

Abstract
Several epidemiological and preclinical studies suggest that non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit cyclooxygenase (COX), reduce the risk of Alzheimer's disease (AD) and can lower β-amyloid (Aβ) production and inhibit neuroinflammation. However, follow-up clinical trials, mostly using selective cyclooxygenase (COX)-2 inhibitors, failed to show any beneficial effect in AD patients with mild to severe cognitive deficits. Recent data indicated that COX-1, classically viewed as the homeostatic isoform, is localized in microglia and is actively involved in brain injury induced by pro-inflammatory stimuli including Aβ, lipopolysaccharide, and interleukins. We hypothesized that neuroinflammation is critical for disease progression and selective COX-1 inhibition, rather than COX-2 inhibition, can reduce neuroinflammation and AD pathology. Here, we show that treatment of 20-month-old triple transgenic AD (3 × Tg-AD) mice with the COX-1 selective inhibitor SC-560 improved spatial learning and memory, and reduced amyloid deposits and tau hyperphosphorylation. SC-560 also reduced glial activation and brain expression of inflammatory markers in 3 × Tg-AD mice, and switched the activated microglia phenotype promoting their phagocytic ability. The present findings are the first to demonstrate that selective COX-1 inhibition reduces neuroinflammation, neuropathology, and improves cognitive function in 3 × Tg-AD mice. Thus, selective COX-1 inhibition should be further investigated as a potential therapeutic approach for AD.
Keywords: 3 × Tg-AD mice, Alzheimer's disease, COX-1, microglia, SC-560
Alzheimer's disease (AD) is an age-related neurodegenerative disease characterized by progressive memory and cognitive decline and accompanied by neuropathological changes such as senile plaques, neurofibrillary tangles, and clusters of activated glial cells (Mattson 2004; Wyss-Coray 2006). Evidence from post-mortem analysis strongly supports an important role for neuroinflammation in the pathogenesis of AD, with activated microglia and reactive astrocytes surrounding the amyloid deposits and increased expression of several inflammatory mediators (McGeer et al. 1987; Haga et al. 1989; Wisniewski et al. 1992). Previous epidemiological and experimental studies provide evidence that long-term use of non-steroidal anti-inflammatory drugs (NSAIDs) may reduce AD risk by inhibiting inflammatory responses (Lim et al. 2000, 2001; McKee et al. 2008). Although several distinct actions have been described, traditional NSAIDs primarily exert an anti-inflammatory effect through inhibition of cyclooxygenase (COX) isoforms.
COX-1 and -2 are key elements of the innate immune system. Because of the classical view of COX-1 being constitutive and thus exerting only homeostatic functions and COX-2 being induced by inflammatory stimuli, COX-2 has been considered the most appropriate pharmacological target to treat neuroinflammation. However, both COX-1 and COX-2 are constitutively expressed in brain, and several recent studies have challenged this prevalent view (Choi et al. 2009; Aid et al. 2010). COX-1 is actively involved in immunoregulation of the central nervous system (CNS) (Garcia-Bueno et al. 2009; Choi et al. 2010; Matousek et al. 2010; Dargahi et al. 2011), and COX-1 gene deletion reduces β-amyloid (Aβ)-induced neuroinflammation and neuronal damage (Choi and Bosetti 2009).
COX-1 is predominantly expressed by microglia in rodent and human brain (Hoozemans et al. 2001), and its brain expression increases with aging (Aid and Bosetti 2007). Based on this combined evidence, COX-1 is likely to be involved in the neuroinflammatory process associated with AD. Indeed, in post-mortem brain from AD patients, increased COX-1-expressing microglia are found surrounding amyloid plaques (Yermakova et al. 1999; Hoozemans et al. 2001). In contrast, COX-2 is not expressed by microglia or astrocytes in the AD brain and shows time-dependent expression changes in neurons, where its expression is increased in the early phase and then decreased in the later phase of the disease (Hoozemans et al. 2008). In this study, we hypothesized that selective COX-1 inhibition would protect the brain against AD-related neuroinflammation and improve cognitive deficits. To test this hypothesis, we administered the COX-1 inhibitor SC-560 to aged triple transgenic AD mice (3 × Tg-AD), which progressively develop extracellular amyloid plaques, intracellular neurofibrillary tangles, and cognitive impairment (Oddo et al. 2003). We found that SC-560 treatment significantly reduces amyloid deposits, tau hyperphosphorylation, and neuroinflammation, and ameliorates cognitive deficits in aged 3 × Tg-AD mice with significant pathology. These results suggest that COX-1 may play an important role in the pathogenesis of AD and a selective COX-1 inhibitor may be therapeutically beneficial in AD.
Materials and methods
Animals and drug treatments
Twenty-month-old male homozygous 3 × Tg-AD mice on a B6/SJV 129 background were used. The 3 × Tg-AD mice express three mutant human genes: PS1M146V knock-in, APPswe, and TauP301L mutations (Oddo et al. 2003). The 3 × Tg-AD mice were derived from founders received from Dr. LaFerla's laboratory (University of California, Irvine). Mice were housed in standard cages in 12 h light/12 h dark cycle and given food and water ad libitum. SC-560 was initially dissolved in dimethylsulfoxide (DMSO) and then diluted with 25% Solutol HS 15 (BASF, Ludwigshafen, Germany) in sterile saline to a final DMSO concentration of 10%. Mice were injected intraperitoneally with SC-560 (30 mg/kg; once a day for 8 day; Cayman Chemical, Ann Arbor, MI, USA). Mice in the vehicle group were injected for eight consecutive days with DMSO/Solutol at the same concentration as was used to dissolve SC-560. SC-560 specifically inhibits COX-1 (Smith et al. 1998; Walker et al. 2001). This treatment regimen was based on previously published data (Choi et al. 2008; Matousek et al. 2010) and significantly decreased brain levels of prostanoids, including prostaglandin E2 (PGE2) and TxB2, in C57BL/6 mice (Choi et al. 2008). There were no significant changes in body weight between or within the groups of mice. All procedures were approved by the National Institutes of Health (NIH) Animal Care and Use Committee in accordance with NIH guidelines on the care and use of laboratory animals.
