

Background: The LSD1 histone demethylase regulates gene expression by demethylating chromatin.
Results: SFMBT1 associates with the LSD1 complex and is essential for its chromatin association, histone demethylation, and induction of EMT.
Conclusion: SFMBT1 is a novel, integral component of the LSD1 complex.
Significance: Understanding how the LSD1 complex is recruited to chromatin may offer new opportunities for therapeutic intervention.
Keywords: Cell Invasion, Chromatin Histone Modification, Epigenetics, Epithelial-to-mesenchymal Transition, Nickel, Transcription Repressor
Abstract
Chromatin readers decipher the functional readouts of histone modifications by recruiting specific effector complexes for subsequent epigenetic reprogramming. The LSD1 (also known as KDM1A) histone demethylase complex modifies chromatin and represses transcription in part by catalyzing demethylation of dimethylated histone H3 lysine 4 (H3K4me2), a mark for active transcription. However, none of its currently known subunits recognizes methylated histones. The Snai1 family transcription factors are central drivers of epithelial-to-mesenchymal transition (EMT) by which epithelial cells acquire enhanced invasiveness. Snai1-mediated transcriptional repression of epithelial genes depends on its recruitment of the LSD1 complex and ensuing demethylation of H3K4me2 at its target genes. Through biochemical purification, we identified the MBT domain-containing protein SFMBT1 as a novel component of the LSD1 complex associated with Snai1. Unlike other mammalian MBT domain proteins characterized to date that selectively recognize mono- and dimethylated lysines, SFMBT1 binds di- and trimethyl H3K4, both of which are enriched at active promoters. We show that SFMBT1 is essential for Snai1-dependent recruitment of LSD1 to chromatin, demethylation of H3K4me2, transcriptional repression of epithelial markers, and induction of EMT by TGFβ. Carcinogenic metal nickel is a widespread environmental and occupational pollutant. Nickel alters gene expression and induces EMT. We demonstrate the nickel-initiated effects are dependent on LSD1-SFMBT1-mediated chromatin modification. Furthermore, in human cancer, expression of SFMBT1 is associated with mesenchymal markers and unfavorable prognosis. These results highlight a critical role of SFMBT1 in epigenetic regulation, EMT, and cancer.
Introduction
Covalent posttranslational modifications of histones are crucial determinants of chromatin structure and gene transcription (1–4). Histone posttranslational modifications may serve as regulatory marks to be recognized by specific “reader” proteins (5, 6), which are typically part of large multisubunit chromatin-modifying or -remodeling complexes. Recruitment of such complexes can in turn further modify the epigenetic landscape and impact gene expression. Histone lysine methylation is associated with gene activation or repression, depending on the site and degree of the modification. In particular, methylation of lysine 4 of histone H3 (H3K4)4 correlates with gene activation. Monomethylation of H3K4 (H3K4me1) is a marker for enhancers, whereas di- and trimethylations of H3K4 (H3K4me2/3) are highly enriched at active promoters. Dynamic H3K4 methylation is critical for transcriptional regulation.
Lysine-specific demethylase 1 (LSD1, also known as KDM1A, AOF2), the first identified histone demethylase, is a flavin adenine dinucleotide (FAD)-dependent amine oxidase that specifically removes methyl groups from H3K4me1/2, thereby repressing transcription (7). As precise gene regulation is instrumental in signal transduction, cell fate determination and differentiation, LSD1 has emerged as a pivotal player in various normal developmental processes, and its deregulation has been involved in diseases such as cancer. The importance of LSD1-dependent transcriptional regulation is reflected by its association with a wide variety of transcription factors. For instance, LSD1 occupies and controls H3K4 methylation at the promoters of a subset of OCT4- and NANOG-repressed developmental genes in human embryonic stem (ES) cells to maintain stem cell pluripotency (8). During ES cell differentiation, LSD1-mediated demethylation of H3K4me1 decommissions enhancers of active genes and facilitates stem cell differentiation (9). LSD1 interacts with orphan nuclear receptor TLX to regulate neural stem cell proliferation (10). In pituitary organogenesis, LSD1 is recruited by the transcription factor ZEB1 to repress its targets (11). During hematopoiesis, LSD1 associates with key hematopoietic regulators GFI1/1B, TAL1, BLIMP1, BCL11A, and nuclear receptors TR2 and TR4 for differentiation of multiple hematopoietic lineages (12–16). In addition, LSD1 binds to the p53 tumor suppressor protein (17, 18). LSD1 also forms complexes with RBPJ to mediate Notch signaling in development and cancer (19, 20).
We and others recently showed that LSD1 functions as a corepressor physically associating with the zinc finger transcription factors Snai1 and Snai2 (21–23), which are crucial inducers of epithelial-to-mesenchymal transition (EMT) (24). EMT is a reprogramming event by which epithelial cells decrease expression of epithelial genes, lose cell adhesion, and acquire enhanced motility and invasiveness (25). It is a vital developmental program but is often reactivated in cancer, adversely contributing to malignant progression (25). Our previous study showed that LSD1 is essential for Snai1-mediated repression of epithelial markers (22). Consistently, LSD1 is overexpressed in advanced cancers, and its elevated expression correlates with tumor progression and unfavorable clinical outcome (26–30).
Although the critical role of LSD1 in a broad spectrum of physiological processes and its involvement in pathogenesis has been well documented, much remains to be understood about its functional regulation. LSD1 was mostly found in a core repressor complex with RCOR1 (CoREST) as well as histone deacetylase 1 and 2 (HDAC1/2) (7, 31–33). Recruitment of chromatin-modifying complexes to particular genomic sites generally requires multivalent interactions, involving combinatorial recognition of DNA sequence by associated sequence-specific transcription factors and local chromatin environment by the reader subunits. Although the LSD1 complex interacts with various transcription factors to regulate distinct target genes, it remains largely elusive how this complex recognizes epigenetic settings, in particular histone posttranslational modifications that may act as docking signals for its recruitment. Except for the catalytic domain of LSD1, no subunits in the complex have been known to recognize modified histone tails.
In the present study we purified the Snai1-LSD1 protein complex and identified a novel associated factor, SFMBT1 (Scm-like with four MBT domains 1) (34). SFMBT1 belongs to the malignant brain tumor (MBT) domain-containing protein family that is critical in chromatin regulation (35). Most MBT proteins characterized so far selectively bind mono- and di-methylated lysine residues on the amino-terminal histone tails. Our subsequent biochemical analysis showed that SFMBT1 associated with the LSD1 complex through physical interaction with the core subunit RCOR1. Unlike other MBT members that bind lower methylated lysines, SFMBT1 recognizes di- and tri-methylated H3K4 in vitro. Moreover, SFMBT1 is indispensable for Snai1-induced recruitment of LSD1 to chromatin, demethylation of H3K4me2, and transcriptional repression. Consistent with these findings, SFMBT1 is essential for induction of EMT by stimuli such as TGFβ and heavy metal nickel.
EXPERIMENTAL PROCEDURES
Affinity Purification of Protein Complexes Associated with Snai1
HEK293 cells were transfected with pcDNA3-Snai1-FLAG and selected by G418. Clones were picked and examined for Snai1 expression by immunoblotting with anti-FLAG antibodies (Sigma). Cells stably expressing Snai1-FLAG were subjected to affinity purification as previously described (36). Briefly, nuclear extracts (300 mg) were prepared and immunoprecipitated with anti-FLAG antibody-conjugated agarose (Sigma). After extensive washing, protein complexes were eluted using a 50 μg/ml FLAG peptide (Sigma) at room temperature for 30 min. The eluted protein samples were separated on the 4–12% NuPAGE Bis-tris gels (Invitrogen) and analyzed by silver staining and LC-MS/MS mass spectrometry at Taplin Biological Mass Spectrometry Facility of Harvard Medical School.
RT-Quantitative PCR
Cells were harvested in TRIzol (Invitrogen) followed by total RNA extraction. Reverse transcription of RNA was conducted using Moloney murine leukemia virus (MuLV) reverse transcriptase with random primers. The expression levels of selected genes were determined by real-time PCR with the SYBR Green PCR kit (Applied Biosystems). Data were normalized against β-actin.
