 primer 2 or SK1 primer 1 and the NEO primer. This was combined with 21 μl of PCR Platinum SuperMix (Invitrogen) for a total reaction volume of 25 μl for PCR. The following primers (10 μm) from Integrated DNA Technologies (Coralville, IA) were used for the detection of the wild-type or knock-out SK1: SK1 primer 1 (5′-GCAGTGACAAAAGCTGCCGAATGCTGATG-3′). PCR was performed on a Biometra Thermocycler T3000 with the following reaction conditions: 95 °C for 0.5 min, 55 °C for 0.5 min, and 72 °C for 0.5 min for 40 cycles.
Immunoblotting
The cells were directly lysed in RIPA buffer (purchased as 10× concentrate from Cell Signaling Technologies) and sonicated. Protein levels were estimated by BCA assay (Pierce), proteins were normalized, and Laemmli buffer was added accordingly. The samples were boiled for 5–10 min, and proteins were separated via SDS-PAGE (4–15%, Tris-HCl) using the Bio-Rad Criterion system. Proteins were transferred to nitrocellulose membranes and blocked for at least 1 h with 5% nonfat milk in PBS/0.1% Tween 20 (PBS-T). The membranes were incubated with primary antibodies diluted 1:1000 or 1:3000 β-actin at 4 °C overnight. Secondary antibody incubation occurred for 1 h at room temperature at a 1:5000 dilution. Proteins were visualized by enhanced chemiluminescence (Pierce).
Quantitative (Real Time) Reverse Transcriptase-PCR (qRT-PCR)
Extraction of RNA from HeLa cells was performed using Qiagen RNeasy kits in combination with Qiashredders (Qiagen). RNA (1 μg) was converted to cDNA using SuperScript III first strand synthesis systems (Invitrogen), and cDNA was diluted 1:10 with molecular biology grade water (Sigma).
Each 25 μl of RT-PCR contained a ratio of 12.5:0.5:0.5:6.5 (SYBR Green:10 μm reverse primer:10 μm forward primer:distilled H2O). Using a Bio-Rad iCycler, reactions detecting expression of human genes were carried out as previously described (16). Briefly, following 3 min at 95 °C, 40 cycles of 10 s of melt at 95 °C, 45 s of annealing at 55 °C, and 45 s of extension at 68 °C were carried out. The following primers sequences were used to detect expression: human β-actin forward (hβ-actin F), 5′-ATTGGCAATGAGCGGTTCC-3′; hβ-actin reverse (hβ-actin R), 5′-GGTAGTTTCGTGGCCACA-3; hSK1 F, 5′-CTGGCAGCTTCCTTGAACCAT-3′; hSK1 R, 5′-TGTGCAGAGACAGCAGGTTCA-3′; hRANTES F, 5′-GCTGTCATCCTCATTGCTACTG-3′; hRANTES R, 5′-TGGTGTAGAAATACTCCTTGATGTG-3′; hCXCL1 F, 5′-CGCCCAAACCGAAGTCATAGCC-3′; hCXCL1 R 5′-TTCCTCCCTTCTGGTCAGTTG-3′; hCCL20 F 5′-AGCAACTTTGACTGCTGTCTTGG-3′; hCCL20 R 5′-AGGAGACGCACAATATATTTCACCC-3′. TaqMan primer/probe mixes for p65, CCXL10, and IL-8 primers were purchased from Invitrogen and were used according to manufacturer protocol. Using Q-Gene software, threshold cycle (Ct) values were normalized to β-actin and displayed as mean normalized expression.
Measurement of RANTES Levels in Media
The ELISA kit for human RANTES was obtained from R&D Systems (Minneapolis, MN). Measurement of secreted RANTES in serum-free DMEM was done according to the manufacturer's protocol. RANTES levels were normalized to the total amount of proteins determined by BCA assay.
Immunofluorescence and Confocal Microscopy
Laser-scanning confocal and immunofluorescence microscopy analysis were carried out as previously described with minor modifications (36). HeLa cells were grown on poly-d-lysine-coated 35-mm confocal dishes (MatTek Corporation). The following day, the cells were deprived of serum overnight prior to being exposed to treatments. The cells were fixed using 3.7% paraformaldehyde, washed with 1× PBS, and permeabilized with 0.1% Triton X-100. The cells were blocked in 2% human serum and exposed to primary antibodies (1:100) in 2% serum overnight. Incubation with secondary antibody was performed according to the manufacturer's protocol. Using a LSM510 META confocal microscope (Carl Zeiss, Inc.), photo images were obtained and then analyzed using free downloadable LSM image browser software.
Cellular Sphingolipids Extraction and Analysis
This procedure was performed as previously described (36). Briefly, adherent cells were directly lysed with 2 ml of 2:3 70% isopropanol/ethyl acetate. After addition of internal standards to the tubes, the samples were centrifuged at 2000 × g. The upper phase was transferred to a new glass tube, and a new round of extraction was performed on the remaining volume. After combining the two extracts, sphingolipids and inorganic phosphates were measured. Sphingolipid species were identified on a Thermo Finnigan TSQ 7000 triple quadrupole mass spectroscopy. Sphingolipid species were normalized to total lipid phosphates present in the cells after a Bligh and Dyer extraction.
Luciferase Assay
Transcriptional activity of NF-κB was measured using an NF-κB consensus sequence upstream of the luciferase reporter gene (Stratagene). Briefly, HeLa cells were plated in 60-mm dishes and co-transfected with NF-κB luciferase (1.5 μg) and constitutively expressing galactosidase (0.5 μg) (Invitrogen) using Xtremegene 9 (Roche Applied Science) according to the manufacturer's protocol. After 6 h of transfection, media were replaced for 1 h prior to incubation with inhibitors and stimulation with TNF as indicated. For siRNA experiments, cells were treated with AStar or SK1 siRNA for 48 h prior to transfection with reporter constructs. Following stimulation, luciferase activity was assessed using the Luciferase reporter kit (Stratagene) according to the manufacturer's protocol. Galactosidase activity was assayed using the High Sensitive B-galactosidase kit (Stratagene) according to the manufacturer's protocol. Measured luciferase activity (NF-κB-dependent) was normalized to measured galactosidase activity (constitutive).
Statistical Analysis
The data are represented as the means ± S.E. Unpaired Student's t test and two-way analysis of variance with Bonferroni post-test statistical analyses were performed using Prism/GraphPad software.
RESULTS
SK1 Knockdown Enhances TNF-induced RANTES in HeLa Cells
We previously reported an increase in RANTES induction upon the loss of SK1 in MCF7 and MEFs (16). However, because MCF7 cells undergo cell death in response to TNF and MEFs are not particularly responsive to TNF, we elected to find an alternative model system. To this end, HeLa cells were selected as an inflammatory model that can withstand TNF in the absence of translational inhibitors. To validate the previous observations, siRNA was used to knock down SK1, and the effects on RANTES and SK1 mRNA were analyzed (Fig. 1). As can be seen, a significant knockdown of SK1 mRNA was observed in siRNA-treated cells compared with all-Star (AStar) negative controls with greater than 80% knockdown (Fig. 1A). Notably, a small but statistically insignificant increase in SK2 mRNA was also observed (data not shown). Importantly, TNF induced RANTES mRNA expression in both AStar- and SK1 siRNA-treated cells. Strikingly, SK1 knockdown significantly enhanced RANTES induction in response to TNF with no statistically significant effect on unstimulated levels (Fig. 1B). Importantly, the changes in RANTES mRNA were also reflected in levels of RANTES protein in the media (Fig. 1B). These results replicate our previous findings in MCF7 cells and MEFs, and they confirm the utility of HeLa cells as a model system to explore regulation of RANTES by SK1.
FIGURE 1.

Loss of SK1 significantly enhances RANTES induction. A, HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h, and SK1 mRNA levels were assessed by qRT-PCR (n = 3). B, HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h and treated with vehicle (PBS) or TNF (20 ng/ml) for 24 h, and the levels of RANTES mRNA were assessed by qRT-PCR. The data represent means ± S.E. from six independent experiments. C, HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h and treated with vehicle (PBS) or TNF (20 ng/ml) for 24 h in serum-free media, and RANTES levels in the media were analyzed by ELISA. The data represent means ± S.E. of two independent experiments performed in duplicate. **, p < 0.01; ***, p < 0.001.