Behavioral tests
Spatial learning and memory was assessed using the Morris Water Maze, as previously described (McKee et al. 2008). Briefly, a circular plastic pool was filled with water (22°C) that was colored with non-toxic white paint to obscure the location of a submerged platform. Three visual cues were placed around the tank to orient the mice, with the platform remaining in a fixed location. The platform location was kept constant for each mouse during training, and it was 1.5 cm beneath the surface of the water. On each day, training consisted of five trials. For each trial, the mouse was place into the pool facing the wall from one of four randomly varied start positions and allowed to swim until finding the platform. If a mouse failed to find the platform within 60 s, the mouse was manually guided to the hidden platform and allowed to stay on the platform for 30 s. Probe trials for retention of spatial training were conducted 1.5 and 24 h after the last training trial. During the probe trials, the platform was removed and mice were free to swim in the pool for 60 s. Mice were monitored with the camera on the ceiling directly over the pool and recorded for subsequent analysis. The escape latency to cross the platform location, the number of platform location crosses, and path length were recorded.
Tissue preparation
Mice were deeply anesthetized and transcardially perfused 50 mL of 0.9% saline. The brain was immediately removed from the cranium and bisected sagitally. One hemisphere was snap frozen and processed for biochemical analysis. The other hemisphere was placed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 48 h and followed by cryoprotection in 30% sucrose at 4°C. Regularly spaced series of sagittal sections (30-μm thick) were collected in cryoprotectant solution and stored at −20°C until processing.
Immunological and histological staining
Immunohistochemistry was achieved using conventional avidinbiotin immunoperoxidase and dual indirect immunofluorescence methods, the latter employing Alexa fluor-conjugated secondary antibodies. Briefly, fixed free-floating sections were rinsed three times with phosphate-buffered saline (PBS) and incubated blocking solution (normal serum and 0.2% Triton X-100 in PBS) for 1 h at 25°C. To quench the endogenous peroxidase activity, sections were incubated for 10 min in 0.3% H2O2. The appropriate primary antibody was applied and sections were incubated overnight at 4°C. Bound antibody was detected with biotinylated secondary antibody. The horseradish peroxidase method (Vectastatin Elite Kit; Vector Laboratories, Burlingame, CA, USA) and 3,3-diaminobenzidine (Sigma-Aldrich, St Louis, MO, USA) were used for visualization of antibody. In double-labeling studies, sections were incubated in the appropriate Alexa fluor 488- or Alexa fluor 594-conjugated secondary antibodies (Life Technologies, Grand Island, NY, USA) at a dilution of 1: 400 in blocking solution for 1 h at 25°C. The following primary antibodies and respective dilutions were used in this study: mouse monoclonal anti-Aβ1-16 6E10 (1: 1000; Covance Research Laboratories, Princeton, NJ, USA), phospho-tau pSer202/pThr205 (1: 200; AT8; Thermo Scientific, Rockford, IL, USA), rat anti-CD11b (1: 200; Serotec, Raleigh, NC, USA), mouse anti-glial fibrillary acidic protein (GFAP) (1: 500; Sigma-Aldrich), rabbit anti-ionized calcium binding adaptor molecule-1 (Iba-1) (1: 200; Wako, Osaka, Japan), rat anti-CD68 (1: 200; AbD Serotec), or mouse anti-CD16/CD32 (1: 200; FcγRIII/II; BD Biosciences, Sparks, MD, USA). Congo red stain was prepared per manufacturer's protocol (Sigma-Aldrich). Serial sections were mounted onto gelatin-coated slides, allowed to dry fully, placed in Congo red stain for 30 min, and dipped in histoclear, then cover-slipped with mounting media (Wilcock et al. 2006). Serial sections of each hemisphere were stained by thioflavin S to identify Aβ plaques (Oddo et al. 2006c). Brain sections were incubated in 1% thioflavin S (Sigma-Aldrich) for 5 min, washed twice in 70% ethanol and water, respectively, and then mounted with fluorescent mounting medium (Vector Laboratories). Bright-field images were acquired using an AX10 microscope (Zeiss, Thornwood, NY, USA) and fluorescent dyes were imaged using a LSM 510 confocal microscope (Zeiss). For quantification, every sixth serially collected section from each mouse hemisphere was analyzed microscopi-cally, and the number of congophilic amyloid plaques and P-tau (AT8) immunoreactive neurons in the hippocampal subiculum region was counted as previously described (Oddo et al. 2006a).
RNA extraction and reverse transcription quantitative real-time PCR
Total RNA was extracted from each hemisphere using RNeasy Lipid Tissue Midi Kit (Qiagen, Valencia, CA, USA), and cDNA was synthesized using the High Capacity cDNA Reverse Transcription (RT) Kit (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer's protocol (Aid et al. 2008; Choi et al. 2008). cDNA samples were used as templates for TaqMan-based quantitative PCR on an ABI 7000 Sequence Detection System (Applied Biosystems) with the TaqMan Gene Expression Master Mix and mouse gene-specific probe-based primers (TaqMan Gene Expression Assays; Applied Biosystems). The following TaqMan Gene Expression Assays were used in this study: tumor necrosis factor (Tnf), Mm00443258_m1; inducible nitric oxide synthase (Nos2), Mm00440502_m1; interleukin 1beta (Il1b), Mm01336189_m1; chitinase 3-like 3 (Chi3l3; Ym1), Mm04213363_u1; arginase 1 (Arg1), Mm00475988_m1; macro-phage scavenger receptor 1 (Msr1), Mm00446214_m1; Phospho-glycerate kinase 1 (Pgk1), Mm00435617_m1. Pgk1 was used as the endogenous control. The quantification and normalization of results were based on computation of specific gene threshold cycle values (Ct) and Pgk1 Ct values. All samples were run in triplicate and at least two independent experiments were repeated.
