

Abstract
Immunoglobulin (Ig)-binding proteins are employed by a variety of organisms to evade the immune system. We now report for the first time that meningococcal strains from several capsular groups exhibit Ig-binding activity that is dependent on human serum factors. A protein mediating Ig binding was identified as T and B cell stimulating protein B (TspB) by immunoprecipitation and by mass spectroscopic analysis of tryptic peptides. Recombinant TspB and derivatives verified Ig binding, with a preference for human IgG2 Fc, and localized the IgG-binding region to a highly conserved subdomain of TspB. Antiserum produced in mice against the conserved subdomain, detected the presence of TspB on the cell surface by flow cytometry when bacteria were grown in the presence of human serum. By fluorescence microscopy, we observed formation of an extracellular matrix having characteristics of a biofilm containing TspB, human IgG, DNA, and large aggregates of bacteria. TspB is encoded by gene ORF6 in prophage DNA, which others have shown is associated with invasive meningococcal strains. Knocking out ORF6 genes eliminated IgG binding and formation of large bacterial aggregates in biofilm. Reintroduction of a wild-type ORF6 gene by phage transduction restored the phenotype. The results show that TspB mediated IgG binding and aggregate/biofilm formation triggered by factors in human serum. As has been observed for other Ig-binding proteins, the activities mediated by TspB may provide protection against immune responses, which is in accordance with the association of prophage DNA carrying ORF6 with invasive meningococcal strains.
Keywords: Ig-binding, prophage, bacterial aggregation, biofilm
Introduction
Pathogenic Neisseria meningitidis (Nm) is a major cause of bacterial meningitis and sepsis (1). Invasive Nm disease is characterized by a host range limited to humans, rapid onset, major debilitating sequelae in approximately 10% of cases (2), and a mortality rate that ranges from 5% to 15% depending on the Nm strain and age of the patient. Most cases of invasive meningococcal disease occur in the semiarid Sahel region of sub-Saharan Africa, where rates of endemic disease are high and periodic outbreaks of epidemic disease are common (3). In the rest of the world, with the exception of localized country-specific epidemics, meningococcal disease is relatively rare. However, meningococcal carriage in the nasopharnyx is common, even in countries with little disease. Rates range from 5% to 20% or more, depending on the population studied (4). Given relatively high carriage rates, it is unclear why invasive disease remains rare. A recent genetic study of carriage versus disease strains showed a correlation between the presence of a particular prophage DNA and invasive disease (5), particularly in young adults (6). However, the reasons why the prophage DNA contributed to invasiveness were unclear. In this report, we show that Nm T and B cell stimulating protein B (TspB), which is encoded by the prophage gene ORF6, is an Ig-binding protein (Igbp) that mediates formation of a biofilm containing TspB, IgG, DNA, and bacteria.
Expression of proteins that bind Ig is a strategy commonly used by human pathogens to facilitate survival during infection. Investigated examples include the Staphylococcus aureus multivalent cell wall-associated Igbps Staphylococcal protein A (SpA) and Sbi (7), Streptococcus groups C and G protein G (SpG) (8), Group A Streptococcal proteins Enn, M, Sib and FcRa (reviewed in (9)), Peptostreptococcus magnus protein L (10), and Histophilus somniIbpA (11). The mechanisms of immune evasion provided by Igbps are complex and variable depending on the particular protein. For example, the S. pyogenes M protein has binding domains for human factor H, serum albumin, and fibrinogen, in addition to IgG3 (9). Sbi binds to human IgG, β(2)-glycoprotein I, and complement protein C3 (12). The Igbps can be associated with the membrane or secreted, or both as alternative forms of the same protein. Secreted Igbps can form aggregates with Ig, which can result in non-productive consumption of complement (12). Igbps generally exhibit selectivity for immunoglobulin class, subclass, and species (13). Different regions of the IgG molecule are recognized including the CH2-CH3 region of Fc (SpA, SpG) and the framework region of the κ chain VL domain. Ig-binding subdomains of Igbps are typically 55–75 amino acids in length.
Data presented here, showing that Nm strains express an Ig-binding protein that promotes formation of Nm aggregates with biofilm-like characteristics and is encoded by a gene linked to invasive meningococcal disease, may have important implications for understanding Nm pathogenesis and the reasons why some Nm strains cause disease while others do not. The data may also provide critical insight for evaluating the ability of candidate meningococcal vaccines to elicit protection against disease.
Materials And Methods
Ethics statement
Donated human blood used in this study was obtained from adult donors under a protocol approved by the Children’s Hospital & Research Center Oakland Institutional Review Board with written informed consent obtained from all donor participants.
All procedures involving animals were performed in the CHORI Animal Research Facility, which is an AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International) accredited facility. The investigators adhered to the “Guide for the Care and Use of Laboratory Animals” prepared by the Committee for the Update of the Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Research Council. Protocols involving the use of animals were approved by the CHORI Institutional Animal Care and Use Committee.
Bacterial strains and culture conditions
Strains 860800 (Y:P1.5–1,10–4:ST29), A22 (W135:P1.18–1,3:ST22), H44/76 (B:P1.7,16:ST32), MC58 (B:P1.7,16–2:ST74) and Z1092 (A:P1.5–2,10:ST1) are described at http://pubmlst.org/perl/bigsdb/bigsdb.pl?db=pubmlst_neisseria_isolates. Nm strain 4243 (C:P1.5,2:ST11) is described in (14), NMB (B:2a:P1.5,2:ST8) in (15), and Ug 13/07 (X:P1.19,26:ST5403) in (16). The bacterial strains were stored as suspensions in 10% milk solution (Carnation non-fat dry milk) or whole human serum (donor 1 or 2; serially cultured bacteria) at −80°C until used. Meningococcal strains were grown to an OD620nm of 0.6–0.8 in chemically defined Catlin 6 Medium (17) with 2 mM glutamine (CDM) or CDM supplemented with 5% HuS. The serum was heat inactivated for 30 min at 56°C and, in some experiments, the HuS was depleted of IgG (dHuS) by passage over a Protein G column (HiTrap Protein G, GE Healthcare Bioscience, Piscataway, NJ). Additionally, for H44/76 tspB mutant strains plates and culture media contained 50 μg/ml kanamycin, 3 μg/ml erythromycin and 2.5 μg/ml chloramphenicol. In experiments shown in Fig. 1, Fig. 6b. and 6c., and Fig. 7 CDM contained 4 mM D, L lactate and 0.5 mg/ml Cohn fraction IV (Sigma-Aldrich, Saint Louis, MO).
FIGURE 1.
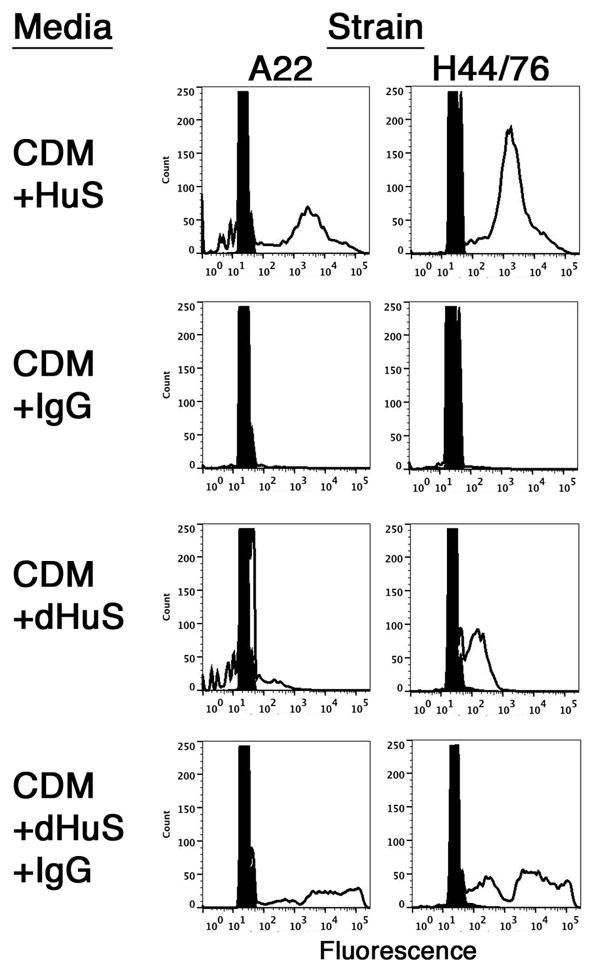
Effect of bacterial culture conditions on immunoglobulin-binding activity of NmW135 strain A22 and NmB strain H44/76 by flow cytometry. Bacteria were cultured in the indicated media. The rightward shift to higher fluorescence indicates bound human IgG (unfilled histograms). Cells cultured in CDM and incubated with secondary antibody only (filled histograms) served as the negative control.
FIGURE 6.