ChIP Analysis
ChIP assay was conducted as previously reported (22). Briefly, cells were cross-linked with 1% formaldehyde for 10 min. The reaction was stopped by the addition of glycine. Cross-linked cells were washed in 1× PBS and collected. Cell pellets were washed with washing buffer (0.25% Triton X-100, 10 mm EDTA, 0.5 mm EGTA, 10 mm Tris, pH 8.0) resuspended in sonication buffer (1 mm EDTA, 0.5 mm EGTA, and 10 mm Tris pH 8.0), mixed with glass beads, and subjected to sonication. The sonicated samples were diluted in ChIP buffer (0.01% SDS 1.0% Triton X-100, 1.0 mm EDTA, 20 mm Tris, pH 8.1, 150 mm NaCl) and incubated with antibody against Snai1 (Santa Cruz), Snai2 (Cell Signaling), SFMBT1 (Bethyl), LSD1 (Upstate Biotechnology), H3K4Me2 (Millipore), and H3K4me3 (Cell Signaling), and protein A slurry (Invitrogen). The immunoprecipitates were subjected to a series of washing steps to remove nonspecific binding material. After reversal of cross-linking, DNA samples were purified, and the enrichment of specific genomic regions was determined by real-time quantitative PCR. Final results represent the percentage of input chromatin.
Histone Peptide Pulldown Assay
Four micrograms of biotinylated histone H3 or H4 peptides (ANASPEC) were incubated with cell lysate from HEK293 cells transfected with FLAG-SFMBT1 in 400 μl of binding buffer (50 mm Tris, pH 7.5, 100 mm NaCl, 0.1% Nonidet P-40) at 4 °C for overnight. The next day 12 μl of streptavidin beads (Promega) were added into the binding buffer and incubated at 4 °C for 1 h with rotation. After three washes with the binding buffer, the beads were boiled in protein loading buffer and subjected to SDS-PAGE and Western blotting with anti-FLAG antibodies.
Generation of Snail-ER Cells and Lentiviral shRNA Knockdown
Cells were infected with lentivirus expressing both GFP and the Snail1-ER fusion protein. GFP-positive cells were sorted out by flow cytometry. These cells were subsequently transduced with lentiviral vector pLKO or shRNA targeting SFMBT1 (OpenBiosystems). After selection with puromycin (2 μg/ml) for 1 week, these cells were subjected to 4-hydroxy-tamoxifen (4HT) (100 nm) treatment for 2 days and followed by RT-quantitative PCR and ChIP assays.
Treatment with TGFβ and Nickel and Transwell Invasion Assay
A549 cells were transduced with lentiviral vector pLKO or shRNA targeting SFMBT1. After selection with puromycin (1 μg/ml) for 1 week, cells were cultured in 6-well plates with or without TGFβ (5 ng/ml) or nickel chloride (1 mm) for 2 days. Morphological changes were observed, and phase contrast images were taken under a microscope. These cells were then subjected to RT-quantitative PCR and Transwell assays. The membranes of Transwell chambers were coated with 20 μg/ml fibronectin at 37 °C overnight. Cells (4 × 104) were resuspended with serum-free medium and added into the top of the insert. Inserts were placed in full medium with serum. After 24 h, cells that migrated to the bottom of the membrane were fixed and stained with 0.1% crystal violet. Pictures were taken under a microscope, and the numbers of cells were counted.
Gene Expression and Disease Association Analysis
To investigate whether SFMBT1 may play a role in breast cancer patient survival, we first chose the GSE25055 dataset (from the NCBI GEO website) as the training dataset and performed a univariate Cox proportional hazards regression analysis to find genes whose expression was potentially associated with patient survival. This gene list was then overlapped with the EMT gene list (Sabiosciences) and further limited by clustering analysis to six genes that are coexpressed with SFMBT1 in breast cancers (CDH2, VIM, and CALD1, which were up-regulated after EMT, and CLDN5, KRT19, and CDH1, which were down-regulated after EMT). We next constructed a multi-gene score using the following algorithm. Normalized probeset log2 values were subtracted with median values from all samples for one particular probeset. The multi-gene score is the averaged sum of the three EMT-up-regulated genes and SFMBT1 minus that of SFMBT1 and three EMT-down-regulated genes. Based on the gene expression heatmap, a cutoff was set to stratify patients into three equal number groups having low, medium, and high seven-gene scores. This effectively separated patients into poor and good prognosis groups.
Statistical Analyses of Experimental Data
Independent Student's t test was used to analyze data from various experiments.
RESULTS
The MBT Domain-containing Protein SFMBT1 Is Associated with Snai1
Snai1 induces EMT primarily through direct repression of epithelial genes. In an effort to better understand Snai1-mediated epigenetic regulation, we employed affinity purification and mass spectrometry to identify Snai1-associated proteins. Because Snai1 protein with an amino-terminal fusion tag fails to interact with LSD1 and repress transcription, we stably expressed human Snai1 with a carboxyl FLAG tag (Snai1-FLAG) in HEK293 cells. Nuclear extracts were prepared from ∼1 × 109 cells and subjected to affinity purification with the anti-FLAG M2 antibody beads followed by gel separation and mass spectrometry analysis (Fig. 1A).
FIGURE 1.

The MBT domain-containing protein SFMBT1 is associated with Snai1. A, identification of Snai1-associated proteins. Left, silver staining of Snai1-associated proteins. Nuclear extracts prepared from cells stably expressing Snai1-FLAG or empty vector (mock) were immunoprecipitated with anti-FLAG-agarose beads. The protein complex was eluted with FLAG peptides after extensive wash and separated by 4–12% gradient SDS-PAGE. Right, a list of potential Snai1-associated proteins was identified by mass spectrometric analysis. B, a phylogenetic tree of fly and human MBT domain-containing proteins derived from multiple sequence alignment. dL(3)MBT, dSCM, and dSFMBT are three MBT proteins in Drosophila. WB, Western blot. C, SFMBT1 interacts with Snai1. HEK293 cells were transiently transfected with Snai1 together with or without FLAG-SFMBT1, lysed, and followed by co-IP with anti-FLAG antibodies and Western blotting with anti-Snai1 and Anti-FLAG antibodies. Snai1 was specifically co-precipitated with SFMBT1. D, SFMBT1 interacts with Snai2. Snai2 and FLAG-SFMBT1 were co-expressed in HEK293 cells and subjected to co-IP analysis similar to B.
Consistent with previous studies that Snai1 interacts with the LSD1 complex, a large number of peptides corresponding to virtually all known subunits of the LSD1 complex, including the core components LSD1, RCOR1, and HDAC1/2, were found to be co-purified with Snai1 (Fig. 1A). By contrast, despite their expression in HEK293 cells, no single peptides of other histone-modifying enzymes (e.g. H3K27 methyltransferase EZH2, H3K9 methyltransferases G9a and Suv39h, JARID family of H3K4 demethylases) or DNA methyltransferases were detected. These results were confirmed by a replicate large scale purification, suggesting that the LSD1 complex is the major, if not the only, chromatin modifying complex that stably associates with Snai1.
In addition to the known components of the LSD1 complex, several peptides derived from p53 and FBXW1B (also known as βTrCP2) were retrieved (Fig. 1A). p53 was previously reported to bind Snai1 as well as LSD1 (17, 18, 37). FBXW1B is highly homologous to FBXW1A (βTrCP), a component of the Skp1-Cul1-F-box-protein (SCF) ubiquitin ligase known to recognize both Snai1 and Snai2 and mediate their polyubiquitination and degradation (23, 38, 39). Identification of the LSD1 complex and other Snai1 interactors validated the affinity purification approach.
Interestingly, seven peptides derived from SFMBT1 (34) were identified in the Snai1 complex (Fig. 1A). SFMBT1 is a member of the MBT protein family (35), which consists of four subgroups (Fig. 1B). MBT domains from the other three subgroups are methyl-lysine readers and interact with histone tails (35). The SFMBT1 subgroup is much less characterized, and it was unclear whether SFMBT1 might function as a histone reader. It was thus appealing that SFMBT1 might contribute to chromatin recognition by the LSD1 complex during its recruitment.
