

Abstract
Recent studies revealed that elk-like S170N/N174T mutation in mouse prion protein (moPrP), which results in an increased rigidity of β2-α2 loop, leads to a prion disease in transgenic mice. Here we characterized the effect of this mutation on biophysical properties of moPrP. Despite similar thermodynamic stabilities of wild type and mutant proteins, the latter was found to have markedly higher propensity to form amyloid fibrils. Importantly, this effect was observed even under fully denaturing conditions, indicating that the increased conversion propensity of the mutant protein is not due to loop rigidity but rather results from greater amyloidogenic potential of the amino acid sequence within the loop region of S170N/N174T moPrP.
Keywords: Prion protein, prion diseases, amyloid, protein structure
Introduction
Prion diseases or transmissible spongiform encephalopathies (TSEs) are a group of fatal neurodegenerative disorders including Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker disease in humans, bovine spongiform encephalopathy in cattle, scrapie in sheep and goat, and chronic wasting disease in cervids [1,2]. The critical molecular event in TSE pathogenesis appears to be the conformational conversion of the normal form of the prion protein, PrPC, to a misfolded aggregated form, PrPSc. It is generally believed that PrPSc itself represents the infectious prion agent, which self-propagates by binding to PrPC and inducing conformational conversion of the latter protein to the PrPSc state. This ‘protein-only’ model is supported by wealth of experimental data [1,2], most notably the recent success in generating infectious prions in vitro from bacterially expressed recombinant prion protein [3-6].
Despite the recent progress in prion research, the molecular mechanisms of prion protein conformational conversion and prion propagation remain poorly understood. This is largely due to challenges in elucidating the structure of the infectious PrPSc conformer. Another major challenge in the field is to understand the molecular basis of barriers for disease transmission between different species. While it appears that prion transmissibility is related to a complex interplay between the structure of donor PrPSc and conformational preferences of host PrPC [7,8], molecular-level details of this interplay are not clear.
High-resolution structures for the native form of PrP from many species have been determined by NMR and X-ray crystallography [9,10]. The native protein consists of two distinct regions: a largely disordered and flexible N-terminal domain and a folded C-terminal domain, each comprising approximately 100 amino acids [11]. The folded domain contains three α-helices, two short β-strands, and loops connecting these secondary structure elements [9,10,12,13]. Intriguingly, native prion proteins from different species appear to have very similar overall three-dimensional structures. The only region showing substantial species-specific differences is the loop (residues 165-175 in huPrP) connecting the second strand of β-sheet to α-helix 2. This β2-α2 loop is rigid in elk and bank vole, where a part of it forms a 310-helix [14,15]. By contrast, in most other mammalian species, the β2-α2 loop is disordered and flexible [13,16,17]. Structural rigidity of β2-α2 loop appears to arise from the presence of Asn at position 170, as indicated by the observation that, in contrast to wild-type mouse prion protein (wt moPrP), NMR structures of moPrP with elk PrP-mimicking S170N/N174T or vole PrP-mimicking S170N amino acid substitutions are both characterized by a rigid β2-α2 loop [14,15]. This loop has recently gained particular interest following the observation that transgenic mice expressing S170N/N174T moPrP spontaneously developed TSE disease, suggesting that the β2-α2 loop rigidity may play an important role in the pathogenesis of prion diseases [18]. To gain insight into the molecular basis of the latter finding, here we undertook biophysical studies aimed at understanding the role of residues within the loop region in prion protein conformational conversion in vitro.
Materials and Methods
Protein purification
Full length wt moPrP and the S170N/N174T variant were expressed in E. coli and purified as previously described [19]. The proteins were stored in 10 mM sodium acetate buffer (pH 4) at -80°C.