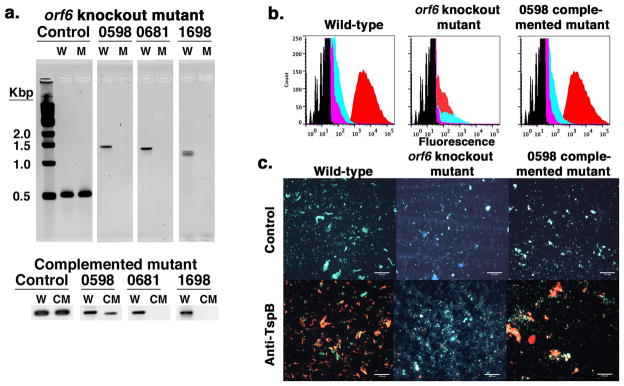
Knocking out ORF6 in NmB strain H44/76 and effect on human Ig binding. Panel a. shows PCR products of wild-type (W) and ORF6 mutant (M) H44/76 genomic DNA amplified with primers specific for a segment just upstream of NMBH4476_0681 (positive control for the presence of genomic DNA) and primers specific for each ORF6 gene. The ORF6 genes include NMBH4476_0598, NMBH4476_0681 and NMBH4476_1698. The expected PCR products for the control, 0598, 0681, and 1698 specific segments are approximately 500 bp, 1370 bp, 1280 bp, and 1180 bp, respectively. The lower panel shows a portion of the gel for the same PCR analysis testing the ORF6 knockout mutant into which wild-type phage DNA was re-introduced by transduction with phage produced in wild-type H44/76 (CM). Panel b. shows binding of human IgG (red histograms), mouse anti-TspB1628 CR (cyan histograms) and irrelevant control mouse (magenta histograms) IgG to wild-type, ORF6 knockout and NMBH4476_0598 complemented mutant H44/76 strains by flow cytometry. Panel c. is a set of fluorescence micrographs of bacteria from the flow cytometry experiment shown in panel b. where human IgG is marked by green fluorescence and mouse IgG by red fluorescence for the indicated strain cultured in CDM+HuS and either irrelevant mouse control serum (upper panels) or mouse anti-TspB1628 CR (lower panels). Blue fluorescence represents DAPI DNA stain. Size bars = 50 μm.
FIGURE 7.
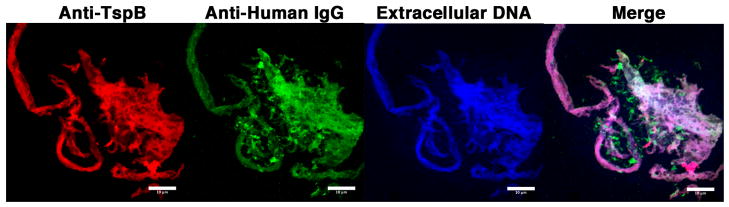
Biofilm containing TspB, IgG, and DNA enveloping aggregates of NmB cultured in the presence of human serum. Laser scanning confocal micrographs (63x) of an aggregate of NmB strain H44/76 cultured in CDM+HuS and mouse anti-TspB1628 CR. Human IgG is marked by green fluorescence, TspB with red fluorescence, and extracellular DNA by blue fluorescence. Size bars = 10 μm.
Serial overnight culture in HuS
Bacteria (−80°C storage) were diluted 1:80,000 in 1 ml of CDM containing 20% whole human serum. The bacteria were incubated at 37°C overnight with 4–5% CO2 in 2 ml Eppendorf tubes under constant rotation. The following day the process was repeated for a total of 3 rounds of culture. Bacteria used for experiments depicted in Figs. 1, 6b., 6c. and 7 were from stocks that had been serially cultured.
Cloning of TspB variants
Genomic DNA was prepared from NmB strain MC58 and NmA strain Z2491 using kits purchased from QIAGEN (Valencia, CA) or 5PRIME (Gaithersburg, MD). DNA sequences encoding TspB CR (for NMB1548, NMB1628, NMB1747, NMA0776 and NMA1797), TspB CRPro and full-length TspB (both for NMB1628 only) were amplified by PCR (see supplemental Table S1 for primers used) and cloned into plasmid pCR2.1 (Topo TA Cloning kit, Invitrogen) according to manufacturer’s instructions. The coding fragments were then excised from the plasmid with the appropriate primer-designed restriction enzymes and ligated into the expression plasmids pQE30 or pQE31 (QIAGEN). Plasmid DNA was prepared from the resulting E. coli transformants and the presence of the correct insert was confirmed by restriction endonuclease digestion and DNA sequencing.
Purification of TspB variants
The TspB variant-encoding plasmid constructs were transformed into E. coli strain BL21(DE3) (Invitrogen) and protein expression was carried out at 37°C for 3h (pQE30/31) after induction with 1 mM IPTG at OD600 of ~0.5. TspB variants were then purified by Ni2+-NTA affinity chromatography (5 ml HisTrap HP column, GE Healthcare) under denaturing conditions, using protocols provided by QIAGEN (The QIAexpressionist handbook). Columns were washed with 5 column volumes of wash buffer (100 mM NaH2PO4, 10 mM Tris•HCl, pH 6.3, 8 M urea) and Elution buffer D (same as Wash buffer, pH 5.9). Finally, the TspB derivative was eluted using buffer E (same as Wash buffer, pH 4.5). Fractions (10× 2 ml each) eluting after 20 ml of buffer E had passed through the column were collected, combined and concentrated using a 10 kDa membrane (Corning, Corning, NY) to a final concentration of 1 mg/ml. The TspB0776 CR derivative required the additional use of 0.2M acetic acid containing 6 M guanidine HCl to completely elute from the column.
IgG binding to Nm strains by flow cytometry
Meningococcal strains were grown under the conditions described above. Binding experiments were performed as described in (18). FITC- or Dylight 488-conjugated anti-Human IgG at 5 μg/ml (Jackson ImmunoResearch Laboratories, West Grove, PA) or FITC-, Dylight 488, or Rhodamine Red-X-conjugated anti-mouse IgG at 5 μg/ml (Jackson ImmunoResearch), were used for detecting bound IgG as indicated. The samples were analyzed using the BD Biosciences FACSCalibur or Fortessa flow cytometers. For flow cytometry binding experiments with mouse anti-TspB CR and control (mice injected with adjuvant only) serum, bacteria were grown the indicated medium only or medium supplemented with a 1:200 dilution of pooled antisera from mice immunized with three 25 μg doses of TspB1628 CR with alum adjuvant, or pooled serum from mice given adjuvant alone. The bacteria were then pelleted, washed, fixed and analyzed as in (18).
Isolation of Nm Igbp
For each culture condition, the cells were grown to an OD620nm of ~0.6. The cells were pelleted, cell pellets were washed with PBS, and aliquots were frozen until used. For the immunoprecipitation experiment, an aliquot of cells was defrosted and washed twice with PBS. After washing, the cell pellet was resuspended in solubilization buffer (10 mM Tris, pH 7.8, containing 10 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.2% sodium deoxycholic acid, and 0.1% sodium dodecylsulfate). Insoluble material was removed by centrifugation and the supernatants were transferred to a column containing Protein A or Protein G Sepharose CL-4B beads (Sigma-Aldrich). The extract was mixed with the beads overnight at 4°C. After washing the beads, proteins retained by the beads were eluted by boiling the beads in SDS-PAGE sample buffer without reducing agent and resolved on 4%–12% SDS-PAGE gel (Invitrogen, Carlsbad, CA). After staining the SDS-PAGE gel with Simply Blue (Invitrogen), the bands of proteins retained by the Protein A or G beads (from both MH and CDM+HuS samples) were excised, proteins in the gel slices were digested with trypsin (Worthington Biochemical Corporation, Lakewood, NJ), and the tryptic peptides were extracted from the gel slices for MALDI-TOF analysis (Bruker Daltonics Autoflex, Billerica, MA). Only a few masses with signals barely above background were observed in the negative control MH samples (data not shown). In contrast, large numbers of mass peaks were obtained for most of the CDM+HuS protein bands. The set of observed peptide masses was analyzed using Mascot (www.matrixscience.com or (19)). Once proteins were identified, expected masses of TspB tryptic peptides were calculated using PeptideMass (20) and additional expected masses were located in the mass spectra.
TspB ELISA
Recombinant TspB derivatives (100 μl/well at a concentration of ~10 μg/ml in PBS) were adsorbed to 96-well Immulon 2HB round-bottom microtiter plates (Dynatech, Chantilly, VA). Negative control wells contained 1% BSA in PBS buffer. After washing with PBS and blocking the plates with PBS containing 1% BSA and 0.1% sodium azide; (pH 7.4), human donor serum, IgG-depleted human serum from the donors 1 and 2, the IgG purified from donor 1 and donor 2 serum, purified human IgG, IgM, IgA, and IgG purchased from Jackson ImmunoResearch, purified human IgG1, IgG2, and IgG3 from Millipore (Billerica, MA), whole mouse serum from Invitrogen, purified mouse IgG1, IgG2a, IgG2b, IgG3 and IgM mAbs obtained from Southern Biotech (Birmingham, AL), purified human albumin, fibrinogen (both from Sigma-Adrich) and human factor H (Complement Technology, Tyler, TX) were added to the wells as serial dilutions in blocking buffer. After an overnight incubation at 4°C, plates were washed again with PBS and the presence of bound antibody was detected using the appropriate anti-human or anti-mouse Ig-alkaline phosphatase-conjugated secondary antibody (Invitrogen). Controls included wells sensitized, instead of the TspB derivative, with BSA or irrelevant E. coli proteins of BL21(DE3) carrying plasmid without tspB from the same fraction of the Ni2+NTA column as that used to purify the TspB derivative, but including antibodies and secondary antibodies. Absorbance values at 405 nm were measured at 60 min after development with p-nitrophenyl phosphate substrate (SIGMAFAST, Sigma-Aldrich). Enzymatic desialylation of IgG (50 μg) was performed using Sialidase A™ (Prozyme, Hayward, CA) under native conditions according to the manufacturer’s instructions. Fab and Fc fragments of human IgG2 (Millipore) were prepared by incubating the mAb (5 mg/ml) with papain conjugated to agarose beads (Pierce Chemical Company, Rockford, IL) in 25 mM sodium phosphate buffer, pH 7, containing 2 mM cysteine at 37°C with constant mixing for 5 days. The reaction was monitored by SDS-PAGE to ensure that it went to completion. The antibody fragments were purified by passing the reaction through a Protein A-agarose column (Sigma-Aldrich). The Fab fragment passed through the column unbound and, after washing the column, the Fc fragment was eluted in 0.2 M glycine, pH 2.5. The eluate was immediately adjusted to pH 7 with Tris base. Both Fab and Fc preparations were dialyzed against PBS and the concentration determined by SDS-PAGE using the whole molecule IgG2 as a standard prior to testing binding to TspB by ELISA. Bound IgG2, Fab and Fc were detected using Zymax™ AP-goat anti-human IgG (H+L) secondary antibody (Invitrogen).