The association between Snai1 and SFMBT1 was subsequently verified by co-immunoprecipitation (co-IP) assays. Plasmids expressing Snai1 and FLAG-tagged SFMBT1 were transiently transfected into HEK293 cells. Whole cell extracts were prepared and immunoprecipitated with anti-FLAG antibodies followed by immunoblotting with anti-Snai1 antibodies. SFMBT1 indeed pulled down Snai1 (Fig. 1C). These reciprocal affinity precipitation studies confirmed the association between SFMBT1 and Snai1. In a parallel co-IP experiment, SFMBT1 also co-immunoprecipitated Snai2 (Fig. 1D).
SFMBT1 Associates with Snai1 through Binding to the LSD1 Complex
The LSD1 complex is known to directly bind to Snai1. The newly discovered SFMBT1-Snai1 interaction raised the possibility that SFMBT1 may interact with the LSD1 complex. When HEK293 cells were transfected with FLAG-SFMBT1, immunoprecipitation of exogenous SFMBT1 with FLAG antibodies was sufficient to enrich endogenous LSD1 (Fig. 2A). Furthermore, in untransfected cells, immunoprecipitation of endogenous SFMBT1 with SFMBT1-specific antibodies also pulled down endogenous LSD1 (Fig. 2B). These results demonstrate the association between SFMBT1 and LSD1 and the existence of an endogenous SFMBT1-LSD1 complex. Consistently, in our large scale purification of SFMBT1-associated proteins, the LSD1 complex was readily identified (40).
FIGURE 2.

SFMBT1 associates with Snai1 through the LSD1 complex. A, exogenous SFMBT1 interacts with endogenous LSD1. HEK293 cells were transfected with vector or FLAG-SFMBT1 and subjected to co-IP analysis with anti-FLAG antibodies followed by immunoblotting (WB) with anti-LSD1 antibodies. B, detection of endogenous SFMBT1-LSD1 complex. HEK293 cellular lysates were subjected to co-IP analysis with IgG (control) or anti-SFMBT1 antibodies followed by immunoblotting with anti-LSD1 antibodies. C, the association between SFMBT1 and Snai1 is dependent on LSD1. HEK293 cells were transduced with a lentiviral vector or two independent shRNAs targeting LSD1 (#1 and #2) (22) and subsequently transfected with Snai1 alone or in combination with FLAG-SFMBT1. Cells were subjected to co-IP with anti-FLAG antibodies followed by immunoblotting with indicated antibodies.
Because of the apparently mutual associations between Snai1, SFMBT1, and the LSD1 complex, we suspected that SFMBT1 might associate with Snai1 through the LSD1 complex. To test this idea, we first depleted LSD1 in HEK293 cells with two independent lentiviral short hairpin RNAs (shRNAs) (22) (Fig. 2C). Control and LSD1-depleted cells were then transiently transfected with Snai1 and FLAG-SFMBT1 followed by co-IP assays. In control cells, immunoprecipitation of SFMBT1 with anti-FLAG antibodies efficiently pulled down both LSD1 and Snai1 (Fig. 2C, second lane from the left); however, in cells depleted of LSD1, the association between SFMBT1 and Snai1 was substantially reduced (Fig. 2C, third and fourth lanes from the left), suggesting that SFMBT1 interacts with Snai1 through the LSD1 complex.
The LSD1 complex consists of many subunits, and it was unclear to which subunit SFMBT1 might bind. We decided to use the GST pulldown assay to address this question. SFMBT1 contains four MBT repeats at the amino terminus, an SPM motif at the carboxyl terminus and a highly conserved domain (DUF3588) with unknown function in the middle (Fig. 3A) (35). Each of these domains was fused to GST, and the resulting fusion proteins were purified from bacteria (Fig. 3A). When these GST fusion proteins were incubated with extracts prepared from HEK293 cells transfected with LSD1, none of them exhibited detectable binding of LSD1 (Fig. 3A), suggesting that SFMBT1 does not directly interact with LSD1. We thus further examined other subunits of the LSD1 complex. When incubated with lysate from cells transfected with RCOR1, a dimerization partner of LSD1 and a core subunit of the complex (31–33), the middle part of SFMBT1 showed strong association with RCOR1 (Fig. 3A), suggesting that SFMBT1 associates with the LSD1 complex through binding to RCOR1.
FIGURE 3.

SFMBT1 interacts with the LSD1 complex subunit RCOR1. A, SFMBT1 interacts with RCOR1. Top, structural diagram of SFMBT1. Indicated regions of SFMBT1 (N, amino acids (aa) 1–479; M, aa 494–699; C, aa 721–866) were fused to GST. The fusion proteins were purified from bacteria and incubated with extracts prepared from HEK293 cells transfected with RCOR1 or LSD1. After extensive wash, the protein complex was subjected to Western blotting (WB) with indicated antibodies. B, the linker region of RCOR1 is responsible for SFMBT1 binding. Top, domains of RCOR1. Different fragments of RCOR1 containing various domains (E+S1, amino acids (aa) 1–293; S1, aa 181–293; L+S2, aa 283–482; S2, aa 381–482) were fused to GST and subjected to pulldown assays with cell lysates expressing FLAG-SFMBT1 or FLAG-L3MBTL1 followed by Western blotting with anti-FLAG antibodies.
The interaction between SFMBT1 and RCOR1 was further validated by a reciprocal pulldown assay. RCOR1 contains several functional domains (32) (Fig. 3B). In this experiment, different domains of RCOR1 were fused to GST (Fig. 3B) and incubated with cellular extract containing FLAG-SFMBT1. The linker region of RCOR1, which forms an elongated structure with the “tower domain” of LSD1 (33), interacted with SFMBT1 (Fig. 3B). As a control, L3MBTL1, another MBT domain protein that does not have the unique middle domain of SFMBT1, did not bind to RCOR1 (Fig. 3B). Collectively, these results suggest that SFMBT1 indirectly associates with Snai1 through binding to the LSD1 complex core subunit RCOR1, resulting in assembly of a larger complex minimally comprising Snai1-LSD1-RCOR1-SFMBT1.
SFMBT1 Is Recruited to Snai1 Target Genes
The LSD1 complex is recruited to epithelial gene promoters by Snai1 (22). Due to its association with the LSD1 complex, SFMBT1 was expected to be recruited to these genes by Snai1 as well. To verify this idea, we employed an inducible form of Snai1, namely Snai1-ER, in which Snai1 is fused to a mutant form of the estrogen receptor ERα ligand binding domain that recognizes the synthetic ligand 4HT (41). The Snai1-ER fusion protein is retained in the cytoplasm and thus unable to repress transcription in the nucleus. Upon the addition of 4HT, the fusion protein translocates into the nucleus and binds and represses Snai1 target genes. This inducible model has been validated in various cultured cell lines and in vivo (41). When MCF7 human breast cancer epithelial cells were transduced with lentivirus expressing Snai1-ER, ChIP analysis with anti-Snai1 antibodies revealed that the binding of the Snai1 fusion protein to the E-cadherin promoter was minimal in cells treated with vehicle (DMSO) but was strongly induced after cells were treated with 4HT (Fig. 4A).
FIGURE 4.
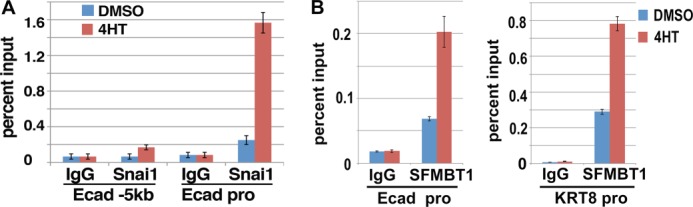
SFMBT1 is recruited to Snai1 target genes. A, validation of Snai1-ER, an inducible form of Snai1. MCF7 cells expressing Snai1-ER were treated with DMSO (vehicle) or 4HT followed by ChIP assay with IgG or anti-Snai1 antibody. Binding of Snai1-ER to the E-cadherin promoter (Ecad-pro) and a 5-kb upstream region was examined. B, binding of SFMBT1 to epithelial gene promoters is induced by Snai1. MCF7 Snai1-ER cells were treated with DMSO or 4HT followed by ChIP assay with IgG (control) or antibodies for SFMBT1. Binding at promoters of E-cadherin and KRT8 was determined. Experiments were conducted in triplicate, and error bars indicate S.D.