Thioflavin T (ThT) assay
The progress of amyloid fibril formation was followed by a fluorimetric ThT assay [20]. The reaction under partially denaturing conditions was performed by adding the protein (0.5 mg/ml) to 50 mM sodium phosphate buffer (pH 6.5) containing 2 M GuHCl (Thermo Scientific) and 40 μM ThT. 200 μl aliquots of this mixture were transferred to individual wells of a 96-well plate (Micro-assay plate, Greiner Bio-one) containing a 3 mm glass bead (Fisher Scientific). The experiment was carried out at 37°C using the Biotek FLx800 plate reader in a moderate shaking mode. The excitation and emission wavelengths were 450 and 480 nm respectively. The reaction under fully denaturing conditions was performed in a manual format as the reaction under these conditions using the automated plate reader format was very slow making such measurements impractical. The protein (0.5 mg/ml) was incubated in the same buffer as above but containing 4 M GuHCl (but no ThT) at 37 °C in 1.5 conical tubes (filled to the volume of 1 ml) with continuous rotation at 8 rpm (Barnstead Thermolyne Labquake rotator). Five μl aliquots were withdrawn at different time points and transferred to a quartz cell containing 495 μl of 10 μM ThT in 50 mM sodium phosphate (pH 6.5). Fluorescence at 482 nm was measured in Fluoromax-3 (Horiba Scientific) spectrofluorometer using the excitation wavelength of 450 nm. In each case, ThT fluorescence curves were fit to the equation [21]:
| (1) |
where yi and yf are the initial and final baselines and t0 is the mid-point of fibrillization. Rate constant and lag phase are given by 1/τ and t0-2τ respectively.
Equilibrium unfolding in GuHCl
The unfolding curves were obtained at 25°C using an automatic titrator (Microlab 500 series) attached to an AVIV 215 circular dichroism spectrometer. In these experiments, native proteins (2 μM) in 50 mM phosphate buffer (pH 6.5) were titrated (in 0.07 M increments) with 8 M buffered GuHCl stock solution containing identical protein concentration. Upon each addition of GuHCl, the mixture was incubated for 30 s and the extent of protein unfolding was monitored by ellipticity at 222 nm. Unfolding curves were analyzed according to the two-state folding model [22].
Atomic force microscopy (AFM)
Sample were deposited on freshly cleaved mica, incubated for 1 min and then washed with pure water followed by drying with nitrogen. All images were recorded in a tapping mode using a Digital Instruments Multimode atomic force microscope equipped with Nanoscope IV controller. Data were processed and heights of fibrils were measured using Nanoscope SPM software package.
Electron microscopy
Fibrils were deposited on Formvar/carbon-coated grids and negatively stained by incubating with 2 % uranyl acetate solution for 30 s. Electron micrographs were obtained using a JEOL 1200EX transmission electron microscope.
H/D exchange mass spectrometry (HXMS)
The HXMS experiments were performed essentially as described previously [23-25]. Briefly, amyloid fibrils were incubated in D2O for 24 h at 25°C. Fibrils were then dissociated into monomers by treatment with ice-cold solution of 7 M GuHCl in 0.1 M phosphate buffer (pH 2.4). These monomers were then diluted 10 times with ice cold 0.05% trifluoracetic acid in water and subjected to pepsin digestion. The resulting peptic fragments were separated on a C18 column and analyzed on-line for deuterium incorporation by Finnigan LTQ mass spectrometer. The extent of deuterium incorporation was calculated as
| (2) |
where %D is the relative amount of amide deuterium atoms incorporated in each peptic fragment, m(t) is the observed centroid mass of the peptide at time point t, m(0%) is the measured mass of an undeuterated reference sample, m(100%) is the observed mass of a fully deuterated reference sample.
Results and discussion
While many factors may affect the pathogenesis of prion diseases in vivo, the finding that rigid loop-inducing amino acid substitutions in moPrP are sufficient to result in a spontaneous development of TSE in transgenic mice [18] suggests that these amino acid substitutions may increase the intrinsic ability of the prion protein to undergo conformational conversion to misfolded aggregated form(s). To test this possibility, we compared the kinetics of amyloid fibril formation in vitro by the recombinant protein corresponding to wt moPrP and the rigid loop S170N/N174T variant. Fibrillar aggregates formed by the recombinant PrP have been previously shown to cause TSE disease in animals [4,26]. However, the infectivity of these preparations is usually much lower compared to brain-derived prions, indicating that the structure of fibrillar aggregates formed in vitro is likely not fully equivalent to that of brain PrPSc. Nevertheless, PrP amyloid fibrils have been frequently used as a convenient model in vitro to study mechanistic aspects of prion protein conformational conversion [27]. They are especially informative when used to assess the effect of species- or disease-specific mutations on the conversion propensity of monomeric PrP to misfolded aggregates [28-30].
