

Summary
The ability to measure human aging from molecular profiles has practical implications in many fields, including disease prevention and treatment, forensics, and extension of life. Although chronological age has been linked to changes in DNA methylation, the methylome has not yet been used to measure and compare human aging rates. Here, we build a quantitative model of aging using measurements at more than 450,000 CpG markers from the whole blood of 656 human individuals, aged 19 to 101. This model measures the rate at which an individual’s methylome ages, which we show is impacted by gender and genetic variants. Furthermore, we show that differences in aging rates help explain epigenetic drift and are reflected in the transcriptome. Our model highlights specific components of the aging process and provides a quantitative read-out for studying the role of methylation in age-related disease.
Introduction
Not everyone ages in the same manner. It is well known that women tend to live longer than men, and lifestyle choices such as smoking and physical fitness can hasten or delay the aging process (Steven N., 2006; Blair et al., 1989). These observations have led to the search for molecular markers of age which can be used to predict, monitor, and provide insight into age-associated physiological decline and disease. One such marker is telomere length, a molecular trait strongly correlated with age (Harley et al., 1990) which has been shown to have an accelerated rate of decay under environmental stress (Epel et al., 2004; Valdes et al.). Another marker is gene expression, especially for genes that function in metabolic and DNA repair pathways which are predictive of age across a range of different tissue types and organisms (Fraser et al., 2005; Zahn et al., 2007; de Magalhães et al., 2009).
A growing body of research has reported associations between age and the state of the epigenome— the set of modifications to DNA other than changes in the primary nucleotide sequence (Fraga and Esteller, 2007). In particular, DNA methylation associates with chronological age over long time scales (Alisch et al., 2012; Christensen et al., 2009; Bollati et al., 2009; Boks et al., 2009; Rakyan et al., 2010; Bocklandt et al., 2011; Bell et al., 2012) and changes in methylation have been linked to complex age-associated diseases such as metabolic disease (Barres and Zierath, 2011) and cancer (Jones and Laird, 1999; Esteller, 2008). Studies have also observed a phenomenon dubbed “epigenetic drift”, whereby the DNA methylation marks in identical twins increasingly differ as a function of age (Fraga et al., 2005; Boks et al., 2009). Thus, the idea of the epigenome as a fixed imprint is giving way to the model of the epigenome as a dynamic landscape that reflects a variety of chronological changes. The current challenge is to determine whether these changes can be systematically described and modeled to detect different rates of human aging, and to tie these rates to related clinical or environmental variables.
The mechanisms that drive changes in the aging methylome are not well understood, although they have been attributed to at least two underlying factors (Vijg and Campisi, 2008; Fraga et al., 2005). First, it is possible that environmental exposure will over time activate cellular programs associated with consistent and predictable changes in the epigenome. For example, stress has been shown to alter gene expression patterns through specific changes in DNA methylation (Murgatroyd et al., 2009). Alternatively, spontaneous epigenetic changes may occur with or without environmental stress, leading to fundamentally unpredictable differences in the epigenome between aging individuals. Spontaneous changes may be caused by chemical agents that disrupt DNA methyl groups or through errors in copying methylation states during DNA replication. Both mechanisms lead to differences between the methylomes of aging individuals, suggesting that quantitative measurements of methylome states may identify factors involved with slowed or accelerated rates of aging.
To better understand how the methylome ages and to determine whether human aging rates can be quantified and compared, we initiated a project to perform genome-wide methylomic profiling of a large cohort of individuals spanning a wide age range. Based on these findings, we constructed a predictive model of aging rate which we show is influenced by gender and specific genetic variants. These data help explain epigenetic drift and suggest that age-associated changes in the methylome lead to changes in transcriptional patterns over time. These findings were replicated in a second large cohort.
Global methylomic profiling over a wide age range
We obtained methylome-wide profiles of two different cohorts (N1 = 482, N2 = 174) sampled from a mixed population of 426 Caucasian and 230 Hispanic individuals, aged 19 to 101. Samples were taken as whole blood and processed using the Illumina Infinium HumanMethylation450 BeadChip assay (Bibikova et al., 2011), which measures the methylation states of 485,577 CpG markers. Methylation was recorded as a fraction between zero and one, representing the frequency of methylation of a given CpG marker across the population of blood cells taken from a single individual. Conservative quality controls were applied to filter spurious markers and samples (Experimental Procedures). For simplicity, we discarded values for markers on sex chromosomes..Association tests revealed that 70,387 (15%) of the markers had significant associations between methylation fraction and age (Figure 1, FDR < 0.05 by F-Test, Experimental Procedures). We were able to verify at a P < 0.05 significance level 53,670 (76%) of these associations using 40 young and old samples recently published by Heyn et al. (Heyn et al., 2012). More detailed accounts of the individual aging markers and their genomic features are presented in the Supplementary Information (Figure S1, Tables S1, S2). The resulting dataset represents the largest and highest-resolution collection of methylation data produced for the study of aging, providing an unprecedented opportunity to understand the role of epigenetics in the aging process. The complete methylation profiles are available at the Gene Expression Omnibus (GSE40279, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40279).
Figure 1. Global data on the aging methylome.
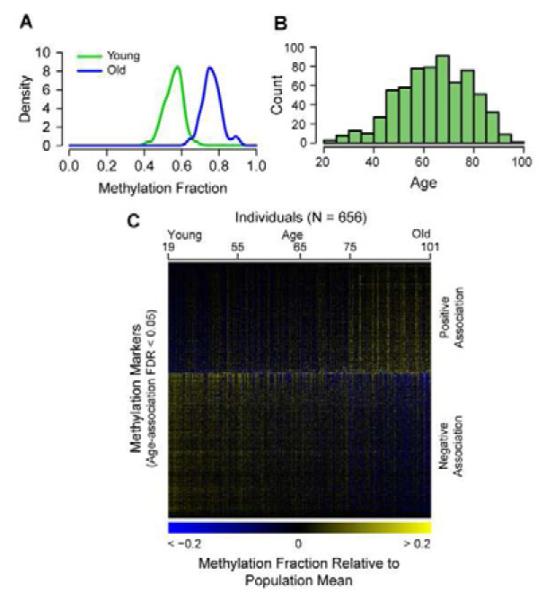
(A) A density plot of methylation fraction values for the marker cg16867657, separated by young (green) and old (blue) individuals. (B) A histogram of the age distribution for all individuals. (C) A heat map of the age-associated methylation markers, sorted by the magnitude of association (regression coefficient). The individuals are ordered youngest to oldest. A specific example of an age-associated region is shown in Figure S1. Annotation coincidence tables are also provided as Tables S1, S2.