We next investigated whether SFMBT1 could be recruited to epithelial genes in a Snai1-dependent manner. MCF7 cells expressing Snai1-ER were treated with DMSO or 4HT and subjected to ChIP assays with IgG or anti-SFMBT1 antibodies. There seemed to be low basal levels of binding of SFMBT1 to the promoters of E-cadherin and keratin 8 (KRT8) genes (Fig. 4B), both of which are established, direct targets of Snai1. Importantly, treatment with 4HT significantly increased the occupancy of SFMBT1 at these promoters (Fig. 4B), suggesting that Snai1 indeed recruits SFMBT1 to its target genes.
SFMBT1 Recognizes Tri- and Dimethylated H3K4
Recruitment of SFMBT1 to the Snai1 target genes prompted us to investigate its role in Snai1-mediated epigenetic regulation. MBT domains characterized to date are readers of post-translational modifications of histones, more specifically, mono- and dimethylated lysine residues (35). SFMBT1 contains four MBT repeats that may potentially bind methyl lysines. Interestingly, none of the previously known subunits of the LSD1 complex recognize methylated histone tails. Therefore, we postulate that SFMBT1 may be a candidate histone methyl-lysine reader of the LSD1 complex.
Before Snai1-initiated repression in epithelial cells, Snai1 target genes (including many epithelial genes) are actively expressed, possessing histone marks associated with active transcription. It is hence conceivable that the putative histone reader may recognize certain active marks and facilitate the recruitment/action of the LSD1 complex. We thus focused on active lysine methylation marks, most notably the H3K4 methylations at various degrees. In addition, monomethylation of H3K9, H3K27, and H4K20 is linked to gene activation (42). Therefore, we evaluated the binding capacity of SFMBT1 with candidate histone species that harbor various forms of lysine methylation in vitro. Biotinylated H3 and H4 peptides were incubated with cellular extract expressing FLAG-SFMBT1. Histone peptides and possible bound SFMBT1 were purified with streptavidin beads. Among the examined peptides, both tri- and dimethylated H3K4 exhibited detectable binding to SFMBT1 (Fig. 5A). H3K4me3 appeared to have a higher affinity toward SFMBT1 than H3K4me2. Unmodified or mono-methylated H3 or H4 tails did not show evident association with SFMBT1 (Fig. 5A). This result suggests that SFMBT1 may recognize specific histone methyl marks, in particular H3K4me2/3, and its binding specificity may differ from that of previously characterized other human MBT proteins that demonstrate selective binding only to mono- and dimethylated histone peptides (35).
FIGURE 5.
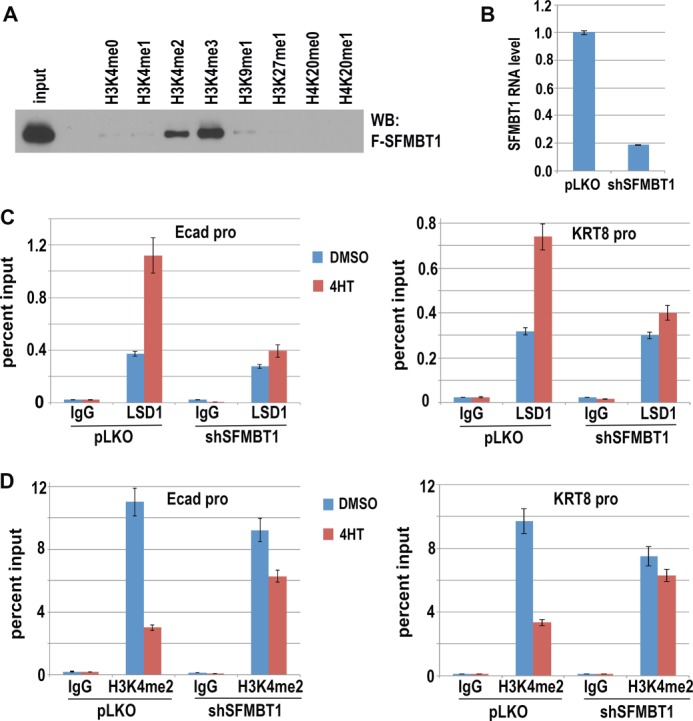
SFMBT1 binds H3K4me2/3 and is required for Snai1-dependent LSD1 recruitment and H3K4 demethylation. A, biotinylated peptides comprising the amino-terminal amino acid residues of histones H3 (residues 1–21, and 21–44) and H4 (residues 8–30) with the methylated lysines indicated on top were incubated with cell extract expressing FLAG-SFMBT1. Streptavidin beads were then used to precipitate peptides. Peptide-bound SFMBT1 was detected by immunoblotting (WB) with anti-FLAG antibodies. B, depletion of SFMBT1. MCF7 cells expressing Snai1-ER were infected with lentiviral vector pLKO or shRNA targeting SFMBT1. SFMBT1 RNA levels were determined by quantitative RT-PCR. C and D, SFMBT1 is essential for Snai1-induced recruitment of LSD1 to epithelial genes and demethylation of H3K4me2. Snai1-ER MCF7 cells were infected with lentiviral vector pLKO or shRNA targeting SFMBT1 followed by treatment with DMSO or 4HT for 2 days. Cells were harvested for ChIP analysis with IgG or antibodies for LSD1 (C) and H3K4me2 (D). Experiments were conducted in triplicate, and error bars represent S.D. Ecad-pro, cadherin promoter.
SFMBT1 Is Required for Snai1-induced Recruitment of LSD1, Demethylation of Dimethyl-H3K4, and Transcriptional Repression
Based on the SFMBT1 ability to bind histone marks and its potential as a histone reader, we next tested whether SFMBT1 contributed to Snai1-dependent chromatin recruitment of LSD1. We used shRNAs (in the pLKO lentiviral vector) to efficiently deplete SFMBT1 in Snai1-ER MCF7 cells (Fig. 5B). Consistent with our previous finding that Snai1 recruited LSD1 to specific genomic sites (22), 4HT treatment stimulated the association of LSD1 to the promoters of epithelial genes E-cadherin and KRT8 in control cells transduced with the empty pLKO vector, but this effect was virtually abolished in SFMBT1-depleted cells (Fig. 5C), demonstrating that SFMBT1 is essential for chromatin association of LSD1 induced by Snai1.
LSD1 specifically demethylates mono- and dimethylated H3K4. We further determined the role of SFMBT1 in the demethylation process using the inducible Snai1-ER cells. Similar to our previous observation (22), induction of Snai1 by 4HT potently reduced the level of H3K4me2 at the epithelial gene promoters in control cells (Fig. 5D). In cells depleted of SFMBT1, however, demethylation of H3K4me2 was greatly impaired (Fig. 5D). LSD1 cannot demethylate trimethylated H3K4. Indeed, this mark was not responsive to 4HT treatment regardless of the SFMBT1 depletion status (Fig. 6A). These results suggest that SFMBT1 is indispensable for Snai1-dependent recruitment of LSD1 to chromatin and subsequent H3K4me2 demethylation.
FIGURE 6.

SFMBT1 is essential for Snai1-mediated transcriptional repression. A, H3K4me3 levels at the E-cadherin promoter are not affected by Snai1. Control (pLKO) or SFMBT1-depleted MCF7 Snai1-ER cells were treated with DMSO or 4HT for 2 days followed by ChIP analysis with antibodies recognizing H3K4me3. Ecad-pro, cadherin promoter. B, SFMBT1 is required for repression of endogenous E-cadherin by Snai1. MCF7 cells stably expressing Snai1-ER were infected with lentiviral vector pLKO or shRNA targeting SFMBT1 followed by treatment with DMSO or 4HT. Cells were subjected to RT-PCR to determine the RNA levels of E-cadherin. C, SFMBT1 is required for Snai1-dependent repression of E-cadherin in MCF10DCIS cells. MCF10DCIS cells were transduced with lentiviral Snai1-ER-IRES-GFP, and GFP+ cells were divided into high and low expression groups. These cells were further infected with lentiviral vector pLKO or shRNA targeting SFMBT1 followed by 4HT treatment. E-cadherin expression was determined by RT-PCR. Results were derived from triplicate experiments, and error bars represent S.D. D, a working model for SFMBT1 in recruiting the LSD1 complex. Left, in epithelial cells without Snai1/2, the LSD1-RCOR1-SFMBT1 complex is present, but it does not stably associate with epithelial gene promoters. Middle, when Snai1/2 are induced, multivalent binding events tether the LSD1 complex to chromatin at epithelial genes. Right, in the absence of SFMBT1, LSD1 fails to steadily bind to chromatin (the Snai1-LSD1 interaction alone appears to be insufficient to anchor the complex on chromatin).
