

Abstract
A series of mercapto-oxo containing reagents [3-mercaptopropionic acid (H2-3MPA), 4-mercaptophenol (H2-4MP), 2-mercaptopyridine-N-oxide (H-2MPO)] was reacted with diethyl zinc (ZnEt2) in hexanes/pyridine (py) to yield {(μ4-3MPA)[Zn(Et)(py)]4}∞ (1), [(py)2(Et)Zn(μ3-4MP)Zn(Et)(py)]2 (2), and (2MPO)Zn(Et)py (3). For polymeric 1, each of the functional sites of the 3MPA was bound to four tetrahedral (Td) coordinated Zn(Et)(py) subunits. The sulfur of the 3MPA bridges two of the ‘Zn(Et)(py)’ subunits, which are also bridged by the two carboxylate oxygens of another 3MPA to propagate the chain. In contrast, 2 forms a discrete tetranuclear species consisting of two Zn(Et)(py) moieties bridged by the oxygens of two 4MP ligands with the thiolate sites of each terminated by Zn(Et)(py)2 moieties. Compound 3 adopts a monomeric species using a chelating 2MPO, a terminal Et, and a bound py to fill the Td coordination of the Zn metal center. Compounds 1 - 3 were then used to generate nanoparticles via solution precipitation and solvothermal routes to determine the effect these precursors have on the morphology and composition of the final materials produced. Compounds 1 - 3 were found to form zincite, zinc metal, or mixed zincite/wurtzite phases from solution precipitation or solvothermal routes; however, no routes yielded the mixed anion (i.e., ZnOxSy) materials.
Keywords: O,S ligands; zinc; zincite; wurtzite; ceramics; nanomaterials
Introduction
Zinc occupies a major role in numerous and diverse research arenas ranging from biology where zinc ions are an essential element in the growth cycle of mammals (and plants) [1, 2] to materials systems where zinc ceramics are an important component for numerous electronic applications [1, 3-12]. For Zn-based ceramic materials, a few of the many applications that employ zincite (ZnO) include transistors, sensing electrodes, rubber products, surface acoustic wave (SAW) devices, and phosphors. [1, 3-12] In addition, other zinc materials, such as the wurtzite phase of zinc sulfite (ZnS), have also found use in a variety of applications including photovoltaic applications, luminescent paints, and flat panel displays. [1, 12] Due to the widespread use of these materials, it is not surprising that the production and utility of Zn-based nanomaterials has become of increased interest with the promise of new properties in this size regime. Typically, the synthesis and resultant properties of Zn-based (ZnO or ZnS) nanoparticles result from very specific precursors and processing conditions. [1, 3-23]
This investigation discusses our efforts to develop single-source precursor that could generate either the ZnO, ZnS, or the mixed anion Zn(OxSy), dependant upon the processing conditions. It was reasoned that if the Zn derivative of mercapto-oxo reagents (see Figure 1), such as 3-mercaptopropionic acid (H2-3MPA), 4-mercaptophenol (H2-4MP), 2-mercaptopyridine-N-oxide (H-2MPO) were developed, the desired product could be preferentially formed by the processing conditions. The development of optimized nanoparticles with an emphasis on the precursor structure-morphology relationship for several different metal oxide systems has been a goal of our research interests. [24-31] To make meaningful connections between the starting materials and the final nanoceramics produced, it is necessary to have a series of well-characterized precursors prior to initiating processing.[24-31] For this study, diethyl zinc (ZnEt2) was modified by a variety of oxo-mercapto ligands (Hn-L) to yield: {(μ4-3MPA)[Zn(Et)(py)]4}∞ (1), [(py)2(Et)Zn(μ3-4MP)Zn(Et)(py)]2 (2), and (2MPO)Zn(Et)(py) (3) where py = pyridine (eq 1). These compounds were then used to generate nanoparticles from solution precipitation and solvothermal routes. The synthesis, characterization, and nanomaterials produced from 1 - 3 under a variety of conditions are discussed below.
| (1) |
Figure 1.
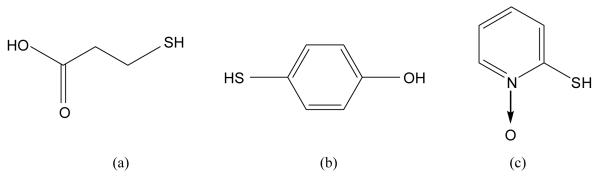
Schematic representation of (a) 3-mercaptopropionic acid (H2-3MPA), (b) 4-mercaptophenol (H2-4MP), (c) 2-mercaptopyridine-N-oxide (H-2MPO).
Results and Discussion
A search [32, 33] of potential Zn alkyl single-source precursors substituted with both an oxygen and sulfur atom revealed that only three crystallographically characterized species were available: [Zn(Et)(μ-SC(NEt)2)O-μ)(μ-ONEt2)]2 [34], [(CH3)Zn(μ3-O(CH2)2S(CH3)(μ-Cl)Zn(CH3)]2 [35], and (THF)(Et)Zn[S(P(μ-N(But))2P(S)(NHBut))N(But)] [36]. The potential residual anions (i.e., Cl and P) of the latter species prohibit their use for production of the Zn-based nanoceramics of interest. Therefore, the synthesis of acceptable precursors was undertaken using the mercapto-oxo ligands shown in Figure 1 (H2-3MPA, H2-4MP, H-2MPO). These ligands were selected based on availability, dual functionality, and general constructs of the pendant chain. A literature search of metal complexes that employ H2-3MPA, [33, 37-42] H2-4MP, [32, 33] or H-2MPO [43-47] ligands, indicated that none of the previous structurally characterized species had demonstrated reactivity at both functional sites of these ligands. Therefore, the modification of these ligands with ZnEt2 was undertaken. ZnEt2 was chosen due to its availability, the ease of conversion to an alkoxy derivative, and the high solubility of alkyl alkoxides. [48] Since transition metal thiolates [33, 49] or oxygen bearing ligands (i.e., carboxylate and alkoxides) [25, 50-60] are often reported to oligomerize, a strong Lewis base (py) was employed to minimize oligomerization and aid in solubility.
Precursor Synthesis
After dissolving the desired mercapto-oxo ligand in py, the reaction mixture was slowly added to a stirring solution of ZnEt2 in hexanes. Upon addition, the reaction mixture turned pale green and a white precipitate formed immediately. The precipitate was easily dissolved upon gentle heating. If more than one equivalent of the ligand was added a precipitate formed that could not be redissolved. Crystals were isolated from slow evaporation of the volatile portion of the reaction mixture at glovebox temperatures. The 2:1 metal:ligand stoichiometry ensures that all of the active sites can be potentially bound by Zn moieties. In order to assist in understanding the products formed, single crystal X-ray studies were undertaken.