SK2 Knockdown and Inhibition Diminish RANTES Production
Our previous study (16) suggested that sphingosine was important for RANTES induction in response to TNF. Thus, it was a possibility that the observed effects may not be specifically due to the loss of SK1. To confirm this, the effects of SK2 siRNA on TNF-stimulated RANTES induction were determined. As can be seen, SK2 significantly decreased SK2 mRNA levels (supplemental Fig. S1A) but had minimal effects on SK1 expression (data not shown). As with SK1, the loss of SK2 had no significant effect on basal RANTES, but surprisingly, in TNF-stimulated cells, SK2 siRNA significantly decreased RANTES induction (supplemental Fig. S1B). Thus, the specific loss of SK1 but not SK2 enhances RANTES production in TNF-stimulated HeLa cells.
The differential effects of SK1 and SK2 prompted us to determine whether pharmacological inhibitors of SKs produced the same effect as the loss of SK1. For this, the generic SK inhibitor SKi-II was utilized. As can be seen, SKi-II had no effect on basal RANTES mRNA or protein yet caused a small but significant decrease in RANTES mRNA and protein levels (supplemental Fig. S2, A and B) in TNF-stimulated cells. Recent studies have suggested that SKi-II preferentially inhibits SK2 (37), and therefore these results are consistent with the observed effect of SK2 siRNA.
RANTES Induction in Response to TNF Is NF-κB-dependent
Several transcription factors have been shown to regulate RANTES transcription including STAT3 (20, 38) and NF-κB (39), depending on the cell type and stimulus. Accordingly, to examine SK1 regulation of RANTES, it was first important to determine the transcription factor primarily responsible for RANTES induction in HeLa cells in response to TNF. To this end, we utilized BAY 11-7082, a well known inhibitor of NF-κB activation that acts by irreversible inhibition of IκB-α phosphorylation (41). To test the efficiency of this inhibitor, localization of the NF-κB subunits p65/RelA and p50/NF-κB was assessed by immunofluorescence and confocal microscopy (Fig. 2). As can be seen in control samples, p65 and p50 primarily reside in the cytosol basally and translocate to the nucleus upon TNF stimulation. Importantly, BAY treatment resulted in a dramatic decrease in nuclear translocation of p65 (Fig. 2A) and p50 (Fig. 2B), thus confirming its efficacy. Next, the effect of BAY treatment on RANTES induction was assessed by RT-PCR. The data demonstrate that basal levels of RANTES were largely unaffected by BAY treatment. Notably, TNF induction of RANTES was inhibited by more than 80% (Fig. 2C). To further verify that RANTES is NF-κB-dependent in the current system, we used siRNA against p65 and achieved greater than 80% knockdown (data not shown). Again, TNF induction of RANTES was similarly abolished (Fig. 2D). Together, these results indicate that RANTES induction in response to TNF is largely NF-κB-dependent in HeLa cells.
FIGURE 2.

RANTES induction is highly NF-κB-dependent. HeLa cells were pretreated with either Me2SO or 5 μm BAY for 30 min prior to treatment with vehicle (PBS) or TNF (20 ng/ml) for 30 min. A and B, cells were fixed and incubated with anti-p65 (A) or anti-p50 antibodies (B). A secondary antibody with a 488 fluorophore was utilized, and cells were visualized by confocal microscopy. C, HeLa cells were treated with vehicle (Me2SO) or BAY (5 μm) for 30 min prior to stimulation with PBS or TNF (20 ng/ml) for 24 h. RNA was extracted, and RANTES mRNA levels were assessed by qRT-PCR. The data represent means ± S.E. of three independent experiments. D, HeLa cells were treated with 20 nm Astar or p65 siRNA for 48 h and treated with vehicle (PBS) or TNF (20 ng/ml) for 24 h, and the levels of RANTES mRNA were assessed by qRT-PCR. The data represent means ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01.
TNF-induced Acute Activation of NF-κB Is SK1- and S1P-independent
Previous research has reported regulation of the NF-κB pathway by SK1 (42) with subsequent studies suggesting that this occurs through S1P-mediated activation of TRAF2 (11, 42). However, because the effects of SK1 on RANTES (knockdown increases RANTES levels) are contradictory to the role of NF-κB in induction of RANTES, it became critical to determine any possible interactions between SK1 and the well known TNF signaling cascade leading to NF-κB activation (43). However, it was first necessary to determine whether TNF increased S1P levels within the time frame of early NF-κB pathway activation and whether this was through SK1 (Fig. 3A). For this, HeLa cells were transfected with AStar or SK1 siRNA and subsequently stimulated with TNF for 2.5 or 10 min prior to lipid extraction. These times were selected to correlate with early activation of NF-κB pathway as described previously (11) (44). As can be seen, HeLa cells treated with AStar siRNA show a significant increase in S1P production at both 2.5 min (approximately 20-fold) and 10 min of TNF stimulation (approximately 15-fold; Fig. 3A). Strikingly, these increases were completely abrogated in HeLa cells treated with SK1 siRNA (Fig. 3A) Thus, acute TNF stimulation leads to an increase in cellular S1P levels that is wholly dependent on SK1.
FIGURE 3.

SK1 is dispensable for TNF-mediated activation of NF-κB. A, HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h prior to treatment with TNF (20 ng/ml) or PBS for 2.5 and 10 min. Cellular lipids were directly extracted in organic solvent extraction, and S1P levels were analyzed by tandem LC/MS mass spectrometry. Lipid levels were normalized to nmol of total. The data represent means ± S.E. of two independent experiments performed in duplicate. B, HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h prior to treatment with TNF (20 ng/ml) for 0, 10, or 30 min. Early downstream signals of TNF including IKKα/β phosphorylation and IκBα degradation were examined via immunoblot. The total levels of NF-κB subunits p65 and p50 were also analyzed. Actin was included as a loading control. Each blot is representative of three independent experiments. C, MEFs derived from WT or SK1−/− mice were treated with 4 nm TNF for 0, 5, 15, 30, or 60 min, and IκBα levels were assessed by immunoblot with actin as a loading control; blots are representative of three independent experiments. D, HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h prior to treatment with various TNF doses as indicated for 10 min 2.5, 5, 10, or 15 ng/ml TNF were added for 10 min. IKKα/β phosphorylation and IκBα degradation were examined via immunoblot with actin as loading controls. Each blot is representative of three independent experiments. *, p < 0.05; **, p < 0.01.
Having confirmed this, the role of SK1 and, by extension, S1P in regulating key components of the TNF signaling pathway were next evaluated (Fig. 3). One of the earliest events in this cascade is phosphorylation of the IKKs. As can be seen, HeLa cells treated with control AStar siRNA exhibited robust increases in phosphorylated IKKα/β (pIKKα/β) at 10 min of TNF treatment, returning to basal levels by 30 min (Fig. 3B). Notably, cells treated with SK1 siRNA also exhibited robust phosphorylation of IKK proteins in response to TNF, which temporally mirrored the control siRNA-treated cells (Fig. 3B). Importantly, there was no difference in total levels of IKKα and IKKβ in the presence or absence of TNF or SK1 siRNA (Fig. 3B).
The next major step downstream of IKK phosphorylation and activation is the phosphorylation and degradation of IκB-α. Consistent with activation of IKKs, cells treated with AStar siRNA or SK1 siRNA showed robust IκBα phosphorylation in response to TNF treatment, which was followed by IκB-α degradation (Fig. 3B). Furthermore, robust degradation of IκB in response to TNF was also observed in A549 cells, a distinct model of TNF-induced inflammatory signaling (supplemental Fig. S3).
The above results suggest that TNF activation of the NF-κB cascade is not affected by the loss of SK1 in A549 or HeLa cells. However, to further evaluate this by genetic means, IκB-α degradation was examined in MEFs derived from WT and SK1−/− mice (Fig. 3C). Similar to the results with knockdown of SK1 in HeLa cells, IκB-α degradation upon TNF treatment was unaffected in SK1 null fibroblasts (Fig. 3C). WT cells treated with TNF displayed IκB-α degradation beginning at 5 min, which returned to basal levels following 1 h of treatment (Fig. 3C). SK1−/− fibroblasts, although expressing less total IκBα, exhibited the same pattern of IκB-α degradation as did WT. To confirm that this was not due to clone-specific effects, these observations were confirmed in a different clone of WT and SK1 MEFs (supplemental Fig. S4). As expected, despite lower basal levels of IκB-α, there was no significant difference in the pattern of IκB-α degradation in SK1−/− fibroblasts compared with WT cells (supplemental Fig. S4). These results are in contrast to those of Alvarez et al. (11), who showed that the NF-κB cascade is totally halted in SK1−/− MEFs upon TNF stimulation.