Western blot analysis
Brain hemispheres were homogenized in ristocetin-induced platelet agglutination buffer (Cell Signaling Technology, Danvers, MA, USA) with complete protease inhibitor tablets (Roche, Indianapolis, IN, USA). Samples (10 or 50 μg) were separated by standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (BioRad, Hercules, CA, USA) and blotted onto polyvinylidene fluoride membranes (Li-Cor Biosciences, Lincoln, NE, USA). These membranes were incubated with blocking buffer (Li-Cor Biosciences) to block non-specific protein binding. Membranes were then incubated in blocking buffer with primary antibodies to AT8 (1: 500; Thermo Scientific), TAU-5 (1: 500; Invitrogen, Camarillo, CA, USA), GFAP (1: 1000; Sigma-Aldrich), phosphoglycogen synthase kinase-3β (P-GSK-3β Ser9) (1: 1000; Cell Signaling), GSK-3β (1: 1000; Cell Signaling), cyclin-dependent kinase 5 (Cdk5) (1: 1000; Cell Signaling), protein phosphatase 2A (PP2A) (1: 1000; Cell Signaling), and β-actin (1: 2000; SigmaAldrich). After primary antibody incubations, membranes were washed three times with Tris-buffered saline containing 0.1% Tween-20 (TBS-T) at 25°C on a bench top shaker. Membranes were incubated with secondary antibodies conjugated to IRDye 800CW and IRDye 680RD (Li-Cor Biosciences) for 1 h at 25°C and in the dark with gentle shaking. After secondary antibody incubations, the membranes were washed three times with TBS-T in the dark, and then briefly rinsed in PBS before scanning. Membranes were scanned and analyzed using an Odyssey Infrared Imaging Scanner and Odyssey Imaging Software 3.0 (Li-Cor Biosciences). For signal quantification, antibody signals were analyzed as integrated intensities of regions defined around the bands of interest in either channel using Odyssey Imaging Software 3.0 (Li-Cor Biosciences). Results are expressed as percent relative response (mean ± standard error of the mean) compared with vehicle-treated controls.
Statistical analyses
All statistical analyses were performed using the Prism 5.0 software (Graphpad Software, La Jolla, CA, USA). Group differences were analyzed using standard unpaired t-test and results expressed as mean ± SEM.
Results
SC-560 treatment alters microglial activation status and increases phagocytic capabilities in 3 × Tg-AD mice
Although the precise role of glial cells in AD pathogenesis remains to be elucidated and characterized, glial cells and more specifically activated microglia are thought to be important and have been shown to cluster around plaques in AD brains and transgenic mouse models (McGeer et al. 1987; Haga et al. 1989; Wisniewski et al. 1992). In previous studies, SC-560 was shown to reduce microglial activation in inflamed mouse brain and cultured adult human microglia (Hoozemans et al. 2002; Choi et al. 2008). To determine whether SC-560 treatment would affect microglial activation and the associated inflammatory response, we examined the expression of surface molecules and inflammatory factors. Immunohistochemical staining with CD11b showed that reactive microglia were abundant and closely associated with Aβ deposits in vehicle-treated 3 × Tg-AD mice (Fig. 1a). In contrast, CD11b immunoreactivity was reduced in SC-560-treated 3 × Tg-AD mice (Fig. 1a). We also found a strong increase in Iba-1 immunoreactivity, another microglial marker, in vehicle-treated 3 × Tg-AD mice (Fig. 1b), which was reduced by SC-560-treatment (Fig. 1b). Moreover, SC-560-treated 3 × Tg-AD mice increased FcγRIII/II and CD68 expression (Fig. 1c and e), suggesting a higher phagocytic activity. The change in microglial activation induced by SC-560 treatment was accompanied by a reduction of in the expression of genes encoding the pro-inflammatory factors TNF-α and iNOS (Fig. 1d), and an increase in the mRNA levels of the microglia/macrophages alternative activation markers Ym1 and scavenger receptor Msr1 (Fig. 1d). Consistent with the mRNA finding, significant increase in protein levels of Ym1 was found in SC-560-treated mice (Fig. 1f–h). There were no significant changes in GFAP immunoreactivity (Fig. 2a–c) and protein levels (Fig. 2a–c).
Fig. 1.
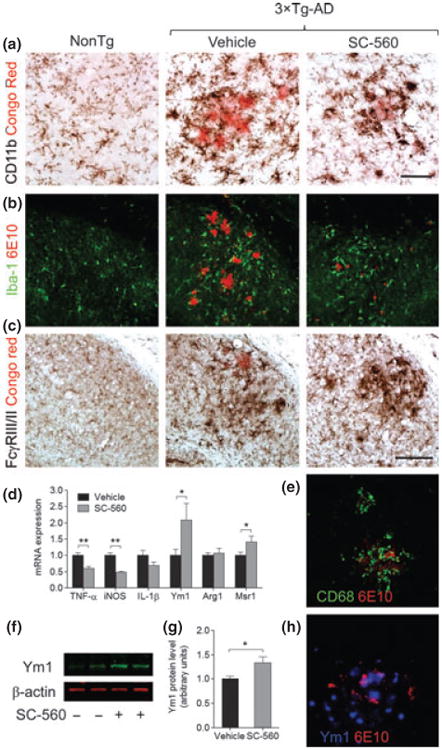
SC-560 treatment modulates microglial activation state. Representative immunostaining of microglial cells in the hippocampal subiculum of Non-Tg and 3 × Tg-AD mice treated with vehicle or SC-560. Double-staining of CD11b (a), Iba-1 (b), FcγRIII/II (c), CD68 (e), Ym1 (h), and amyloid deposits (Congo red or 6E10) showing reactive microglial cells surrounding amyloid plaques. Scale bar: a, e, h, 50 μm; b, c, 100 μm. (d) Expression of mRNA for pro-inflammatory factors and alternative activation markers in the brains of 3 × Tg-AD mice measured by quantitative real-time PCR. (f) Representative western blot of Ym1 in the brain homogenates 3 × Tg-AD mice treated with vehicle or SC-560. β-actin was used as loading control. (g) Quantification of Ym1 expression. Data shown are the means ± SEM (n = 6 mice per group). *p < 0.05 versus vehicle-treated 3 × Tg-AD mice; **p < 0.01 versus vehicle-treated 3 × Tg AD mice.
Fig. 2.
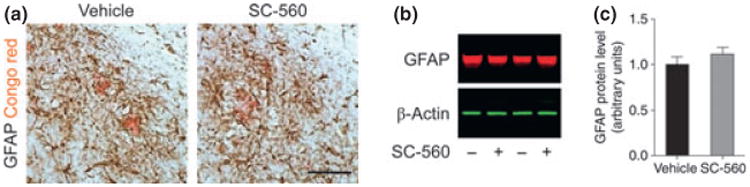
Effect of SC-560 treatment on astrocyte activation. (a) Representative immunostaining of astrocytes in the hippocampal subiculum of 3 × Tg-AD mice treated with vehicle (left) or SC-560 (right). Double-staining of glial fibrillary acidic protein (GFAP) and amyloid deposits (Congo red) showing reactive astrocyte surrounding amyloid plaques. Scale bar, 100 μm. (b) Representative western blot of GFAP in the brain homogenates of 3 × Tg-AD mice treated with vehicle or SC-560. β-actin was used as loading control. (c) Quantification of GFAP expression. Data shown are the means ± SEM (n = 6 mice per group).