As in previous studies [28,29,31], fibrillization experiments were performed under partially denaturing conditions (2 M GuHCl) that facilitate spontaneous fibrillization of PrP in vitro. The reaction was followed by the ThT fluorescence assay in an automated 96-well plate format. Consistent with previous studies [32], wt moPrP forms amyloid fibrils through a process characterized by a distinct lag period (corresponding to nuclei formation), followed by a relatively rapid growth phase. As shown in Fig. 1, the same characteristics are observed for the mutant protein. However, the reaction for the latter protein is much faster as compared to wt moPrP, with the lag phases of 15.4 ± 1.0 h and 4.8 ± 0.47 h for wt moPrP and S170N/N174T moPrP, respectively (Fig. 1). Thus, elk-like PrP mutation that results in increased loop rigidity greatly increases the propensity of prion protein to form amyloid fibrils under these experimental conditions. It should be noted that the final ThT fluorescence intensity was substantially stronger for wt PrP fibrils than for the mutant PrP fibrils (kinetic traces in Fig. 1 are intensity normalized). While this could potentially suggest some degree of differences in structural organization of fibrils formed by the two proteins, to the best of our knowledge, no correlation has yet been established between ThT fluorescence intensity and specific structural features of amyloid fibrils.
Fig 1.
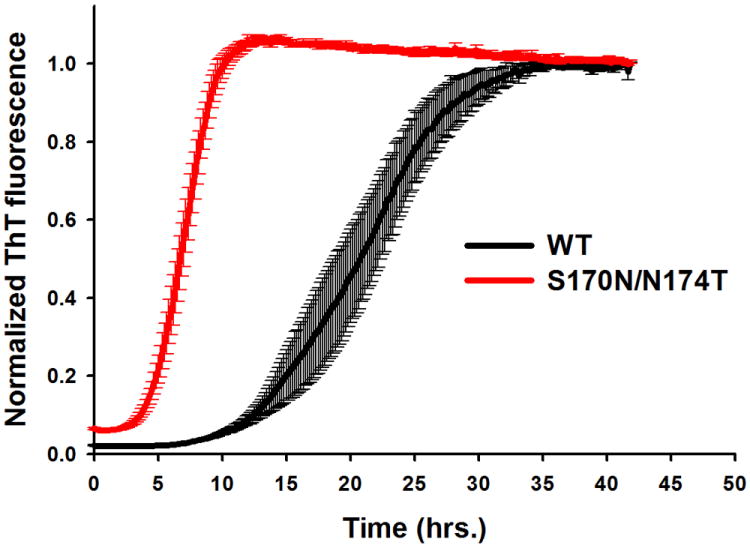
Time-course of amyloid fibril formation for wild type moPrP and S170N/N174T moPrP under partially denaturing conditions. Experiments were performed in the presence of 2 M GuHCl. Each curve represents an average of at least three independent experiments, with six replicates in each experiment. Error bars represent standard error. For clarity of the figure, curves for wt and mutant proteins were normalized to the same ThT fluorescence intensity, even though the ThT response for wt moPrP was approximately three times stronger compared to that for the mutant protein.
It has been shown that the familial mutations within the folded domain of human prion protein often result in thermodynamic destabilization of the protein, and this reduced thermodynamic stability leads to an increased propensity of PrP to undergo a conversion to amyloid fibrils and other oligomeric β-sheet forms in vitro [28,29]. Therefore, we tested whether similar thermodynamic arguments could be used to explain the observed increase in fibrillization rate of the rigid loop moPrP variants as compared to the wt protein. To this end, thermodynamic stability of the proteins was probed by equilibrium unfolding in GuHCl using ellipticity at 222 nm as a measure of the degree of protein unfolding. Fig. 2 shows the unfolding curves for the wt moPrP and the S170N/N174T variant. Surprisingly, these curves appeared identical, indicating very similar thermodynamic properties of the two proteins. This was confirmed by the fitting analysis according to a two-state unfolding model [22], as both the midpoint unfolding GuHCl concentrations (Cm) and free energy differences between the native and unfolded state (ΔGu) were essentially identical for wt moPrP (Cm = 2.33 ± 0.02 M, ΔGu = 5.0 ± 0.06 kcal/mol) and S170N/N174T moPrP (Cm = 2.33 ± 0.03 M, ΔGu = 4.86 ± 0.09 kcal/mol). Therefore, greater fibrillization propensity of the rigid loop variant is not due to the effect of amino acid substitutions on the thermodynamic stability of the prion protein.