A predictive model for the aging methylome
We built a predictive model of aging on the primary cohort using a penalized multivariate regression method known as Elastic Net (Zou and Hastie, 2005), combined with bootstrap approaches (Experimental Procedures). The model included both methylomic and clinical parameters such as gender and Body Mass Index (BMI) (Figure 2A). The optimal model selected a set of 71 methylation markers which were highly predictive of age (Figure 2A, Table S3). The accuracy of the model was high, with a correlation between age and predicted age of 96% and an error of 3.9 years (Figure 2B). Nearly all markers in the model lay within or near genes with known functions in aging-related conditions including Alzheimer’s disease, cancer, tissue degradation, DNA damage, and oxidative stress. By way of example, two markers lay within the gene somatostatin (SST), a key regulator of endocrine and nervous system function (Yacubova and Komuro, 2002). SST is known to decline with age and has been linked to Alzheimer’s disease (Saito et al., 2005). As a second example, six model markers lay within the transcription factor KLF14, which has been called a ‘master regulator’ of obesity and other metabolic traits (Small et al., 2011). Given the links between aging, longevity, and metabolic activity (Lane et al., 1996; Tatar et al., 2003), it is not surprising that several of our model markers are implicated in obesity and metabolism.
Figure 2. Model predictions and clinical variables.

(A) A flow chart of the data (green boxes) and analyses (red ovals) used to generate aging predictions (blue boxes). (B) A comparision of predicted and actual ages for all individuals based on the aging model. (C) Out-of-sample predictions for individuals in the validation cohort. (D) Apparent methylomic aging rate (AMAR) for each individual, based on the aging model without clinical variables. The distribution of aging rates shows faster aging for men than women. A table of the markers used in the aging model are provided as Table S3.
We validated this model on the secondary cohort, consisting of an additional 174 independent samples. These samples were processed in the same manner as the primary cohort, then used to predict age based on the original model (i.e., as trained on the original cohort). The predictions were highly accurate, with a correlation between age and predicted age of 91% and an error of 4.9 years (Figure 2C). The significance of the aging model was also confirmed by the dataset presented in Heyn et al., verifying the age association of 70 of the 71 markers (Heyn et al., 2012). Furthermore, the model was able to fully separate old and young individuals in the Heyn et al. study, even for profiles obtained using bisulfate sequencing rather than the bead-chip technology used in this study (Figure S2).
Methylome aging rate and its associations
While the aging model is able to predict the age of most individuals with high accuracy, it is equally valuable as a tool for identifying individual outliers who do not follow the expectation. For example, Figure 2B highlights two individuals whose age is vastly over- or under-predicted based on their methylation data. To examine whether these differences reflect true biological differences in the state of the individual (i.e., versus measurement error or intrinsic variability), we used the aging model to quantify each individual’s apparent methylomic aging rate (AMAR), defined as the ratio of the predicted age, based on methylation data, to the chronological age. We then tested for associations between AMAR and possibly relevant clinical factors, including gender and BMI. Analysis of ethnicity and diabetes status was not possible due to correlations with batch variables (Figure S3). We found that gender, but not BMI had significant contributions to aging rate (F-test, P = 6×10−6, P > 0.05, Experimental Procedures). The methylome of men appeared to age approximately 4% faster than that of women (Figure 2D), even though the overall distributions of age were not significantly different between the men and women in the cohort (P > 0.05, KS-test). Likewise, the validation cohort confirmed the increased aging rate for men (P < 0.05), but was inconclusive for BMI (P > 0.05). This complements a previous finding of an epigenetic signal for BMI that does not change with age (Feinberg et al., 2010).
As genetic associations have been previously reported with human longevity and aging phenotypes (Atzmon et al., 2006; Suh et al., 2008; Willcox et al., 2008; Wheeler et al., 2009), we examined whether the model could distinguish aging rates for individuals with different genetic variants. For this purpose, we obtained whole-exome sequences for 252 of the individuals in our methylome study at 15x coverage. After sequence processing and quality control, these sequences yielded 10,694 common single nucleotide variants across the population (Experimental Procedures). As a negative control, we confirmed that none of the genetic variants were significant predictors of age itself, which is to be expected since the genome sequence is considered to be relatively static over the course of a lifetime. On the other hand, one might expect to find genetic variants that modulate the methylation of age-associated markers, i.e. methylation quantitative-trait loci or meQTLs (Bell et al., 2011). Testing each genetic variant for association with the top age-associated methylation markers, we identified 303 meQTLs (Experimental Procedures, FDR < 0.05, Figure 3A). For validation, we selected 8 genetic variants (corresponding to 14 meQTLs) to test in a validation cohort of 322 individuals from our methylation study. This analysis found that 7 of 8 genetic variants (corresponding to 7 meQTLs) remained highly significant in the validation cohort (FDR < 0.05, Table S4). While all of these variants acted in cis with their meQTLs (within 150 kbp), we confirmed that none directly modified the CpG site or associated probe sequence of the associated methylation marker.
Figure 3. Genetic effects on methylomic aging.

(A) We surveyed genomic variants for an association with age-associated methylation markers. 8 genetic variants, corresponding to 14 meQTLs, were chosen for validation. Of these, 7 were significant in the validation cohort and two showed an association with AMAR. (B) A plot of the trend between the methylation marker cg27367526 (STEAP2) and age. The state of variant rs42663 (GTPBP10) causes an offset in this relationship. (C) A second example for cg18404041 and rs2230534 (ITIH1, NEK4). A table of confirmed genetic associations is provided as Table S4.