SC-560 treatment reduces amyloid deposits and tau hyperphosphorylation in 3 × Tg-AD mice
We next sought to determine whether changes in microglial activation and inflammatory response correlated with improvement of amyloid and tau pathology following SC-560 treatment. The number of Congophilic amyloid plaques in Subiculum was significantly reduced in SC-560-treated 3 × Tg-AD mice compared with vehicle-treated mice (Fig. 3a and c). Similarly, thioflavin S-positive plaques in the Subiculum were decreased in SC-560-treated 3 × Tg-AD mice compared with vehicle-treated mice (Fig. 3b). To determine whether SC-560 treatment decreases amyloid plaques by changing amyloid precursor protein (APP) processing, we analyzed the levels of full-length APP (APP-FL) and APP C-terminal fragments (CTFs). We found no significant difference in the levels of APP-FL by western blot analysis between untreated and treated 3 × Tg-AD mice (Fig. 3d and e). In addition, no significant difference was apparent in CTF-α and -β levels (Fig. 3d). Therefore, these results suggest that SC-560 treatment does not modulate APP processing, but rather, it promotes plaque clearance.
Fig. 3.
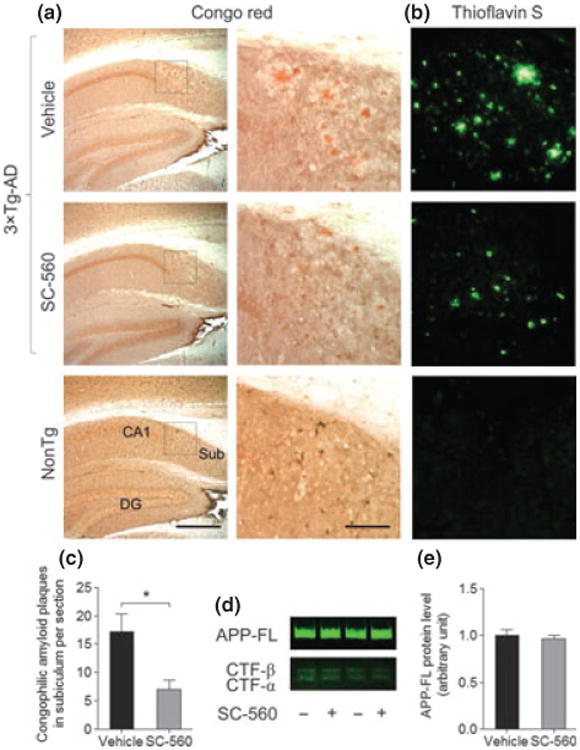
SC-560 treatment reduces amyloid deposits. (a, b) Representative images of Congo red (a) and thioflavin S staining (b) in hippocampal subiculum in 3 × Tg-AD mice treated with vehicle or SC-560. Scale bar, 100 μm. Boxed regions (left) indicate the areas magnified (right), respectively. (c) Quantification of Congophilic amyloid deposits in the subiculum. Data are means ± SEM (n = 6 per group). *p < 0.05 versus vehicle-treated 3 × Tg-AD mice. (d) Representative western blot of full-length APP (APP-FL) and APP C-terminal fragments (CTFs) in the brain homogenates of 3 × Tg-AD mice treated with vehicle or SC-560. (e) Quantification of APP-FL expres-sion. Data shown are the means ± SEM (n = 6 mice per group).
We further investigated the effect of SC-560 on tau hyperphosphorylation by counting the number of cells immunoreactive to the AT8 antibody, which recognizes pathological phosphorylation of tau that is associated with AD (Goedert et al. 1995). Intensely stained AT8-immunoreactive cells were observed in the subiculum in vehicle-treated 3 × Tg-AD mice (Fig. 4a). SC-560 treatment significantly decreased the number of AT8-immunoreactive cells in 3 × Tg-AD mice compared with vehicle-treated mice (Fig. 4a and b). We also found that the protein levels of phosphorylated tau (AT8 and TAU-5) were significantly reduced in SC-560-treated compared with vehicle-treated 3 × Tg-AD mice (Fig. 4c and d).
Fig. 4.
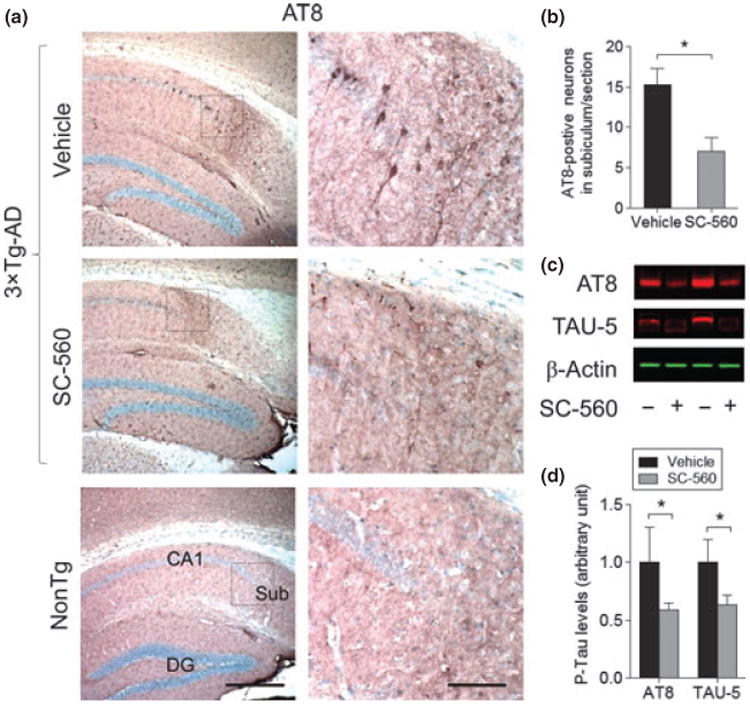
SC-560 treatment reduces phosphorylated tau. (a) Representative images of AT8 staining (a) in hippocampal subiculum in 3 × Tg-AD mice treated with vehicleorSC-560. Scale bar, 100 μm. Boxed regions (left) indicate the areas magnified (right), respectively. (b) Quantification of AT8-positive cells in the subiculum. Data are means ± SEM (n = 6 per group). *p < 0.05 versus vehicle-treated 3 × Tg-AD mice. (c) Representative western blot of phosphorylated tau (AT8) and total tau (TAU-5) in the brain homogenates of 3 × Tg-AD mice treated with vehicle or SC-560. β-actin was used as loading control. (d) Quantification of AT8 and TAU-5 expression. Data shown are the means ± SEM (n = 6 mice per group). *p < 0.05 versus vehicle-treated 3 × Tg-AD mice.