X-ray structure
The crystal structures of 1 - 3 were successfully solved and their structure plots are shown in Figures 2 - 4, respectively. For all three compounds, each functional site on the specific ligand was bound by a Zn(Et)(py)x moiety. The nuclearity of the final product appears to be controlled by the oxo portion of the ligand coupled with the necessary binding of py to fill empty coordination sites. The metrical data of 1 - 3 were found to be consistent with those reported in the literature. [33-36, 38, 39, 43-48] Select angles and bond distances are given in Table 1.
Figure 2.

Structure plot of 1. Thermal ellipsoid of the heavy atoms are drawn at the 30% level. Carbon atoms are drawn as ball and stick figures and hydrogen atoms omitted for clarity.
Figure 4.

Structure plot of 3. Thermal ellipsoid of the heavy atoms are drawn at the 30% level. Carbon atoms are drawn as ball and stick figures and hydrogen atoms omitted for clarity.
Table 1.
Analytical data for 1 - 3.
| Comp’d | Bond Distance (Å) | Bond Angles (deg) | 13C MAS (ppm) |
|---|---|---|---|
|
| |||
| 1 | Zn(1)-O(1) 2.018 Zn(1)-S(1) 2.371 Zn(1)-N(1) 2.214 Zn(1)-C(2) 2.116 |
N(1)-Zn(1)-O(1) 93.23 N(1)-Zn(1)-C(2) 105.99 O(1)-Zn(1)-C(2) 117.49 |
180.1, 43.3, 24.2, (O2CCH2CH2S) 147.4, 140.3, 125.4 (py) 14.3, −2.1(CH2CH3) |
|
| |||
| 2 | Zn(1)-O(1) 2.014 Zn(2)-S(1) 2.286 Zn(1)-N(1) 2.110 Zn(1)-C(2) 1.980 |
N(1)-Zn(1)-O(1) 99.78 N(1)-Zn(1)-C(1) 116.35 O(1)-Zn(1)-C(1) 127.38 |
147.5, 139.5, 125.1 (py) 133.6, 117.9 (SC6H4O) 13.9, −0.2 (CH2CH3) |
|
|
|||
| 3 | Zn(1)-O(1) 1.996 Zn(1)-S(1) 2.353 Zn(1)-N(2) 2.121 Zn(1)-C(6) 1.985 |
C(6)-Zn(1)-O(1) 123.24 N(2)-Zn(1)-C(6) 111.21 N(2)-Zn(1)-S(1) 103.94 |
161.2, 131.0, 128.6, 117.9 (O(C5H4)S) 148.4, 137.7, 125.3 (py) 12.6, −3.6 (CH2CH3) |
For 1 (Figure 2), the 3MPA ligand was found to act as a bridge between four Zn(Et)(py) moieties. Each tetrahedrally (Td) bound Zn atom was bridged by the μ-S atom of one 3MPA and the μ-O2CR functionality of another 3MPA, which propagates the chain, forming a symmetrical molecule. Several crystal structures with the 3MPA bound to a transition metal have been previously reported. [37-39, 41, 42, 61] In this set of compounds, the 3MPA was found to act as a terminal ligand binding through the thiolate [41] or through the oxide [61], chelating through a single oxygen and thiolate [37, 42], or bridging as noted for (C5Me4Et)2Zr[(μ-O2C(CH2)2S)Au(P(C6H5)(CH3)2]2 [38] or [(CO)9Os3]2(μ-3MPA) [39]. As can be discerned, the polymeric nature that the 3MPA ligand displays in 1 has not been previously reported. [33]
Switching to the 4MP ligand, a tetranuclear species was isolated as 2 (Figure 3) with a Zn(Et)(py)x (x = 1 or 2) moiety bound to each functional atom. For 2, the oxygen of the two 4MP ligands bridge between two Td bound Zn(Et)(py) moieties followed by binding two Zn(Et)(py)2 moieties to the para S atoms of each 4MP ligand. This forms a centrosymmetric D2h complex. The preferential orientation of the oxo ligands to the central Zn atoms allows for the thiolate S to be terminated by a doubly solvated Zn(Et)(py)2 center. While numerous transition metal compounds are available that employ the 4MP ligand, [32, 33] 2 is the first that is substituted by a transition metal at both functional sites.
Figure 3.
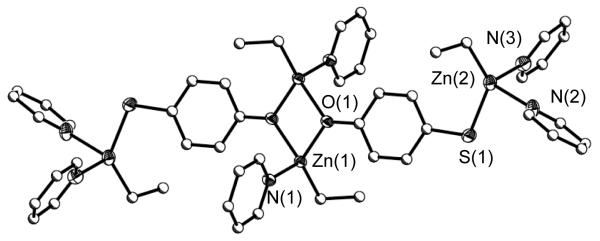
Structure plot of 2. Thermal ellipsoid of the heavy atoms are drawn at the 30% level. Carbon atoms are drawn as ball and stick figures and hydrogen atoms omitted for clarity.
Using the 1,2 substituted 2MPO ligand, a monomeric Td bound Zn species was isolated as 3 (Figure 4). In 3, each 2MPO ligand chelates to the Td bound Zn metal center with an additional Et and py molecule to complete its coordination. The 2MPO ligand (or methyl derivatives) has been reported to chelate to numerous metal centers, [32, 33] with 6 of these utilize Zn [43-47]. Previously reported 2MPO-Zn species were found to be disubstituted chelated monomers [43, 46, 47] or chelating unidentate bridged dinuclear [44, 45] species. Coupled with the coordination of py, the chelating aspect of the 2MPO ligand prevents polymer formation and is in-line with what was observed for the other 2MPO-Zn [43-47] species; however, these previously reported compounds were homoleptically substituted by 2MPO ligands. Compound 3 is the first Zn alkyl 2MPO substituted compound reported. Due to the arrangement of the different atoms of the ligands bound to the metal center, the Zn metal centers are considered chiral and both the R- and S-enantiomer are observed in the same unit cell.
Bulk solid state
To elucidate if the bulk powder was consistent with the single crystal structures solved for 1 - 3, additional analytical analyses were undertaken. FTIR data of each of the free ligands clearly shows the oxo stretches and bends associated with the respective functional groups, however, the thiolate stretches are much less pronounced and difficult to discern. Therefore, most sample analysis focused on changes in the oxo moiety stretches. For the 3MPA ligand and compound 1, the carboxylate stretches shift from 1709 to 1564 cm−1, which is consistent with a bridging carboxylate ligand. [25, 50, 52, 53] Compounds 1 and 2 exhibit no stretches associated with the −OH of either ligand indicating complete exchange. For compound 3, there was a significant shift in the stretches of the 2MPO ligand upon complexation; however, unequivocal identification of the N O stretch could not be determined. Solid-state 13C CPMAS NMR spectra were collected for dried crystalline powders of 1 - 3 to further establish the bulk material’s identity. All samples displayed the appropriate number and type of chemical shifts that would be consistent with the single crystal structure and are tabulated in Table 1 and shown in Figure 5. Elemental analyses of 1 was found to be consistent with the crystal structures. For 2, the resultant elemental data were consistent with the single crystal structure if a solvent molecule of py was removed from the calculations. Compound 3 was found to be slightly lower in C content than the expected value, which may possibly be due to loss of the bound py. Combined these data indicate that the bulk materials of 1 - 3 are consistent with the crystal structures.