Fig 2.
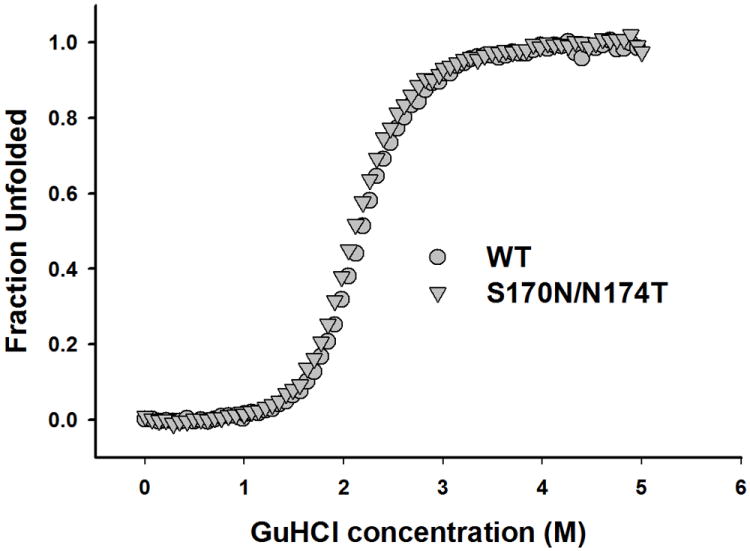
GuHCl-induced equilibrium unfolding for wild type moPrP and the S170N/N174T variant as monitored by ellipticity at 222 nm. The curves represent the average of three experiments.
Next, we asked the question whether the rigid loop-promoting amino acid substitutions affect the final structure of PrP amyloid fibrils. Formation of different types of fibrils could potentially explain differences in fibrillization kinetics of wild type and mutant proteins. However, when examined by atomic force microscopy, fibrils formed from wt moPrP and the S170N/N174T variant appeared morphologically very similar. In both cases, fibrils were of variable length, and AFM images indicated the presence of both individual protofilaments as well as thicker (higher) structures resulting from lateral association of these protofilaments (Fig. 3 A and B). Height measurements in the regions corresponding to individual protofilaments revealed no difference between structures formed by wt and mutant protein (4.3 ± 0.1 and 4.3 ± 0.2 nm for wt and mutant PrP, respectively). Fibrils were further examined by electron microscopy. Again, no measurable differences were found between the structures formed by the two proteins. In both cases, most fibrils appeared to consist of at least two protofilaments that self-associate into irregularly twisted structures similar to those observed in previous studies with wt mouse PrP [31]. Thus, despite differences in ThT fluorescence intensity (see above), no morphological differences could be detected between the two fibril types, at least within the resolution limits of current AFM and EM measurements.
Fig 3.
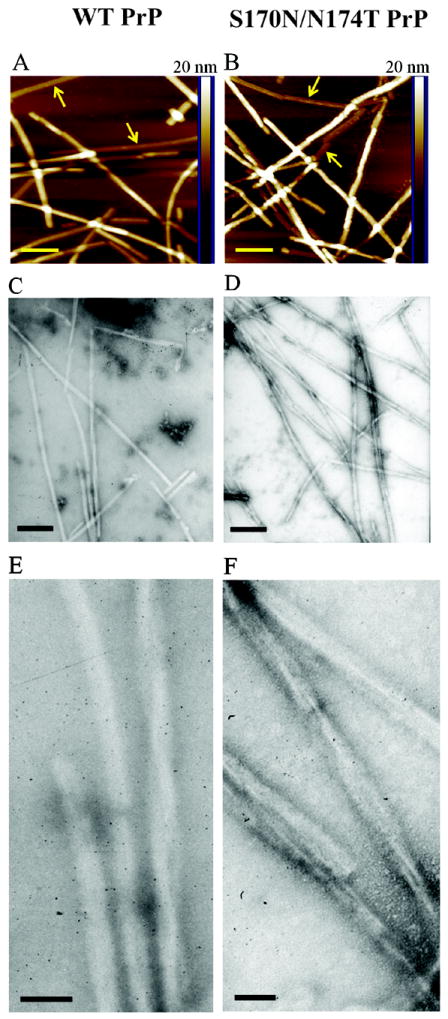
Morphology of fibrillar structures formed by wt moPrP (left panels) and the S170N/N174T variant (right panels). (A and B) Atomic force microscopy images in height mode. The horizontal bar corresponds to 200 nm, and the vertical color bar at the right side of each image represents height scale. Examples of individual (non-associated) protofilaments are marked with arrows. (C-F) Electron micrographs of the two fibril types. Scale bars in panels C and D correspond to 200 nm and those in panels E and F correspond to 50 nm. Fibrils were prepared in 2 M GuHCl.