Finally, to be sure that the dose of TNF used is not overwhelming the response, thus masking any differences that could otherwise be seen, a dose response with TNF in HeLa cells treated with AStar or SK1 siRNA was performed, and IKK phosphorylation and IκBα degradation were assessed. Even at very low doses, no differences were seen between the control and the SK1 knockdown cells (Fig. 3D). Taken together, these results demonstrate that SK1 is not required for this early signaling event.
Loss of SK2 Does Not Affect NF-κB Pathway Activation
Because the loss of SK2 resulted in a decrease in RANTES, we also considered that SK2 might affect NF-κB activation in this system. As can be seen, loss of SK2 somewhat decreased total IKK levels, yet IKK phosphorylation remained intact at both time points compared with AStar-treated cells (supplemental Fig. S1C). Moreover, IκB-α degradation was comparable in SK2-treated cells compared with AStar controls (supplemental Fig. S1C). Collectively, these results show that SK2 is also not required for acute activation of the NF-κB cascade.
Nuclear Translocation of NF-κB Subunits Does Not Require SK1
Although the early activation of the NF-κB pathway by TNF appears to be independent of SK1, it is also possible that subsequent steps, such as translocation of NF-κB subunits to the nucleus, an essential step for NF-κB-mediated gene expression (45), may require SK1. Initially, the levels of the NF-κB subunits p50/NFkB1 and p65/RelA were assessed in the presence and absence of SK1 (Fig. 3B), with results indicating no significant difference. Accordingly, the translocation of both subunits to the nucleus was assessed by immunofluorescence and confocal microscopy (Fig. 4). As seen in Fig. 4A, the p65 subunit localized to the cytoplasm under basal conditions and upon treatment with TNF translocated to the nucleus (blue, Draq5) (Fig. 4A). Strikingly, this occurred in both AStar- and SK1-siRNA-treated cells. To confirm this, TNF-mediated p65 nuclear translocation was assessed in MEFs derived from WT or SK1−/− mice. Importantly, as with siRNA-treated HeLa cells, TNF induced comparable translocation of p65 to the nucleus in both WT and SK1−/− cells (Fig. 4B). Taken together, these results indicate that SK1 is not required for nuclear translocation of NF-κB subunits in response to TNF.
FIGURE 4.
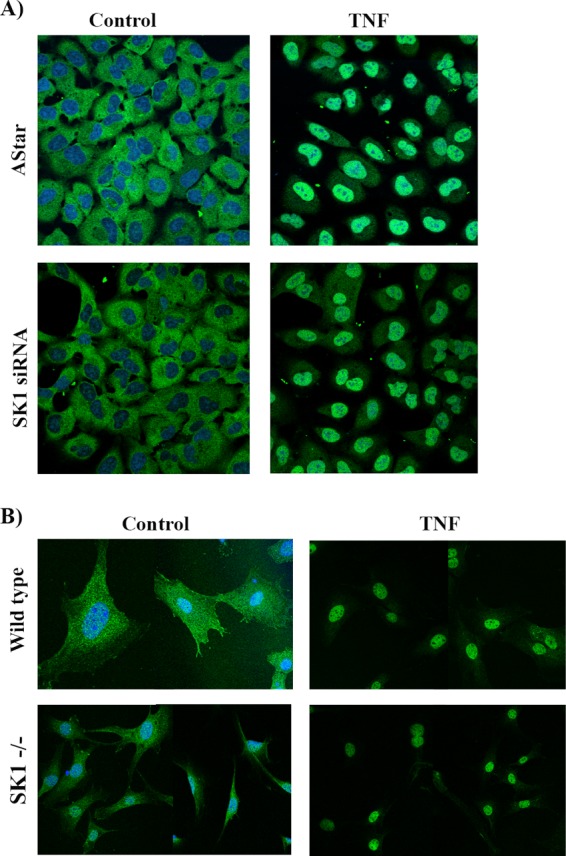
SK1 is not required for TNF-induced nuclear translocation of p65/RelA. A, HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h prior to treatment with vehicle (PBS) or TNF (20 ng/ml) for 30 min. The cells were fixed, permeabilized, and probed with p65 antibody. p65 localization was examined by confocal microscopy. B, WT or SK1−/− MEFs were exposed to TNF (50 ng/ml) for 30 min. The cells were fixed, permeabilized, and probed with p65 antibody. p65 localization was examined using confocal microscopy. Images are representative of at least five fields taken from at least two independent experiments.
SK1 Is Not Required for TNF-mediated Activation of NF-κB Reporter Activity
Although nuclear translocation of NF-κB subunits remained intact, it is also possible that the loss of SK1 may affect NF-κB-mediated transcription following translocation of NF-κB to the nucleus (e.g., promoter binding, subunit phosphorylation, or interactions with other factors). To probe this, an NF-κB luciferase reporter assay was performed (Fig. 5). The results demonstrated that TNF strongly increases NF-κB transcriptional activity (25-fold over vehicle) in negative control siRNA-transfected cells. Strikingly, loss of SK1 had no significant effect on basal reporter activity and did not prevent TNF-mediated NF-κB activity. Indeed, loss of SK1 tended to increase NF-κB reporter activity compared with negative control cells, although this did not reach statistical significance (Fig. 5). Taken together with the results above, this indicates that SK1 is not required for NF-κB-mediated gene transcription in response to TNF.
FIGURE 5.
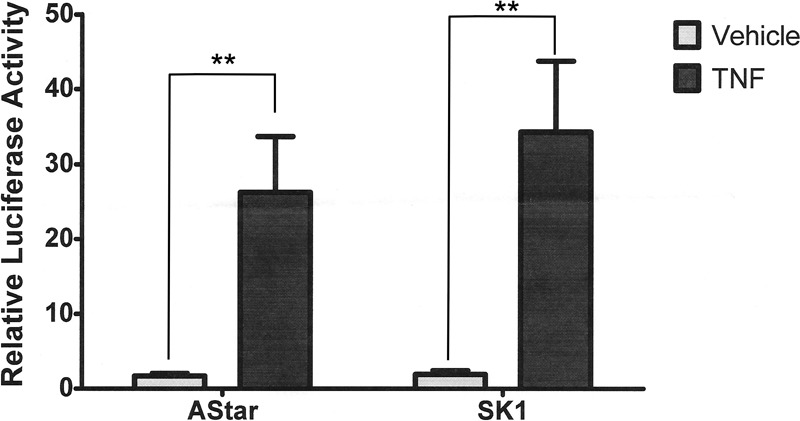
SK1 is not required for transcriptional NF-κB activity following TNF treatment. HeLa cells were treated with 20 nm Astar or SK1 siRNA for 24 h prior to co-transfection with LacZ and NF-κB-Luciferase constructs. After 6 h of transfection, the cells were treated with vehicle (PBS) or TNF (20 ng/ml) for 18 h. Luciferase and galactosidase activities were extracted and assayed as described under “Experimental Procedures,” and the measured luciferase activity was normalized to measured galactosidase activity. The data are presented as means ± S.E. of three independent experiments (**, p < 0.01).
p38 MAPK Requires SK1 to Regulate RANTES Expression
Because SK1 did not appear to significantly affect the NF-κB pathway, it was necessary to consider alternative pathways that might regulate RANTES expression in an SK1-dependent manner. The p38 MAPK pathway is a well known component of TNF signaling (46, 47) and can also be regulated by SK1 (48–50). Furthermore, this pathway can also affect chemokine expression, although the effects on RANTES vary depending on the cell type and stimulus (34, 51, 52). Consequently, as a first step, the role of p38 MAPK in RANTES regulation was determined utilizing the p38 MAPK inhibitor BIRB 796 (53). As shown in Fig. 6A, inhibition of p38 MAPK resulted in a significant increase in both RANTES mRNA and protein levels upon TNF stimulation (2.5-fold compared with vehicle) (Fig. 6, A and B).
FIGURE 6.

Inhibition of p38 MAPK enhances RANTES induction. A, HeLa cells were treated with vehicle (Me2SO) or the p38 MAPK inhibitor BIRB796 (10 μm) for 1 h prior to treatment with PBS or TNF (20 ng/ml) for 24 h. Subsequently, RANTES mRNA levels were assessed by qRT-PCR. The data are presented as means ± S.E. of three independent experiments. B, HeLa cells in serum-free media were treated with vehicle (Me2SO) or the p38 MAPK inhibitor BIRB796 (10 μm) for 1 h prior to treatment with PBS or TNF (20 ng/ml) for 24 h. RANTES protein levels in the media were assessed by ELISA. The data are shown as means ± S.E. from two independent experiments performed in duplicate. C, HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h prior to treatment with vehicle (PBS) or TNF (20 ng/ml) for 10 min. Phospho-p38 and total p38 levels were assessed by immunoblotting with actin as loading. The data are representative of three independent experiments. D, the ratio of phospho-p38/total p38 in TNF-stimulated samples was quantified using ImageJ software. The data are shown as means ± S.E. from three independent experiments. **, p < 0.01; ****, p < 0.0001.