Figure 5.

Solid state 13C CP MAS NMR spectra of (a) 1, (b) 2, and (c) 3. * denotes spinning sidebands.
Solution state
Since these compounds are dissolved prior to nanoparticle production, it is of interest to elucidate, if possible, the solution structural aspects of these compounds. Dried crystalline materials of 1 - 3 were dissolved separately in py-d5 under an argon atmosphere. While the solubilities of 1 - 3 were too low to obtain meaningful 13C NMR spectra, the 1H NMR data offered some insights into the solution behavior of these compounds.
The spectrum of compound 1 displayed two triplet resonances for the protons of the 3MPA (δ= 3.21 and 3.12) ligand, along with a quartet and a triplet for the Et ligands, consistent with basic constructs of the polymer. The bound py peaks are present but overlap with the py-d5 resonances. The NMR data collected for 1 indicates that the central core found in the crystal structure was retained in solution. It was reasoned that a smaller component of the chain is the solubilized species, since it is unlikely that the polymer would remain intact. For 2, the expected two aryl protons (δ = 7.80 and 7.04) are present in the NMR spectrum, along with the resonances associated with the Et groups and several overlapping py resonances. The expected quartet and triplet proved to be more complicated multiplets due to overlapping of the slightly varied Et ligand. However, the spectrum collected appears to be consistent with the solid state structure. For 3, the monomeric species was observed in solution as represented by the observed 1:1 ratio of 2MPO:Et resonance with overlapping py resonances. The monomer would be favored with the coordinated solvent and bidentate 2MPO ligand. Based on the simple spectrum noted this is most likely the solution state structure of 3.
Nanoparticles
Using the well-characterized 1 - 3 precursors, thermal analyses of these materials were undertaken to determine if they were amenable to nanoparticle processing. TGA/DTA investigations under argon revealed that the first weight was lost prior to 300°C for each of these compounds [compound: [°C (% total wt loss)] where 1: 100 °C(3%), 200(40), 300(50), and 450(55); 2: 100(15), 200(40) and 500(55); 3: 100(30), 300(45), and 400(55)]. This low temperature decomposition of a significant portion of the organic ligands suggested that these precursors would be acceptable for our routes to nanoparticle materials. [31] It is of note that 1 - 3 continued to lose weight as temperatures increased, never attaining a stable weight up to 1000 °C. Powder XRD analyses of the resulting TGA/DTA calcined powders, proved to be phase pure zincite (PDF# 36-1451). While this was discouraging for the production of the materials of initial interest [i.e., Zn(OxSy)], the temperatures used in this experiment were significantly higher than the routes proposed and surfactants were not present, which could assist in stabilizing the mixed phases.
Solvothermal Route
Initial experiments focused on generating Zn-based nanoparticles under solvothermal conditions using 1 - 3 dissolved in py. Table 2 summarizes the different systems used and the products formed. The green colored precursor solutions for each sample were sealed in a Parr digestion bomb setup under argon and then heated to 200 °C for 24h. After workup, the resulting powder was analyzed by PXRD (Figure 6 a-c) where 1 was found to have formed zincite, 2 was reduced to yield Zn°, and 3 possessed mixed phases of zincite and wurtzite (PDF# 36-1450) with some extraneous peaks attributed to the presence of excess ligand. In order to further characterize these materials, TEM images were obtained. Figure 7a-c show the images recorded for these as-synthesized nanomaterials. For 1, >10 nm sized zincite dots were observed; 2 surprisingly, yielded wires of Zn° where the aspect ratio of one of the individual wires approached 45 (80 nm × 3600 nm); 3 presented irregularly shaped nanomaterials of zincite and wurtzite that were >10 nm in size. While the zincite and wurtzite phases noted for 1 and 3 are commonly reported for solvothermal and solution route processes, we have not been able to identify any previous solution routes that generate Zn° nanowires as noted for 2; however it is of note that Rataboul et al. formed nanoparticles of Zn° from a ZnR2 R = C6H11 precursor and anisole.[62] Alternative routes to Zn° typically use vapor process at high temperature of Zn°, ZnO and ZnS [63-70] anodic processes [71-74], reduction of ZnO or ZnS under reducing atmospheres [75, 76], and nebulized spray of Zn(O2CCH3)2 [77].
Table 2.
Nanoparticle Data for 1 - 3 under different processing conditions.
| Solvo | Initial Phasea |
av Particle size (nm) Expb; Calcd a |
Calcinced Phaseb |
av Particle size (nm) Expb; Calcd a |
|---|---|---|---|---|
| 1 | zincite | >10 ; 8.7(1) | zincite | 10-40 ; 25.5(1) |
| 2 | zinc metal | 80 × 3600 ; 27.5 (1) | zincite | 100 × 1600 ; 15.4(1) |
| 3 | zincite (maj) wurtzite (min) |
>10 ; ----- | zincite (maj) wurtzite (min) |
rods 200 × 1800, elliptical rods 100 × 200 ; 34.3 (9) |
| MeIm/H2O Soltn PPT |
Initial Phase |
av Particle size (nm) Expb; Calcd a |
Calcinced Phase |
av Particle size (nm) Expb; Calcd a |
|---|---|---|---|---|
| 1 | zincite | 5-7 ; 6.5 (1) | ----- | ----- ; ----- |
| 2 | zincite | 7-10 ; 6.8 (1) | ----- | ----- ; ----- |
| 3 | zincite | 13-23 × <100 ; 12.7(1) |
----- | ----- ; ----- |
| HAD Soltn PPT |
Initial Phase |
av Particle size (nm) Expb; Calcd a |
Calcinced Phase |
av Particle size (nm) Expb; Calcd a |
|---|---|---|---|---|
| 1 | zincite | ----- ; 9.2(2) | zincite | 25 ; 16.7(1) |
| 2 | zinc metal | ----- ; 43.6(25) | zincite | 20 ; 15.9(1) |
| 3 | zincite (maj) wurtzite (min) |
----- ; ----- | zincite (maj) wurtzite (min) |
25 ; 14.2(1) |
----- data not obtained;
Phase/Size determined by PXRD analyses;
Size determined by TEM analyses
Figure 6.

PXRD pattern of initial nanoparticles generated by the solvothermal route using (a) 1 (b) 2, and (c) 3 and calcined particles from (d) 1, (e) 2, and (f) 3.
Figure 7.

TEM of initial nanoparticles generated by the solvothermal route using (a) 1 (b) 2, and (c) 3 and calcined particles from (d) 1,(e) 2, and (f) 3.