The methylation marker cg27193080 was one of those found to be significantly associated with age (P < 10−17), and its methylation fraction was found to be influenced by the Single Nucleotide Polymorphism (SNP) variant rs140692 (P < 10−21) (Figure 3B). This meQTL was particularly interesting as both the SNP and the methylation marker mapped to the gene methyl-CpG binding domain protein 4 (MBD4, with the SNP in an intron and the methylation marker just upstream of the coding region), one of the few known genes encoding a protein that can bind to methylated DNA. This meQTL thus captures a cis-relationship in which rs140692 influences the methylation state of MBD4. That MBD4 plays a role in human aging is supported by previous work linking MBD4 to DNA repair, as well as work showing that mutations and knock-downs of MBD4 lead to increased genomic instability (Bellacosa et al., 1999; Bertoni et al., 2009).
Of the seven validated meQTLs, three were identified that had a statistically-significant association not only with age but also with aging rate (AMAR, FDR < 0.05, Figure 3B,C). One is the genetic marker rs2230534, which is a synonymous mutation in the gene NEK4, and has a cis association with the methylation marker cg18404041. The NEK family of kinases plays a key role in cell cycle regulation and cancer (Moniz et al., 2011). The second variant is rs2818384, which is a synonymous mutation in the gene JAKMIP3, and has a cis association with the methylation marker cg05652533. Copy-number variants in JAKMIP3 have been previously associated with glioblastoma (Xiong et al., 2010). The final variant found to influence AMAR is rs42663, which is a missense mutation in the gene GTPBP10, and associates with cg27367526 in the gene STEAP2. STEAP2 is known to play a role in maintenance of iron and copper homeostasis— metals which serve as essential components of the mitochondrial respiratory chain (Ohgami et al., 2006). Studies have shown that perturbations of iron concentrations can induce DNA damage through oxidative stress in mammalian cells (Hartwig and Schlepegrell, 1995; Karthikeyan et al., 2002). These meQTLs represent genetic variants that appear to broadly influence the aging methylome and may be good candidates for further age-associated disease and longevity research.
A Multi-tissue Diagnostic
Our aging model was derived from whole blood, which is advantageous in the design of practical diagnostics and for testing samples collected from other studies. To investigate whether our aging model was representative of other tissues, we obtained DNA methylation profiles for 368 individuals in the control category of The Cancer Genome Atlas (TCGA) (Collins and Barker, 2007), including 83 breast, 183 kidney, 60 lung, and 42 skin samples. An aging model based on both our primary and validation cohorts demonstrated strong predictive power for chronological age in these samples (expected value R = 0.72), although each tissue had a clear linear offset (intercept and slope) from the expectation (Figure 4A). This offset was consistent within a tissue, even across different batches of the TCGA data. We adjusted for each tissue trend using a simple linear model, producing age predictions with an error comparable to that found in blood (Figure 4B). Furthermore, predicted AMARs in each tissue supported the effect of men appearing to age more quickly than women (P < 0.05). Thus, computation of aging rate (AMAR) from blood samples reflects trends that are not specific to blood and may be common throughout many tissues of the human body. Furthermore, this analysis provides evidence that the observed methylomic changes are intrinsic to the methylome and not due primarily to cell heterogeneity, i.e. changing cell-type composition of whole blood with age. In this regard, this study is consistent with a prior analysis of purified CD4+ T-cells and CD14+ monocytes, in which the age-associated epigenetic modifications were found to be similar to the changes observed in whole blood (Rakyan et al, 2010).
Figure 4. Multi-tissue support.
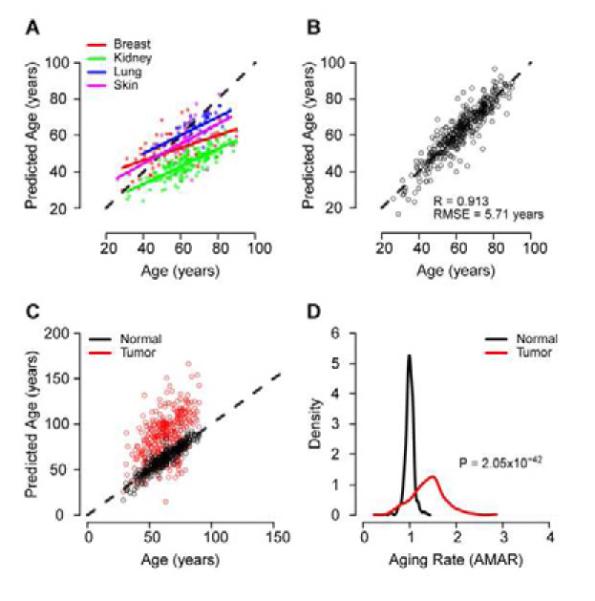
(A) Predictions of age made by the full aging model on the TCGA control samples. There is a high correlation between chronological and predicted age, but each tissue has a different linear intercept and slope. (B) After adjusting the intercept and slope of each tissue, the error of the model is similar to that of the original whole blood data. Age predictions made on cancer samples are presented as Figure S2. (C) Age predictions made on matched normal and tumor samples from TCGA. Predictions are adjusted for the linear offset of the parent tissue (breast, kidney, lung, skin). (D) Tumor samples show a significant increase in AMAR.
To investigate the similarities and differences between the tissues, we built age models de novo for breast, kidney, and lung tissues (Table S5; the skin cohort had too few samples to build a model). Most of the markers in the models differed, though, all of these models and the primary model share the markers cg23606718 and cg16867657. These markers are both annotated to the gene ELOVL2, which has been linked to the photoaging response in human skin (Kim et al., 2010).
The TCGA dataset also contains methylome profiles representing a total of 319 tumors and matched normal tissue samples (breast, kidney, lung, skin). Interestingly, use of our aging model indicated that tumors appear to have aged 40% more than matched normal tissue from the same individual (Wilcox test, P < 10−41, Figure 4C,D). Accelerated tumor aging was apparent regardless of the primary tissue type. We investigated whether this was the result of broad shifts in global methylation levels by examining all 70,387 age-associated markers, of which 44% tend to increase and 56% tend to decrease with age. Methylation fraction values in matched tumor and normal samples supported the finding that tumors coincide with older values for 74% of the markers regardless of the trending direction (Binomial P ~ 0). Furthermore, separate aging models built in the matched normal and tumor samples confirm the apparent aging effect (Figure S4).