To compare the structural properties of fibrils formed by the two proteins at the molecular level, we employed the method of hydrogen/deuterium exchange coupled with mass spectrometry. By measuring the extent of deuterium labeling for individual peptic fragments derived from fibrillized proteins, this method allows the identification of systematically H-bonded β-sheet core region of fibrillar aggregates [23-25]. Previous studies mapped this amyloid core region in fibrils formed by the recombinant human and Syrian hamster PrP to residues ~169-221 and ~163-222, respectively [23-25]. The β-sheet core region for fibrils formed by moPrP appears to be generally similar, as peptic fragments showing protection from deuterium labeling are limited to the ~160-223 region (Fig. 4). Importantly, no significant difference could be detected between the ‘protection maps’ for fibrils formed from wt moPrP and the S170N/N174T variant, indicating essentially identical core regions for both fibril types (Fig. 4).
Fig 4.
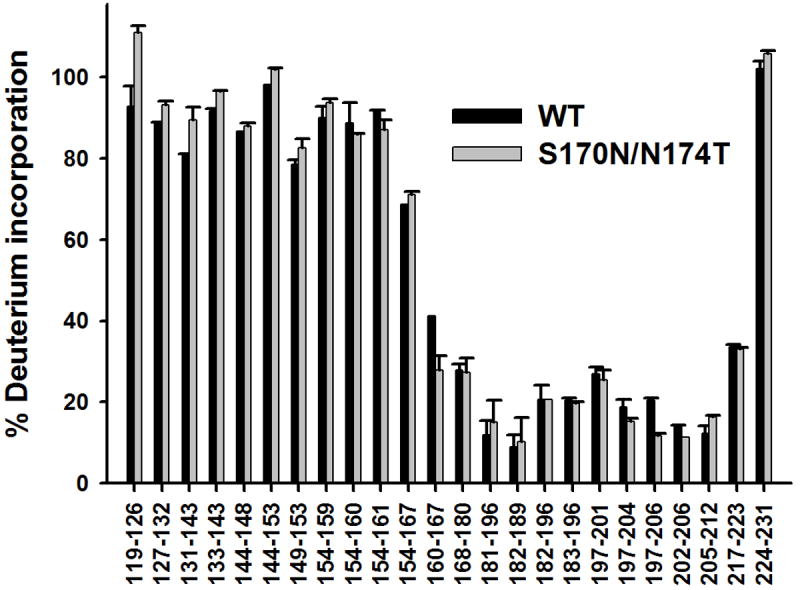
Deuterium incorporation for peptic fragments derived from amyloid fibrils formed by wild type moPrP and S170N/N174T moPrP. Fibrils formed in 2 M GuHCl were incubated for 24 h in D2O.
Very similar core regions for amyloids formed by wt moPrP and the rigid loop variant do not rule out the possibility of subtle packing differences within these core regions (that could potentially account for differences in ThT fluorescence intensity). Indeed, differences in sheet-to-sheet interfaces have been previously shown in crystal structures of six-residue peptide fragments corresponding to the loop region (residues 170-175) in human/mo and elk PrP [33-35]. These crystal structures show that the peptide derived from elk PrP sequence forms a stronger and more prototypical steric zipper than that formed by the corresponding sequence in human/mo PrP. However, high-resolution crystallographic studies are limited to short peptide fragments only, and it is not known whether the packing of these peptides in crystals faithfully represents the structural properties of corresponding sequences in amyloid fibrils formed by the full-length prion protein.