Because this result strongly mimics the effect of SK1 loss on RANTES levels (Fig. 1B), it next became important to assess the effect of SK1 knockdown on p38 MAPK activation. Following 10 min of TNF stimulation, phosphorylation of p38 MAPK was notably increased in control cells. Strikingly, TNF activation of p38 MAPK was significantly blunted following treatment with SK1 siRNA (Fig. 6C). Quantification of the levels of phospho-p38 shows a significant decrease in p38 MAPK activation (Fig. 6D). These results suggest that induction of p38 MAPK by TNF serves as a negative regulator of induction of RANTES. Moreover, the results suggest that SK1 is required for full activation of p38 MAPK in TNF-stimulated HeLa cells.
Finally, because loss of SK2 suppressed RANTES and appeared to be independent of NF-κB activation, we also speculated that loss of SK2 might also affect p38 MAPK. Interestingly, cells treated with SK2 siRNA showed significantly higher p38 MAPK phosphorylation compared with AStar-treated cells (supplemental Fig. S1B). Together with the above results, this provides further evidence that p38 MAPK activation attenuates RANTES production in TNF-stimulated HeLa cells.
Interactions of the p38 MAPK and NF-κB Pathways
Previous studies have suggested that p38 MAPK and NF-κB may function as complementary pathways in response to some stimuli (54, 55). Here, because SK1 appears to be acting through regulation of p38 MAPK but independent of the NF-κB pathway, it was important to determine whether these pathways are functioning independently of each other. To determine the role of the NF-κB pathway in the p38 MAPK pathway, the effect of the NF-κB inhibitor BAY 11-7082 on p38 MAPK phosphorylation was assessed. Strikingly, BAY treatment resulted in a significant increase in the phosphorylation of p38 MAPK both at base line and following TNF stimulation (Fig. 7A). Similarly, following pretreatment with the p38 MAPK inhibitor BIRB 796, there was a significant increase in the transcriptional activity of NF-κB following TNF treatment (Fig. 7B). Therefore, in HeLa cells, the p38 MAPK pathway and the NF-κB pathway exert dual inhibition on each other following TNF treatment.
FIGURE 7.
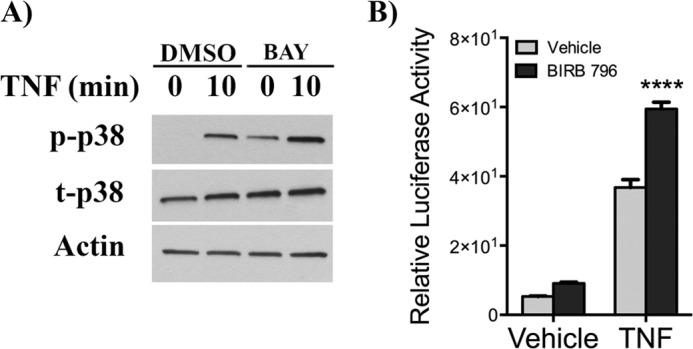
p38 MAPK and NF-κB pathways cross-talk in the TNF response. A, HeLa cells were treated with dimethyl sulfoxide (DMSO) or BAY (5 μm) for 30 min prior to stimulation with vehicle (PBS) or TNF (20 ng/ml) for 10 min. Phospho-p38 and total p38 levels were assessed by immunoblotting with actin as loading control. The data are shown as means ± S.E. from three independent experiments. B, HeLa cells were co-transfected with LacZ and NF-κB-Luciferase constructs for 6 h and preincubated with vehicle (dimethyl sulfoxide) or BIRB796 (10 μm) for 1 h prior to stimulation with PBS or TNF (20 ng/ml) for 18 h. Luciferase and galactosidase activities were extracted and assayed as described under “Experimental Procedures,” and measured luciferase activity was normalized to measured galactosidase activity. The data are presented as means ± S.E. of three independent experiments. ****, p < 0.0001.
SK1 Knockdown Affects Multiple Cytokines and Chemokines in a Manner Similar to the Effects on RANTES
The results thus far suggest that SK1 regulates RANTES independently of NF-κB, despite this pathway being a major regulatory factor for RANTES in HeLa cells. Moreover, the observation that loss of SK1 enhanced RANTES induction is somewhat counterintuitive because SK1 is thought to be pro-inflammatory (1). This led us to question what are the effects of SK1 on other chemokines and cytokines beyond RANTES. To this end, a quantitative PCR array that evaluates the expression of 84 key-secreted cytokines and chemokines central to the immune response was utilized. As expected, RANTES levels were enhanced in SK1 siRNA cells treated with TNF, consistent with the preceding results. Interestingly, SK1 knockdown dramatically increased TNF induction of multiple other chemokines including CXCL1, CCL20, CXCL10, and the interleukins IL-8 and IL-6 when compared with TNF-stimulated negative control siRNA cells (supplemental Table 1). Moreover, real time PCR analysis of some of these mediators with independent primers confirmed the array results indicating that a loss of SK1 significantly enhanced TNF induction of numerous chemokines in HeLa cells including CXCL1, CXCL10, IL-8, and CCL20 (Fig. 8). Collectively, these results suggest that SK1 has a broad role in regulation of TNF-mediated chemokine transcription and, together with the results above, suggest that SK1 might be a negative regulator of NF-κB activation in the coordination of the TNF-induced inflammatory response in HeLa cells.
FIGURE 8.
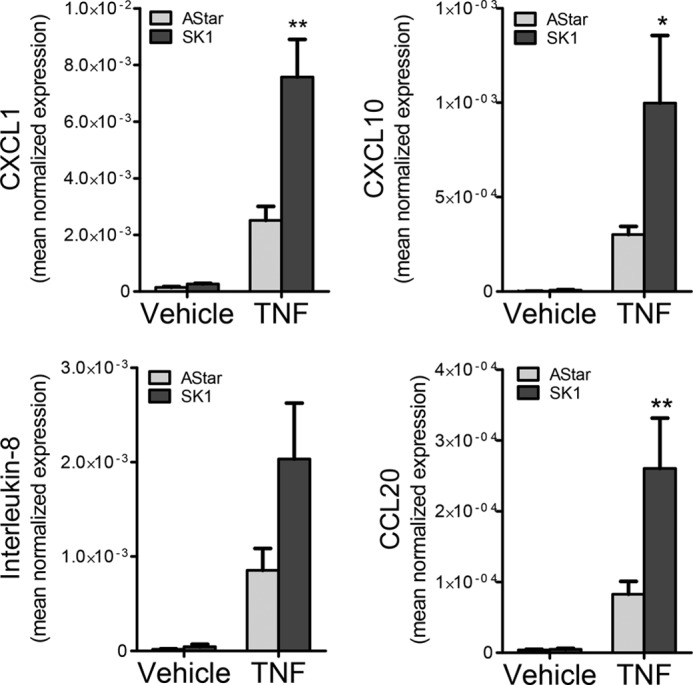
Loss of SK1 enhances the levels of multiple chemokines and cytokines. HeLa cells were treated with 20 nm Astar or SK1 siRNA for 48 h prior to treatment with vehicle (PBS) or TNF (20 ng/ml) for 24 h. RNA was extracted, and the levels of CXCL1, CXCL10, IL-8, and CCL20 mRNA were assessed by qRT-PCR. The data are presented as means ± S.E. from three independent experiments. *, p < 0.05; **, p < 0.01.
DISCUSSION
SK1 is a key enzyme of sphingolipid metabolism, regulating the levels of the pro-growth lipid S1P with the pro-apoptotic lipids ceramide and sphingosine. In the current study, we report that loss of SK1 significantly increases levels of the pro-inflammatory chemokine RANTES in TNF-stimulated HeLa cells and that SK1 exerts these effects through regulation of the p38 MAPK pathway. Notably, SK1 was not required for TNF-stimulated activation, nuclear translocation, or transcriptional activity of NF-κB subunits. Finally, we also show that loss of SK1 has similar effects on multiple chemokines. These results suggest that SK1 can have an anti-inflammatory role in the TNF response that mechanistically occurs through p38 MAPK.