Different aging rates lead to divergent methylomes
If individuals indeed age at different rates, it might be expected that their individual methylomes should diverge over time. This is based on the premise that the methylomes of the very young share certain similarities, and that these similarities diminish as individuals accumulate changes over time. This effect, called epigenetic drift, has been observed in monozygotic twins (Fraga et al., 2005) but few specific hypothesis have been put forth to account for it. To examine epigenetic drift in our samples, we computed the deviance of each methylation marker value as its squared distance from the expected population mean (Figure 5A, Experimental Procedures). Then, in addition to testing for markers whose methylation fraction changes with age (Figures 5B,C), we were able to test for markers whose deviance changes with age (Figures 5D,E) (Breusch and Pagan, 1979). Increasing deviance was a widespread phenomenon—we identified 27,800 markers for which the deviance was significantly associated with age (FDR < 0.05), of which 27,737 (99.8%) represented increased rather than decreased deviance (Figure 5E, Figure S5). For any given individual, especially high or low methylome deviance was a strong predictor of aging rate (R = 0.47, P ~ 0), suggesting that differences in aging rates account for part of methylome heterogeneity and epigenetic drift.
Figure 5. Age associations for methylation fraction and deviance.

(A) Methylation fraction values for are shown for the marker cg24724428. Over any subset of the cohort, we consider two group methylation statistics: the mean and variance. Marker variance is a measure of the mean methylation deviance, which is defined as the squared difference between an individual’s methylation fraction and their expected methylation fraction. (B) A density plot showing the change in mean methylation with age for the marker cg24724428. Young and old groups are based on the top and bottom 10%. (C) A histogram of the significance of association between the methylation fraction of all markers and age. P-values are signed such that positive values represent an increase of methylation with age. Markers which exceeded the FDR < 0.05 threshold are grouped into the most extreme bins. (D) A density plot showing the change in methylation deviance with age for the marker cg24724428. (E) A histogram in the same form as ‘D’, of the significance of association between the methylation deviance of all markers and age. Aging trends are mapped for CpG islands in Figure S3.
Another way to examine epigenetic drift is in terms of Shannon Entropy, or loss of information content in the methylome over time (Shannon and Weaver, 1963). An increase in entropy of a CpG marker means that its methylation state becomes less predictable across the population of cells, i.e. its methylation fraction tends towards 50% (Experimental Procedures). Indeed, over all markers associated with a change in methylation fraction in the sample cohort, 70% tended towards a methylation fraction of 50% (Figure 6A, Binomial P ~ 0, Table S2). Consequently, we observed a highly significant increase in methylome entropy over the sample cohort (R = 0.21, P < 10−7). Furthermore, extreme methylome entropy for an individual was highly correlated with accelerated aging rate based on AMAR (R = 0.49, P ~ 0, Figure 6B).
Figure 6. Methylome-wide trends with age.
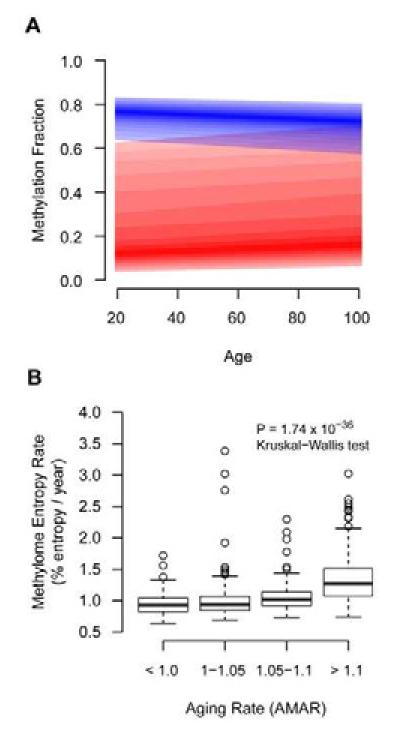
(A) Aggregate regression lines for all methylation markers that increased with age (red) and decreased with age (blue). The darkest color represents the median regression line and the bounds represent the 25% and 75% quantile. Both increasing and decreasing markers trend toward moderate methylation fraction values. (B) An entropy aging rate was calculated as the mean Shannon entropy of age-associated methylation markers divided by chronological age. This was strongly associated with AMAR.
Aging rates and the transcriptome
As changes in methylation have been directly linked to changes in gene expression (Sun et al., 2011), we were interested in whether these changes in the aging methylome were mirrored on a functional level in the human transcriptome and reflected differences in aging rates. For this purpose, we obtained and analyzed publicly-available gene expression profiles from the whole blood of 488 individuals spanning an age range of 20 to 75 (Emilsson et al., 2008). We found strong evidence for genes whose expression associates with age (326 genes, FDR < 0.05) and for genes with increasing expression deviance (Binomial P < 10−276, Experimental Procedures). Strikingly, we found that genes with age-associated expression profiles were more likely to have nearby age-associated methylation markers in our data (P < 0.01, Table S6). We used this information to build a model of aging based on the expression of genes that were associated with age in the methylome (Figure 7A, Table S7, Experimental Procedures). This model demonstrated a clear ability to measure aging rate using expression data, reproducing our finding of increased aging rates for men as compared to women (Figure 7B, 11% difference, P < 10−4). The gender effect was not present in a model built using all available genes rather than those associated with age-related changes in the methylome (P > 0.05). Thus, age-associated changes to the methylome are indicative of functional changes in gene expression patterns.
Figure 7. Transcription aging model.
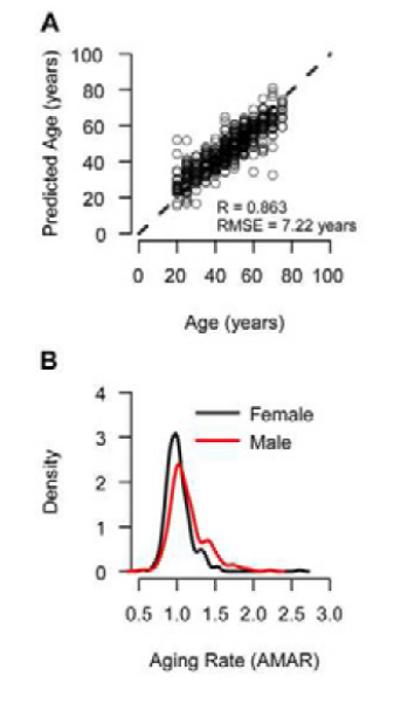
(A) We built an aging model using mRNA expression data for genes which showed an aging trend in the methylome. It’s standard error (RMSE = 7.22 years) is increased due to the rounding of ages to the nearest five-year interval in the dataset. (B) Similar to the methylome, the transcriptome shows an increased aging rate for men as compared to women (P < 10−4).