As mentioned, extraneous peaks were present in the PXRD pattern, which were attributed to the presence of excess ligand for 1 - 3. The material was therefore processed using a Lindberg/Blue M single-zone tube furnace at 600 °C under a flowing argon atmosphere to assist in removing volatile organic species. Figures 6d-f show the PXRD of the heat treated nanoceramics. As can be observed, the residual organics had been removed, wherein the nanoparticles generated from 1 and 3 retain their original phase(s), but 2 was converted to zincite. It was of interest to determine the effect that heating had had on the final morphology and the resultant TEM images are shown in Figures 7d-f. Not surprisingly for 1, since the passivating organic ligands had been removed, the particles had sintered into substantially larger particles and were polydispersed in both size and shape. For 2, the rod shape was maintained but the wires proved to be thicker, shorter, and coarser then the original wires. A closer inspection of the calcined wires show that they are now made up of smaller particles templated along the original wire morphology. For 3, the original dots transformed into long rods (200 × 1800 nm) or small elliptical rods with an aspect ratio of about 2 (100 × 200 nm). EDS analysis of the longer rods showed these to be zincite (ZnO) and the smaller ellipsoids were identified as wurtzite (ZnS). It is of note that only 3 showed any evidence of S present. This indicates that the other species have the sulfur remaining in solution as a soluble Zn-S complex or as a decomposed organic precursor. Table 2 summarizes the morphology observed for 1 - 3 for this process. 1-Methyl Imidazole Solution Route. Since mixed anion species were not observed in the solvothermal route (vide infra), an alternative approach was undertaken using solution precipitation methods. While a wide variety of solution synthesis routes to nanoparticles are available, the use of a methyl imidazole/water (MeIm/H2O) solvent system was previously successful for the production of morphologically varied ZnO from structurally diverse Zn(R)(OR)(solv)x precursors. [48] Therefore, the green colored precursor solution of 1 - 3 in py were injected rapidly into a solution of MeIm/H2O at reflux temperatures. For each sample, the solution immediately generated a white precipitate. The reaction was heated for an additional half an hour and then allowed to cool to room temperature. After separation by centrifugation, a PXRD pattern was obtained and TEM images were collected to characterize these nanomaterials.
Powder XRD patterns of the precipitates generated from 1 - 3 were found to be in agreement with the zincite phase (Figures 8a-c). Based on the Scherrer equation the particles were estimated to be ~6.5(1), 6.8(1), and 12.7(1) nm for 1 - 3, respectively. Interestingly, no ZnS was observed in any of the patterns noted most likely due to the added oxidant water favoring the harder oxo ligation. To further investigate the size and morphology of these particles, TEM images were obtained (Figure 9a-c, Table 2). EDS did not indicate the presence of any S atoms. For polymeric 1, 5 - 7 nm dots were obtained; 2 yielded 7-10 nm square-like particles; for 3, rods on the order of 13 - 23 nm in width by >100 nm in length were isolated. These sizes are in agreement with the Scherer calculated values with the exception of 3 where the calculated value is close to the width of the rods.
Figure 8.

X-ray powder diffraction of process powder generated from MeIm/H2O solution precipitation route using (a) 1, (b) 2, (c) 3.
Figure 9.

TEM image of nanoparticles generated by the MeIm/H2O solution precipitation route using (a) 1, (b) 2, (c) 3.
Hexadecylamine Solution Route
Alternative solution routes were also investigated that did not use water as the oxidant. Amine based routes that employed hexadecylamine (HDA) were selected based on previous successes with these systems in producing nanomaterials. [24, 27] For these reactions, 0.25 g of 1 - 3 was dissolved in 0.8 g oleylamine (OA) and then injected into a solution (2.41 g) of HDA at 300 °C. PXRD patterns of the final material isolated from 1 - 3 are shown in Figure 10a-c. It was found that zincite, zinc metal (PDF# 04-0831) and mixed phases of zincite and wurtzite were formed from 1 - 3, respectively. Calculations indicate that 1 and 2 had formed nanoparticles of >10 and >44 nm, respectively. For compound 3 the PXRD pattern was found to have two phases with the major one being identified as zincite. Due to the difficulty in distinguishing definitive independent peaks for profile measurements, crystallite size determination via the Scherrer equation could not be meaningfully undertaken. The nanoparticles from 1 - 3 could not be adequately dispersed which meant TEM images could not obtained of the “as precipitated materials”. Upon heating to 600 °C, samples 1 and 2 were found to form zincite (Figure 10d-f), with 3 maintaining its mixed phase and these materials were easily dispersed in toluene. TEM images (Figure 11a-c) show that each formed nanodots of materials (Table 2), ranging in size from 14-17 nm for 1 - 3.
Figure 10.

PXRD of initial nanoparticles generated by the solution route using HDA and (a) 1, (b) 2, and (c) 3 and calcined nanoparticles from (d) 1, (e) 2, and (f) 3.
Figure 11.

TEM of (a) 1 calcined, (b) 2 calcined, and (c) 3 calcined generated by the HDA route.
Previously, a connection between the precursor structure and the final nanoparticle formation, termed the precursor structure affect (PSA) [24-31, 48] was established. Therefore, in trying to understand the decomposition of the molecules and the formation of the resultant nanoparticles it is important to address the initial reactions that may occur upon dissolution and the subsequent residual organic moieties generated. The metal ligand bonds are expected to be the weak link in these compounds with bond enthalpies reported for C-Zn (ZnEt2), S-Zn, O-Zn at ~47.5, 49, and 67.9 kJ/mol, respectively.[78] These data indicate that the Zn-O bond will be the last broken in 1 - 3. Therefore only ZnO building blocks that are different for each species and capped with the oxo moiety and a thiolate terminus should be considered in evaluating the final nanomorphology. The fate of the alkyl is assumed to be elimination as the volatile ethylene gas with subsequent coordination by the amine or hydroxides dependant on the different processing schemes. The solvothermal and HDA routes allow for ligand effects to play a major role in final observed morphology versus the MeIm/H2O with hydrolysis as a competing pathway. While a great deal more data is required, some preliminary concepts of the potential effects these ligands impart are discussed below.
For 1, based on the Zn-O bond being the last to decompose, the 3MPA ligand would therefore act like a carboxylate ligand yielding similar results observed for other zinc carboxylate nanoparticle morphologies. [79-81] Therefore, the isolation of nanodots for all three routes was in line with previous reports.
For 2, the MeIm/H2O route yielded square-like nanomaterials of the oxide. In general, hydrolysis of 2 should remove the Zn-C and Zn-S bonds and then most of the growth would necessarily come from the dinuclear Zn2O2 template, yielding the zincite phase. This is consistent with the similar structural growth template/nanoparticle formation noted for the Zn(OR)(R)(solv)x system. [48] In contrast, the solvothermal and HDA route of 2 were found to yield Zn° wires. The reduction of the Zn+2 metal center must be attributed to two factors: the amine solvent and the 4MP decomposition. Previously, using alkoxide precursors and amine solvent systems, we have generated Ge° nanoparticles. Similar chemistry is expected between the two systems.[24, 27] In addition, the 4MP ligand can be envisioned to easily oxidize to a quinolate-like product while reducing the Zn+2 center. Further, the 4MP contains the basic make-up of the precursors (Zn-O or Zn-S) used by the literature reports for the synthesis of Zn° wires.[63-77] Combined the formation of Zn° could be anticipated; however, it appears that this is the first solution route that successfully yielded Zn° nanowires.