The lack of any effect of rigid loop-inducing amino acid substitutions on the global thermodynamic stability of PrP and apparently similar structural organization of fibrillar aggregates formed by wt and mutant PrP are intriguing, raising questions regarding the mechanism by which these mutations could increase the propensity of prion protein for conformational conversion to misfolded aggregates such as amyloid fibrils. When considering this question, one should note that prion protein fibrillization in vitro requires at least partial destabilization/unfolding of the native protein structure. This is typically accomplished by studying the reaction in the presence of chemical denaturants such as urea or GuHCl [28,30,32]. The experiments shown in Fig. 1 were performed in a buffer containing 2 M GuHCl. Under these conditions, the monomeric PrP exists as a mixture of different conformers including a natively folded protein, a fully unfolded protein and, likely, a partially structured folding intermediate [36]. Therefore, it is not clear whether the increased amyloidogenic propensity of the S170N/N174T moPrP variant in 2 M GuHCl is indeed due to the presence of rigid loop in the native PrP structure or rather it simply reflects an increased amyloidogenic potential of the amino acid sequence containing these amino acid substitutions, regardless of the specific PrP secondary structure. To probe this question, we repeated the fibrillization experiments in the presence of 4 M GuHCl, i.e., under the conditions in which PrP is fully unfolded, with no natively folded protein present (see the unfolding curves in Fig. 2). Since these measurements were impractical in the plate reader format (refer to Materials and Methods), they were performed in a manual format. Under these conditions, the reactions for wt moPrP and the S170N/N174T variant were characterized by lag phases of 22.5 ± 2.5 h and 13.8 ± 2.0 h, respectively. Thus, it appears that also under fully denaturing conditions, the S170N/N174T moPrP variant has higher fibrillization propensity than the wt protein.
Since side chain of Asn170 (present in rigid loop elk PrP and S170N/N174T moPrP) has well defined orientation and protrudes from PrPC, whereas Ser170 (present in wt moPrP) is disordered [14,37], it was previously suggested that the increased conversion propensity of S170N/N174T moPrP to PrPSc as observed in animal studies may be due to the ability of Asn170 to engage in long-range intermolecular bonds with complementary sequences, as such interactions could facilitate formation of steric zippers [18]. However, the present observation that S170N/N174T moPrP is more amyloidogenic also under the denaturing conditions (i.e., when native rigid loop is no longer present) strongly suggests that this increased amyloidogenicity is not necessarily due to loop rigidity per se (or PrP secondary structure) but rather results from greater intrinsic amyloidogenic potential of the amino acid sequence within the loop region of S170N/N174T moPrP. This finding in vitro is likely of physiological relevance as available data suggest that the direct monomeric precursor of PrPSc is not the natively folded PrPC but rather the unfolded form of the protein [38] or a loosely structured folding intermediate [39,40]. Indeed, our present observations and conclusions are consistent with recent data on the role of the β2-α2 loop in interspecies prion transmission in experimental animals [41]. The latter studies indicate that it is the identity in the primary structure of the loop, particularly at position 170, and not loop rigidity as such that influences cross-species seeding of prion protein conversion to PrPSc in vivo.
Conclusions
In summary, we have characterized biophysical properties of prion disease-causing S170N/N174T variant of moPrP, finding that this variant has substantially higher intrinsic amyloidogenic potential as compared to wt moPrP. Despite the differences in fibrillization kinetics, amyloid fibrils formed by the wt and mutant proteins appear to be structurally similar, at least at the level of overall morphology and the identity of amyloid core region. It was previously shown that, in contrast to wt moPrP, the native structure of S170N/N174T moPrP is characterized by rigid β2-α2 loop. However, our present results argue that the increased conversion propensity of the S170N/N174T variant is not necessarily due to loop rigidity per se (or PrP secondary structure) but it reflects greater amyloidogenic potential of the stretch of amino acids within the 165-175 region of S170N/N174T moPrP.
Fig 5.