SK1 Regulation of RANTES
RANTES promotes the chemotaxis of multiple cell types to sites of inflammation and is essential for the physiological inflammatory response. Here, we find that a loss of SK1 in a TNF-stimulated model of inflammation markedly increases RANTES mRNA and protein, corroborating and extending our previous study (16). Importantly, this was specific to SK1 because the loss of SK2 produced the opposite effect, attenuating RANTES induction by TNF. Notably, this latter effect differs from our previous observations in MEFs and MCF-7 cells (16). This suggests cell-specific roles for SK2 in the TNF response and, consistent with this, the effects of TNF on acute S1P levels were wholly SK1-dependent. Thus, the effect of SK2 could be due to basal effects on cellular sphingolipids rather than a specific signaling role in the TNF response. Studies exploring this further are ongoing in our laboratory.
Because RANTES mRNA is reported to be extremely stable (28), efforts to further explore SK1-RANTES connections focused on transcriptional regulation. Although regulated by multiple transcription factors such as STAT3 (20), NF-AT, and activator protein 1 (31), RANTES induction in the current system was highly NF-κB-dependent (Fig. 2). Despite this, SK1 does not appear to affect RANTES through the NF-κB pathway; indeed, loss of SK1 was able to enhance RANTES levels even in the presence of NF-κB inhibitors (data not shown), thereby identifying a novel pathway by which SK1 regulates gene expression. Because RANTES is primarily regulated at the transcriptional level, we can surmise that the effects of SK1 must also occur transcriptionally, potentially through transcription factors downstream of the p38 MAPK pathway. Alternatively, this may occur through non-promoter-mediated effects such as regulation of histone deacetylases. Indeed, because SK2 is reported to increase gene transcription through inhibiting histone deacetylases (56), we initially speculated that loss of SK1 could increase substrate availability for SK2, thereby up-regulating RANTES through prolonged inhibition of histone deacetylases. However, when we explored this possibility, we found that the histone deacetylase inhibitor trichostatin A significantly blunted RANTES induction in our system (data not shown). Clearly, the transcriptional mechanisms by which SK1 affects RANTES require further study.
SKi-II and the RANTES Response
Despite the use of genetic tools, assessing the effects of SK inhibitors on RANTES induction was also important yet, unexpectedly, experiments with the generic inhibitor SKi-II showed an inhibition of RANTES induction. A primary reason for this relates to specificity of the SKi-II inhibitor for SK isoforms because recently published data reports that this inhibitor more favorably targets SK2 rather than SK1 (37). Consistent with this, results with this inhibitor more closely match the effects of SK2 siRNA in the HeLa system (supplemental Fig. S1B). A second, related reason, relates to the more general possibility of off target effects; indeed, SKi-II is reported to also inhibit PI3K, ERK, and PKCα (57). In the absence of strong specific SK1 inhibitors, we acknowledge this as a limitation of the current study. The testing of newer, more specific SK1 inhibitors remains an ongoing endeavor in our lab; moreover, because SK1 is considered a strong therapeutic target for the treatment of various pathologies (1), future studies determining whether post-translational inhibition of SK1 can adversely induce chemokines are of paramount importance.
SK1 and the NF-κB Pathway
A major finding of this study, mechanistically, is that SK1 and SK1-derived S1P are completely dispensable for activation of the NF-κB pathway and, in fact, can enhance gene expression mediated by this pathway. This is shown by a number of lines of evidence. First, TNF induced an acute increase in S1P levels that was completely prevented by loss of SK1. Second, loss of SK1 did not prevent phosphorylation of IKK, IκB, or the subsequent degradation of IκB in response to TNF. Importantly, this was not an effect of siRNA or cell type because similar effects were observed in SK1−/− fibroblasts as well as in A549 cells. Third, loss of SK1 did not have effects further downstream in the NF-κB pathway because total levels of p65 and p50 were unchanged, and their translocation into the nucleus remained intact (also observed in both siRNA-treated cells and SK1−/− fibroblasts). Finally, transcriptional NF-κB activity, as assessed by a luciferase reporter assay, remained intact and indeed was slightly increased (albeit not significantly) following loss of SK1. This was also true for endogenous genes, with loss of SK1 enhancing the induction of the highly NF-κB-dependent RANTES, rather than inhibiting it as would be expected if NF-κB activation were compromised. Furthermore, loss of SK1 had similar effects even in the presence of NF-κB pathway inhibition (data not shown). Taken together, this clearly suggests an NF-κB-independent mechanism for SK1 and, by extension, SK1-derived S1P in this system. Strikingly, this sharply contrasts with previous studies reporting SK1 as essential for NF-κB activation through effects on TRAF2 activation and subsequent activation of IKK and degradation of IκB (11).The reasons for this discrepancy are unclear. It cannot be explained by the doses of TNF utilized, because similar effects were observed at a wide range of TNF doses (Fig. 3D). It is also possible that SK1 requirement for NF-κB activation is cell type-specific; however, comparable effects were seen across three different cell types including MEFs that were previously reported to require SK1 for activation of the TNF cascade (11). A primary possibility to consider was that TNF did not induce an acute generation of S1P in these systems, given that S1P was reported to be key for activation of TRAF2 and subsequent IκB degradation (11). However, as seen above (Fig. 2A), this was not the case, because TNF induced a clear increase in intracellular S1P that was wholly SK1-dependent. Looking beyond sphingolipids, an alternative reason somewhat related to cell type is that TRAF5 could compensate for TRAF2 (presuming that is inactive in the absence of S1P) in the current system. Consistent with this, NF-κB activation remains intact in TRAF2 knock-out mice because of TRAF5 compensation, whereas only double knock-out mice for TRAF2 and TRAF5 have defective NF-κB activation (58). Although exploring this possibility is beyond the scope of the current study, our data nonetheless suggest that SK1 and intracellular S1P regulation of NF-κB and gene transcription in the cytokine response may be more complex than currently thought.
SK1 Regulation of p38 MAPK Activation and the Interconnection with the NF-κB Pathway
The role of p38 MAPK in the inflammatory response is well documented, and it has been reported to affect the levels of multiple cytokines and chemokines including IL-12, IL-1B, IL10, and IL-6 (59, 60); notably, however, this regulation is both cell type- and stimulus-dependent. Here, in TNF-stimulated HeLa cells, inhibition of p38 MAPK resulted in a marked up-regulation of RANTES mRNA and protein, results that were concordant with a study in smooth muscle cells stimulated with LPS and IFN-Y (34). Consequently, the fact that SK1 knockdown significantly reduces p38 MAPK activity strongly suggests that the effects of SK1 on RANTES are mediated by activation of the p38 MAPK pathway. Corroborating evidence for this inhibitory role of p38 MAPK on RANTES was also provided by experiments with SK2 siRNA. As with loss of SK1, the effects of SK2 siRNA on RANTES appear to be largely independent of the NF-κB pathway yet led to augmented phosphorylation of p38 MAPK.
The p38 MAPK pathway cross-talks with several other pathways such as the NF-κB, Akt, and Wnt/β-catenin pathways (61). Most reports state that p38 MAPK can positively regulate NF-κB activity, albeit through various mechanisms (62–64). In contrast, inhibition of p38 MAPK in our system caused a significant increase in NF-κB activity in HeLa cells. This is consistent with two previous studies in IL-1-stimulated HeLa and HEK293 cells (65) and in KB epithelial cells (66). Although TNF is reported to activate p38 MAPK independently of the NF-κB pathway (67), it was also important to determine whether there was reciprocal cross-talk between NF-κB and p38 MAPK in the current system. Indeed, inhibition of the NF-κB pathway also enhanced p38 MAPK phosphorylation both basally and upon stimulation with TNF, although it should be noted that the magnitude of p38 MAPK activation was comparable. Nonetheless, this does raise the possibility that the reduced induction of RANTES seen upon NF-κB inhibition is a result of dual mechanisms; namely, both decreased NF-κB-dependent transcription and an active suppression of RANTES mediated by p38 MAPK. These mechanisms, and their relationship to SK1, are currently under further investigation in our laboratory. The interconnections and the complexity of SK1 regulation of the RANTES response are summarized in Fig. 9.
FIGURE 9.