Generating tspB deletion mutants
The tspB triple mutants were generated in NmB strain H44/76 by replacing the genes NMBH4476_0598, NMBH4476_0681 and NMBH4476_1698 with kanamycin (Kan), erythromycin (Erm) and chloramphenicol (Cm) resistance cassettes, respectively. Sequences of approximately 400 bp flanking gene NMBH4476_0681, amplified using primer sets 3 and 4, were cloned up- and downstream of the ErmR cassette into plasmid pGEM-Erm (pGEM3Zf(+) carrying ErmR cassette in SmaI site). To generate the NMBH4476_0598 and NMBH4476_1698 deletion constructs, each ORF together with ~400 bp of up- and downstream sequence (amplified using primer sets 1 and 5, respectively) was first cloned into plasmid pGEM-T Easy (Promega, Madison, WI) according to manufacturer’s instructions. Second, through inverse PCR, the flanking regions of NMBH4476_0598 and NMBH4476_1698, connected via the plasmid backbone, were amplified using primer sets 2 and 6 (6R1 and 6F1 or 6R2 and 6F2 to allow for subsequent insertion of the CmR cassette in either orientation), respectively. The antibiotic resistance cassettes KanR (isolated from plasmid pUC4K, GE Healthcare, via BamHI restriction digest) and CmR (isolated from pCOM (21) derivative via restriction digest with XbaI and BamHI) were subsequently cloned into the resulting PCR products cut with the corresponding restriction enzymes, thus replacing the tspB genes in the religated plasmid. With regards to the 5′–3′ direction of the replaced tpsB genes, the NMBH4476_0598 construct contained KanRcassette in reverse orientation, the NMBH4476_0681 construct ErmR cassette in the forward orientation, while the NMBH4476_1698 construct was generated with the CmR cassette in either orientation. The deletion constructs were sequentially introduced into strain H44/76 as described previously (22) and the targeted tspB genes were replaced via homologous recombination. All deletion constructs contained primer-designed DNA-uptake sequences (DUS) (23) at the distal ends of the tspB flanking regions to facilitate DNA incorporation. Transformants were selected on chocolate agar containing the appropriate antibiotic for selection (50 μg/ml kanamycin, 2.5 μg/ml erythromycin and/or 2.5 μg/ml chloramphenicol). To avoid re-introduction of wild-type prophage DNA by phage transduction, individual colonies were cultured under selection in CDM+HuS+antibiotics as described above and tested for reactivity with anti-human IgG and anti-TspB by flow cytometry. Selected clones that were negative for human IgG/anti-TspB binding were streaked on chocolate agar with antibiotics and the process of selecting colonies, culturing in media with HuS, and analysis by flow cytometry was repeated a total of three times. Successful deletion of the tspB genes was verified by PCR using genomic DNA prepared from the mutant clones. Internal verification PCR, as seen in Fig. 6a., was carried out using a primer set specific for a ~500 bp segment just upstream of NMBH4476_0681 (positive control), primers Ver3 and Ver4 to specifically detect presence of the NMBH4476_0598 ORF (expected product size ~1370 bp), primers Ver4 and Ver5 to detect presence of the NMBH4476_0681 ORF (expected product size ~1280 bp) and primers Ver6 and Ver7 to detect presence of the NMBH4476_ 1698 ORF (expected product size ~1180 bp). Once a gene replacement has been successful, its verification PCR should not yield the expected PCR product. All primer sequences are listed in supplemental Table S1.
Re-introducing prophage DNA into the ORF6 knockout mutant by transduction with wild-type phage
NmB strain H44/76 was grown in MH media supplemented with 2.5% glucose (100 ml) overnight at 37°C in an incubator/shaker with 5% CO2. The bacteria were removed by centrifugation and phage present in the supernatant were isolated using a protocol described in (24). The resulting phage stock was heated to 70°C for 30 minutes, treated with DNase I and RNase A (25 μg/ml) at 37°C for 1 hr, re-precipitated with PEG/NaCl and suspended in sterile PBS before use. Ten ORF6 knockout mutant colonies were selected from a chocolate agar plate containing antibiotics (Kan, Erm and Cm) and combined with 10 μl of phage stock in 1 ml of MH and left standing at 37°C for 2 hrs. 5 μl of the culture was streaked on a chocolate agar plate containing antibiotics. After overnight incubation at 37°C in 5% CO2, 10 individual colonies were cultured in 0.5 ml of CDM+5% HuS+antibiotics and tested for human IgG/anti-TspB binding by flow cytometry as described above. Four of the 10 were positive for human IgG binding and reactivity with anti-TspB. The procedure was repeated for the positive clones. Finally, a complemented mutant was selected and grown in CDM+5% human serum (not heat inactivated)+antibiotics. Surviving colonies were used to prepare frozen milk stocks as described above. PCR was performed using primers specific for each ORF6 gene to verify reinsertion and to identify which of the three possible genes was reinserted.
Immunizing mice with TspB variants
Recombinant TspB1628 CR, expressed in E. coli BL21(DE3) and purified by Ni2+-NTA affinity chromatography under denaturing conditions as described above and dialyzed against 2× 4L of 0.9% NaCl was used to immunize groups of 10 mice with three injections of either 0 μg, 1 μg, 5 μg or 25 μg of protein. The protein was given with alum (Alhydrogel 1.0% (w/v), Superfos Biosector, Frederikssund, Denmark) or Freund’s (Pierce) adjuvant. Serum of each mouse was tested for reactivity with recombinant TspB1628 CR by ELISA after three immunizations and pooled according to the antigen used.
Fluorescence Microscopy
An aliquot of the bacteria used for flow cytometry experiments described above that had been labeled with Dylight 488-conjugated anti-human IgG or Rhodamine Red-X-conjugated anti-mouse IgG (Jackson ImmunoResearch) were incubated with 0.5 μg/ml DAPI (to stain bacterial intracellular DNA) or Hoechst 33258 (to stain extracellular DNA as indicated) for 5 minutes at ambient temperature, washed in PBS-BSA and resuspended in PBS containing 0.5% formaldehyde. An aliquot was spotted onto a cover slip and allowed to dry in the dark. The sample was fixed with 4% paraformaldehyde (Sigma) for 5 minutes at ambient temperature, washed with PBS and mounted on a slide with a drop of Fluoro-gel with Tris Buffer (Electronic Microscopy Sciences, Hatfield, PA). Images were obtained at 20x, 40x, and 100x magnification using a Zeiss Axioplan fluorescence microscope with Q-Imaging Micropublisher 3.3 RTV attachment. Confocal images were obtained using a Zeiss LSM710 (CHORI) laser scanning confocal microscope (63× magnification) and were analyzed using ImageJ Software [41] and co-localization with JACoP [35].
Results
Detection of human IgG bound to Nm strains by flow cytometry
When Nm bacteria were grown in Catlin 6 chemically defined medium (CDM) (25) supplemented with heat-inactivated human serum (HuS), genetically diverse Nm strains from several capsular groups were observed to bind human IgG by flow cytometry. For example, the results of measuring IgG binding to NmW135 strain A22 and NmB strain H44/76 cultured under different conditions are shown in Fig. 1. When cells were grown in CDM supplemented with 5% HuS, both strains showed binding of human IgG (Fig. 1, first row). In contrast, bacteria grown in CDM supplemented with the same amount of IgG purified from the donor serum (donor 2), no IgG binding was observed (Fig. 1, second row). Also, cells grown in the presence of serum that had been depleted of IgG (CDM+dHuS) by Protein G affinity chromatography showed little or no IgG binding (Fig. 1, third row). IgG binding was restored when the removed IgG was added back to the serum (Fig. 1, fourth row). The results suggest that bacteria cultured in CDM alone did not express antigens reactive with IgG antibodies purified from the human donor serum, but that an unknown factor present in human serum stimulated expression of antigens with immune reactivity to IgG present in the donor serum or, alternatively, activated or induced expression of a mediator of non-immune binding.