With the bidentate 2MPO ligand in 3, the metal center is protected allowing only one side to participate in the early growth process. This would favor very controlled growth with the 2MPO protecting particular growth faces and dots and rods are observed in every route investigated. The mixed phases must be a result of the bidentate nature of the 2MPO maintaining the Zn-S interaction to a greater extent in comparison to the ligands of 1 and 2.
Summary and Conclusion
We have generated a new set of well-characterized Zn mercapto-oxo ligated species (1 - 3) with novel structural arrangements in comparison to existing literature crystallographically characterized species. Based on their physical properties, compounds 1 - 3 were used for nanoparticle synthesis. For 1, independent of the processing route, zincite nanodots were formed. In contrast, converting 2 under solvothermal or HDA solution precipitation route generated Zn° wires; whereas, using MeIm/H2O, the zincite phase was obtained. For 3, only mixed phases of zincite and wurtzite (ZnS) of varied shapes and sizes were isolated. Compound 3 was the only species to yield any sulfide phase, which was attributed to the bidentate nature of the 2MPO ligand. Additional development of a single-source precursor that can make either the oxide or sulfide phase with chelating oxo-thiolate ligands, for a number of other systems are underway.
Experimental
All reactions were performed under a dry, inert atmosphere using standard Schlenk line and glovebox techniques. The following chemicals were purchased from Aldrich and used without further purification: ZnEt2 (1.0 M solution in hexanes), H2-4MP, H2-3MPA, H-2MPO, oleylamine (OA), hexadecylamine (HDA), and chloroform (CHCl3). Anhydrous py and 1-methyl imidazole (MeIm) were used as received in Sure/Seal™ bottles.
FT-IR spectral data were obtained on a Bruker Vector 22 spectrometer using KBr pellets pressed under an argon atmosphere and handled under an atmosphere of flowing nitrogen. Elemental analyses were performed on a Perkin-Elmer 2400 CHN-S/O elemental analyzer. Solution NMR data were obtained on a Bruker DRX400 NMR spectrometer with 1 - 3 dissolved in py-d5 and referenced to residual protonated solvent signal. The 1H and 13C NMR spectra were obtained on a using a 5 mm broadband probe and collected at 400.1 and 100.1 MHz, respectively. The py resonances associated with 1 - 3 could not be resolved from the py-d5 solvent signals and are not included in the spectral write-up. Solid-state 13C NMR spectra were obtained on a Bruker Avance 400 using a 4 mm probe spinning between 5 and 10 kHz. Chemical shifts were referenced to a secondary external glycine reference (δ = 176 ppm) with respect to TMS (δ = 0 ppm). Simultaneous thermogravimetric/differential thermal analysis (TGA/DTA) experiments were performed on an STD 2960 under an atmosphere of argon at a ramp-rate of 5 oC/min. to 650 °C. Powder XRD data were collected on a Panalytical X’Pert Pro using a zero-background holder.
General Precursor Synthesis
Due to the similarity of the synthesis (1 - 3), a general description is provided with specific details described for each compound. The appropriate ligand (Figure 1) was dissolved in ~10 mL of py and slowly added to a stirring solution of 1.0 M ZnEt2 in hexanes. A precipitate immediately formed which dissolved upon gentle heating, resulting in a clear green solution. After stirring for 12h, the reaction mixture was set aside to allow the volatile portion of the reaction to slowly evaporate yielding X-ray quality crystals. All analytical analyses were performed on vacuum dried crystalline material.
{(μ4-3MPA)[Zn(Et)(py)]4}∞ (1)
Used ZnEt2 (4.00 g, 5.51 mmol), H2-3MPA (0.298 g, 2.75 mmol) in py (~10 mL). Yield: 78.9% (0.979 g). FTIR (KBr pellet, cm−1): 2852(m), 1602(m), 1564(w), 1485(m), 1446(m), 1410(m), 1212(s), 1070(s), 1039(s), 1011(s), 756(s), 697(m), 674(s), 630(s), 593(s), 487(s), 420(s). 1H NMR (400.1 MHz, py-d5) δ 3.21 (1.0H, t, SCH2CH2CO2, JH-H = 5.6 Hz), 3.12 (1.0H, t, SCH2CH2CO2, JH-H = 5.6 Hz), 1.61 (1.9H, t, CH2CH3, JH-H = 8.4 Hz), 0.72 (2.3H, q, CH2CH3, JH-H = 8.4 Hz). Elemental analysis for C17H24N2O2SZn2 (451.18): calcd. C 45.25, H 6.21, N 5.38; found C 45.65, H 6.22, N 5.21.
[(py)2(Et)Zn(μ3-4MP)Zn(Et)(py)]2 (2)
Used ZnEt2 (4.00 g, 5.51 mmol), H2-4MP (0.347 g, 2.76 mmol) in py (~10 mL). Yield: 66.5% (1.08 g). FTIR (KBr pellet, cm−1): 2848(m), 1602(m), 1583(m), 1480(w), 1446(w), 1265(w), 1215(m), 1169(s), 1068(m), 1039(m), 1012(s), 824(m), 753(s), 697(m), 657(m), 600(s), 513(s), 419(s). 1H NMR (400.1 MHz, py-d5) δ 7.80 (1.1H, d, SC6H4O), 7.04 (1.0H, bs, SC6H4O), 1.60 (2.7H, m, CH2CH3) 0.780 (2.0H, m, CH2CH3). Elemental analysis for C55H63N7O2S2Zn4 (1179.72): calcd. C 55.98, H 5.38, N 8.31; C50H58N6O2S2Zn4 (−1 py, 1100.76): calcd. C 54.56, H 5.31, N 7.63; found C 54.12, H 5.04, N 7.66.
(2MPO)Zn(Et)py (3)
Used ZnEt2 (4.00 g, 5.51 mmol), H-2MPO (0.701 g, 5.51 mmol) in py (~10 mL). Yield: 80.5% (1.33 g). FTIR (KBr pellet, cm−1): 2849(m), 1600(m), 1542(m), 1459(w), 1415(s), 1264(s), 1200(w), 1149(w), 1088(m), 1066(m), 1037(m), 831(m), 755(w), 702(w), 631(s), 597(s), 497(s). 1H NMR (400.1 MHz, py-d5) δ 8.42 (1.1H, m, C5H4NOS), 7.80 (1.0H, d, C5H4NOS), 7.00 (1.4H, m, C5H4NOS), 6.71 (1.4H, m, C5H4NOS), 1.66 (4.0H, t, CH2CH3, JH-H = 8.0Hz) 0.815 (3.20H, q, CH2CH3, JH-H = 8.0 Hz). Elemental analysis for C12H14N2OSZn (299.68): calcd. C 48.09, H 4.71, N 9.35; found C 47.45, H 4.66, N 9.18.