SC-560 treatment increases GSK-3β phosphorylation
Although the molecular mechanisms are not fully understood, several kinases and phosphatases, such as GSK-3β, c-Jun, N-terminal kinases, Cdk5, and PP2A, have been implicated in the hyperphosphorylation of tau (Atzori et al. 2001; Iqbal et al. 2005). We further determined a possible role of SC-560 treatment in regulating GSK-3β in aged 3 × Tg-AD mice. We found that SC-560 treatment increased phosphorylation of GSK-3β at Ser9 residue (Fig. 5a and b). As phosphorylation of Ser9 residue in GSK-3β leads to inhibition of its activity (Sutherland et al. 1993), SC-560 treatment may inhibit GSK-3β activity. In addition, we found that SC-560 treatment did not change the levels of Cdk5 and PP2A (Fig. 5a).
Fig. 5.
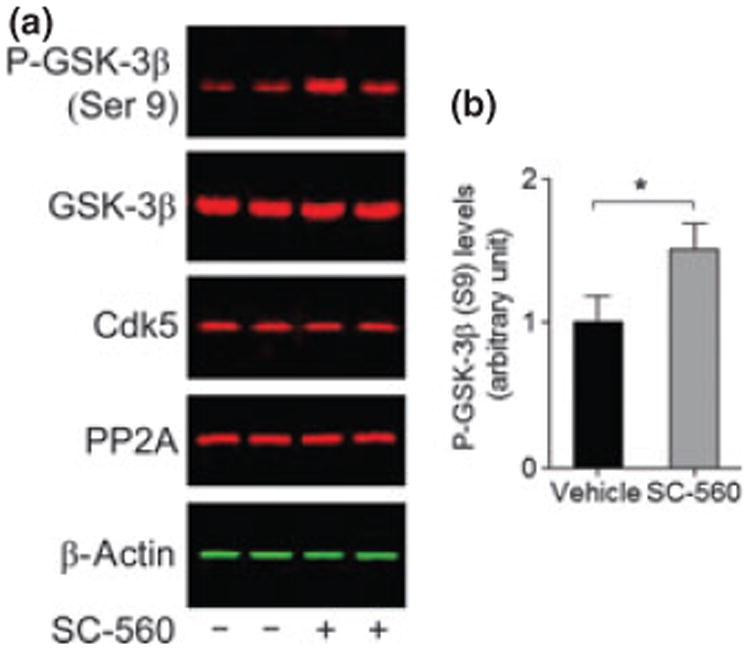
SC-560 treatment increases phosphorylated glycogen syn-thase kinase-3β (GSK-3β). (a) Representative western blot of GSK-3β phosphorylated at Ser 9, total GSK-3β, cyclin-dependent kinase 5 (CDK5), and protein phosphatase 2A (PP2A) in the brain homogenates of 3 × Tg-AD mice treated with vehicle or SC-560. (b) Quantification of phosphorylated GSK-3β (Ser 9) expression. Data shown are the means ± SEM (n = 6 mice per group). *p < 0.05 versus vehicle-treated 3 × Tg-AD mice.
SC-560 treatment ameliorates the spatial learning and memory deficits in 3 × Tg-AD mice
The 3 × Tg-AD mice develop progressive accumulation of plaques and tangles, which are associated with age-dependent cognitive impairment (Oddo et al. 2008). We next sought to determine whether changes in amyloid and tau pathology correlated with improvement of cognitive deficits following SC-560 treatment. We found that both vehicle- and SC-560-treated 3 × Tg-AD mice did not reach criterion after 5 day of training trials as determined by an escape latency < 20 s (n = 6 per group); nevertheless, the escape latency at training day 5 was significantly lower for SC-560-treated 3 × Tg-AD mice (Fig. 6b). In contrast, vehicle-treated 3 × Tg-AD mice were impaired in learning, as indicated by slower improvements in the escape latency across consecutive trials (Fig. 6b). Furthermore, we found that SC-560 treatment not only promoted learning during the training trials but also improved memory retention during the probe trials, as demonstrated by the significant decrease in the latency to cross the platform location (Fig. 6c) and by the significant increase in the number of platform location crosses (Fig. 6d).
Fig. 6.
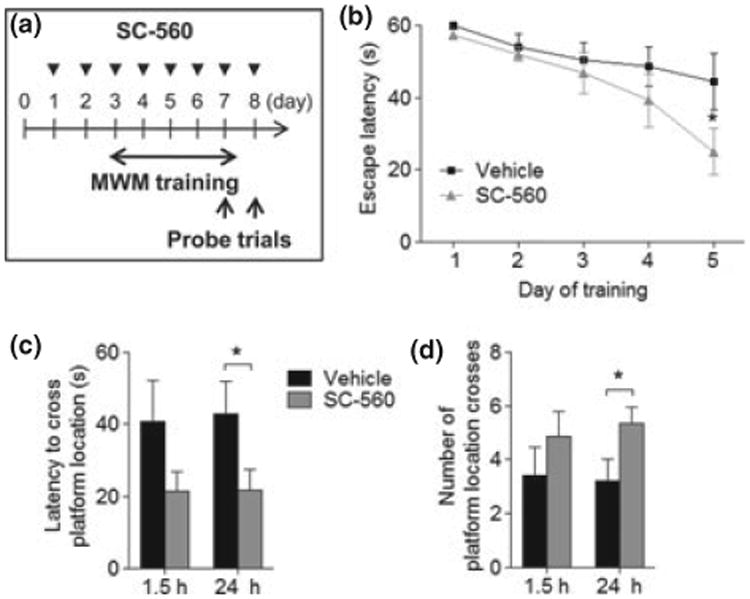
SC-560-treated aged 3 × Tg-AD mice show improved spatial learning and memory. (a) Schematic representation of experimental design. The arrowheads represent the injections for the SC-560 or vehicle. (b) Acquisition of spatial learning in the Morris Water Maze hidden-platform task. Escape latency represents time taken to escape to the hidden platform. (c and d) Probe trial without platform. Probe analysis was performed on day 5 (1.5 h after training day 5) and day 6 (24 h after training day 5). Latency is measured as the time to cross the exact platform location (c). Platform crossing is calculated as the number of crosses over the exact location of the hidden platform (d). Data are means ± SEM (n = 6 per group). *p < 0.05 versus vehicle-treated 3 × Tg-AD mice.