Growing Nm strains in the presence of active human complement increases IgG-binding activity
The results shown in Fig. 1 for strain A22 utilized bacteria taken from a particular frozen milk stock. However, we found that bacteria taken from stocks prepared from different A22 colonies exhibited variable degrees of IgG binding. The variability was not limited to strain A22. Fig. 2a. shows the results of IgG binding experiments with genetically and antigenically diverse strains representing capsular groups A (Z1092), B (NMB), C (4243), X (Ug13/0) and Y (860800) in addition to group W135 strain A22 from a frozen stock different from that used for experiments shown in Fig. 1. The experiments used HuS from two donors (donors 1 and 2) who lack intrinsic bactericidal activity against any of the test strains and showed no Ig reactivity with the strains when they were cultured in plain CDM or MH media (data not shown). The data presented in Fig. 2a shows that HuS-dependent stimulation of IgG binding was not unique to a specific donor or strain. Within the group of strains tested, IgG binding was variable but, regardless of which strain or donor serum was used, IgG binding to the bacteria was observed to some degree.
FIGURE 2.
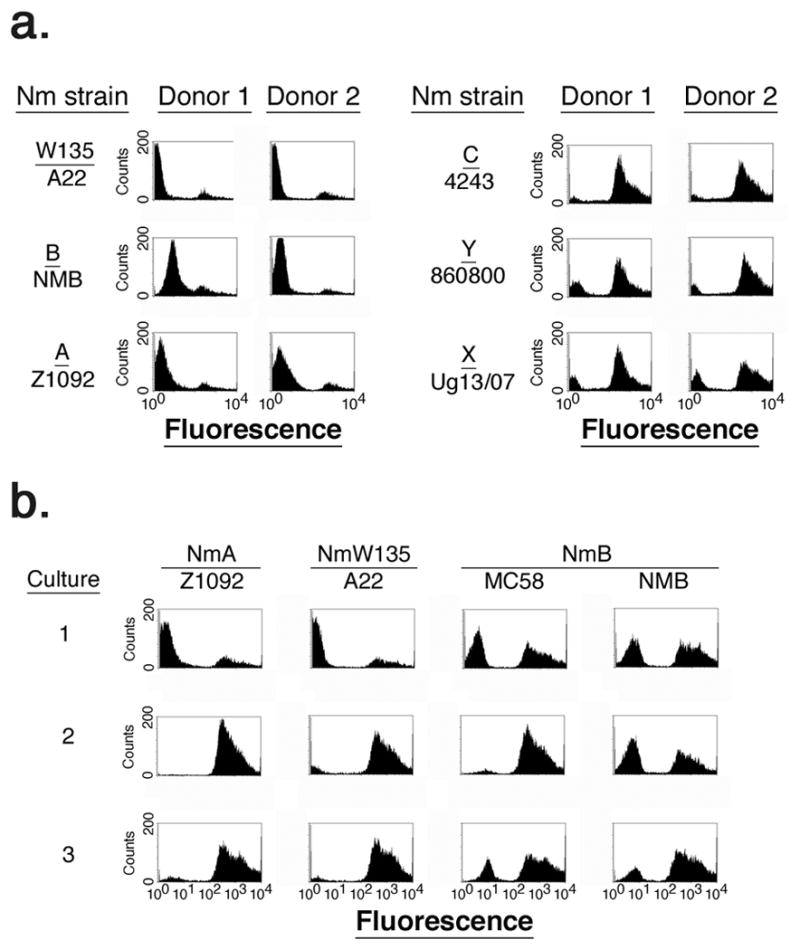
Immunoglobulin-binding protein activity exhibited by diverse Nm strains. Panel a. shows IgG binding to diverse Nm strains with serum from two donors. Panel b. shows changes in IgG binding to Nm strains after each of three rounds of serial growth (indicated by the number on the left) of bacteria in human serum with active complement.
As shown in Fig. 2b, when Nm strains showing low Ig-binding activity (i.e. strains A22, NMB, and Z1092) were sequentially diluted and re-cultured three times (each round of culture indicated by the numbers at the left of Fig. 2b) in CDM containing 20% human serum with active complement (serum that was not heat-inactivated) prepared from donor 2, an increase in the population of cells exhibiting IgG-binding activity was observed. After three rounds of culture the IgG-positive fraction of cells for each strain tested was increased compared to the first round of culture. Thus, IgG-binding activity was variable for particular strains, but serially culturing bacteria in the presence of serum with active human complement led to an increase in the population of IgG-binding bacteria.
Although we have not yet determined the identity of the factor in human serum that triggers IgG binding, in data not shown (BA Flitter. MK-W Cheng and GR Moe, unpublished), we observed that the factor was sensitive to boiling and proteolysis by trypsin or proteinase K. Also, reactivity was lost over time when the serum was stored at 4°C (>24 hrs) or incubated at 37°C for several hours. The factor was retained by a 100 kDa filter, and when human serum was fractionated using the Cohn method (26), the factor stimulating IgG binding was present in Cohn fraction IV. Lipids extracted from serum or Cohn fraction IV did not stimulate IgG binding by Nm strains. Commercially prepared Cohn fraction IV at a concentration of 0.5 mg/ml added to CDM consistently activated IgG binding in several Nm strains and, therefore, was used as a supplement in experiments with mutants described below to ensure activation of IgG-binding activity.
Recovery of IgG-binding protein by elution from Protein A or G and identification by mass finger print analysis
To identify the protein mediating binding of IgG to Nm strains when grown in the presence of human serum, an immunoprecipitation experiment was performed using Protein A- or Protein G-modified beads. IgG and associated proteins were isolated from solubilized bacteria (strain A22) cultured in CDM+HuS or MH (negative control). Fig. 3 shows the proteins retained by Protein A and G beads resolved on an SDS-PAGE gel and visualized with Simply Blue. Protein A or G beads retained only trace amounts of proteins when the cells were grown in MH (right portion of the gel) but there were several additional bands present in the samples prepared from cells cultured in CDM supplemented with human serum (left portion of the gel).
FIGURE 3.
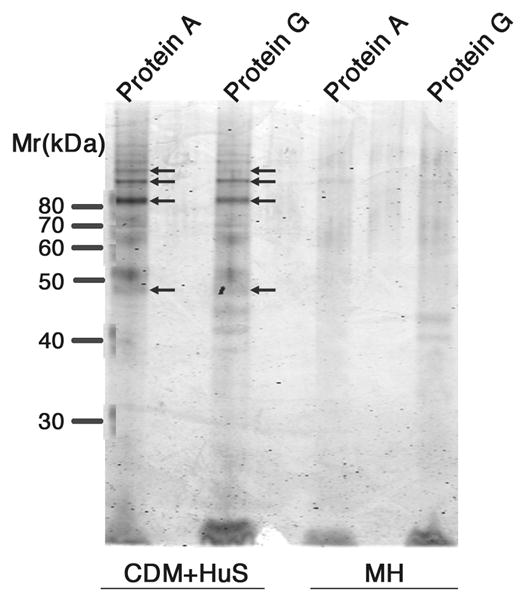
Isolation of Nm proteins binding to human IgG by precipitation with Protein A or G beads. NmW135 strain A22 was cultured in CDM+HuS or MH as indicated. Unmarked lanes were intentionally left unloaded. The arrows indicate bands that produced a common set of tryptic peptides.
Although the appearance of several bands on the gel suggested multiple IgG-interacting proteins, mass spectroscopic analysis revealed a common set of tryptic peptides was observed for 4 of 9 bands analyzed. The bands are indicated by arrows in Fig. 3. The protein corresponding to the common set of tryptic peptides identified by mass fingerprint analysis using Mascot (19) was most similar to the product of gene NMA0776 from NmA strain Z2491 (sequences could not be matched to strain A22 since the genome has not been sequenced). The common set of tryptic peptides producing major peaks in the mass spectra resulted in a Mowse score of 64 (values >50 are significant) and an expectation value of 0.0026 (values <0.05 are significant). NMA0776 is one of two related genes encoded by the genome of NmA strain Z2491, which share sequence homology with a gene coding for T and B cell stimulating protein B (TspB) identified by Ala’Aldeen and coworkers (27), as well as a prophage gene, ORF6, identified by Bille et al. in invasive meningococci (5, 6). The second tspB/ORF6-related gene present in Z2491 is NMA1797. Once the candidate Ig-binding protein was identified, we re-analyzed the mass spectrum for the presence of lower abundance masses expected either for a protein encoded by NMA1797 or additional TspB related proteins encoded by genomes of other sequenced Nm strains. However, the set of tryptic peptide masses corresponded only to a protein encoded by NMA0776. Although the protein appeared as several bands on the gel shown in Fig. 3, including those with apparent masses larger than the predicted mass of TspB (~58 kDa), the set of tryptic peptides corresponded to most of the predicted full-length protein. Therefore, it appeared that TspB might have been aggregated even after boiling in sample buffer or modified in a manner that hinders mobility in an SDS-PAGE gel.