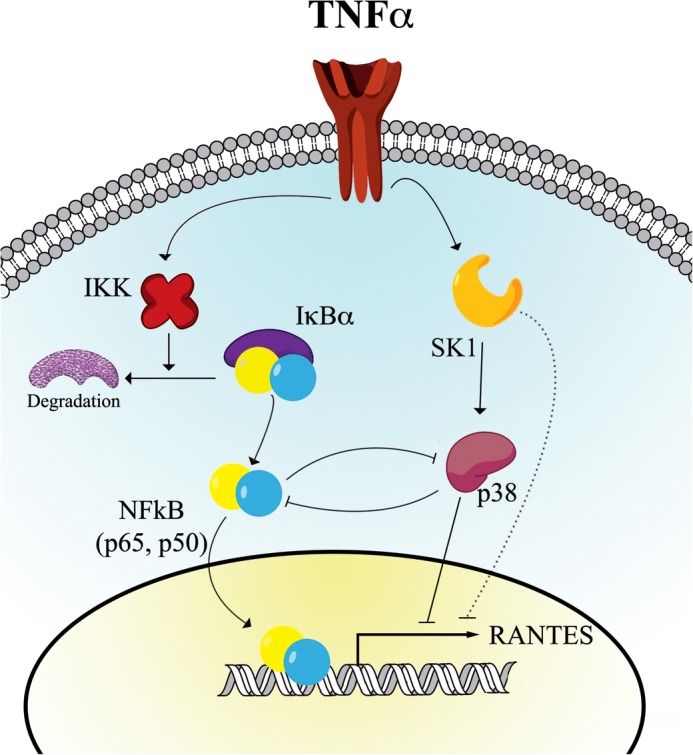
Proposed model of RANTES regulation by SK1. TNF stimulation leads to activation of SK1 and S1P generation that, in turn, mediates activation of p38 MAPK. Activation of this pathway suppresses RANTES transcription through an as yet unknown repressor factor. Concurrently, TNF activates the NF-κB pathway independently of SK1, which causes transcriptional up-regulation of RANTES expression and a simultaneous inhibition of p38 MAPK activation. It should also be noted that we cannot rule out the possibility that SK1 has effects on RANTES transcription independently of the p38 MAPK pathway.
The Sphingolipid Regulator of RANTES
The current study also returns us to the question of the identity of specific sphingolipid regulator(s) of RANTES induction in the TNF response. In our previous study, data suggested that sphingosine derived from secretory sphingomyelinase and acid ceramidase was important for RANTES induction in MCF-7 cells, and this was further supported by the enhancing effects of SK1 and SK2 siRNA on RANTES induction in that system (16). In contrast, in HeLa cells, no significant increases in ceramide or sphingosine were observed upon acute TNF stimulation. Moreover, no differences in either lipid was observed in SK1 siRNA-treated cells, suggesting that enhancement of RANTES upon loss of SK1 is not through a general increase in sphingosine (supplemental Fig. S5).
As noted above, the time frame of S1P generation and its coincidence with p38 MAPK activation leads us to conclude that likely the effects of SK1 in this system are mediated through S1P activation of p38 MAPK activation. That being said, all lipid measurements here were at very short time points to detect S1P production as documented by Alvarez et al. (11) and Pettus et al. (44). Thus, we cannot explicitly rule out a role for ceramide or sphingosine in regulating RANTES production at later time points. Indeed, in our previous study, TNF-induced increases in secretory sphingomyelinase activity and ceramides occurred at later times (>12 h) of stimulation (16). In addition, although we could not detect any changes in total sphingosine or ceramide levels at early time points, we also cannot rule out the possibility of local changes in lipid levels occurring in specific cellular compartments that may impact RANTES induction. Finally, it is a distinct possibility that multiple sphingolipids can regulate RANTES induction through distinct mechanisms. Clearly, given the importance of RANTES in physiological inflammation, and its role in a variety of pathologies (21–27), further research in this area could yield a number of potential therapeutic targets.
SK1 Regulation of the Chemokine Response
Beyond RANTES, the loss of SK1 also had a broader effect on inflammatory cytokine and chemokine production in tumor cells, having a comparable effect on CCL20, CXCL1, and CXCL10, among others. Importantly, this offers additional evidence arguing against a role for SK1 in regulating NF-κB in this system; induction of CCL20 and CXCL10 is also partially NF-κB-dependent, whereas CXCL1 induction is NF-κB-independent (data not shown). Although the biological consequences of SK1 loss and subsequent chemokine up-regulation are unclear, many possibilities are suggested by their known roles. For example, CXCL10 overexpression leads to anti-tumor effects by blocking angiogenesis as seen in cervical carcinoma (68). Notably, SK1 is reported to be strongly pro-angiogenic (69); thus, regulation of CXCL10 may be a potential mechanism by which inhibiting SK1 blocks angiogenesis. CCL20 expression in the colon recruits dendritic cells to combat infections (70), important for recognition of cancer cells on one hand, but also the source of an exacerbated inflammatory response that would set the ground for cancer; thus, targeting SK1 in this context could be a double-edged sword. Finally, CXCL1 has been considered an oncogene in human melanoma (71), and so targeting SK1 in this context might exacerbate its oncogenic effects. Collectively, the up-regulation of any of these chemokines in the tumor environment could be detrimental to cancer patients or could promote tumor progression in some cancers. Consequently, although studies have suggested that SK1 is an attractive therapeutic target for many cancers (1), further research is clearly required to ensure that undesirable side effects, which could potentially promote tumor progression, do not occur.
Conclusions
In summary, the current study suggests a novel anti-inflammatory function of SK1 and intracellular S1P in the regulation of chemokine production in the TNF response. Importantly, SK1 seemed to exert its effects through modulation of the p38 MAPK pathway and was not required for activation of the canonical NF-κB pathway; indeed, NF-κB activity was somewhat enhanced by loss of SK1. Taken together, this suggests a novel mechanism downstream of p38 MAPK by which SK1 can negatively regulate gene expression in the TNF response. Because SK1 is considered an attractive therapeutic target for many cancers and other disorders, a cautious approach may be required to ensure that SK1 inhibition does not trigger a similar pro-inflammatory chemokine response. Indeed, such an effect could have deleterious effects on disease progression and patient health.
Supplementary Material
Acknowledgments
We thank all the Obeid and Hannun lab members for constructive comments during the presentation of this work. We also thank the Medical University of South Carolina (MUSC) Cell and Molecular Imaging Core for use of the confocal microscope and the Stony Brook Lipidomics/Proteomics Core for analysis of cellular sphingolipids.
This work was supported, in whole or in part, by National Institutes of Health Grants PO1-CA97132 (to L. M. O.) and R01-GM062887 (to L. M. O.). This work was also supported by MERIT Award BX000156 (to L. M. O.) from the Office of Research and Development, Department of Veterans Affairs, Northport Veterans Affairs Medical Center (Northport, NY).

This article contains supplemental Table S1 and Figs. S1–S5.
- SK
- sphingosine kinase
- CCL
- chemokine (C-C motif) ligand
- CXCL
- chemokine (CXC motif) ligand
- IKK
- IκB kinase
- MEF
- mouse embryonic fibroblasts
- RANTES
- regulated on activation, normal T cell expressed and secreted
- S1P
- sphingosine-1-phosphate
- TRAF
- TNF receptor-associated factor
- qRT-PCR
- quantitative (real time) RT-PCR.