Discussion
In this study, we demonstrated that treatment of 20-month-old 3 × Tg-AD mice with the COX-1 inhibitor SC-560 significantly improved memory deficits and reduced amyloid deposition and tau phosphorylation in the hippocampus. We also found that SC-560 treatment alters the phenotype of activated microglia, promoting their phagocytic capability, reducing the expression of pro-inflammatory factors while increasing the expression of markers of alternative activation. The altered microglial phenotype may play a role in the reduction of amyloid and tau pathology and in rescuing impaired memory in aged 3 × Tg-AD mice.
Although both COX-1 and COX-2 are normally and constitutively expressed in the brain, in the AD brain clusters of COX-1-expressing microglia are detected around Aβ plaques (Yermakova et al. 1999) and could be involved in the neuroinflammatory response (Hoozemans et al. 2008). A critical role of COX-1 in the neuroinflammatory response is supported by our previous studies in COX-1 -/- mice, which show reduced neuronal damage and inflammatory response in response to lipopolysaccharide or Aβ (Choi and Bosetti 2009). Importantly, a reduction in activated microglia was observed along with a concomitant decrease in pro-inflammatory markers, including PGE2, associated with the classically activated microglial phenotype. In addition, several COX-1 inhibition strategies in animal models of AD have demonstrated a marked reduction in both amyloid burden and cognitive deficits. Recently, Coma and colleagues reported that triflusal, a platelet anti-aggregant and irreversible COX-1 inhibitor, could rescue cognitive deficits by reducing the dense-core amyloid plaque load, associated glial cell activation, and pro-inflammatory cytokine levels (Coma et al. 2010). An average of two years after the ADAPT trial was stopped; follow-up results suggest that after exclusion individuals with baseline cognitive syndromes, naproxen (slightly more selective for COX-1 than COX-2) cuts the risk of developing AD (Breitner et al. 2011). In the 117 ADAPT patients from whom CSF was collected 21 to 41 months after treatment was terminated, the tau-to-Aβ42 ratio was reduced by more than 40 percent in the group originally assigned to naproxen compared with placebo. In addition, NSAIDs with preferential COX-1 selectivity, such as NCX-2216 and indomethacin, also reduced amyloid burden (Jantzen et al. 2002; Sung et al. 2004). Another study suggested that SC-560 treatment could reduce neurotoxicity of synthetic Aβ in vitro (Bate et al. 2003). These data support a central role for COX-1 in microglia modulation and Aβ clearance in AD.
More than 10 studies of NSAIDs in a transgenic mouse model of AD have documented their effects on amyloid load and inflammation (McGeer and McGeer 2007), but Kotilinek and colleagues proposed a third possible mechanism by which NSAIDs may improve Aβ-induced deficits in memory and long-term plasticity independently of reducing inflammation or lowering Aβ42, and the improved memory function was inversely related to PGE2 response at synapse (Kotilinek et al. 2008). Although it is a matter of debate whether neuroinflammation is an underlying cause or a resulting condition in AD, targeting this process is a promising therapeutic option (Wyss-Coray 2006). Experimentally and clinically, AD is accompanied by a chronic neuroinflammation characterized by the activation of resident glial cells, primarily microglia. Activated microglia secrete a variety of pro-inflammatory and neurotoxic factors that are believed to induce and/or exacerbate neurodegeneration (Block et al. 2007). However, beneficial aspects of microglial activation and cytokines have also been identified (Bruce et al. 1996; Albensi and Mattson 2000; Hickman et al. 2008; Jimenez et al. 2008). These distinct effects may be explained by the induction of microglial response that is both phenotypically and functionally heterogeneous. It seems that, as AD progresses, microglia change toward a pro-inflammatory phenotype and lose their Aβ-clearing activities, resulting in reduced Aβ uptake and degradation (Jimenez et al. 2008). SC-560-treated 3 × Tg-AD mice presented different microglial activation, more similar to resting microglia, and increased expression of CD16/CD32 (FcγRIII/II), CD68, and Msr1, which suggests a higher phagocytic activity. This change in microglial activation was accompanied by an increase in the expression of Ym1, a marker for alternative activation (Jimenez et al. 2008). Fcγ receptors are among the best characterized phagocytic receptors and are detected in amyloid plaques and on ramified microglia, throughout the cortex and white matter of AD subjects (Peress et al. 1993). PGE2 has been reported to be an endogenous suppressor of macrophage FcγR phagocytosis through an EP2 receptor dependent signaling pathway (Aronoff et al. 2004). The pro-inflammatory milieu-induced suppression of Aβ phagocytosis can be relieved by NSAID ibuprofen or an EP2 receptor antagonist, suggesting that the production of PGE2 could lead to its receptor-dependent inhibition of microglial clearance of Aβ and plaques (Koenigsknecht-Talboo and Landreth 2005). In addition, gene deletion of EP2 receptor reduces oxidative damage and amyloid burden in a mouse model of AD (APPswe-PS1ΔE9:EP2-/-), indicating that PGE2 signaling via the EP2 receptor promotes age-dependent oxidative damage and increased Aβ accumulation (Liang et al. 2005). Similarly, reduction in cerebral cortical amyloid burden was achieved with transplantation of EP2-null bone marrow, suggesting that part of the cerebral Aβ lowering effect observed in APPswe-PS1ΔE9 mice that lacked EP2-/- receptors derived from enhanced phagocytosis by EP2-/- microglia (Keene et al. 2010). Interestingly, the inhibition of PGE2 production in macrophages by acetylsalicylic acid would lead to an increase of macrophage scavenger receptor CD36 expression, which plays a key role in the phagocytosis (Vinals et al. 2005). Increased expression of the macrophage scavenger receptor CD36 has been associated with up-regulation of markers of the alternative phenotype Ym1 and Fizz1 in Alzheimer's transgenic mice (Escribano et al. 2010). Overall, our data together with published observations suggest that COX-1-derived PGE2 and its downstream signaling pathway can modulate microglial phenotype and PGE2-related changes in functional microglial activation may be stopped by inhibiting COX-1 or specific downstream PGE2 receptors.