Sequence analysis of TspB proteins
Querying 27 genomes of Neisseriaceae species available at NCBI for similarity to the translated protein sequence of NMA0776 using BLAST sequence homology search, we found homologous protein sequences in invasive meningococcal group A, B, and C and Neisseria gonorrhoeae strains as well as commensal strains. The number of genes encoding putative complete TspB proteins found in any given strain ranged from 2 to 4, with most genomes containing additional partial sequences homologous to portions of ORF6. When the identified protein (translated nucleotide) sequences from Nm group A, B, and C strains were compared, a pattern emerged. As shown schematically in Fig. 4a., the sequences of the amino and carboxyl terminal ends of the proteins are variable but there is a core region of about 326 amino acids (indicated by the black, light grey and grey ovals in Fig. 4a.) that is relatively conserved (~80% identity). Most of the variability between sequences within this “conserved region” or CR is located in a 64 residue segment (position indicated by the light grey oval in Fig. 4a) that connects 2 subdomains of 107 and 82 residues, respectively (black and grey ovals in Fig. 4a), that are >90% identical among all sequences currently available. There are three sequences for the connecting peptide among all the TspB proteins in the database. The CR is followed by a variable proline-rich segment of approximately 77 amino acids (indicated by hexagons in Fig. 4a). Such proline-rich segments are known to adopt extended helical structures (28). The variable sequences at the C- terminal end of the protein contain hydrophobic segments (position indicated by black rectangle in Fig. 4a) of sufficient length to span the membrane as a-helices and, thus, may represent membrane anchor sequences.
FIGURE 4.
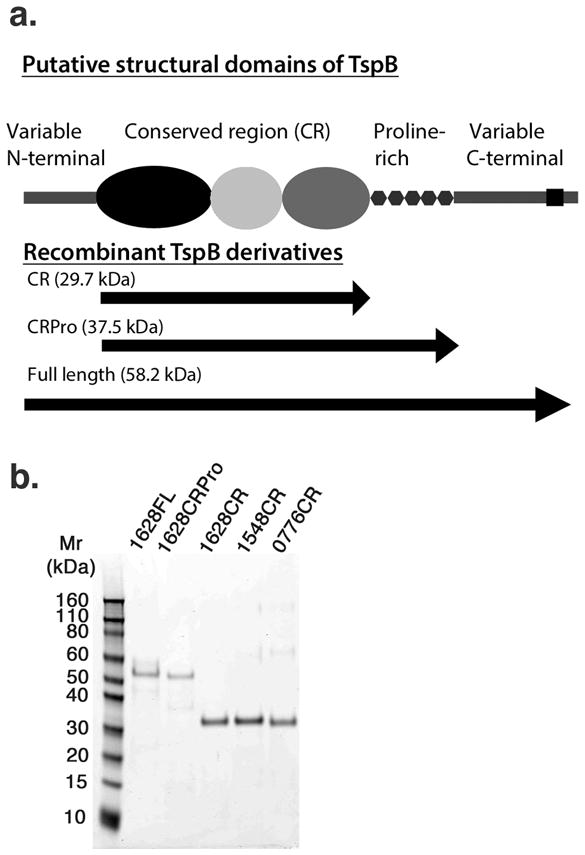
Schematic representation of TspB subdomains determined by sequence analysis and SDS-PAGE of purified recombinant TspB derivatives. In panel a., the length of each illustrated object corresponds to the relative number of amino acids in each segment based on the sequence of NMB1628. The putative structural domains comprising each recombinant protein are indicated by arrows in the lower portion of panel a. Predicted molecular masses for the recombinant proteins based on the sequence of NMB1628 are indicated in parentheses. Panel b. shows a 10% SDS-PAGE gel of purified recombinant proteins corresponding to full length TspB1628 (1628FL), TspB1628 CRPro (1628CRPro), TspB1628 CR (1628CR), TspB1548 CR (1548CR), and TspB0776 CR (0776CR) that were used in binding studies shown in Fig. 5.
Cloning, expression, purification and functional activity of the TspB conserved region (TspB CR)
We cloned nucleotide sequences encoding all conserved TspB domains from both NmB strain MC58 (NMB1548, NMB1628 and NMB1747) and NmA strain Z2491 (NMA0776 and NMA1797) (hereafter referred to as ‘TspB(gene number) CR’) as well as the full length protein and the CR plus the proline-rich segment (CRPro; Fig. 4a) encoded by NMB1628 from strain MC58. The CRs coded by NMB1628, NMB1548 and NMA0776 represent the range of sequence diversity of 32 TspB CRs sequences for N. meningitidis available at NCBI, with NMB1628 being the most common and NMA0776 the most diverse of these sequences. The CR domains of NMB1628 and NMB1528 differ at only 3 positions in the variable peptide segment.
The resulting tspB constructs, encoding TspB CR, CRPro as well as full length TspB (segments encompassed by each construct indicated by the arrows in the lower portion of Fig. 4a) were expressed in E. coli strain BL21(DE3) as an amino-terminal His6-tagged protein and purified under denaturing conditions in 8 M urea. All the recombinant TspB derivatives bound strongly to the Ni2+NTA affinity column in 8 M urea, requiring relatively large volumes (4 or more column volumes) of pH 4.5 buffer to elute the proteins from the column. As shown in Fig. 4b, when resolved by SDS-PAGE, the purified TspB CRs appeared as single bands corresponding to their expected masses (expected masses indicated in Fig. 4a), while full length TspB1628 and TspB1628 CRPro appeared as one major band with fainter bands higher and lower than the expected mass, which was similar to what was observed for TspB isolated from bacteria by immunoprecipitation (Fig. 3). Bands corresponding to a dimer and tetramer were apparent in the preparation of TspB0776 CR even after boiling in SDS sample buffer. Taken together, the purification data suggest that TspB CR may be polymeric even in solutions containing 8 M urea and, in the case of TspB0776 CR, after boiling in SDS sample buffer.
The TspB derivatives were evaluated for the ability to bind Ig from donor and commercial sources of human serum by ELISA with the purified, recombinant TspB derivative serving as the solid-phase antigen. A representative example is shown in Fig. 5a for IgG binding to each of the TspB derivatives (filled circles, full length (FL) TspB1628; filled squares, TspB1628 CRPro; filled triangles, TspB1628 CR; filled inverted triangles, TspB1548 CR; filled diamonds, TspB0776 CR) using serum from donor 2 as the source of IgG. Each binding experiment was performed in duplicate with wells on the plate not containing TspB derivatives as the negative control (open circles, dashed line). All of the TspB CRs, TspB1628 CRPro and full length TspB1628 exhibited similar reactivity with human serum IgG by ELISA, even when the recombinant proteins were purified under denaturing conditions and used directly without refolding (Fig. 5a). Unlike some other Ig-binding proteins (29), the TspB constructs were not reactive with human IgG on Western blots (data not shown), suggesting that they retained native structure in urea that was lost when boiling the protein in SDS-PAGE sample buffer.
FIGURE 5.
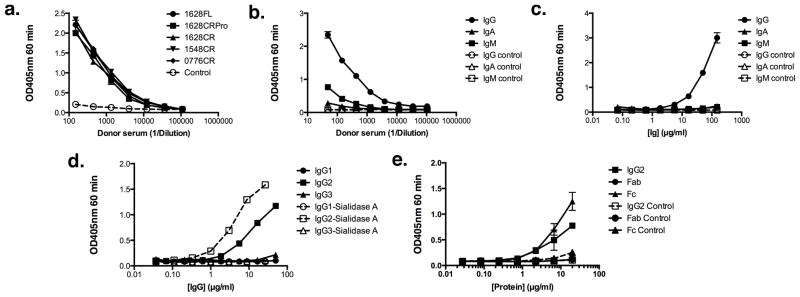
Immunoglobulin-binding activity of recombinant TspB derivatives by ELISA. Panel a. shows IgG-binding activity in whole serum from donor 2 of full length (FL) TspB1628 (filled circles), TspB1628 CRPro (filled squares), TspB1628 CR (filled triangles), TspB1548 CR (inverted triangles), and TspB0776 CR (filled diamonds) recombinant proteins. The control (open circles, dashed line) was serum diluted into wells sensitized with BSA only and detection with secondary antibody. Panel b. shows binding of IgG (filled circles), IgA (filled triangles) and IgM (filled squares) in serum from donor 2 with recombinant TspB1628 CR. The controls (open symbols, dashed lines) were the same as in 5a. except for detection using the indicated anti-IgG, anti-IgM and anti-IgA secondary antibody. Panel c. shows binding of purified IgG (filled circles), IgA (filled triangles) and IgM (filled squares) with recombinant TspB1628 CR. The controls (open symbols, dashed lines) were the same as in b. Panel d. shows binding of purified human IgG1 (filled circles), IgG2 (filled squares), or IgG3 (filled triangles) paraproteins with recombinant TspB1628 CR not treated or treated with Sialidase A™ (corresponding open symbols, dashed lines). Panel e. shows binding of purified human IgG2 (filled squares), IgG2 Fab (filled circles), and IgG2 Fc (filled triangles). The control in panel e. (open symbols, dashed lines) was irrelevant E. coli protein from strain BL21(DE3) transformed with a plasmid lacking tspB and purified by Ni2+-NTA using the same method as the recombinant TspB1628 CR derivative. Each point in the graphs represents the average of duplicate determinations and the error bars the SEM.