Removal of H3K4me2 by LSD1 is required for Snai1-mediated transcriptional repression (22); we hence expected that SFMBT1 was also essential for Snai1 repressive activity. In control pLKO-transduced MCF7 Snai1-ER cells, treatment with 4HT robustly inhibited endogenous E-cadherin RNA expression, but this inhibition was significantly compromised when SFMBT1 was depleted (Fig. 6B).
We noticed that depletion of SFMBT1 did not completely abolish Snai1-mediated repression of E-cadherin and wondered whether this might be related to the expression levels of Snai1. We transduced another transformed mammary epithelial cell line, MCF10DCIS, with lentiviral Snai1-ER-IRES-GFP. GFP-positive cells were sorted and, based on signal intensity, grouped as GFP-low (i.e. expressing lower levels of Snai1-ER) and GFP-high (i.e. expressing higher levels of Snai1-ER). Each group of cells was further infected with lentiviral vector pLKO or shRNA targeting SFMBT1. In both groups treatment of cells with 4HT decreased RNA levels of endogenous E-cadherin, and the repression was more robust in cells expressing higher levels of GFP (i.e. Snai1-ER) (Fig. 6C). As expected, depletion of SFMBT1 weakened the 4HT-induced repression on E-cadherin expression in both GFP-low and -high groups (Fig. 6C). But in GFP-high cells, de-repression of E-cadherin by SFMBT1 depletion is less robust than that in GFP-low cells (Fig. 6C). These observations suggest that SFMBT1 is required for Snai1-mediated transcriptional repression and may also imply that high levels of Snai1 may possess SFMBT1-independent repressive activity.
Based on these findings, we propose the following working model (Fig. 6D): SFMBT1 generally exists as a part of the LSD1 corepressor complex (at least consisting of LSD1-RCOR1-SFMBT1). This complex is present in epithelial cells but is not particularly enriched at epithelial genes. When Snai1-like factors are induced, Snai1 recruits this complex to its genomic targets through physical interaction with LSD1. Binding of SFMBT1 to histone marks facilitates this recruitment and/or further stabilizes the association of the complex with chromatin. In the absence of SFMBT1, Snai1 alone appears to be insufficient to tether the complex to chromatin.
SFMBT1 Is Essential for Induction of EMT by TGFβ
The aforementioned experimental settings mostly involve ectopic expression of Snai1. We decided to investigate the role of SFMBT1 in more physiological EMT models. TGFβ is a potent driver of EMT, tumor progression, and metastasis (43). Treatment with TGFβ is often sufficient to induce EMT in various epithelial cells. Depending on the context, TGFβ up-regulates endogenous Snai1 and/or Snai2, which are essential for TGFβ-induced EMT phenotype (44–46). Snai1 and Snai2 share the nearly identical SNAG repressive domain, which is sufficient for binding LSD1 (21, 22). The Snai2-LSD1 interaction was confirmed experimentally (23).
TGFβ treatment of A549 human lung cancer epithelial cells activated expression of Snai2 (Fig. 7A). To define the role of SFMBT1 in TGFβ-induced EMT, we depleted SFMBT1 in A549 cells (Fig. 7B). Upon TGFβ treatment, control A549 cells displayed decreased expression of epithelial genes, such as E-cadherin and KRT8 (Fig. 7, C and D) and increased expression of the mesenchymal marker N-cadherin (Fig. 7D). By contrast, in SFMBT1-depleted cells down-regulation of epithelial markers by TGFβ was significantly impaired, whereas induction of N-cadherin was not affected (Fig. 7, C and D).
FIGURE 7.

SFMBT1 is required for TGFβ-induced repression of epithelial genes and EMT. A, induction of Snai2 by TGFβ. A549 epithelial lung cancer cells were treated with TGFβ, and RNA levels of Snai1/2 were subsequently determined by RT-PCR. B, depletion of SFMBT1 in A549 cells by lentiviral shRNA was verified by RT-PCR. C and D, SFMBT1 is required for down-regulation of epithelial markers by TGFβ. A549 cells were transduced with lentiviral vector pLKO or shRNA targeting SFMBT1 followed by TGFβ treatment. Cells were harvested to measure E-cadherin and KRT8 RNA levels by RT-PCR (C) as well as E-cadherin and N-cadherin protein levels by Western blotting (D). Error bars indicate S.D. from triplicate experiments. E, SFMBT1 is required for TGFβ-induced morphological changes in A549 cells. Phase-contrast images of control (pLKO) and SFMBT1-depletd A549 cells treated with TGFβ for 2 days. F, depletion of SFMBT1 blocked TGFβ-induced cell invasion. Control and SFMBT1-depleted A549 cells were treated with TGFβ for 2 days and plated into Transwell (pre-coated with fibronectin). One day later cells invaded through the membranes were counted.
During EMT, epithelial cells lose cell-cell adhesion and acquire a mesenchymal, fibroblast-like morphology. A549 cells display a typical epithelial phenotype; cells are cohesive and clustered with extensive cell-cell contact (Fig. 7E). After treatment with TGFβ, these cells became elongated and dissociated from each other (Fig. 7E), which is characteristic of EMT and consistent with the changes in gene expression. In contrast, SFMBT1-depleted cells largely resisted the “scattering” effect of TGFβ (Fig. 7E).
By promoting morphological changes, EMT confers increased invasiveness on cells. To evaluate whether SFMBT1 regulates cell invasion, we performed Transwell invasion assays. Transwell cell-permeable membranes were coated with fibronectin to mimic the extracellular matrix that cells encounter during migration and invasion. A549 cells had limited capacity to transverse the membrane. However, after TGFβ treatment, control cells exhibited a dramatically enhanced invasive property as more cells seen invaded through the membrane (Fig. 7F). Depletion of SFMBT1 essentially abolished TGFβ-induced invasion (Fig. 7F). Taken together, these results suggest that SFMBT1 is required for TGFβ-induced EMT and cell invasion.
SFMBT1 Is Essential for Nickel-induced EMT
Many environmental pollutants (including some heavy metals) are carcinogenic. Exposure to nickel and its compounds is fairly extensive in both environmental and occupational settings. The metal carcinogenicity is in large part attributed to their epigenetic effect (47), although the underlying mechanism remains poorly understood. Carcinogenic nickel compounds induce EMT and promote malignant progression during carcinogenesis (48). Exposure of A549 cells to nickel chloride (NiCl2) strongly up-regulated Snai2 and concomitantly down-regulated epithelial markers including E-cadherin (Fig. 8A).
FIGURE 8.
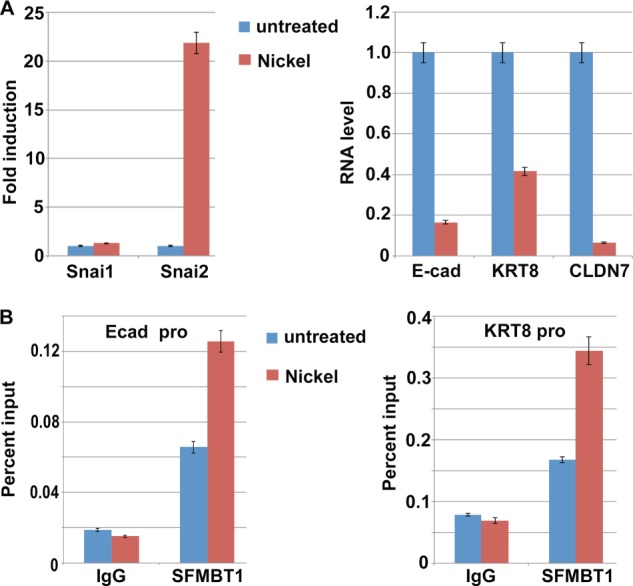
Nickel treatment up-regulates Snai2, down-regulates epithelial markers, and enhances binding of SFMBT1 to epithelial gene promoters. A, induction of Snai2 and repression of epithelial genes E-cadherin (Ecad), KRT8, claudin 7 (CLDN7) by nickel treatment. A549 epithelial lung cancer cells were treated with nickel for 2 days, and RNA levels of the indicated genes were subsequently determined by RT-PCR. Error bars indicate S.D. from triplicate experiments. B, enhanced binding of SFMBT1 to epithelial gene E-cadherin and KRT8 promoters by nickel treatment. A549 cells were treated with nickel for 2 days followed by ChIP assay with IgG (control) or antibodies for SFMBT1.