One of the key outcomes of this study is that treatment of 3 × Tg AD mice with SC-560 for 8 days reduces the amyloid deposition in the hippocampal subiculum. Although this effect appears dramatic considering that it takes months to reach AD neuropathological hallmarks, other interventions have demonstrated a rapid reduction of AD-like pathology in animal models. Heneka et al. reported that oral treatment of APP-overexpressing mice with ibuprofen for 7 days resulted in a significant reduction of amyloid deposits in the hippocampus (Heneka et al. 2005). More recently, Cramer et al. reported that oral administration of the retinoid × receptors agonist bexarotene to 11-month-old APP/PS1 mice resulted in a reduction of amyloid plaques more than 50% within just 3 days and restoration of memory and cognition (Cramer et al. 2012). Another important aspect of this study is that hyperphosphorylated tau is reduced by SC-560 treatment. Although it remains to be determined whether SC-560 can reduce tau pathology directly in the absence of reduced Aβ accumulation, previous studies in 3 × Tg-AD mice support a causal relationship between Aβ and tau. Ibuprofen reduces Aβ, hyperphosphorylated tau, and memory deficits in 3 × Tg-AD mice (McKee et al. 2008). Aβ immunotherapy in 3 × Tg-AD mice clears not only Aβ pathology but also clears somatodendritic tau (Oddo et al. 2006a). Furthermore, single immunization with anti-oligomeric specific antibody reduces tau as well as Aβ pathology (Oddo et al. 2006b).
There are some limitations in our study that need to be acknowledged. First, despite SC-560 selectivity toward COX-1 and previously published evidence that SC-560 at 30 mg/kg given i.p. to mice for 7 days significantly attenuated lipopolysaccharide-induced increase in PGE2, PGD2, PGF2α, and TxB2, the effect on prostaglandin levels in the 3 × Tg-AD mice have not been determined. Therefore, despite the improvement in AD pathology by COX-1 inhibition, a direct relationship between COX-1-derived prostaglandin production and memory impairment cannot be inferred. Second, our short-term treatment and the relatively small group size still leave some open questions on whether the effects of the inhibitor are acute and reversible upon discontinuation of the treatment. Further study will be needed to determine whether the beneficial action of SC-560 is long-term and persists after discontinuation of the treatment. Third, the question remains regarding a possible role of SC-560 treatment in regulating tau phosphorylation in 3 × Tg-AD mice. It has been shown that several kinases and phosphatases regulate tau phosphorylation (Atzori et al. 2001; Iqbal et al. 2005). Previous findings have shown that c-Jun N-terminal kinase may be activated by accumulation of unfolded damaged proteins, and, conversely, that activation of c-Jun N-terminal kinase itself may stimulate the formation of insoluble bodies (Atzori et al. 2001; Sherman and Goldberg, 2001). Additional studies in transgenic mouse models expressing only mutant tau genes are required to determine whether SC-560 directly reduces hyperphosphorylated tau or indirectly, as a consequence of reduced Aβ accumulation.
Although the exact mechanisms by which SC-560 affects tau pathology and microglial phenotypic changes remain to be determined, selective COX-1 inhibition alters microglial phenotype and it may account for the reduction of amyloid and tau pathology and for the rescue of impaired memory in aged 3 × Tg-AD mice. These results suggest that COX-1 may play an important role in AD pathogenesis, and that selective COX-1 inhibitors may be therapeutically beneficial in AD.
Acknowledgments
We thank S.M. Jin for assistance with confocal microscopy, Catherine Spong and Daniel Abebe for providing the Water Maze apparatus and for helpful technical advice, Elkhansa Sidahmed, Mohamed Mughal for their help with the animal work. The authors declare no competing interests. This study was supported by the Intramural Research Program of the National Institute on Aging and National Institute of Neurological Disorders and Stroke, National Institutes of Health, and in part by National Institutes of Health grant R01 AG026478.