REFERENCES
- 1. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling. Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 2. Deutschman D. H., Carstens J. S., Klepper R. L., Smith W. S., Page M. T., Young T. R., Gleason L. A., Nakajima N., Sabbadini R. A. (2003) Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am. Heart J. 146, 62–68 [DOI] [PubMed] [Google Scholar]
- 3. Yung L. M., Wei Y., Qin T., Wang Y., Smith C. D., Waeber C. (2012) Sphingosine kinase 2 mediates cerebral preconditioning and protects the mouse brain against ischemic injury. Stroke 43, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park S. W., Kim M., Brown K. M., D'Agati V. D., Lee H. T. (2012) Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 23, 266–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selvam S. P., Ogretmen B. (2013) Sphingosine kinase/sphingosine 1-phosphate signaling in cancer therapeutics and drug resistance. Handb. Exp. Pharmacol. 216, 3–27 [DOI] [PubMed] [Google Scholar]
- 6. Pitson S. M., Moretti P. A., Zebol J. R., Lynn H. E., Xia P., Vadas M. A., Wattenberg B. W. (2003) Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 22, 5491–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strub G. M., Paillard M., Liang J., Gomez L., Allegood J. C., Hait N. C., Maceyka M., Price M. M., Chen Q., Simpson D. C., Kordula T., Milstien S., Lesnefsky E. J., Spiegel S. (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hisano Y., Kobayashi N., Yamaguchi A., Nishi T. (2012) Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS One 7, e38941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nijnik A., Clare S., Hale C., Chen J., Raisen C., Mottram L., Lucas M., Estabel J., Ryder E., Adissu H., Sanger Mouse Genetics Project, Adams N. C., Ramirez-Solis R., White J. K., Steel K. P., Dougan G., Hancock R. E. (2012) The role of sphingosine-1-phosphate transporter Spns2 in immune system function. J. Immunol. 189, 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aarthi J. J., Darendeliler M. A., Pushparaj P. N. (2011) Dissecting the role of the S1P/S1PR axis in health and disease. J. Dent. Res. 90, 841–854 [DOI] [PubMed] [Google Scholar]
- 11. Alvarez S. E., Harikumar K. B., Hait N. C., Allegood J., Strub G. M., Kim E. Y., Maceyka M., Jiang H., Luo C., Kordula T., Milstien S., Spiegel S. (2010) Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sawada M., Kiyono T., Nakashima S., Shinoda J., Naganawa T., Hara S., Iwama T., Sakai N. (2004) Molecular mechanisms of TNF-α-induced ceramide formation in human glioma cells. p53-mediated oxidant stress-dependent and -independent pathways. Cell Death Differ. 11, 997–1008 [DOI] [PubMed] [Google Scholar]
- 13. Osawa Y., Banno Y., Nagaki M., Brenner D. A., Naiki T., Nozawa Y., Nakashima S., Moriwaki H. (2001) TNF-α-induced sphingosine 1-phosphate inhibits apoptosis through a phosphatidylinositol 3-kinase/Akt pathway in human hepatocytes. J. Immunol. 167, 173–180 [DOI] [PubMed] [Google Scholar]
- 14. Clarke C. J., Cloessner E. A., Roddy P. L., Hannun Y. A. (2011) Neutral sphingomyelinase 2 (nSMase2) is the primary neutral sphingomyelinase isoform activated by tumour necrosis factor-α in MCF-7 cells. Biochem. J. 435, 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clarke C. J., Truong T. G., Hannun Y. A. (2007) Role for neutral sphingomyelinase-2 in tumor necrosis factor α-stimulated expression of vascular cell adhesion molecule-1 (VCAM) and intercellular adhesion molecule-1 (ICAM) in lung epithelial cells. p38 MAPK is an upstream regulator of nSMase2. J. Biol. Chem. 282, 1384–1396 [DOI] [PubMed] [Google Scholar]
- 16. Jenkins R. W., Clarke C. J., Canals D., Snider A. J., Gault C. R., Heffernan-Stroud L., Wu B. X., Simbari F., Roddy P., Kitatani K., Obeid L. M., Hannun Y. A. (2011) Regulation of CC ligand 5/RANTES by acid sphingomyelinase and acid ceramidase. J. Biol. Chem. 286, 13292–13303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levy J. A. (2009) The unexpected pleiotropic activities of RANTES. J. Immunol. 182, 3945–3946 [DOI] [PubMed] [Google Scholar]
- 18. Maune S., Berner I., Sticherling M., Kulke R., Bartels J., Schröder J. M. (1996) Fibroblasts but not epithelial cells obtained from human nasal mucosa produce the chemokine RANTES. Rhinology 34, 210–214 [PubMed] [Google Scholar]
- 19. Casola A., Henderson A., Liu T., Garofalo R. P., Brasier A. R. (2002) Regulation of RANTES promoter activation in alveolar epithelial cells after cytokine stimulation. Am. J. Physiol. Lung Cell Mol. Physiol. 283, L1280–L1290 [DOI] [PubMed] [Google Scholar]
- 20. Kovacic J. C., Gupta R., Lee A. C., Ma M., Fang F., Tolbert C. N., Walts A. D., Beltran L. E., San H., Chen G., St Hilaire C., Boehm M. (2010) STAT3-dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J. Clin. Invest. 120, 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krohn R., Raffetseder U., Bot I., Zernecke A., Shagdarsuren E., Liehn E. A., van Santbrink P. J., Nelson P. J., Biessen E. A., Mertens P. R., Weber C. (2007) Y-box binding protein-1 controls CC chemokine ligand-5 (CCL5) expression in smooth muscle cells and contributes to neointima formation in atherosclerosis-prone mice. Circulation 116, 1812–1820 [DOI] [PubMed] [Google Scholar]
- 22. Charni F., Friand V., Haddad O., Hlawaty H., Martin L., Vassy R., Oudar O., Gattegno L., Charnaux N., Sutton A. (2009) Syndecan-1 and syndecan-4 are involved in RANTES/CCL5-induced migration and invasion of human hepatoma cells. Biochim. Biophys. Acta 1790, 1314–1326 [DOI] [PubMed] [Google Scholar]
- 23. Karnoub A. E., Dash A. B., Vo A. P., Sullivan A., Brooks M. W., Bell G. W., Richardson A. L., Polyak K., Tubo R., Weinberg R. A. (2007) Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449, 557–563 [DOI] [PubMed] [Google Scholar]
- 24. Herder C., Illig T., Baumert J., Müller M., Klopp N., Khuseyinova N., Meisinger C., Poschen U., Martin S., Koenig W., Thorand B. (2008) RANTES/CCL5 gene polymorphisms, serum concentrations, and incident type 2 diabetes. Results from the MONICA/KORA Augsburg case-cohort study, 1984–2002. Eur. J. Endocrinol. 158, R1–R5 [DOI] [PubMed] [Google Scholar]
- 25. Krensky A. M., Ahn Y. T. (2007) Mechanisms of disease. Regulation of RANTES (CCL5) in renal disease. Nat. Clin. Pract Nephrol. 3, 164–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boiardi L., Macchioni P., Meliconi R., Pulsatelli L., Facchini A., Salvarani C. (1999) Relationship between serum RANTES levels and radiological progression in rheumatoid arthritis patients treated with methotrexate. Clin. Exp. Rheumatol. 17, 419–425 [PubMed] [Google Scholar]
- 27. Tyner J. W., Uchida O., Kajiwara N., Kim E. Y., Patel A. C., O'Sullivan M. P., Walter M. J., Schwendener R. A., Cook D. N., Danoff T. M., Holtzman M. J. (2005) CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat. Med. 11, 1180–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hao S., Baltimore D. (2009) The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 10, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Homma T., Matsukura S., Hirose T., Ohnishi T., Kimura T., Kurokawa M., Ieki K., Odaka M., Suzuki S., Watanabe S., Sato M., Kawaguchi M., Schleimer R. P., Adachi M. (2010) Cooperative activation of CCL5 expression by TLR3 and tumor necrosis factor-α or interferon-γ through nuclear factor-κB or STAT-1 in airway epithelial cells. Int. Arch. Allergy Immunol. 152, 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsuzaki S., Ishizuka T., Hisada T., Aoki H., Komachi M., Ichimonji I., Utsugi M., Ono A., Koga Y., Dobashi K., Kurose H., Tomura H., Mori M., Okajima F. (2010) Lysophosphatidic acid inhibits CC chemokine ligand 5/RANTES production by blocking IRF-1-mediated gene transcription in human bronchial epithelial cells. J. Immunol. 185, 4863–4872 [DOI] [PubMed] [Google Scholar]
- 31. Ammit A. J., Lazaar A. L., Irani C., O'Neill G. M., Gordon N. D., Amrani Y., Penn R. B., Panettieri R. A., Jr. (2002) Tumor necrosis factor-α-induced secretion of RANTES and interleukin-6 from human airway smooth muscle cells. Modulation by glucocorticoids and β-agonists. Am. J. Respir. Cell Mol. Biol. 26, 465–474 [DOI] [PubMed] [Google Scholar]
- 32. Kumar D., Hosse J., von Toerne C., Noessner E., Nelson P. J. (2009) JNK MAPK pathway regulates constitutive transcription of CCL5 by human NK cells through SP1. J. Immunol. 182, 1011–1020 [DOI] [PubMed] [Google Scholar]