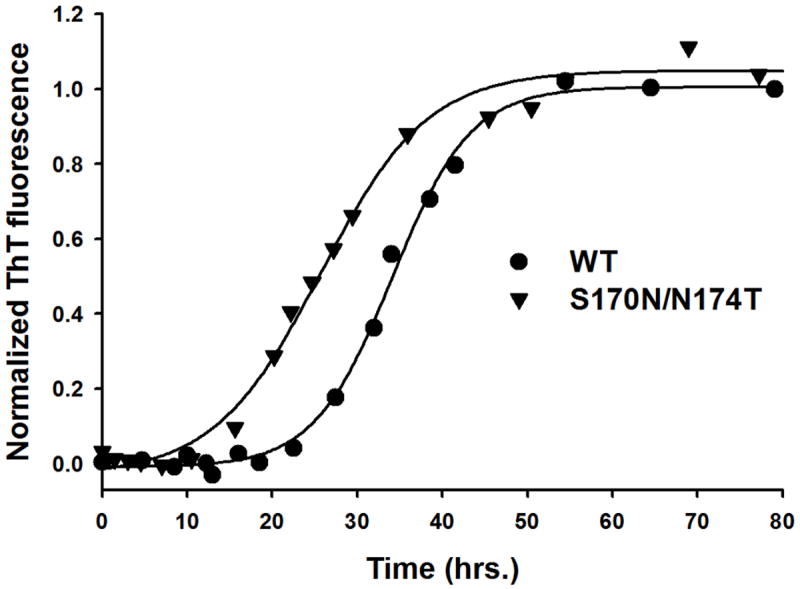
Time-course of amyloid fibril formation for wild type moPrP and S170N/N174T moPrP under fully denaturing conditions. Experiments were performed in the presence of 4 M GuHCl. Solid lines show best fits to equation [1]. The lag phases are 22.5 ± 2.5 h and 13.8 ± 2.0 h for wild type moPrP and S170N/N174T moPrP respectively. The lag phases were determined by fitting individual ThT response curves to equation [1], and these individual values from 3-4 experiments were used to calculate the mean value and associated standard errors.
Highlights.
Rigid loop variant of mouse prion protein shows increased amyloidogenicity
Amyloid fibrils of wild-type and rigid loop PrP variant are structurally similar
The present data in vitro help explain previous observations in transgenic mice
Acknowledgments
This work was supported in whole by National Institutes of Health Grant NS 44158. The authors thank Dr. Krystyna Surewicz for providing mouse prion protein used in some experiments and help with electron microscopy studies.
Abbreviations used
- PrP
prion protein
- GuHCl
guanidine hydrochloride
- ThT
Thioflavin T
- wt
wild type
- moPrP
mouse prion protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;93:337–48. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 2.Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry. 2009;48:2574–85. doi: 10.1021/bi900108v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–5. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–6. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 5.Kim JI, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285:14083–7. doi: 10.1074/jbc.C110.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A. 2012;109:8546–51. doi: 10.1073/pnas.1204498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones EM, Surewicz WK. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell. 2005;121:63–72. doi: 10.1016/j.cell.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 8.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–6. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 9.Pastore A, Zagari A. A structural overview of the vertebrate prion proteins. Prion. 2007;1:185–97. doi: 10.4161/pri.1.3.5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wuthrich K, Riek R. Three-dimensional structures of prion proteins. Adv Protein Chem. 2001;57:55–82. doi: 10.1016/s0065-3233(01)57018-7. [DOI] [PubMed] [Google Scholar]
- 11.Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231) FEBS Lett. 1997;413:282–8. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 12.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121-231) Nature. 1996;382:180–2. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 13.Zahn R, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97:145–50. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci U S A. 2005;102:646–50. doi: 10.1073/pnas.0409008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christen B, Perez DR, Hornemann S, Wuthrich K. NMR structure of the bank vole prion protein at 20 degrees C contains a structured loop of residues 165-171. J Mol Biol. 2008;383:306–12. doi: 10.1016/j.jmb.2008.08.045. [DOI] [PubMed] [Google Scholar]
- 16.Lopez Garcia F, Zahn R, Riek R, Wuthrich K. NMR structure of the bovine prion protein. Proc Natl Acad Sci U S A. 2000;97:8334–9. doi: 10.1073/pnas.97.15.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lysek DA, et al. Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc Natl Acad Sci U S A. 2005;102:640–5. doi: 10.1073/pnas.0408937102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigurdson CJ, et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci U S A. 2009;106:304–9. doi: 10.1073/pnas.0810680105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morillas M, Swietnicki W, Gambetti P, Surewicz WK. Membrane environment alters the conformational structure of the recombinant human prion protein. J Biol Chem. 1999;274:36859–65. doi: 10.1074/jbc.274.52.36859. [DOI] [PubMed] [Google Scholar]