General X-ray Crystal Structure Information
[32, 33] Crystals were mounted onto a thin glass fiber from a pool of Fluorolube™ and immediately placed under a cold N2 vapor stream, on a Bruker AXS diffractometer with SMART 1000 CCD detector. The radiation used was graphite monochromatized MoKα radiation (λ = 0.7107 Å). Lattice determination and data collection were carried out using SMART Version 5.054 software. Data reduction was performed using SAINTPLUS Version 6.01 software. The data were corrected for absorption using the SADABS program within the SAINT software package.
Structures were solved using direct methods that yielded the heavy atoms, along with a number of the lighter atoms. Subsequent Fourier syntheses yielded the remaining lightatom positions. The hydrogen atoms were fixed in positions of ideal geometry and refined using SHELXS software. The final refinement of each compound included anisotropic thermal parameters for all non-hydrogen atoms. It is of note that crystal structures of that contain alkoxide ligands often contain disorder within the atoms of the ligand chain, causing higher than normal final correlations. [55-60] All CIF files were checked at http://www.iucr.org/. Data collection parameters for 1 - 3 are given in Table 3.
Table 3.
Single Crystal Data Parameters for 1 - 3.
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| chemical formula | C17H24N2O2SZn2 | C55H63N7O2S2Zn4 | C12H14N2O1S1Zn1 |
| formula weight | 451.18 | 1179.72 | 299.68 |
| Temperature (K) | 203 | 203 | 203 |
| space group | Orthorhombic Pbca |
Monoclinic C2/c |
Triclinic P1 |
| a (Å) | 13.778(2) | 15.3387(11) | 7.5512(7) |
| b (Å) | 14.427(2) | 9.6531(7) | 8.1605(7) |
| c (Å) | 19.658(3) | 39.213(3) | 12.0073(11) |
| α (deg) | 77.0120(10) | ||
| β (deg) | 99.8690(10) | 73.7450(10) | |
| γ (deg) | 67.1230(10) | ||
| V (Å3) | 3907.8(11) | 5720.1(7) | 648.81(10) |
| Z | 8 | 4 | 2 |
| Dcalcd(mg/m3) | 1.534 | 1.370 | 1.534 |
| μ,(Mo, Kα) (mm−1) | 2.573 | 1.775 | 2.038 |
| R1a (%) (all data) | 8.26 (12.31) | 4.92 (6.05) | 2.52 (2.56) |
| wR2b (%) (all data) | 16.31 (18.58) | 12.01 (13.01) | 6.22 (6.26) |
R1 = Σ ∣ ∣Fo∣ - ∣Fc∣ ∣ / Σ ∣Fo∣ × 100
wR2 = [Σ w (Fo2 - Fc2)2 / Σ (w∣Fo∣2)2]1/2 × 100
Nanoparticle synthesis
Two routes were used to generate nanoparticles (A) solvothermal and (B) solution precipitation. (A) The solvothermal route involved the dissolution of the desired precursor in py (0.5 g / 20 mL) under an atmosphere of argon, sealing it in a Teflon™-lined Parr acid digestion bomb, and heating it to 200 °C for 24 h. After cooling to room temperature each reaction mixture was transferred from the liner to centrifuge tubes and the mother liquor decanted from the precipitate. The precipitate was then washed three times with ethanol (EtOH) and the resulting powder was allowed to dry at room temperature. (B) The solution precipitation route involved either (i) dissolution of the desired precursor (0.5 g / 5 mL) in py followed by injection into a mixture of MeIm:water (14.5 mL: 0.5 mL) at reflux temperatures or (ii) dissolution of the precursor (0.25 g / 0.8 g) in OA followed by injection into a solution (2.41 g) of HDA at 300 °C. After heating for 30 min, the reaction was allowed to cool to room temperature. The precipitate was dissolved in CHCl3, precipitated with MeOH, and collected by centrifugation.
For transmission electron microscopy (TEM) analyses, the desired powder was slurried in toluene and then a small aliquot was placed directly onto a holey-carbon copper coated TEM grid (300 mesh from Electron Microscopy Sciences) and allowed to dry overnight. The resultant particles were studied using a Philips CM 30 TEM operating at 300 kV accelerating voltage. In addition, the powder was characterized using powder XRD on a PANalytical X’Pert Pro using CuKα radiation with step size 0.0167 degree, with 0.152 °/sec dwelling time. Note: The appropriate personal protection (i.e., rubber gloves, glasses, working in well-ventilated areas, etc) should be employed when handling the final nanopowders since freshly formed ZnO is reported to sometimes produce a disorder referred to as oxide shakes or zinc chills. [82]
Acknowledgements
This work was partially funded by the Office of Basic Energy Sciences of the Department of Energy as well as the National Institutes of Health through the NIH Roadmap for Medical Research, Grant #1 R21 EB005365-01 -Information on this RFA (Innovation in Molecular Imaging Probes) can be found at http://grants.nih.gov/grants/guide/rfa-files/RFA-RM-04-021.html. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy under contract DE-AC04-94AL85000.