For further characterization, we focused on TspB1628 CR since the presence of the proline rich region and additional segments in the full-length derivative did not appear to enhance reactivity with IgG (Fig. 5a). In an ELISA using whole serum from donor 2 (Fig. 5b) or commercially obtained preparations of purified Ig (Fig. 5c), recombinant TspB1628 CR showed a preference for binding to IgG (filled circles) compared to IgA (filled squares) or IgM (filled triangles). When irrelevant purified human IgG subclass paraproteins were tested (Fig. 5d), TspB1628 CR appeared to show a preference for binding human IgG2 (filled squares) compared to IgG1 (filled circles) or IgG3 (filled triangles). Treatment with Sialidase A™ further increased binding to IgG2 (open squares, dashed line) but had no effect on binding to IgG1 (open circles, dashed line) or IgG3 (open triangles, dashed line). De-glycosylation with N and/or O glycanases did not increase binding further (data not shown). The IgG2 protein was treated with papain conjugated to agarose beads to generate Fab and Fc fragments. By ELISA, only whole molecule IgG2 (Fig. 5e, filled squares) and the purified Fc fragment (Fig. 5e, filled triangles) showed binding to TspB1628 CR. When other human myeloma IgGs were tested by ELISA, we found that some IgG1 antibodies also bound to the TspB 1628CR preparation. However, when irrelevant proteins from the same fraction (TspB elution fraction) of a mock purification of an E. coli BL21(DE3) extract were used as a control, antibodies from the other subclasses that appeared to show binding to TspB1628 CR exhibited similar or greater binding to the irrelevant E. coli proteins. This indicated that the antibodies were non-specifically binding to irrelevant proteins. Therefore, the irrelevant E. coli proteins were used as a control for specificity in Fig. 5e, to show that the IgG2 antibody and corresponding Fab and Fc fragments did not bind to these E. coli proteins and that TspB specifically reacted with IgG2. Similarly, there was no binding of purified mouse IgG subclass myeloma paraproteins tested for binding to TspB1628 CR (data not shown). Finally, Igbps can be multifunctional proteins with domains that bind to other serum proteins (12). We tested whether TspB1628 CR or full length TspB1628 could bind to purified human albumin, factor H or fibrinogen by ELISA. In data not shown, binding of the serum proteins by TspB1628 CR was not different from binding to the bovine serum albumin negative control at protein concentrations of 100 μg/ml.
Knocking out ORF6 in NmB strain H44/76 and effect on human Ig binding
To establish that TspB is responsible for the immunoglobulin-binding activity exhibited by Nm strains, the ORF6 genes from NmB strain H44/76 were knocked out by homologous recombination with plasmids containing 5′ and 3′ portions of each targeted gene, separated by DNA encoding one of three different antibiotic resistance genes. The H44/76 genome contains 3 ORF6 genes (NMBH4476_0598, NMBH4476_0681 and NMBH4476_1698), as well as 2 truncated genes with DNA sequences homologous to the N-terminal CR subdomain of NMBH4476_1698 (CR subdomain shown schematically in Fig. 4a). Strain H44/76 was chosen for several reasons: the TspB CR domains coded by the three ORF6 genes have sequences nearly identical to the recombinant TspB1628 CR (strain MC58) used to characterize Ig binding, the H44/76 strain is readily transformable, we have a collection of H44/76 mutant strains that could be used for comparing the effects of ORF6 knockouts on serum resistance with other genes known to be important, the strain was an isolate from a fatal case of meningitis and septicemia and was the master strain for the production of an outer membrane vesicle vaccine used in humans (30).
Twelve triple antibiotic resistant mutants were analyzed by PCR to determine the presence or absence of the targeted genes. For these reactions primers were used that are specific for each of the three ORF6 genes (0598, 0681 and 1698), to amplify internal DNA fragments that would be eliminated after successful replacement by resistance cassettes. As a positive control, primers were chosen to amplify a ~500 bp sequence just upstream of NMBH4476_0681 that should be unaffected by the deletion. Fig. 6a shows the resulting PCR products for one representative triple mutant resolved on agarose gels from reactions using genomic DNA from wild-type (indicated by W above each lane) and the ORF6 knockout mutant strains (indicated by M above each lane). PCR products of the expected size were obtained for the genomic DNA positive control sequence and all three ORF6 genes from the wild-type DNA template but only the positive control primers produced a PCR product when the ORF6 knockout mutant DNA was used, showing that the three ORF6 genes were no longer present.
Single colonies from mutants in which the three ORF6 genes were PCR verified to be deleted (9 of 12) were tested for reactivity with anti-TspB1628 CR and human IgG binding by flow cytometry and fluorescence microscopy. This verification process was repeated two more times with 5 single colonies from one of the original mutants to ensure that the final isolate obtained was not contaminated with wild-type phage that could re-introduce an ORF6 gene into a strain with a triple antibiotic resistant background (see complementation experiment below).
By flow cytometry (Fig. 6b), human IgG binding (red histograms) by the ORF6 knockout mutant grown in CDM supplemented with heat-inactivated human serum and Cohn fraction IV was nearly eliminated, as indicated by the ~100-fold decrease in mean fluorescence compared to the wild-type strain. Similarly, binding of anti-TspB (cyan histograms) and irrelevant control mouse serum IgG (magenta histograms) by the ORF6 knockout mutant was reduced compared to wild-type H44/76.
When the cells were examined by fluorescence microscopy (Fig. 6c), most cells reactive with anti-TspB (red fluorescence in lower panels of Fig. 6c) were in large aggregates. In contrast, individual cells as well as aggregates were marked by anti-human IgG (green fluorescence, upper and lower panels). As control (upper panels) bacteria were grown in CDM+HuS and serum from mice that had been injected with adjuvant only. Formation of large aggregates for cells grown in the presence of human serum was not unique to strain H44/76. Similar results were obtained for NmB strains NMB and MC58, NmW135 strain A22 and NmA strain Z2491 (data not shown). Aggregates were also present in fluorescence micrographs of the ORF6 knockout mutant strain, but the characteristics were markedly different, as the aggregates were generally smaller and were not reactive with anti-TspB (Fig. 6c., lower middle micrograph). Furthermore, aggregates reactive with anti-TspB had a particular flat, film-like structure as discussed in greater detail below with regard to Fig. 7.
Complementation of the ORF6 knockout mutant with wild-type phage
To confirm that the differences observed between the wild-type and the ORF6 knockout mutant resulted from the loss of tspB expression, we generated a mutant in which an ORF6 gene was re-introduced into the ORF6 knockout mutant by transduction with phage produced in wild-type H44/76. PCR analysis of the DNA from the phage preparation used for transduction detected the presence of NMBH_0598 and NMBH_1698 (data not shown). NMBH_0681 was detected by PCR only in un-diluted phage DNA, suggesting that phage carrying NMBH_0681 were a minor fraction of phage in the preparation. Phage proteins resolved by SDS-PAGE were not reactive with anti-TspB on Western blots. The presence of phage proteins, however, was verified by staining a parallel-run gel with Simply Blue (data not shown). Therefore, TspB was not present in phage released into the culture medium. After transduction of ORF6 knockout mutant bacteria with phage, cultures were grown in CDM+HuS+antibiotics from 10 individual triple antibiotic-resistant colonies selected from a chocolate agar plate, and were tested for human IgG binding and reactivity with anti-TspB by flow cytometry. All transductants were positive for both antigens compared to the ORF6 knockout mutant, but varied in the ability to bind human IgG and reactivity with anti-TspB. The procedure was repeated with an isolate showing the greatest human IgG/anti-TspB reactivity and finally, from that isolate, clones were isolated from a chocolate agar plate containing antibiotics. DNA was isolated from one of the clones for PCR analysis as described above for the ORF6 knockout strain. As shown in Fig. 6a., lower panel, the complemented mutant was positive for the presence of NMBH_0598, but negative for NMBH_0681 and NMBH_1698, showing that a single ORF6 gene was reintroduced into the ORF6 knockout strain. The 0598-complemented mutant showed human IgG and anti-TspB binding by flow cytometry that was essentially the same as wild-type H44/76 (Fig. 6b., right panel). The complemented mutant also produced large cell aggregates that were reactive with anti-TspB as shown in the fluorescence micrograph in the lower right panel of Fig. 6c. Thus, the phenotype characterized by human IgG binding, surface exposure of TspB and aggregate formation was restored by re-introducing one of three wild-type ORF6 genes, confirming that the phenotype was dependent on expression of ORF6.
TspB, IgG and DNA form a biofilm with bacterial aggregates
By fluorescence microscopy, anti-TspB and anti-human IgG reactivity in bacteria with a functional ORF6 gene, appeared to be localized largely to an extracellular matrix surrounding the bacteria. This can be seen in the laser scanning confocal micrograph of a wild-type H44/76 aggregate stained with anti-mouse IgG (red fluorescence) to detect binding of anti-TspB and anti-human IgG (green fluorescence) shown in Fig. 7. Bacteria, which were not stained in this experiment (an example of a DAPI-stained bacteria aggregate is shown in Fig. 6c), appear as black spots in the micrographs with most of the anti-TspB and human IgG reactivity located in the extracellular spaces between the bacteria. On close examination, the TspB/IgG extracellular matrix has a fibrous appearance. This can be seen in the extended structure on the left side of Fig. 7 marked with anti-TspB (red fluorescence) or anti-human IgG (green fluorescence).