Induction of Snai2 prompted us to determine whether nickel-induced EMT might involve LSD1-SFMT1-dependent epigenetic regulation. ChIP analysis showed that after nickel treatment, the binding of SFMBT1 and LSD1 to epithelial gene promoters was potently increased (Figs. 8B and 9A, respectively), whereas H3K4me2 levels at these promoters significantly decreased (Fig. 9B). Importantly, these effects were negated by depletion of SFMBT1 (Fig. 9, A and B), demonstrating that SFMBT1 is required for nickel-induced recruitment of LSD1 to epithelial genes and subsequent histone demethylation. Consistent with the epigenetic changes, expression of E-cadherin persisted in SFMBT1-depleted cells under nickel treatment (Fig. 9C). Although control cells acquired a mesenchymal cell morphology after exposure to nickel, SFMBT1-depleted cells maintained the epithelial phenotype (Fig. 9D). These observations suggest that SFMBT1 is necessary for induction of EMT by nickel.
FIGURE 9.

SFMBT1 is essential for nickel-induced recruitment of LSD1 to epithelial genes, H3K4me2 demethylation, and EMT. A and B, SFMBT1 is indispensable for nickel-induced recruitment of LSD1 to E-cadherin and KRT8 gene promoters and demethylation of H3K4me2. Control (pLKO) and SFMBT1-depleted A549 cells were treated with nickel for 2 days. Cells were harvested for ChIP assay with IgG or antibodies for LSD1 (A) and H3K4me2 (B). Ecad-pro, cadherin promoter. C and D, SFMBT1 is required for down-regulation of E-cadherin and induction of EMT by nickel. Control and SFMBT1-depleted A549 cells were exposed to nickel for 2 days followed by Western blotting for E-cadherin (C). Cell morphology was recorded as phase-contrast images (D).
SFMBT1 Is Associated with Mesenchymal Markers and Poor Prognosis in Human Cancer
EMT is believed to facilitate tumor cell dissemination and contribute to carcinoma progression and metastasis. EMT inducers Snai1 and Snai2 are associated with tumor metastasis and an adverse clinical outcome in a variety of human cancers (24). Given the role of SFMBT1 in transcriptional repression by Snai1 and induction of EMT, we sought to investigate its potential relevance in human cancer. We chose a publicly available gene expression data set GSE25055, which represents a relatively large cohort (comprising 310 pretreatment breast cancer biopsies) with information of distant-relapse-free survival (49). We analyzed the correlation of SFMBT1 with established epithelial and mesenchymal markers. Consistent with its role in EMT, SFMBT1 expression positively correlated with mesenchymal markers including N-cadherin and vimentin and negatively correlated with epithelial markers including E-cadherin in human breast cancer (Fig. 10A).
FIGURE 10.
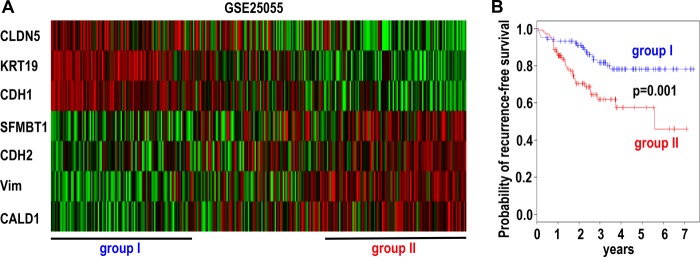
Expression of SFMBT1 in human cancer. A, SFMBT1 is associated with mesenchymal markers (CDH2 (N-cadherin), Vim (vimentin), CALD1 (caldesmon 1)), and inversely with epithelial markers (CDH1 (E-cadherin), CLDN5 (claudin 5), KRT19 (Keratin-19)) in human breast cancer cells (GSE25055). Expression of a 7-gene signature in 310 breast cancer patients is shown in the heatmap. B, Kaplan-Meier distant recurrence-free survival curve based on expression of the 7-gene (including SFMBT1) signature.
Using SFMBT1 and the associated epithelial/mesenchymal markers as a gene signature, the cancer samples were clustered accordingly (Fig. 10A). Patients grouped into the high expression tertile of the mesenchymal markers (including SFMBT1) (group II) had significantly shorter recurrence-free survival than the low expression group (group I) (p = 0.001) (Fig. 10B), suggesting this gene signature has prognostic value. The findings implicate SFMBT1 in EMT and tumor progression in human cancer.
DISCUSSION
The Snail family transcription factors are central regulators of EMT and are implicated in tumor progression and metastasis (24). To gain insights into the epigenetic mechanism underlying their transcriptional repression, we used a proteomic approach to purify protein complexes associated with Snai1. The LSD1 demethylase complex was found to be the predominant Snai1-associated chromatin modifying complex. Interestingly, an MBT domain-containing protein, SFMBT1, was identified as a new Snai1-associated factor. Subsequent biochemical study suggests that SFMBT1 is a component of the LSD1 complex via binding to the RCOR1 core subunit, which is consistent with recent reports (40, 50). MBT domains are methyl lysine recognition readers. We found that SFMBT1 may preferentially bind H3K4me2/3 histone marks highly enriched at active promoters. Our results suggest that SFMBT1 is a candidate reader subunit that helps Snai1 to tether the LSD1 complex to its target gene promoters. SFMBT1 is indeed essential for the chromatin recruitment of the Snai1-LSD1 complex, demethylation of H3K4me2, and transcription repression as well as induction of EMT and an invasive phenotype. EMT promotes carcinoma invasion and metastasis. Expression of Sna1/2 and LSD1 significantly correlates with tumor progression (24, 26–30). SFMBT1 is also associated with a mesenchymal phenotype in human cancer (Fig. 10A). These observations reveal a critical LSD1-SFMBT1-dependent chromatin regulatory program exploited by Snai1 that induces EMT and contributes to cancer progression and metastasis.
Chromatin remodeling and modifying complexes are recruited to chromatin primarily through interactions with sequence-specific DNA binding transcription factors. Such complexes typically contain reader subunits that recognize specific posttranslational modifications of histone tails, which are docking signals independent of underlying DNA sequence. Successful loading of chromatin regulatory complexes at specific genomic loci requires multivalent bindings of transcription factors to specific DNA sequence as well as histone readers to local chromatin environment. Cooperation among these multiple weak interactions not only enhances overall binding strength but also achieves avid specificity. Based on the current study, a multivalent engagement model assembled by Snai1-LSD1-RCOR1-SFMBT1 is expected to be critical for recruiting the complex to chromatin template during Snai1-induced epigenetic reprogramming (Fig. 6D): before Snai1's action, epithelial genes are actively transcribed in epithelial cells and possess di- and trimethyl H3K4 at their promoters. The LSD1 complex is present in these cells but may only have limited affinity toward the epithelial genes (possibly due to recognition of these marks by SFMBT1). When Snai1 is induced in epithelial cells, it recognizes binding sites at many actively transcribed epithelial gene promoters. Snai1 recruits the LSD1 complex to these genomic sites via physical interaction with LSD1 (22); RCOR1 dimerizes with LSD1 and tethers it to nucleosome by contacting DNA backbone (31–33); SFMBT1 associates with RCOR1 as well as binds to the pre-existing active marks H3K4me2/3 present at the promoters of target genes. All these interactions cooperatively contribute to the initial recruitment of the LSD1 repressor complex to Snai1 target genes and/or stabilization of the complex on chromatin and are required for LSD1-mediated H3K4 demethylation. LSD1 specifically demethylates H3K4me2 to an unmethylated form, generating a recognition surface for BHC80 (another subunit) that stabilizes the LSD1 complex on chromatin and may maintain an unmethylated H3K4 status and the repressed state (51). The H3K4me3 mark persists during the process (Ref. 22 and Fig. 6A) and may continue to serve as an anchor for SFMBT1 binding and help retain the complex on chromatin. These events lead to an activation-to-repression transition at the Snai1 target promoters and reset the chromatin state at these loci. The LSD1 complex can be recruited to a variety of target genes through interactions with many different transcription factors. It is likely that SFMBT1 participates in most, if not all, LSD1-mediated epigenetic regulation.