- 33. Wong C. K., Tsang C. M., Ip W. K., Lam C. W. (2006) Molecular mechanisms for the release of chemokines from human leukemic mast cell line (HMC)-1 cells activated by SCF and TNF-α. Roles of ERK, p38 MAPK, and NF-κB. Allergy 61, 289–297 [DOI] [PubMed] [Google Scholar]
- 34. Liu H., Ning H., Men H., Hou R., Fu M., Zhang H., Liu J. (2012) Regulation of CCL5 expression in smooth muscle cells following arterial injury. PLoS One 7, e30873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heffernan-Stroud L. A., Helke K. L., Jenkins R. W., De Costa A. M., Hannun Y. A., Obeid L. M. (2012) Defining a role for sphingosine kinase 1 in p53-dependent tumors. Oncogene 31, 1166–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Canals D., Jenkins R. W., Roddy P., Hernández-Corbacho M. J., Obeid L. M., Hannun Y. A. (2010) Differential effects of ceramide and sphingosine 1-phosphate on ERM phosphorylation. Probing sphingolipid signaling at the outer plasma membrane. J. Biol. Chem. 285, 32476–32485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neubauer H. A., Pitson S. M. (2013) Roles, regulation and inhibitors of sphingosine kinase 2. FEBS J., in press [DOI] [PubMed] [Google Scholar]
- 38. McLoughlin R. M., Jenkins B. J., Grail D., Williams A. S., Fielding C. A., Parker C. R., Ernst M., Topley N., Jones S. A. (2005) IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc. Natl. Acad. Sci. U.S.A. 102, 9589–9594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Werts C., le Bourhis L., Liu J., Magalhaes J. G., Carneiro L. A., Fritz J. H., Stockinger S., Balloy V., Chignard M., Decker T., Philpott D. J., Ma X., Girardin S. E. (2007) Nod1 and Nod2 induce CCL5/RANTES through the NF-κB pathway. Eur. J. Immunol. 37, 2499–2508 [DOI] [PubMed] [Google Scholar]
- 40.Deleted in proof
- 41. Pierce J. W., Schoenleber R., Jesmok G., Best J., Moore S. A., Collins T., Gerritsen M. E. (1997) Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 272, 21096–21103 [DOI] [PubMed] [Google Scholar]
- 42. Xia P., Wang L., Moretti P. A., Albanese N., Chai F., Pitson S. M., D'Andrea R. J., Gamble J. R., Vadas M. A. (2002) Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-α signaling. J. Biol. Chem. 277, 7996–8003 [DOI] [PubMed] [Google Scholar]
- 43. Verhelst K., Carpentier I., Beyaert R. (2011) Regulation of TNF-induced NF-κB activation by different cytoplasmic ubiquitination events. Cytokine Growth Factor Rev. 22, 277–286 [DOI] [PubMed] [Google Scholar]
- 44. Pettus B. J., Bielawski J., Porcelli A. M., Reames D. L., Johnson K. R., Morrow J., Chalfant C. E., Obeid L. M., Hannun Y. A. (2003) The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-α. FASEB J. 17, 1411–1421 [DOI] [PubMed] [Google Scholar]
- 45. Dejardin E. (2006) The alternative NF-κB pathway from biochemistry to biology. Pitfalls and promises for future drug development. Biochem. Pharmacol. 72, 1161–1179 [DOI] [PubMed] [Google Scholar]
- 46. Zhou F. H., Foster B. K., Zhou X. F., Cowin A. J., Xian C. J. (2006) TNF-α mediates p38 MAP kinase activation and negatively regulates bone formation at the injured growth plate in rats. J. Bone Miner Res. 21, 1075–1088 [DOI] [PubMed] [Google Scholar]
- 47. Kilpatrick L. E., Sun S., Mackie D., Baik F., Li H., Korchak H. M. (2006) Regulation of TNF mediated antiapoptotic signaling in human neutrophils. Role of δ-PKC and ERK1/2. J. Leukocyte Biol. 80, 1512–1521 [DOI] [PubMed] [Google Scholar]
- 48. Schenten V., Melchior C., Steinckwich N., Tschirhart E. J., Bréchard S. (2011) Sphingosine kinases regulate NOX2 activity via p38 MAPK-dependent translocation of S100A8/A9. J. Leukocyte Biol. 89, 587–596 [DOI] [PubMed] [Google Scholar]
- 49. Kozawa O., Niwa M., Matsuno H., Tokuda H., Miwa M., Ito H., Kato K., Uematsu T. (1999) Sphingosine 1-phosphate induces heat shock protein 27 via p38 mitogen-activated protein kinase activation in osteoblasts. J. Bone Miner. Res. 14, 1761–1767 [DOI] [PubMed] [Google Scholar]
- 50. Cheon S., Song S. B., Jung M., Park Y., Bang J. W., Kim T. S., Park H., Kim C. H., Yang Y. H., Bang S. I., Cho D. (2008) Sphingosine kinase inhibitor suppresses IL-18-induced interferon-γ production through inhibition of p38 MAPK activation in human NK cells. Biochem. Biophys. Res. Commun. 374, 74–78 [DOI] [PubMed] [Google Scholar]
- 51. Kim M. O., Suh H. S., Brosnan C. F., Lee S. C. (2004) Regulation of RANTES/CCL5 expression in human astrocytes by interleukin-1 and interferon-β. J. Neurochem. 90, 297–308 [DOI] [PubMed] [Google Scholar]
- 52. Melchjorsen J., Paludan S. R. (2003) Induction of RANTES/CCL5 by herpes simplex virus is regulated by nuclear factor κB and interferon regulatory factor 3. J. Gen Virol. 84, 2491–2495 [DOI] [PubMed] [Google Scholar]
- 53. Iwano S., Asaoka Y., Akiyama H., Takizawa S., Nobumasa H., Hashimoto H., Miyamoto Y. (2011) A possible mechanism for hepatotoxicity induced by BIRB-796, an orally active p38 mitogen-activated protein kinase inhibitor. J. Appl Toxicol. 31, 671–677 [DOI] [PubMed] [Google Scholar]
- 54. Baeza-Raja B., Muñoz-Cánoves P. (2004) p38 MAPK-induced nuclear factor-κB activity is required for skeletal muscle differentiation. Role of interleukin-6. Mol. Biol. Cell 15, 2013–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Craig R., Larkin A., Mingo A. M., Thuerauf D. J., Andrews C., McDonough P. M., Glembotski C. C. (2000) p38 MAPK and NF-κB collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J. Biol. Chem. 275, 23814–23824 [DOI] [PubMed] [Google Scholar]
- 56. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. French K. J., Schrecengost R. S., Lee B. D., Zhuang Y., Smith S. N., Eberly J. L., Yun J. K., Smith C. D. (2003) Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 63, 5962–5969 [PubMed] [Google Scholar]
- 58. Tada K., Okazaki T., Sakon S., Kobarai T., Kurosawa K., Yamaoka S., Hashimoto H., Mak T. W., Yagita H., Okumura K., Yeh W. C., Nakano H. (2001) Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-κB activation and protection from cell death. J. Biol. Chem. 276, 36530–36534 [DOI] [PubMed] [Google Scholar]
- 59. Beyaert R., Cuenda A., Vanden Berghe W., Plaisance S., Lee J. C., Haegeman G., Cohen P., Fiers W. (1996) The p38/RK mitogen-activated protein kinase pathway regulates interleukin-6 synthesis response to tumor necrosis factor. EMBO J. 15, 1914–1923 [PMC free article] [PubMed] [Google Scholar]
- 60. Glass C. K., Witztum J. L. (2001) Atherosclerosis. The road ahead. Cell 104, 503–516 [DOI] [PubMed] [Google Scholar]
- 61. Cuadrado A., Nebreda A. R. (2010) Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417 [DOI] [PubMed] [Google Scholar]
- 62. Kefaloyianni E., Gaitanaki C., Beis I. (2006) ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-κB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 18, 2238–2251 [DOI] [PubMed] [Google Scholar]
- 63. Vermeulen L., De Wilde G., Van Damme P., Vanden Berghe W., Haegeman G. (2003) Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 22, 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Saccani S., Pantano S., Natoli G. (2002) p38-dependent marking of inflammatory genes for increased NF-κB recruitment. Nat. Immunol. 3, 69–75 [DOI] [PubMed] [Google Scholar]
- 65. Yagasaki Y., Sudo T., Osada H. (2004) Exip, a splicing variant of p38α, participates in interleukin-1 receptor proximal complex and downregulates NF-κB pathway. FEBS Lett. 575, 136–140 [DOI] [PubMed] [Google Scholar]
- 66. Cheung P. C., Campbell D. G., Nebreda A. R., Cohen P. (2003) Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J. 22, 5793–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Au P. Y., Yeh W. C. (2007) Physiological roles and mechanisms of signaling by TRAF2 and TRAF5. Adv. Exp. Med. Biol. 597, 32–47 [DOI] [PubMed] [Google Scholar]
- 68. Liu M., Guo S., Stiles J. K. (2011) The emerging role of CXCL10 in cancer (Review). Oncol. Lett. 2, 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Anelli V., Gault C. R., Snider A. J., Obeid L. M. (2010) Role of sphingosine kinase-1 in paracrine/transcellular angiogenesis and lymphangiogenesis in vitro. FASEB J. 24, 2727–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ghadjar P., Rubie C., Aebersold D. M., Keilholz U. (2009) The chemokine CCL20 and its receptor CCR6 in human malignancy with focus on colorectal cancer. Int. J. Cancer 125, 741–745 [DOI] [PubMed] [Google Scholar]
- 71. Dhawan P., Richmond A. (2002) Role of CXCL1 in tumorigenesis of melanoma. J. Leukocyte Biol. 72, 9–18 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.