Conclusions
In this study we have shown that genome-wide methylation patterns represent a strong and reproducible biomarker of biological aging rate. These patterns enable a quantitative model of the aging methylome which demonstrates high accuracy and an ability to discriminate relevant factors in aging, including gender and genetic variants. Moreover, our ability to apply this model in multiple tissues suggests the possibility of a common molecular clock, regulated in part by changes in the methylome. It remains to be seen whether these changes occur on an intracellular level uniformly across a population of cells, or reflect consistent changes in tissue composition over time.
The ability to predict age from whole blood may permit a wider analysis in longitudinal studies such as the Framingham Study, the Women’s Health Initiative, blood samples collected on neonatal Guthrie cards and other longitudinal studies with rich annotation of biometric and disease traits. Aging trends could emerge from such studies with many potential practical implications, from health assessment and prevention of disease to forensic analysis. Similar to the effect of gender in this study, the identification of additional biometric or environmental factors that influence AMAR, such as smoking, alcohol consumption, or diet, will permit quantitative assessments of their impacts on health and longevity. A useful example would be to periodically assess the rate of aging of an individual using AMAR and determine if diet or environmental factors can accelerate or retard the aging process and diseases such as age related macular degeneration. As models of human aging improve, it is conceivable that biological age, as measured from molecular profiles, might one day supersede chronological age in the clinical evaluation and treatment of patients.
Experimental Procedures
Sample collection and test procedures
This study was approved by the institutional review boards of the University of California, San Diego, the University of Southern California and West China Hospital. All participants signed informed consent statements prior to participation. Blood was drawn from a vein in the patient’s arm into blood collection tubes containing the anticoagulant acid citrate dextrose. Genomic DNA was extracted from the whole blood using a Qiagen FlexiGene DNA Kit and stored at −20 degrees Celsius. Methylation fraction values for the autosomal chromosomes were measured using the Illumina Infinium HumanMethylation450 BeadChip (Bibikova et al., 2011). This procedure uses bisulfate-treated DNA and two site-specific probes for each marker, which bind to the associated methylated and un-methylated sequences. The intensity of the methylated probe relative to the total probe intensity for each site represents the fractional level of methylation at that site in the sample. These values were adjusted for internal controls using Illumina’s Genome Studio software. Methylation fraction values with a detection p-value greater than 0.01 were set to ‘missing’. One patient sample and 830 markers were removed as they had greater than 5% missing values. The remaining missing values were imputed with the KNN approach (10 nearest markers) using the R “impute” package (Troyanskaya et al., 2001). We performed exome sequencing on 258 of these samples, using a solution hybrid selection method to capture DNA followed by parallel sequencing on an Illumina HiSeq platform. Genotype calls were made using the SOAP program (Li et al., 2008). Calls with a quality score less than twenty were set as missing. Only variants which had fewer than 10% missing calls, were within Hardy-Weinberg equilibrium (P <= 10−4), and of a common frequency (> 5%) were retained (10,694). Individuals with less than 20% missing calls (252) were retained. Additional genotyping was done with multiplex PCR followed by MALDI-TOF mass spectrometry analysis using the iPLEX/MassARRAY/Typer platform.
Methylation quality control
We used principal component (PC) analysis to identify and remove outlier samples. We converted each sample into a z-score statistic, based on the squared distance of its 1st PC from the population mean. The z-statistic was converted to a false-discovery rate using the Gaussian cumulative distribution and the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). Samples falling below an FDR of 0.2 were designated at outliers and removed. This filtering procedure was performed iteratively until no samples were determined to be an outlier. A total of 24 samples were removed in this manner.
Association testing
Association tests for trends in methylation fraction and deviance were performed using nested linear models and the F-test. As methylation levels may be sensitive to a number of factors, we included several covariates, including gender, BMI, diabetes status, ethnicity, and batch. Tests for whole-methylome changes in deviance were computed using the binomial test, based on the number of markers with a positive rather than negative coefficient. Markers were annotated as having support from the TCGA data if the coefficient of aging was the same sign and the significance was better than P < 0.05.
Annotation enrichment
Methylation marker annotations for CpG islands and GO terms were obtained from the IlluminaHumanMethylation450k.db database from Bioconductor (Gentleman et al., 2004). Annotation enrichment tests were performed using the two-sided Fisher’s exact test.
Aging model
The diagnostic model of age was made using a multivariate linear model approach based on the Elastic Net algorithm implemented in the R package ‘glmnet’ (Jerome Friedman, Trevor Hastie, 2010). This approach is a combination of traditional Lasso and ridge regression methods, emphasizing model sparsity while appropriately balancing the contributions of correlated variables. It is ideal for building linear models in situations where the number of variables (markers) greatly outweighs the number of samples. Optimal regularization parameters were estimated using ten-fold cross-validation. We employed bootstrap analysis, sampling the dataset with replacement 500 times and building a model for each bootstrap cohort. We included in the final model only markers that were present in more than half of all bootstraps. The covariates gender, BMI, diabetes status, ethnicity, and batch were included in the model and were exempted from penalization (regularization). P-values are based on a least-squares model built using the same terms and drop-one F-tests. As BMI was strongly associated with age, the term was first adjusted for age before computing significance in the model. AMAR was computed using the aging model, but without the variables gender, BMI, and diabetes status. The coefficients were not changed. AMAR was then taken as an individual’s predicted age divided by her or his actual age.
Genetic variant associations
Each genetic variant was tested for association in an additive model with the top aging associated methylation markers using nested linear models and the F-test. We included covariates for gender, BMI, diabetes status, ethnicity, and batch. Variant positions were based on the human reference build GRCh37 and gene annotations were based on chromosomal proximity within 20kbp.