It was unexpected that SFMBT1 was able to bind tri-methyl H3K4. There are nine MBT members in human and most of them can be evolutionary linked to one of three Drosophila orthologs: dL(3)MBT (the founding member of the MBT family), dSCM, and dSFMBT (Fig. 1B). Despite the nomenclature, human SFMBT1 and related SFMBT2 form a rather distant subgroup without an obvious ortholog in fly (Fig. 1B). Different groups of MBT proteins exist in discrete large protein complexes and display divergent biological functions (35). Only SFMBT1 forms a complex with LSD1. MBT domain is a structural motif consisting of ∼100 amino acids, and its overall structure is highly conserved. MBT domains form a narrow and deep cavity of aromatic residue-lined pocket that specifically recognizes low methylated lysines (35). However, in SFMBT1, such methyl-lysine recognition residues are not all conserved, making it possible to form a larger pocket to accommodate trimethyl lysine. Interestingly, the Caenorhabditis elegans MBT protein LIN-61 was shown to preferentially bind trimethyl H3K9 (52). Future structural studies are warranted to delineate the methyl lysine binding features of SFMBT1.
Epigenetic therapies conventionally target the catalytic activities of chromatin regulatory complexes. Chromatin readers recognize histone posttranslational modifications and critically interpret their biological significance. The protein-protein interactions between the reader modules and histone modifications are amenable to small molecule inhibitors. Given their importance in epigenetic signaling and chemical tractability, chromatin readers have emerged as new therapeutic targets (53, 54). Small-molecule MBT antagonists that interfere with the MBT-histone interaction have been under development (54, 55). LSD1 is deregulated in cancer (26–30). The LSD1 complex contains two enzymatic subunits, LSD1 and HDAC1/2, that are currently explored as therapeutic targets (54, 56). SFMBT1 may represent another epigenetic vulnerability of this complex that can be exploited for pharmacological intervention of EMT-dependent cancer progression.
Acknowledgments
We thank M. Angela Nieto (Instituto de Neurociencias, Consejo Superior de Investigaciones Científicas-Universidad Miguel Hernández, Spain) for the Snail-ER construct and Jorg Bungert for critically reading the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01CA137021 (NCI; to J. L.). This work was also supported by the Florida Bankhead-Coley Cancer Research Program (09BN-12-23092 and 2BT01).
- H3K4
- lysine 4 of histone H3
- LSD1
- lysine-specific demethylase 1
- EMT
- epithelial-to-mesenchymal transition
- SFMBT1
- Scm-like with four MBT domains 1
- MBT
- malignant brain tumor
- Bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- KRT8
- keratin 8
- 4HT
- 4-hydroxy-tamoxifen
- ER
- endoplasmic reticulum
- IP
- immunoprecipitation.
REFERENCES
- 1. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 2. Li B., Carey M., Workman J. L. (2007) The role of chromatin during transcription. Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 3. Berger S. L. (2007) The complex language of chromatin regulation during transcription. Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 4. Campos E. I., Reinberg D. (2009) Histones. Annotating chromatin. Annu. Rev. Genet. 43, 559–599 [DOI] [PubMed] [Google Scholar]
- 5. Taverna S. D., Li H., Ruthenburg A. J., Allis C. D., Patel D. J. (2007) How chromatin-binding modules interpret histone modifications. Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Musselman C. A., Lalonde M. E., Côté J., Kutateladze T. G. (2012) Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi Y., Lan F., Matson C., Mulligan P., Whetstine J. R., Cole P. A., Casero R. A., Shi Y. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 [DOI] [PubMed] [Google Scholar]
- 8. Adamo A., Sesé B., Boue S., Castaño J., Paramonov I., Barrero M. J., Izpisua Belmonte J. C. (2011) LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat. Cell Biol. 13, 652–659 [DOI] [PubMed] [Google Scholar]
- 9. Whyte W. A., Bilodeau S., Orlando D. A., Hoke H. A., Frampton G. M., Foster C. T., Cowley S. M., Young R. A. (2012) Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun G., Alzayady K., Stewart R., Ye P., Yang S., Li W., Shi Y. (2010) Histone demethylase LSD1 regulates neural stem cell proliferation. Mol. Cell. Biol. 30, 1997–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J., Scully K., Zhu X., Cai L., Zhang J., Prefontaine G. G., Krones A., Ohgi K. A., Zhu P., Garcia-Bassets I., Liu F., Taylor H., Lozach J., Jayes F. L., Korach K. S., Glass C. K., Fu X. D., Rosenfeld M. G. (2007) Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446, 882–887 [DOI] [PubMed] [Google Scholar]
- 12. Saleque S., Kim J., Rooke H. M., Orkin S. H. (2007) Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 27, 562–572 [DOI] [PubMed] [Google Scholar]
- 13. Hu X., Li X., Valverde K., Fu X., Noguchi C., Qiu Y., Huang S. (2009) LSD1-mediated epigenetic modification is required for TAL1 function and hematopoiesis. Proc. Natl. Acad. Sci. U.S.A. 106, 10141–10146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Su S. T., Ying H.-Y., Chiu Y.-K., Lin F.-R., Chen M.-Y., Lin K.-I. (2009) Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation. Mol. Cell. Biol. 29, 1421–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu J., Bauer D. E., Kerenyi M. A., Vo T. D., Hou S., Hsu Y. J., Yao H., Trowbridge J. J., Mandel G., Orkin S. H. (2013) Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc. Natl. Acad. Sci. U.S.A. 110, 6518–6523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui S., Kolodziej K. E., Obara N., Amaral-Psarris A., Demmers J., Shi L., Engel J. D., Grosveld F., Strouboulis J., Tanabe O. (2011) Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic β-type globin promoters in differentiated adult erythroid cells. Mol. Cell. Biol. 31, 3298–3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang J., Sengupta R., Espejo A. B., Lee M. G., Dorsey J. A., Richter M., Opravil S., Shiekhattar R., Bedford M. T., Jenuwein T., Berger S. L. (2007) p53 is regulated by the lysine demethylase LSD1. Nature 449, 105–108 [DOI] [PubMed] [Google Scholar]
- 18. Tsai W. W., Nguyen T. T., Shi Y., Barton M. C. (2008) p53-targeted LSD1 functions in repression of chromatin structure and transcription in vivo. Mol. Cell. Biol. 28, 5139–5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mulligan P., Yang F., Di Stefano L., Ji J.-Y., Ouyang J., Nishikawa J. L., Toiber D., Kulkarni M., Wang Q., Najafi-Shoushtari S. H., Mostoslavsky R., Gygi S. P., Gill G., Dyson N. J., Näär A. M. (2011) A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Mol. Cell 42, 689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yatim A., Benne C., Sobhian B., Laurent-Chabalier S., Deas O., Judde J. G., Lelievre J. D., Levy Y., Benkirane M. (2012) NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol. Cell 48, 445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin Y., Wu Y., Li J., Dong C., Ye X., Chi Y. I., Evers B. M., Zhou B. P. (2010) The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 29, 1803–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin T., Ponn A., Hu X., Law B. K., Lu J. (2010) Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 29, 4896–4904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Z. Q., Li X. Y., Hu C. Y., Ford M., Kleer C. G., Weiss S. J. (2012) Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic breast cancer 1, early onset (BRCA1) repression. Proc. Natl. Acad. Sci. U.S.A. 109, 16654–16659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peinado H., Olmeda D., Cano A. (2007) Snail, Zeb, and bHLH factors in tumor progression. An alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428 [DOI] [PubMed] [Google Scholar]