Footnotes
Supplementary Information. CCDC-701768-701770 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
- [1].Nguyen T, Panda A, Olmstead MM, Richards AF, Stender M, Brynda M, Power PP. J. Am. Chem. Soc. 2005;127:8545–8552. doi: 10.1021/ja042958q. [DOI] [PubMed] [Google Scholar]
- [2].Ibrahim MM, Seebacher J, Steinfeld G, Vahrenkamp H. Inorg. Chem. 2005;44:8531–8538. doi: 10.1021/ic0508270. [DOI] [PubMed] [Google Scholar]
- [3].Wagner JF. Thin Solid Films. 2008;516:1755–1784. [Google Scholar]
- [4].Nakanotani H, Yahior M, Adachi C, Yano K. Appl. Phys. Lett. 2007;90:26104/26101–26103. [Google Scholar]
- [5].Moon Y-K, -H. KS, Moon D-Y, Kim W-S, Park J-W. J. Korean Phys. Soc. 2007;51:1732–1735. [Google Scholar]
- [6].Redinger D, Subramanian V. IEEE Trans. Elect. Dev. 2007;54:1301–1307. [Google Scholar]
- [7].Fortunato E, Barquinha P, Goncalvez G, Pereira L, Martins R. Solid State Electronics. 2008;52:443–448. [Google Scholar]
- [8].Lee S-Y, Umar A, Suh D, Park J-E, Hahn Y-B, Ahn J-Y, Lee S-K. Physica E. 2008;40:866–872. [Google Scholar]
- [9].Logothetidis S, Laskarakis A, Kassavetis S, Lousinian S, Gravalidis C, kiriakidis G. Thin Solid Films. 2008;15:1345–1349. [Google Scholar]
- [10].Park SHK, Hwang CS, Jeong HY, Chu HY, Cho KI. Electrochem. Sol. St. Lett. 2008;11:H10–H14. [Google Scholar]
- [11].Ain Y, Wang XD, Wang ZL. Nature. 2008;451:809–U805. doi: 10.1038/nature06601. [DOI] [PubMed] [Google Scholar]
- [12].Li X, Wang X, Xiong Q, Eklund PC. Nano Letters. 2005;5:1982–1986. doi: 10.1021/nl0513885. [DOI] [PubMed] [Google Scholar]
- [13].Monge M, Kahn ML, Maisonnat A, Chaudret BACI. 42:5321–5324. doi: 10.1002/anie.200351949. [DOI] [PubMed] [Google Scholar]; Angew. Chem. IEEE. 2003;42:5321–5324. doi: 10.1002/anie.200351949. [DOI] [PubMed] [Google Scholar]
- [14].Cozzoli DP, Kornowski A, Weller H. J. Phys. Chem. B. 2005;109:2638–2644. doi: 10.1021/jp0457139. [DOI] [PubMed] [Google Scholar]
- [15].Pacholski C, Kornowski A, Weller H. Angew. Chem. IEEE. 2004;43:4774–4777. doi: 10.1002/anie.200453880. [DOI] [PubMed] [Google Scholar]
- [16].Byrappa K, Ohara S, Adschiri T. Adv. Drug Del. Rev. 2008;60:299–327. doi: 10.1016/j.addr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- [17].Afzaal M, Malik MA, O’Brien P. New J. Chem. 2007;31:2029–2040. [Google Scholar]
- [18].Ganguli AK, Vaidya S, Ahmad T. Bull. Mater. Sci. 2008;31:415–419. [Google Scholar]
- [19].Mezy A, Gerardin C, Tichit D, Ravot D, Suwanboon S, Tedenac JC. J. Cer. Soc. Jpn. 2008;116:369–373. [Google Scholar]
- [20].Ahmadi M, Javadpour S, Khosravi A, Gharavi A. Jpn. J. Appl. Phys. 2008;47:5089–5092. [Google Scholar]
- [21].Lau BLT, Hsu-Kim H. Env. Sci. Tech. 2008;42:7236–7241. doi: 10.1021/es801360b. [DOI] [PubMed] [Google Scholar]
- [22].Luo JW, Mo RF, Jin GQ, Guo XY. Chin. J. Inorg. Chem. 2008;24:1084–1088. [Google Scholar]
- [23].Gerion D, Pinaud F, Williams SC, Parak WJ, Zanchet D, Weiss S, Alivisatos AP. J. Phys. Chem. B. 2001;105:8861–8871. [Google Scholar]
- [24].Gerung H, Boyle TJ, Tribby LJ, Bunge SD, Brinker CJ, Han SM. J. Am. Chem. Soc. 2006;128:5244–5250. doi: 10.1021/ja058524s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Boyle TJ, Ottley LAM, Rodriguez MA. Polyhedron. 2005;24:1727–1738. [Google Scholar]
- [26].Bunge SD, Krueger KM, Boyle TJ, Rodriguez MA, Headley TJ, Colvin VL. J. Mater. Chem. 2003;13:1705–1709. [Google Scholar]
- [27].Gerung H, Bunge SD, Boyle TJ, Brinker CJ, Han SM. Chem. Commun. 2005;14:1914–1916. doi: 10.1039/b416066e. [DOI] [PubMed] [Google Scholar]
- [28].Boyle TJ, Hernandez-Sanchez BA, Baros CM, Brewer LN, Rodriguez MA. Chem. Mater. 2007;19:2016–2026. [Google Scholar]
- [29].Boyle TJ, Ottley LAM, Daniel-Taylor SD, Tribby LJ, Bunge SD, Costello AL, Alam TM, Gordon JC, McCleskey TM. Inorg. Chem. 2007;46:3705–3713. doi: 10.1021/ic070253u. [DOI] [PubMed] [Google Scholar]
- [30].Boyle TJ, Bunge SD, Alam TM, Holland GP, Headley TJ, Avilucea G. Inorg. Chem. 2005;44:1309–1318. doi: 10.1021/ic0485155. [DOI] [PubMed] [Google Scholar]
- [31].Hernandez-Sanchez BA, Boyle TJ, Baros CM, Brewer LN, Headley TJ, Tallant DR, Rodriguez MA, Tuttle BA. Chem. Mater. 2007;19:1459–1471. [Google Scholar]
- [32].Allen FH. Acta Cryst. 2002;B58:380–388. [Google Scholar]
- [33].Evans WJ, Gummersheimer TS, Boyle TJ, Ziller JW. Organometallics. 1994;13:1281–1284. [Google Scholar]
- [34].Chunggaze M, Malik MA, O’Brien P, White AJP, Williams DJ. J. Chem. Soc., Dalton Trans. 1998:3839–3844. [Google Scholar]
- [35].Lewinski J, Marciniak W, Ochal Z, Lipkowski J, Justyniak I. Eur. J. Inorg. Chem. 2003;15:2753–2755. [Google Scholar]
- [36].Lief GR, Moser DF, Stahl L, Staples RJ. J. Organomet. Chem. 2004;689:1110–1121. [Google Scholar]
- [37].Schwab JJ, Wilkinson EC, Wilson SR, Shapley PA. J. Am. Chem. Soc. 1991;113:6124–6129. [Google Scholar]
- [38].Wenzel B, Lonnecke P, Hey-Hawkins E. Eur. J. Inorg. Chem. 2002:1761–1764. [Google Scholar]
- [39].Ainschough EW, Brodie AM, Coll RK, Mair AJA, Waters JM. J. Organomet. Chem. 1996;509:259–264. [Google Scholar]