Abbreviations
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- Aβ
β-amyloid
- COX
cyclooxygenase
- DMSO
dimethylsulfoxide
- GFAP
glial fibrillary acidic protein
- GSK-3β
glycogen synthase kinase-3β
- PBS
phosphate-buffered saline
- PGE2
prostaglandin E2
- PP2A
protein phosphatase 2A
References
- Aid S, Bosetti F. Gene expression of cyclooxygenase-1 and Ca(2+)-independent phospholipase A(2) is altered in rat hippocampus during normal aging. Brain Res Bull. 2007;73:108–113. doi: 10.1016/j.brainresbull.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aid S, Langenbach R, Bosetti F. Neuroinflammatory response to lipopolysaccharide is exacerbated in mice genetically deficient in cyclooxygenase-2. J Neuroinflammation. 2008;5:17. doi: 10.1186/1742-2094-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aid S, Silva AC, Candelario-Jalil E, Choi SH, Rosenberg GA, Bosetti F. Cyclooxygenase-1 and -2 differentially modulate lipopolysaccharide-induced blood-brain barrier disruption through matrix metalloproteinase activity. J Cereb Blood Flow Metab. 2010;30:370–380. doi: 10.1038/jcbfm.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173:559–565. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, Mirra SS, Migheli A. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropathol Exp Neurol. 2001;60:1190–1197. doi: 10.1093/jnen/60.12.1190. [DOI] [PubMed] [Google Scholar]
- Bate C, Veerhuis R, Eikelenboom P, Williams A. Neurones treated with cyclo-oxygenase-1 inhibitors are resistant to amyloid-beta1-42. NeuroReport. 2003;14:2099–2103. doi: 10.1097/00001756-200311140-00018. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Breitner JC, Baker LD, Montine TJ, et al. ADAPT Research Group. Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7:402–411. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- Choi SH, Bosetti F. Cyclooxygenase-1 null mice show reduced neuroinflammation in response to beta-amyloid. Aging (Albany NY) 2009;1:234–244. doi: 10.18632/aging.100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Langenbach R, Bosetti F. Genetic deletion or pharmacological inhibition of cyclooxygenase-1 attenuate lipopolysaccharide-induced inflammatory response and brain injury. FASEB J. 2008;22:1491–1501. doi: 10.1096/fj.07-9411com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Aid S, Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol Sci. 2009;30:174–181. doi: 10.1016/j.tips.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Aid S, Choi U, Bosetti F. Cyclooxygenases-1 and -2 differentially modulate leukocyte recruitment into the inflamed brain. Pharmacogenomics J. 2010;10:448–457. doi: 10.1038/tpj.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coma M, Sereno L, Da Rocha-Souto B, et al. Triflusal reduces dense-core plaque load, associated axonal alterations and inflammatory changes, and rescues cognition in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2010;38:482–491. doi: 10.1016/j.nbd.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargahi L, Nasiraei-Moghadam S, Abdi A, Khalaj L, Moradi F, Ahmadiani A. Cyclooxygenase (COX)-1 activity precedes the COX-2 induction in Abeta-induced neuroinflammation. J Mol Neurosci. 2011;45:10–21. doi: 10.1007/s12031-010-9401-6. [DOI] [PubMed] [Google Scholar]
- Escribano L, Simon AM, Gimeno E, et al. Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35:1593–1604. doi: 10.1038/npp.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bueno B, Serrats J, Sawchenko PE. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. J Neurosci. 2009;29:12970–12981. doi: 10.1523/JNEUROSCI.2373-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Haga S, Akai K, Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol. 1989;77:569–575. doi: 10.1007/BF00687883. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, et al. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJ, Rozemuller AJ, Janssen I, De Groot CJ, Veerhuis R, Eikelenboom P. Cyclooxygenase expression in microglia and neurons in Alzheimer's disease and control brain. Acta Neuropathol. 2001;101:2–8. doi: 10.1007/s004010000251. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Veerhuis R, Janssen I, van Elk EJ, Rozemuller AJ, Eikelenboom P. The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: implications for Alzheimer's disease. Brain Res. 2002;951:218–226. doi: 10.1016/s0006-8993(02)03164-5. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Rozemuller JM, van Haastert ES, Veerhuis R, Eikelenboom P. Cyclooxygenase-1 and -2 in the different stages of Alzheimer's disease pathology. Curr Pharm Des. 2008;14:1419–1427. doi: 10.2174/138161208784480171. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez S, Baglietto-Vargas D, Caballero C, et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene CD, Chang RC, Lopez-Yglesias AH, et al. Suppressed accumulation of cerebral amyloid beta peptides in aged transgenic Alzheimer's disease mice by transplantation with wild-type or prostaglandin E2 receptor subtype 2-null bone marrow. Am J Pathol. 2010;177:346–354. doi: 10.2353/ajpath.2010.090840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, et al. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008;131:651–664. doi: 10.1093/brain/awn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. J Neurosci. 2005;25:10180–10187. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, et al. Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001;22:983–991. doi: 10.1016/s0197-4580(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Matousek SB, Hein AM, Shaftel SS, Olschowka JA, Kyrkanides S, O'Banion MK. Cyclooxygenase-1 mediates prostaglandin E(2) elevation and contextual memory impairment in a model of sustained hippocampal interleukin-1beta expression. J Neurochem. 2010;114:247–258. doi: 10.1111/j.1471-4159.2010.06759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2007;28:639–647. doi: 10.1016/j.neurobiolaging.2006.03.013. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- McKee AC, Carreras I, Hossain L, et al. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008;1207:225–236. doi: 10.1016/j.brainres.2008.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM. A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol. 2006a;168:184–194. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006b;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006c;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tseng B, Cheng D, Vasilevko V, Cribbs DH, LaFerla FM. Blocking Abeta42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Abeta and tau pathology. J Neurosci. 2008;28:12163–12175. doi: 10.1523/JNEUROSCI.2464-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peress NS, Fleit HB, Perillo E, Kuljis R, Pezzullo C. Identification of Fc gamma RI, II and III on normal human brain ramified microglia and on microglia in senile plaques in Alzheimer's disease. J Neuroimmunol. 1993;48:71–79. doi: 10.1016/0165-5728(93)90060-c. [DOI] [PubMed] [Google Scholar]
- Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Zhang Y, Koboldt CM, et al. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci USA. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung S, Yang H, Uryu K, Lee EB, Zhao L, Shineman D, Trojanowski JQ, Lee VM, Pratico D. Modulation of nuclear factor-kappa B activity by indomethacin influences A beta levels but not A beta precursor protein metabolism in a model of Alzheimer's disease. Am J Pathol. 2004;165:2197–2206. doi: 10.1016/s0002-9440(10)63269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296(Pt 1):15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinals M, Bermudez I, Llaverias G, Alegret M, Sanchez RM, Vazquez-Carrera M, Laguna JC. Aspirin increases CD36, SR-BI, and ABCA1 expression in human THP-1 macrophages. Cardiovasc Res. 2005;66:141–149. doi: 10.1016/j.cardiores.2004.12.024. [DOI] [PubMed] [Google Scholar]
- Walker MC, Kurumbail RG, Kiefer JR, Moreland KT, Koboldt CM, Isakson PC, Seibert K, Gierse JK. A three-step kinetic mechanism for selective inhibition of cyclo-oxygenase-2 by diarylheterocyclic inhibitors. Biochem J. 2001;357:709–718. doi: 10.1042/0264-6021:3570709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Morgan D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat Protoc. 2006;1:1591–1595. doi: 10.1038/nprot.2006.277. [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer's disease. Acta Neuropathol. 1992;84:117–127. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Yermakova AV, Rollins J, Callahan LM, Rogers J, O'Banion MK. Cyclooxygenase-1 in human Alzheimer and control brain: quantitative analysis of expression by microglia and CA3 hippocampal neurons. J Neuropathol Exp Neurol. 1999;58:1135–1146. doi: 10.1097/00005072-199911000-00003. [DOI] [PubMed] [Google Scholar]