Computing methylation deviance
Methylation deviance was computed using the following approach: First, we removed the methylation trends due to all given variables, including age, gender, and BMI by fitting a linear model for each marker and acting only on the residuals. Next, we identified and removed highly non-normal markers based on the Shapiro-Wilk test (P < 10−5). To allow for naturally occurring extreme deviations in the normality test, we first estimated the outliers of each marker based on a Grubb’s statistic, choosing the inclusion threshold based on the Benjamini-Hochberg FDR (Benjamini and Hochberg, 1995). If any samples had an FDR less than 0.4, we ignored them and repeated the outlier detection until no outliers were detected. Finally, the deviance of each remaining marker was computed as the square of its adjusted methylation value.
Entropy analysis
Entropy statistics were computed on methylation data adjusted for covariates and filtered for normality (see computing methylation deviance). We computed the normalized Shannon entropy (Shannon and Weaver, 1963) of an individual’s methylome according to the formula:
where MFi is the methylation fraction of the ith methylation marker and N is the number of markers.
Mapping CpG islands
Genomic positions and marker annotations for 27,176 CpG islands were obtained from the IlluminaHumanMethylation450k.db database from Bioconductor (Gentleman et al., 2004). We obtained the positions for markers within each island with at least four markers (25,028), as well as the nearest 100 markers upstream and downstream. These positions were then combined with the marker value of interest (i.e. methylation fraction, aging coefficient, deviance) to produce a genomic map for each island and the surrounding region. After normalizing each map to the center of the island, we averaged the values at each relative genomic point across all islands to produce a common map.
Supplementary Material
Highlights.
Human aging rates can be quantified from the methylome
Aging rates are affected by both gender and genetics
Different aging rates account for part of epigenetic drift
Methylome aging rates correspond with patterns of transcription
Acknowledgements
We thank Janusz Dutkowski, Kumar Sharma and Mariano Alvarez for critical discussions and Daniel O’Conner for reviewing the manuscript. L.Zhao, L. Z and K.Z. were supported by grants from National Basic Research Program of China (973 Program, 2013CB967504), NSFC (Grant 81130017), NEI/NIH grants EY014428, EY018660, EY019270, EY021374, VA Merit Award, the Research to Prevent Blindness, and the Burroughs Wellcome Fund Clinical Scientist Award in Translational Research. G.Ha., M.C., and T.I. are supported by NIH grants P50GM085764 and R01E5014811. This work is a product of the Sage Federation, a consortium of research labs whose goal is to encourage greater openness and sharing of biomedical data and analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers The complete methylation profiles have been deposited in NCBI’s Gene Expression Omnibus under accession number GSE40279.
Supplemental Information Supplemental information includes 7 tables and 5 figures and can be found with this article online at ###.
Author Contributions L.Zhao, L.Zhang, G.Hu., S.S, and Y.G. collected and processed samples with guidance from K.Z.; B.K., M.B. and J.F. performed the methylation assays; L.Zhao, L.Zhang and K. Z. performed exome sequencing and genotyping; G.Ha. and J.G. performed the principal statistical analyses with guidance from T.I., R.D., M.C., and S.F. I.R. discussed the entropy metric. G.Ha., J.G., T.I., Y.G., and K.Z. wrote the manuscript.
Author Information B.K, M.B, and J.F. work for Illumina Inc.
REFERENCES
- Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. [Accessed May 17, 2012];Age-associated DNA methylation in pediatric populations. Genome Research. 2012 22:623–632. doi: 10.1101/gr.125187.111. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22300631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzmon G, Rincon M, Schechter CB, Shuldiner AR, Lipton RB, Bergman A, Barzilai N. [Accessed January 18, 2012];Lipoprotein Genotype and Conserved Pathway for Exceptional Longevity in Humans. PLoS Biol. 2006 4:e113. doi: 10.1371/journal.pbio.0040113. Available at: UR http://dx.doi.org/10.1371/journal.pbio.0040113,http://dx.doi.org/10.1371/journal.pbio.00401 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres R, Zierath JR. [Accessed January 18, 2012];DNA methylation in metabolic disorders. The American Journal of Clinical Nutrition. 2011 doi: 10.3945/ajcn.110.001933. Available at: http://www.ajcn.org/content/early/2011/02/02/ajcn.110.001933.abstract. [DOI] [PubMed] [Google Scholar]
- Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK. [Accessed January 31, 2012];DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biology. 2011 12:R10. doi: 10.1186/gb-2011-12-1-r10. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21251332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JT, Tsai P-C, Yang T-P, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al. [Accessed May 17, 2012];Epigenome-Wide Scans Identify Differentially Methylated Regions for Age and Age-Related Phenotypes in a Healthy Ageing Population. PLoS Genet. 2012 8:e1002629. doi: 10.1371/journal.pgen.1002629. Available at: http://dx.doi.org/10.1371/journal.pgen.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. [Accessed November 3, 2011];MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proceedings of the National Academy of Sciences of the United States of America. 1999 96:3969–3974. doi: 10.1073/pnas.96.7.3969. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=22404&tool=pmcentrez&rendert ype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. [Accessed January 18, 2012];Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995 57:289–300. Available at: http://www.jstor.org/stable/2346101. [Google Scholar]
- Bertoni C, Rustagi A, Rando TA. [Accessed July 8, 2011];Enhanced gene repair mediated by methyl-CpG-modified single-stranded oligonucleotides. Nucleic acids research. 2009 37:7468–7482. doi: 10.1093/nar/gkp757. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2794159&tool=pmcentrez&rend ertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. [Accessed March 1, 2012];High density DNA methylation array with single CpG site resolution. Genomics. 2011 98:288–295. doi: 10.1016/j.ygeno.2011.07.007. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21839163. [DOI] [PubMed] [Google Scholar]