- 25. Thiery J. P., Acloque H., Huang R. Y., Nieto M. A. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 [DOI] [PubMed] [Google Scholar]
- 26. Kahl P., Gullotti L., Heukamp L. C., Wolf S., Friedrichs N., Vorreuther R., Solleder G., Bastian P. J., Ellinger J., Metzger E., Schüle R., Buettner R. (2006) Androgen receptor coactivators lysinespecific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 66, 11341–11347 [DOI] [PubMed] [Google Scholar]
- 27. Schulte J. H., Lim S., Schramm A., Friedrichs N., Koster J., Versteeg R., Ora I., Pajtler K., Klein-Hitpass L., Kuhfittig-Kulle S., Metzger E., Schüle R., Eggert A., Buettner R., Kirfel J. (2009) Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma. Implications for therapy. Cancer Res. 69, 2065–2071 [DOI] [PubMed] [Google Scholar]
- 28. Lim S., Janzer A., Becker A., Zimmer A., Schüle R., Buettner R., Kirfel J. (2010) Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31, 512–520 [DOI] [PubMed] [Google Scholar]
- 29. Serce N., Gnatzy A., Steiner S., Lorenzen H., Kirfel J., Buettner R. (2012) Elevated expression of LSD1 (lysine-specific demethylase 1) during tumour progression from pre-invasive to invasive ductal carcinoma of the breast. BMC Clin. Pathol. 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jie D., Zhongmin Z., Guoqing L., Sheng L., Yi Z., Jing W., Liang Z. (2013) Positive expression of LSD1 and negative expression of E-cadherin correlate with metastasis and poor prognosis of colon cancer. Dig. Dis. Sci. 58, 1581–1589 [DOI] [PubMed] [Google Scholar]
- 31. Lee M. G., Wynder C., Cooch N., Shiekhattar R. (2005) An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437, 432–435 [DOI] [PubMed] [Google Scholar]
- 32. Shi Y.-J., Matson C., Lan F., Iwase S., Baba T., Shi Y. (2005) Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 19, 857–864 [DOI] [PubMed] [Google Scholar]
- 33. Yang M., Gocke C. B., Luo X., Borek D., Tomchick D. R., Machius M., Otwinowski Z., Yu H. (2006) Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 23, 377–387 [DOI] [PubMed] [Google Scholar]
- 34. Wu S., Trievel R. C., Rice J. C. (2007) Human SFMBT is a transcriptional repressor protein that selectively binds the N-terminal tail of histone H3. FEBS Lett. 581, 3289–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonasio R., Lecona E., Reinberg D. (2010) MBT domain proteins in development and disease. Semin. Cell Dev. Biol. 21, 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ogawa H., Ishiguro K., Gaubatz S., Livingston D. M., Nakatani Y. (2002) A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296, 1132–1136 [DOI] [PubMed] [Google Scholar]
- 37. Lee S. H., Lee S.-J., Jung Y. S., Xu Y., Kang H. S., Ha N.-C., Park B.-J. (2009) Blocking of p53-Snail binding, promoted by oncogenic K-Ras, recovers p53 expression and function. Neoplasia 11, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou B. P., Deng J., Xia W., Xu J., Li Y. M., Gunduz M., Hung M.-C. (2004) Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6, 931–940 [DOI] [PubMed] [Google Scholar]
- 39. Yook J. I., Li X. Y., Ota I., Fearon E. R., Weiss S. J. (2005) Wnt-dependent regulation of the Ecadherin repressor snail. J. Biol. Chem. 280, 11740–11748 [DOI] [PubMed] [Google Scholar]
- 40. Lin S., Shen H., Li J. L., Tang S., Gu Y., Chen Z., Hu C., Rice J. C., Lu J., Wu L. (2013) Proteomic and functional analyses reveal the role of chromatin reader SFMBT1 in regulating epigenetic silencing and the myogenic gene program. J. Biol. Chem. 288, 6238–6247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Frutos C. A., Dacquin R., Vega S., Jurdic P., Machuca-Gayet I., Nieto M. A. (2009) Snail1 controls bone mass by regulating Runx2 and VDR expression during osteoblast differentiation. EMBO J. 28, 686–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 43. Xu J., Lamouille S., Derynck R. (2009) TGF-β-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vincent T., Neve E. P., Johnson J. R., Kukalev A., Rojo F., Albanell J., Pietras K., Virtanen I., Philipson L., Leopold P. L., Crystal R. G., de Herreros A. G., Moustakas A., Pettersson R. F., Fuxe J. (2009) A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 11, 943–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu Y.-N., Abou-Kheir W., Yin J. J., Fang L., Hynes P., Casey O., Hu D., Wan Y., Seng V., Sheppard-Tillman H., Martin P., Kelly K. (2012) Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor β-initiated prostate cancer epithelial-mesenchymal transition. Mol. Cell. Biol. 32, 941–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Batlle R., Alba-Castellón L., Loubat-Casanovas J., Armenteros E., Francí C., Stanisavljevic J., Banderas R., Martin-Caballero J., Bonilla F., Baulida J., Casal J. I., Gridley T., de Herreros A. G. (2013) Snail1 controls TGF-β responsiveness and differentiation of mesenchymal stem cells. Oncogene 32, 3381–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chervona Y., Costa M. (2012) The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic. Biol. Med. 53, 1041–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu C. H., Tang S. C., Wang P. H., Lee H., Ko J. L. (2012) nickel-induced epithelial-mesenchymal transition by reactive oxygen species generation and E-cadherin promoter hypermethylation. J. Biol. Chem. 287, 25292–25302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hatzis C., Pusztai L., Valero V., Booser D. J., Esserman L., Lluch A., Vidaurre T., Holmes F., Souchon E., Wang H., Martin M., Cotrina J., Gomez H., Hubbard R., Chacón J. I., Ferrer-Lozano J., Dyer R., Buxton M., Gong Y., Wu Y., Ibrahim N., Andreopoulou E., Ueno N. T., Hunt K., Yang W., Nazario A., DeMichele A., O'Shaughnessy J., Hortobagyi G. N., Symmans W. F. (2011) A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 305, 1873–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang J., Bonasio R., Strino F., Kluger Y., Holloway J. K., Modzelewski A. J., Cohen P. E., Reinberg D. (2013) SFMBT1 functions with LSD1 to regulate expression of canonical histone genes and chromatin-related factors. Genes Dev. 27, 749–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lan F., Collins R. E., De Cegli R., Alpatov R., Horton J. R., Shi X., Gozani O., Cheng X., Shi Y. (2007) Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 448, 718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Koester-Eiserfunke N., Fischle W. (2011). H3K9me2/3 binding of the MBT domain protein LIN-61 is essential for Caenorhabditis elegans vulva development. PLoS Genet. 7, e1002017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dawson M. A., Kouzarides T., Huntly B. J. (2012) Targeting epigenetic readers in cancer. N. Engl. J. Med. 367, 647–657 [DOI] [PubMed] [Google Scholar]
- 54. Arrowsmith C. H., Bountra C., Fish P. V., Lee K., Schapira M. (2012) Epigenetic protein families. A new frontier for drug discovery. Nat. Rev. Drug Discov. 11, 384–400 [DOI] [PubMed] [Google Scholar]
- 55. James L. I., Barsyte-Lovejoy D., Zhong N., Krichevsky L., Korboukh V. K., Herold J. M., MacNevin C. J., Norris J. L., Sagum C. A., Tempel W., Marcon E., Guo H., Gao C., Huang X. P., Duan S., Emili A., Greenblatt J. F., Kireev D. B., Jin J., Janzen W. P., Brown P. J., Bedford M. T., Arrowsmith C. H., Frye S. V. (2013) Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat. Chem. Biol. 9, 184–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang Y., Greene E., Murray Stewart T., Goodwin A. C., Baylin S. B., Woster P. M., Casero R. A., Jr. (2007) Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc. Natl. Acad. Sci. U.S.A. 104, 8023–8028 [DOI] [PMC free article] [PubMed] [Google Scholar]