Nm strains are known to form complex biofilms on surfaces and the biofilms have been shown to contain extracellular DNA (31, 32). To determine whether the TspB/IgG matrix also contained extracellular DNA, after fixing the bacteria, the samples were stained with a fluorescent DNA dye that does not stain bacterial intracellular DNA. As shown by the blue fluorescence in Fig. 7, the matrix contained DNA in addition to TspB and human IgG. The Manders’ coefficient estimating DNA fluorescence overlapping anti-human IgG or anti-mouse IgG (anti-TspB) fluorescence was 0.999, indicating that the matrix was composed of a mixture of at least the three components. A series of micrographs recorded successively from the bottom to the top of several aggregates (z-stack) provided an estimate of aggregate depth, which was ~2 μm. Thus, the bacterial aggregates enveloped in an extracellular matrix formed biofilms as defined by their large surface area but small volume dimension. It is unclear at this point whether the biofilms observed formed on the surfaces of the culture tube or at the air-water interface of the culture mixture.
The smaller aggregates observed in micrographs of the ORF6 knockout mutant did not contain TspB as shown in Fig. 6c (middle micrographs) or extracellular DNA (data not shown), suggesting that in this case aggregation was mediated by another mechanism. The serum used to supplement the culture medium did not contain DNA that could be measured by fluorometry. Therefore, the likely source of DNA in the aggregates was from lysed bacteria (31, 32) or membrane blebs such as the DNA-containing membrane blebs released by Neisseria gonorrhoeae (33).
Discussion
Bille et al. used whole genome comparisons between large collections of invasive and carriage strains to identify genes associated with strains that cause meningococcal disease (5). Their analysis led to the identification of prophage DNA associated with invasive strains. The association appeared to be causal and most clearly linked with strains causing disease in young adults aged 13–28 years (5, 6). Based on the association with disease, they referred to the prophage DNA as the meningococcal disease associated island or MDA (5). However, the reasons why the MDA might promote the pathogenic potential of Nm strains were not clear. Deletion of the MDA or specific MDA genes appeared to have no effect in various models of pathogenic potential, including survival in rabbit serum, adherence to cells and survival in a mouse model of meningococcal bacteremia (5). In this report we have shown that TspB, which is encoded by prophage gene ORF6, is an IgG-binding protein. Extracellular release of TspB, IgG binding, and the formation of biofilm in which TspB, IgG and DNA envelop aggregated bacteria depended specifically on human serum factors. Bacteria cultured in rich media containing non-human components or CDM supplemented with mouse serum did not exhibit IgG binding. The IgG binding was mediated by a highly conserved segment of TspB, which we designated as the CR. Furthermore, IgG binding by particular Nm strains was heterogeneous, but was increased by serially culturing the bacteria in human serum with active complement. Since it is not common to culture Nm strains in media supplemented with human serum, the phenotype associated with ORF6 would not be generally apparent. We have not identified the factor in human serum that activates IgG binding by Nm strains. However, it appears to be a protein and to be concentrated in Cohn fraction IV. TspB protein could not be detected in phage particles. Therefore, release of TspB from the bacteria must occur by another mechanism such as secretion via PilQ (5) or sacrificial lysis of a small fraction of cells. The latter is consistent with the presence of, presumably, bacterial DNA in the extracellular matrix of aggregates.
The striking characteristics of aggregation, IgG binding with TspB and biofilm formation raise the question of what purpose they might serve in meningococcal pathogenesis. Mobile DNA, including plasmids, transposons, and bacteriophages, are well-known elements of bacterial evolution that provide a source of new functionalities, facilitating survival in a wide range of niches inhabited by bacteria (reviewed in (34)). Such genetic elements are also associated with human disease, where they may encode factors important for pathogenicity. Mobile genetic elements might encode molecules important for colonization, nutrient acquisition and evasion of innate and adaptive immune responses. Parallel to what we have found for Nm TspB, E. coli genes encoding Igbps (eib genes) have been identified to be carried by prophage DNA and E. coli strains can carry multiple copies (35). For example, the genome of strain ECOR-9 contains 4 distinct copies of eib. As described here for TspB, expression of E. coli Igbps is highly dependent on culture conditions (35). E. coli Igbps form stable multimeric structures that impart resistance to human serum complement as a result of binding human IgA and the Fc segment of IgG (29). As suggested earlier (see Results), TspB CR appears to be polymeric, even in 8M urea, and specifically binds the Fc portion of human IgG2.
By analogy to the examples given above, TspB may facilitate cell aggregation, therefore generating a protective barrier through the formation of polymeric structures containing TspB, IgG and DNA. Aggregated IgG located far from the bacterial surface resulting from interaction with TspB released from bacteria may also serve to non-productively activate complement. The functional role of aggregation (36) in Nm pathogenesis is unclear, but one can speculate that the aggregates could facilitate adhesion to tissue surfaces and provide protection from complement-dependent bacteriolysis and opsonophagocytosis, as the aggregates can be considerably larger than macrophages. The large size of the aggregates may also result in occlusion of capillaries suggesting a possible mechanism for producing purpura, which are a characteristic clinical presentation of meningococcal bacteremia.
Nm strains are known to form complex biofilms on a variety of surfaces that develop in several stages (32). Neisserial biofilms have also been shown to contain DNA and to produce DNases that modulate characteristics of the biofilm (31, 32). It is unclear how the TspB/Ig/DNA biofilm observed in this study may relate to biofilms formed on biological surfaces. Typically, Neisseria spp. organisms form biofilms under conditions of external stress and forces induced by flow over surfaces (31, 32). Nm biofilm formation has been shown to be inhibited by expression of capsular polysaccharide (37) and to involve regulated expression of specific proteins over a time course of several hours (37, 38). In contrast, the TspB/IgG/DNA biofilm observed here must have formed on the culture tube or air-water interface. Also, the bacteria were encapsulated and the biofilm was produced during a few hours of culture in which the time frame for gene regulation would be relatively short.
In summary, we have identified an Nm phenotype characterized by formation of large bacterial aggregates in biofilm containing TspB, IgG and DNA, which was dependent on factors present specifically in human serum and a functional copy of the TspB-encoding prophage gene ORF6. Further studies will be needed to determine what form of TspB is present in biofilm, the mechanism of surface exposure/release from bacteria, the mechanism of biofilm formation and the potential role of TspB in protecting against bacteriolysis and opsonophagocytosis. The altered physical and antigenic characteristics of Nm strains cultured in HuS and their effects on bactericidal activity mediated by anticapsular, anti-porin and anti-factor H binding protein antibodies (data not shown) suggest, that in vitro and in vivo assays commonly used to evaluate protection against Nm strains mediated by vaccine-elicited antibodies in the absence of human serum factors may overestimate their functional activity. Comparative studies with wild-type and ORF6 knockout mutant bacteria cultured in the presence of HuS with those grown in rich media containing non-human supplements will be needed to evaluate this possibility. Finally, the IgG-binding segment of TspB is highly conserved among meningococcal strains that cause disease. Thus, vaccines based on TspB that elicit antibodies having the ability to block Ig-binding activity and/or biofilm formation, such as those described for Staphylococcus aureus protein A (39), may be useful in preventing meningococcal disease.
Supplementary Material
Acknowledgments
We thank Nina E. Moe for performing the initial cloning, expression, purification, and characterization of TspB1628 CR.