- 20.LeVine H., 3rd Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–10. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pedersen JS, Christensen G, Otzen DE. Modulation of S6 fibrillation by unfolding rates and gatekeeper residues. J Mol Biol. 2004;341:575–88. doi: 10.1016/j.jmb.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 22.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 23.Lu X, Wintrode PL, Surewicz WK. Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Natl Acad Sci U S A. 2007;104:1510–5. doi: 10.1073/pnas.0608447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol. 2011;18:504–6. doi: 10.1038/nsmb.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smirnovas V, Kim JI, Lu X, Atarashi R, Caughey B, Surewicz WK. Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J Biol Chem. 2009;284:24233–41. doi: 10.1074/jbc.M109.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177–87. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Surewicz WK, Apostol MI. Prion protein and its conformational conversion: a structural perspective. Top Curr Chem. 2011;305:135–67. doi: 10.1007/128_2011_165. [DOI] [PubMed] [Google Scholar]
- 28.Apetri AC, Vanik DL, Surewicz WK. Polymorphism at residue 129 modulates the conformational conversion of the D178N variant of human prion protein 90-231. Biochemistry. 2005;44:15880–8. doi: 10.1021/bi051455+. [DOI] [PubMed] [Google Scholar]
- 29.Vanik DL, Surewicz WK. Disease-associated F198S mutation increases the propensity of the recombinant prion protein for conformational conversion to scrapie-like form. J Biol Chem. 2002;277:49065–70. doi: 10.1074/jbc.M207511200. [DOI] [PubMed] [Google Scholar]
- 30.Lewis PA, Tattum MH, Jones S, Bhelt D, Batchelor M, Clarke AR, Collinge J, Jackson GS. Codon 129 polymorphism of the human prion protein influences the kinetics of amyloid formation. J Gen Virol. 2006;87:2443–9. doi: 10.1099/vir.0.81630-0. [DOI] [PubMed] [Google Scholar]
- 31.Sun Y, Makarava N, Lee CI, Laksanalamai P, Robb FT, Baskakov IV. Conformational stability of PrP amyloid fibrils controls their smallest possible fragment size. J Mol Biol. 2008;376:1155–67. doi: 10.1016/j.jmb.2007.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc) J Mol Biol. 2005;346:645–59. doi: 10.1016/j.jmb.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 33.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 34.Wiltzius JJ, et al. Molecular mechanisms for protein-encoded inheritance. Nat Struct Mol Biol. 2009;16:973–8. doi: 10.1038/nsmb.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apostol MI, Wiltzius JJ, Sawaya MR, Cascio D, Eisenberg D. Atomic structures suggest determinants of transmission barriers in mammalian prion disease. Biochemistry. 2011;50:2456–63. doi: 10.1021/bi101803k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosszu LL, et al. Definable equilibrium states in the folding of human prion protein. Biochemistry. 2005;44:16649–57. doi: 10.1021/bi051277k. [DOI] [PubMed] [Google Scholar]
- 37.Gorfe AA, Caflisch A. Ser170 controls the conformational multiplicity of the loop 166-175 in prion proteins: implication for conversion and species barrier. FASEB J. 2007;21:3279–87. doi: 10.1096/fj.07-8292com. [DOI] [PubMed] [Google Scholar]
- 38.Hosszu LL, Baxter NJ, Jackson GS, Power A, Clarke AR, Waltho JP, Craven CJ, Collinge J. Structural mobility of the human prion protein probed by backbone hydrogen exchange. Nat Struct Biol. 1999;6:740–3. doi: 10.1038/11507. [DOI] [PubMed] [Google Scholar]
- 39.Apetri AC, Maki K, Roder H, Surewicz WK. Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J Am Chem Soc. 2006;128:11673–8. doi: 10.1021/ja063880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Apetri AC, Surewicz K, Surewicz WK. The effect of disease-associated mutations on the folding pathway of human prion protein. J Biol Chem. 2004;279:18008–14. doi: 10.1074/jbc.M313581200. [DOI] [PubMed] [Google Scholar]
- 41.Bett C, Fernandez-Borges N, Kurt TD, Lucero M, Nilsson KP, Castilla J, Sigurdson CJ. Structure of the beta2-alpha2 loop and interspecies prion transmission. FASEB J. 2012;26:2868–76. doi: 10.1096/fj.11-200923. [DOI] [PMC free article] [PubMed] [Google Scholar]