- Blair SN, Kohl HW, 3rd, Paffenbarger RSJ, Clark DG, Cooper KH, Gibbons LW. [Accessed January 18, 2012];Physical fitness and all-cause mortality. A prospective study of healthy men and women. JAMA: The Journal of the American Medical Association. 1989 262:2395–2401. doi: 10.1001/jama.262.17.2395. Available at: http://www.ncbi.nlm.nih.gov/pubmed/2795824. [DOI] [PubMed] [Google Scholar]
- Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E. [Accessed March 3, 2012];Epigenetic Predictor of Age. PLoS ONE. 2011 6:e14821. doi: 10.1371/journal.pone.0014821. Available at: http://dx.plos.org/10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, Kahn RS, Ophoff RA. [Accessed December 6, 2011];The Relationship of DNA Methylation with Age, Gender and Genotype in Twins and Healthy Controls. PLoS ONE. 2009 4:e6767. doi: 10.1371/journal.pone.0006767. Available at: http://dx.doi.org/10.1371/journal.pone.0006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A. Decline in Genomic DNA Methylation through Aging in a Cohort of Elderly Subjects. Mechanisms of ageing and development. 2009;130:234–239. doi: 10.1016/j.mad.2008.12.003. Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2956267/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breusch TS, Pagan AR. [Accessed March 3, 2012];A Simple Test for Heteroscedasticity and Random Coefficient Variation. Econometrica. 1979 47:1287. Available at: http://www.jstor.org/stable/1911963?origin=crossref. [Google Scholar]
- Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al. [Accessed December 6, 2011];Aging and Environmental Exposures Alter Tissue-Specific DNA Methylation Dependent upon CpG Island Context. PLoS Genet. 2009 5:e1000602. doi: 10.1371/journal.pgen.1000602. Available at: http://dx.doi.org/10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Barker AD. [Accessed May 16, 2012];Mapping the cancer genome. Pinpointing the genes involved in cancer will help chart a new course across the complex landscape of human malignancies. Scientific American. 2007 296:50–57. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17348159. [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. [Accessed January 27, 2010];Genetics of gene expression and its effect on disease. Nature. 2008 452:423–428. doi: 10.1038/nature06758. Available at: http://dx.doi.org/10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. [Accessed December 6, 2011];Accelerated telomere shortening in response to life stress. Proceedings of the National Academy of Sciences of the United States of America. 2004 101:17312–17315. doi: 10.1073/pnas.0407162101. Available at: http://www.pnas.org/content/101/49/17312.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in Cancer. The New England Journal of Medicine. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. Available at: http://www.nejm.org/doi/full/10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Irizarry RA, Fradin D, Aryee MJ, Murakami P, Aspelund T, Eiriksdottir G, Harris TB, Launer L, Gudnason V, et al. [Accessed May 17, 2012];Personalized Epigenomic Signatures That Are Stable Over Time and Covary with Body Mass Index. Science Translational Medicine. 2010 2:49ra67–49ra67. doi: 10.1126/scitranslmed.3001262. Available at: http://stm.sciencemag.org/content/2/49/49ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, et al. [Accessed January 18, 2012];Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America. 2005 102:10604–10609. doi: 10.1073/pnas.0500398102. Available at: http://www.pnas.org/content/102/30/10604.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Esteller M. [Accessed February 10, 2012];Epigenetics and aging: the targets and the marks. Trends in Genetics: TIG. 2007 23:413–418. doi: 10.1016/j.tig.2007.05.008. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17559965. [DOI] [PubMed] [Google Scholar]
- Fraser HB, Khaitovich P, Plotkin JB, Pääbo S, Eisen MB. [Accessed January 18, 2012];Aging and Gene Expression in the Primate Brain. PLoS Biol. 2005 3:e274. doi: 10.1371/journal.pbio.0030274. Available at: UR http://dx.doi.org/10.1371/journal.pbio.0030274,http://dx.doi.org/10.1371/journal.pbio.00302 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. [Accessed January 20, 2012];Bioconductor: open software development for computational biology and bioinformatics. Genome Biology. 2004 5:R80. doi: 10.1186/gb-2004-5-10-r80. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15461798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. [Accessed December 6, 2011];Telomeres shorten during ageing of human fibroblasts. Nature. 1990 345:458–460. doi: 10.1038/345458a0. Available at: http://dx.doi.org/10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Hartwig A, Schlepegrell R. [Accessed December 7, 2011];Induction of oxidative DNA damage by ferric iron in mammalian cells. Carcinogenesis. 1995 16:3009–3013. doi: 10.1093/carcin/16.12.3009. Available at: http://carcin.oxfordjournals.org/cgi/content/abstract/16/12/3009. [DOI] [PubMed] [Google Scholar]
- Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, et al. [Accessed August 12, 2012];Distinct DNA methylomes of newborns and centenarians. Proceedings of the National Academy of Sciences. 2012 109:10522–10527. doi: 10.1073/pnas.1120658109. Available at: http://www.pnas.org/content/109/26/10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome Friedman, Trevor Hastie RT. Regularization Paths for Generalized Linear Models via Coordinate Descent. Journal of Statistical Software. 2010;33 [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Laird PW. [Accessed February 29, 2012];Cancer epigenetics comes of age. Nature Genetics. 1999 21:163–167. doi: 10.1038/5947. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9988266. [DOI] [PubMed] [Google Scholar]
- Karthikeyan G, Lewis LK, Resnick MA. [Accessed December 7, 2011];The mitochondrial protein frataxin prevents nuclear damage. Human molecular genetics. 2002 11:1351–1362. doi: 10.1093/hmg/11.11.1351. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12019217. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Kim M-K, Jin X-J, Oh J-H, Kim JE, Chung JH. [Accessed August 12, 2012];Skin Aging and Photoaging Alter Fatty Acids Composition, Including 11,14,17-eicosatrienoic Acid, in the Epidermis of Human Skin. Journal of Korean Medical Science. 2010 25:980–983. doi: 10.3346/jkms.2010.25.6.980. Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2877242/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Baer DJ, Rumpler WV, Weindruch R, Ingram DK, Tilmont EM, Cutler RG, Roth GS. [Accessed January 18, 2012];Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proceedings of the National Academy of Sciences. 1996 93:4159–4164. doi: 10.1073/pnas.93.9.4159. Available at: http://www.pnas.org/content/93/9/4159.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Kristiansen K, Wang J. [Accessed January 19, 2012];SOAP: short oligonucleotide alignment program. Bioinformatics (Oxford, England) 2008 24:713–714. doi: 10.1093/bioinformatics/btn025. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18227114. [DOI] [PubMed] [Google Scholar]