- [40].John E, Bharadwaj PK, Potenza JA, Schugar HJ. Inorg. Chem. 1986;25:3065–3069. [Google Scholar]
- [41].Carrell HL, Glusker JP, Job R, Bruice TC. J. Am. Chem. Soc. 1977;99:3683–3690. doi: 10.1021/ja00453a028. [DOI] [PubMed] [Google Scholar]
- [42].Yonemura T, Shibuya K, Okamoto KI, Ama T, Kawaguchi H, Yasui T. Inorg. Chim. Acta. 1997;260:119–128. [Google Scholar]
- [43].West DX, Brown CA, Jasinski JP, Jasinski JM, Heathwaite RM, Fortier DG, Staples RJ, Butcher RJ. J. Chem. Cryst. 1998;28:853–860. [Google Scholar]
- [44].Niu D-Z, Sun B-W, Lu Z-S, Song B-L. Chin. J. Struct. Chem. 2001;20:108–111. [Google Scholar]
- [45].Barnett BL, Kretschmar HC, Hartman FA. Inorg. Chem. 1977;16:1834–1838. [Google Scholar]
- [46].Xiong R-G, Song B-L, You X-Z, Mak TCW, Zhou Z-Y. Polyhedron. 1996;15:991–996. [Google Scholar]
- [47].Bond AD, Benevelli F, Jones W. J. Mater. Chem. 2002;12:324–332. [Google Scholar]
- [48].Boyle TJ, Bunge SD, Andrews NL, Matzen LE, Sieg K, Rodriguez MA, Headley TJ. Chem. Mater. 2004;16:3279–3288. [Google Scholar]
- [49].Raper ES. Coord. Chem. Rev. 1997;165:475–567. [Google Scholar]
- [50].Boyle TJ, Andrews NL, Alam TM, Rodriguez MA, Santana JM, Scott BL. Polyhedron. 2002;21:2333–2345. [Google Scholar]
- [51].Coker EN, Boyle TJ, Rodriguez MA, Alam TM. Polyhedron. 2004;23:1739–1747. [Google Scholar]
- [52].Boyle TJ, Tyner RP, Alam TM, Scott BL, Ziller JW, Potter BG. J. Am. Chem. Soc. 1999;121:12104–12112. [Google Scholar]
- [53].Boyle TJ, Alam TM, Tafoya CJ, Scott BL. Inorg. Chem. 1998;37:5588–5594. doi: 10.1021/ic980601f. [DOI] [PubMed] [Google Scholar]
- [54].Bradley DC. Chem. Rev. 1989;89:1317–1322. [Google Scholar]
- [55].Bradley DC, Mehrotra RC, Gaur DP. Metal Alkoxides. Academic Press; New York: 1978. [Google Scholar]
- [56].Caulton KG, Hubert-Pfalzgraf LG. Chem. Rev. 1990;90:969–995. [Google Scholar]
- [57].Chandler CD, Roger C, Hampden-Smith MJ. Chem. Rev. 1993;93:1205–1241. [Google Scholar]
- [58].Hubert-Pfalzgraf LG. New. J. Chem. 1987;11:663–675. [Google Scholar]
- [59].Bradley DC, Mehrotra RC, Rothwell IP, Singh A. Alkoxo and Aryloxo Derivatives of Metals. Academic Press; New York: 2001. [Google Scholar]
- [60].Turova NY, Turevskaya EP, Kessler VG, Yanovskaya MI. The Chemistry of Metal Alkoxide. Kluwer Academic Publishers; Boston: 2002. [Google Scholar]
- [61].Tekeste T, Vahrenkamp H. Inorg. Chem. 2006;45:10799–10806. doi: 10.1021/ic061667a. [DOI] [PubMed] [Google Scholar]
- [62].Rataboul F, Nayral C, Casanove MJ, Masonnat A, Chaudret B. J. Organomet. Chem. 2002:643–644. 307–317. [Google Scholar]
- [63].Heremans JP, Thrush CM, Morelli DT, Wu MC. Phys. Rev. Lett. 2003;91:076804/076801–076804. doi: 10.1103/PhysRevLett.91.076804. [DOI] [PubMed] [Google Scholar]
- [64].Gonzalez JC, Rodrigues V, Bettini J, Rego LGC, Rocha AR, Coura PZ, Dantas SO, Sato F, Galvao DS, Ugarte D. Phys. Rev. Lett. 2004;93:126103/126101–126104. doi: 10.1103/PhysRevLett.93.126103. [DOI] [PubMed] [Google Scholar]
- [65].Yan Y, Liu P, Romero MJ, Al-Jassim MM. J. Appl. Phys. 2003;93:4807–4809. [Google Scholar]
- [66].Kang B-S, Xu YJ, Peng J-H, Wu D-S, Chen X-T, Hu Y-H, Hong M-C, Lu JX. Polyhedron. 1993;12:871–878. [Google Scholar]
- [67].Liu J, Zhang Z, Su X, Zhao YJ. Appl. Phys. Lett. 2005;38:1068–1071. [Google Scholar]
- [68].Kar S, Ghoshal T, Chaudhuri S. Chem. Phys. Lett. 2006;419:174–178. [Google Scholar]
- [69].Peng XS, Zhang LD, Meng GW, Yuan XY, Lin Y, Tian YTJ. J. Phys. D (Appl. Phys) 2003;36:L35–38. [Google Scholar]
- [70].Kast M, Schroeder P, Hyun YJ, Pongratz P, Bruckl H. Nano Letters. 2007;7:2540–2544. doi: 10.1021/nl070934m. [DOI] [PubMed] [Google Scholar]
- [71].Tian M, Kumar N, Xu S, Wang J, Kurtz JS, Chan MHW. Phys. Rev. Lett. 2005;95:076802/076801–076804. doi: 10.1103/PhysRevLett.95.076802. [DOI] [PubMed] [Google Scholar]
- [72].Chang SS, Yoon SO, Park HJ, Sakai A. Mater. Lett. 2002;53:432–436. [Google Scholar]
- [73].Pan H, Sun H, Poh C, Feng YP, Lin JY. Nanotech. 2005;16:1559–1564. [Google Scholar]
- [74].Wang JG, Tian M, Kumar N, Mallouk TE. Nano Letters. 2005;5:1247–1253. doi: 10.1021/nl050918u. [DOI] [PubMed] [Google Scholar]
- [75].Chen YJ, Chi B, Zhang HZ, Chen H, Chen Y. Mater. Lett. 2007;61:144–147. [Google Scholar]
- [76].Li Q, Kwong KW, Ozkaya D, Cockayne DJH. Phys. Rev. Lett. 2004;92:186102/186101–186104. doi: 10.1103/PhysRevLett.92.186102. [DOI] [PubMed] [Google Scholar]
- [77].Vivekchand SRC, Gundiah G, Govindaraj A, Rao CNR. Adv. Mater. 2004;16:1842–1845. [Google Scholar]
- [78].Weast RC, Astle MJ, editors. Crc Handbook of Chemistry and Physics. CRC Press, Inc; Boca Raton, Florida: 1980-1981. [Google Scholar]
- [79].Rodriguez-Gattorno G, Santiago-Jacinto P, Rendon-Vazquez L, Nemeth J, Dekany I, Diaz D. J. Phys. Chem. B. 2003;107:12597–12604. [Google Scholar]
- [80].Seo DU, Lu CZ, Chang HJ, Joo SW. Mater. Sci. Forum. 2004:449–452. 1133–1136. [Google Scholar]
- [81].Bell NS, Tallant DR, Raymond R, Boyle TJ. J. Mater. Res. 2008;23:529–535. [Google Scholar]
- [82].Boyle TJ, Tyner RP, Alam TM, Scott BL, Ziller JW, Potter BG. J. Am. Chem. Soc. 1999;121:12104–12112. [Google Scholar]