Abbreviations
- CDM
Catlin 6 chemically defined medium
- CR
conserved region
- CRPro
conserved and proline-rich regions
- HuS
normal human serum
- dHuS
IgG-depleted human serum
- Igbp
immunoglobulin-binding protein
- MH
Mueller-Hinton medium
- Nm
Neisseria meningitidis
- TspB
T and B cell stimulating protein B
Footnotes
Funding was provided by NIH NIAID grant number AI064314 (www.niaid.nih.gov/Pages/default.aspx), NIH NCRR grant numbers CO6 RR-16226 and S10RR025472 (www.ncrr.nih.gov), Children’s Hospital Branches, Inc. (www.childrenshospitalbranches.org), and the Jennifer Leigh Wells Family (www.moonlight4meningitis.com/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Rosenstein NE, Perkins BA, Stephens DS, Popovic T, Hughes JM. Meningococcal disease. N Engl J Med. 2001;344:1378–1388. doi: 10.1056/NEJM200105033441807. [DOI] [PubMed] [Google Scholar]
- 2.Edmond K, Clark A, Korczak VS, Sanderson C, Griffiths UK, Rudan I. Global and regional risk of disabling sequelae from bacterial meningitis: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:317–328. doi: 10.1016/S1473-3099(10)70048-7. [DOI] [PubMed] [Google Scholar]
- 3.Sultan B, Labadi K, Guegan JF, Janicot S. Climate drives the meningitis epidemics onset in west Africa. PLoS Med. 2005;2:e6. doi: 10.1371/journal.pmed.0020006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yazdankhah SP, Caugant DA. Neisseria meningitidis: an overview of the carriage state. J Med Microbiol. 2004;53:821–832. doi: 10.1099/jmm.0.45529-0. [DOI] [PubMed] [Google Scholar]
- 5.Bille E, Zahar JR, Perrin A, Morelle S, Kriz P, Jolley KA, Maiden MC, Dervin C, Nassif X, Tinsley CR. A chromosomally integrated bacteriophage in invasive meningococci. J Exp Med. 2005;201:1905–1913. doi: 10.1084/jem.20050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bille E, Ure R, Gray SJ, Kaczmarski EB, McCarthy ND, Nassif X, Maiden MC, Tinsley CR. Association of a bacteriophage with meningococcal disease in young adults. PLoS One. 2008;3:e3885. doi: 10.1371/journal.pone.0003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atkins KL, Burman JD, Chamberlain ES, Cooper JE, Poutrel B, Bagby S, Jenkins AT, Feil EJ, van den Elsen JM. S. aureus IgG-binding proteins SpA and Sbi: host specificity and mechanisms of immune complex formation. Mol Immunol. 2008;45:1600–1611. doi: 10.1016/j.molimm.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 8.Raeder R, Boyle MD. Properties of IgG-binding proteins expressed by Streptococcus pyogenes isolates are predictive of invasive potential. J Infect Dis. 1996;173:888–895. doi: 10.1093/infdis/173.4.888. [DOI] [PubMed] [Google Scholar]
- 9.Cleary P, Retnoningrum D. Group A streptococcal immunoglobulin-binding proteins: adhesins, molecular mimicry or sensory proteins? Trends Microbiol. 1994;2:131–136. doi: 10.1016/0966-842x(94)90600-9. [DOI] [PubMed] [Google Scholar]
- 10.Housden NG, Harrison S, Roberts SE, Beckingham JA, Graille M, Stura E, Gore MG. Immunoglobulin-binding domains: Protein L from Peptostreptococcus magnus. Biochem Soc Trans. 2003;31:716–718. doi: 10.1042/bst0310716. [DOI] [PubMed] [Google Scholar]
- 11.Tagawa Y, Sanders JD, Uchida I, Bastida-Corcuera FD, Kawashima K, Corbeil LB. Genetic and functional analysis of Haemophilus somnus high molecular weight-immunoglobulin binding proteins. Microb Pathog. 2005;39:159–170. doi: 10.1016/j.micpath.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Burman JD, Leung E, Atkins KL, O’Seaghdha MN, Lango L, Bernado P, Bagby S, Svergun DI, Foster TJ, Isenman DE, van den Elsen JM. Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein: indications of a novel mechanism of complement evasion by Staphylococcus aureus. J Biol Chem. 2008;283:17579–17593. doi: 10.1074/jbc.M800265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tashiro M, Montelione GT. Structures of bacterial immunoglobulin-binding domains and their complexes with immunoglobulins. Curr Opin Struct Biol. 1995;5:471–481. doi: 10.1016/0959-440x(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 14.Beernink PT, Leipus A, Granoff DM. Rapid genetic grouping of factor h-binding protein (genome-derived neisserial antigen 1870), a promising group B meningococcal vaccine candidate. Clinical and vaccine immunology : CVI. 2006;13:758–763. doi: 10.1128/CVI.00097-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephens DS, Swartley JS, Kathariou S, Morse SA. Insertion of Tn916 in Neisseria meningitidis resulting in loss of group B capsular polysaccharide. Infect Immun. 1991;59:4097–4102. doi: 10.1128/iai.59.11.4097-4102.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beernink PT, Caugant DA, Welsch JA, Koeberling O, Granoff DM. Meningococcal factor H-binding protein variants expressed by epidemic capsular group A, W-135, and X strains from Africa. J Infect Dis. 2009;199:1360–1368. doi: 10.1086/597806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fossa da Paz M, Baruque-Ramos J, Hiss H, Vicentin MA, Leal MB, Raw I. Polysaccharide production in batch process of Neisseria meningitidis serogroup C comparing Frantz, modified Frantz and Catlin 6 cultivation media. Brazil J Micro. 2003;34:27–32. [Google Scholar]
- 18.Flitter BA, Ing JY, Moe GR. Effect of human serum on de-N-acetyl sialic acid epitope expression and antibody activity against N. meningitidis group B. Vaccine. 2010;28:5967–5972. doi: 10.1016/j.vaccine.2010.06.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 20.Wilkins MR, Lindskog I, Gasteiger E, Bairoch A, Sanchez JC, Hochstrasser DF, Appel RD. Detailed peptide characterization using PEPTIDEMASS--a World-Wide-Web-accessible tool. Electrophoresis. 1997;18:403–408. doi: 10.1002/elps.1150180314. [DOI] [PubMed] [Google Scholar]
- 21.Koeberling O, Giuntini S, Seubert A, Granoff DM. Meningococcal outer membrane vesicle vaccines derived from mutant strains engineered to express factor H binding proteins from antigenic variant groups 1 and 2. Clin Vaccine Immunol. 2009;16:156–162. doi: 10.1128/CVI.00403-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moe GR, Zuno-Mitchell P, Lee SS, Lucas AH, Granoff DM. Functional activity of anti-Neisserial surface protein A monoclonal antibodies against strains of Neisseria meningitidis serogroup B. Infect Immun. 2001;69:3762–3771. doi: 10.1128/IAI.69.6.3762-3771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodman SD, Scocca JJ. Identification and arrangement of the DNA sequence recognized in specific transformation of Neisseria gonorrhoeae. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:6982–6986. doi: 10.1073/pnas.85.18.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonnycastle LLC, Menendez A, Scott JK. General Phage Methods. In: Barbas CF, Burton DR, Scott JK, Silverman GJ, editors. Phage Display. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. pp. 15.17–15.19. [Google Scholar]
- 25.Catlin BW. Nutritional profiles of Neisseria gonorrhoeae, Neisseria meningitidis, and Neisseria lactamica in chemically defined media and the use of growth requirements for gonococcal typing. J Infect Dis. 1973;128:178–194. doi: 10.1093/infdis/128.2.178. [DOI] [PubMed] [Google Scholar]
- 26.Cohn EJ, Strong LE, et al. Preparation and properties of serum and plasma proteins; a system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids. Journal of the American Chemical Society. 1946;68:459–475. doi: 10.1021/ja01207a034. [DOI] [PubMed] [Google Scholar]
- 27.Kizil G, Todd I, Atta M, Borriello SP, Ait-Tahar K, Ala’Aldeen DA. Identification and characterization of TspA, a major CD4(+) T-cell- and B-cell-stimulating Neisseria-specific antigen. Infect Immun. 1999;67:3533–3541. doi: 10.1128/iai.67.7.3533-3541.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butcher DJ, Nedved ML, Neiss TG, Moe GR. Proline pipe helix: structure of the tus proline repeat determined by 1H NMR. Biochemistry. 1996;35:698–703. doi: 10.1021/bi952419l. [DOI] [PubMed] [Google Scholar]
- 29.Sandt CH, Hill CW. Nonimmune binding of human immunoglobulin A (IgA) and IgG Fc by distinct sequence segments of the EibF cell surface protein of Escherichia coli. Infect Immun. 2001;69:7293–7303. doi: 10.1128/IAI.69.12.7293-7303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fredriksen JH, Rosenqvist E, Wedege E, Bryn K, Bjune G, Froholm LO, Lindbak AK, Mogster B, Namork E, Rye U, et al. Production, characterization and control of MenB-vaccine “Folkehelsa”: an outer membrane vesicle vaccine against group B meningococcal disease. NIPH annals. 1991;14:67–79. discussion 79–80. [PubMed] [Google Scholar]
- 31.Lappann M, Vogel U. Biofilm formation by the human pathogen Neisseria meningitidis. Med Microbiol Immunol. 2010;199:173–183. doi: 10.1007/s00430-010-0149-y. [DOI] [PubMed] [Google Scholar]
- 32.Neil RB, Apicella MA. Clinical and laboratory evidence for Neisseria meningitidis biofilms. Future Microbiol. 2009;4:555–563. doi: 10.2217/fmb.09.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorward DW, Garon CF, Judd RC. Export and intercellular transfer of DNA via membrane blebs of Neisseria gonorrhoeae. Journal of bacteriology. 1989;171:2499–2505. doi: 10.1128/jb.171.5.2499-2505.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tinsley CR, Bille E, Nassif X. Bacteriophages and pathogenicity: more than just providing a toxin? Microbes Infect. 2006;8:1365–1371. doi: 10.1016/j.micinf.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 35.Sandt CH, Hopper JE, Hill CW. Activation of prophage eib genes for immunoglobulin-binding proteins by genes from the IbrAB genetic island of Escherichia coli ECOR-9. J Bacteriol. 2002;184:3640–3648. doi: 10.1128/JB.184.13.3640-3648.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jain A, Agarwal A. Biofilm production, a marker of pathogenic potential of colonizing and commensal staphylococci. J Microbiol Methods. 2009;76:88–92. doi: 10.1016/j.mimet.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 37.Yi K, Rasmussen AW, Gudlavalleti SK, Stephens DS, Stojiljkovic I. Biofilm formation by Neisseria meningitidis. Infection and immunity. 2004;72:6132–6138. doi: 10.1128/IAI.72.10.6132-6138.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neil RB, Apicella MA. Role of HrpA in biofilm formation of Neisseria meningitidis and regulation of the hrpBAS transcripts. Infection and immunity. 2009;77:2285–2293. doi: 10.1128/IAI.01502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim HK, Emolo C, DeDent AC, Falugi F, Missiakas DM, Schneewind O. Protein A-specific monoclonal antibodies and prevention of Staphylococcus aureus disease in mice. Infection and immunity. 2012;80:3460–3470. doi: 10.1128/IAI.00230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.