- de Magalhães JP, Curado J, Church GM. [Accessed January 18, 2012];Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009 25:875–881. doi: 10.1093/bioinformatics/btp073. Available at: http://bioinformatics.oxfordjournals.org/content/25/7/875.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniz L, Dutt P, Haider N, Stambolic V. [Accessed May 22, 2012];Nek family of kinases in cell cycle, checkpoint control and cancer. Cell Division. 2011 6:18. doi: 10.1186/1747-1028-6-18. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22040655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OFX, Spengler D. [Accessed February 10, 2012];Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nature Neuroscience. 2009 12:1559–1566. doi: 10.1038/nn.2436. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19898468. [DOI] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, McDonald A, Fleming MD. [Accessed December 7, 2011];The Steap proteins are metalloreductases. Blood. 2006 108:1388–1394. doi: 10.1182/blood-2006-02-003681. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1785011&tool=pmcentrez&rend ertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang T-P, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. [Accessed December 6, 2011];Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Research. 2010 doi: 10.1101/gr.103101.109. Available at: http://genome.cshlp.org/content/early/2010/03/09/gr.103101.109.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Iwata N, Tsubuki S, Takaki Y, Takano J, Huang S-M, Suemoto T, Higuchi M, Saido TC. [Accessed January 18, 2012];Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation. Nature Medicine. 2005 11:434–439. doi: 10.1038/nm1206. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15778722. [DOI] [PubMed] [Google Scholar]
- Shannon CE, Weaver W. The Mathematical Theory of Communication. University of Illinois Press; Champaign, IL: 1963. [Google Scholar]
- Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A, Thorsteindottir U, Shin S-Y, Richards HB, Soranzo N, et al. [Accessed July 19, 2011];Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nature genetics. 2011 43:561–564. doi: 10.1038/ng.833. Available at: http://dx.doi.org/10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven N. A. [Accessed January 18, 2012];Why women live longer than men: Sex differences in longevity. Gender Medicine. 2006 3:79–92. doi: 10.1016/s1550-8579(06)80198-1. Available at: http://www.sciencedirect.com/science/article/pii/S1550857906801981. [DOI] [PubMed] [Google Scholar]
- Suh Y, Atzmon G, Cho M-O, Hwang D, Liu B, Leahy DJ, Barzilai N, Cohen P. [Accessed January 18, 2012];Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proceedings of the National Academy of Sciences. 2008 105:3438–3442. doi: 10.1073/pnas.0705467105. Available at: http://www.pnas.org/content/105/9/3438.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Asmann YW, Kalari KR, Bot B, Eckel-Passow JE, Baker TR, Carr JM, Khrebtukova I, Luo S, Zhang L, et al. Integrated Analysis of Gene Expression, CpG Island Methylation, and Gene Copy Number in Breast Cancer Cells by Deep Sequencing. 2011 doi: 10.1371/journal.pone.0017490. Available at: http://www.plosone.org/article/fetchObjectAttachment.action?uri=info:doi/10.1371/journal.p one.0017490&representation=PDF. [DOI] [PMC free article] [PubMed]
- Tatar M, Bartke A, Antebi A. [Accessed October 12, 2011];The endocrine regulation of aging by insulin-like signals. Science (New York, N.Y.) 2003 299:1346–1351. doi: 10.1126/science.1081447. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12610294. [DOI] [PubMed] [Google Scholar]
- Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, Altman RB. [Accessed January 20, 2012];Missing value estimation methods for DNA microarrays. Bioinformatics (Oxford, England) 2001 17:520–525. doi: 10.1093/bioinformatics/17.6.520. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11395428. [DOI] [PubMed] [Google Scholar]
- Valdes A, Andrew T, Gardner J, Kimura M, Oelsner E, Cherkas L, Aviv A, Spector T. Obesity, cigarette smoking, and telomere length in women. The Lancet. 366:662–664. doi: 10.1016/S0140-6736(05)66630-5. Available at: http://www.sciencedirect.com/science/article/pii/S0140673605666305. [DOI] [PubMed] [Google Scholar]
- Vijg J, Campisi J. [Accessed December 6, 2011];Puzzles, promises and a cure for ageing. Nature. 2008 454:1065–1071. doi: 10.1038/nature07216. Available at: http://dx.doi.org/10.1038/nature07216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler HE, Metter EJ, Tanaka T, Absher D, Higgins J, Zahn JM, Wilhelmy J, Davis RW, Singleton A, Myers RM, et al. [Accessed April 6, 2011];Sequential Use of Transcriptional Profiling, Expression Quantitative Trait Mapping, and Gene Association Implicates MMP20 in Human Kidney Aging. PLoS Genet. 2009 5:e1000685. doi: 10.1371/journal.pgen.1000685. Available at: http://dx.doi.org/10.1371/journal.pgen.1000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD. FOXO3A genotype is strongly associated with human longevity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2544566/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M, Dong H, Siu H, Peng G, Wang Y, Jin L. Genome-Wide Association Studies of Copy Number Variation in Glioblastoma. 2010:1–4. Available at: http://ieeexplore.ieee.org/ielx5/5513048/5514659/05516437.pdf?tp=&arnumber=5516437&isnumber=5514659.
- Yacubova E, Komuro H. [Accessed January 18, 2012];Stage-specific control of neuronal migration by somatostatin. Nature. 2002 415:77–81. doi: 10.1038/415077a. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11780120. [DOI] [PubMed] [Google Scholar]
- Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, et al. [Accessed January 18, 2012];AGEMAP: A Gene Expression Database for Aging in Mice. PLoS Genet. 2007 3:e201. doi: 10.1371/journal.pgen.0030201. Available at: UR http://dx.plos.org/10.1371/journal.pgen.0030201,http://dx.plos.org/10.1371/journal.pgen.00 30201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Hastie T. [Accessed January 18, 2012];Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2005 67:301–320. Available at: http://onlinelibrary.wiley.com/doi/10.1111/j.1467-9868.2005.00503.x/abstract. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.