

Abstract
Microglia-neuron interactions play a crucial role in several neurological disorders characterized by altered neural network excitability, such as epilepsy and neuropathic pain. While a series of potential messengers have been postulated as substrates of the communication between microglia and neurons, including cytokines, purines, prostaglandins, and nitric oxide, the specific links between messengers, microglia, neuronal networks, and diseases have remained elusive. Brain-derived neurotrophic factor (BDNF) released by microglia emerges as an exception in this riddle. Here, we review the current knowledge on the role played by microglial BDNF in controlling neuronal excitability by causing disinhibition. The efforts made by different laboratories during the last decade have collectively provided a robust mechanistic paradigm which elucidates the mechanisms involved in the synthesis and release of BDNF from microglia, the downstream TrkB-mediated signals in neurons, and the biophysical mechanism by which disinhibition occurs, via the downregulation of the K+-Cl− cotransporter KCC2, dysrupting Cl−homeostasis, and hence the strength of GABAA- and glycine receptor-mediated inhibition. The resulting altered network activity appears to explain several features of the associated pathologies. Targeting the molecular players involved in this canonical signaling pathway may lead to novel therapeutic approach for ameliorating a wide array of neural dysfunctions.
1. Introduction
Once simply considered as the “guardians” of the central nervous system (CNS), microglia have more recently emerged as key players in regulating neuronal network excitability. Indeed, physical and chemical alterations in the extracellular environment promote the synthesis and release of several microglia-derived molecules which, in turn, shape neuronal circuit function. The effects of such microglia-neuron interactions were found to be critical in the course of different central disorders, and in particular seminal studies provided significant evidence for a role of microglia in the pathogenesis of seizures (for review see [1, 2]) which was associated with increased glutamatergic transmission through the potentiation of NMDA receptor-mediated activity [3]. However, from a theoretical point of view, raising network excitability can be equally achieved through increasing excitatory inputs or removing inhibitory ones. In fact, unmasking silent interconnections can be better achieved through disinhibition than enhanced excitation. Furthermore, disinhibition has been shown as an upstream substrate of activity-dependent enhancement of excitation in several plasticity paradigms [4–7]. Thus, in addition to the glutamatergic hypothesis, it can be postulated that microglia alter neuronal excitability by affecting synaptic inhibition. This hypothesis has been explored during the last decade and the results of several investigations have uncovered molecular mechanisms underlying microglia-mediated disinhibition.
Synaptic inhibition in central neurons is mediated by γ-amino-butyric acid (GABA) and glycine (Gly) which activate ionic channels (GABAAR and GlyR) permeable to anions, namely, chloride (Cl−) and bicarbonate (HCO3 −). Under physiological conditions, Cl− flows inwardly and HCO3 − outwardly, along with their electrochemical gradient. Cl− contribution is by far the more conspicuous, and, consequently, the reversal potential of GABA/glycine (EGABA/Egly) in adult neurons is set below the resting potential (Vr) near the Cl− equilibrium (ECl). It follows that when GABAAR/GlyR are activated, Cl− produces a net hyperpolarization. Although in principle correct, this brief summary of the ionic mechanisms of synaptic inhibition provides a quite static representation of GABA/Gly-mediated transmission and importantly does not take into account that, EGABA/EGly and Vr being only few millivolts apart, even a small change in anion concentrations may have a profound functional impact [8, 9]. In this respect, the critical variable is represented by the intracellular Cl− concentration and the critical property is the capacity of the cell to maintain this concentration low. In the event that intracellular Cl− rises, it follows that (i) EGABA/EGly shifts toward or beyond Vr; (ii) the Cl− gradient across the membrane collapses; and (iii) the previously negligible depolarizing HCO3 − current becomes more relevant. On the whole, an increase in the intracellular Cl− concentration weakens the strength of GABA/Gly-mediated inhibition or, in the extreme case, turns it into paradoxical excitation [10].
How do neurons control intracellular Cl− concentration? Chloride homeostasis in cells is maintained by a group of membrane carriers known as cation-chloride cotransporters (CCCs [11, 12]). The K+-Cl− cotransporter 2, KCC2, is the main CCC isoform expressed in central neurons [13, 14]. KCC2 extrudes Cl− following the K+ gradient, and its activity typically maintains a low intracellular Cl− concentration, which is the prerequisite for an effective GABA/Gly-mediated inhibition. Now, KCC2 activity is not static, but it can be profoundly modulated by different physiological or pathological challenges. The most spectacular example of such plasticity has been extensively described during development [14–16]. KCC2 is little expressed in prenatal and early postnatal brains, but during maturation it undergoes a developmental increase, which parallels the switch in GABA/Gly-mediated transmission from excitatory to inhibitory [14]. These mechanisms are thought not only to play a pivotal role in the activity-dependent development of central synapses during CNS maturation [17] but also to favor a proper wiring by triggering spontaneous rhythmic activity in motor networks [18] and to promote synaptic integration of new born neurons in those area of the brain in which adult neurogenesis occurs [19, 20]. On the other hand, reduction of KCC2 activity has been associated with several neurological diseases and conditions, originally epilepsy and neuropathic pain [10], and more recently motor spasticity [21], stress [22], and schizophrenia [23, 24]. Several lines of evidence accumulated during the last decade have indeed demonstrated that the increase in excitability in these pathological conditions can be largely explained by a loss of inhibition, and KCC2 has been recognized as a key molecular target underlying this loss [25, 26].
The findings in recent years that KCC2 can be dynamically modulated by several intercellular signaling pathways have been particularly interesting [4], the most prevalent being brain-derived neurotrophic factor (BDNF) signaling onto neuronal TrkB receptors [27–31]. Even more intriguing is the finding that, in certain conditions, BDNF in the CNS is not only released by neurons but also by microglia [32]. In this review we summarize and discuss the more relevant findings supporting the role of microglia in conditioning KCC2 function, as well as consequently inhibitory neurotransmission, through the release of BDNF. Several convergent findings uncover a canonical signaling mechanism by which the immune system can control neuronal network excitability by regulating the strength of inhibition.
2. Role of BDNF in the Control of KCC2 Function
BDNF is a neurotrophin with important functions in neuronal survival and differentiation. However, beyond its classical neurotrophic role, BDNF is directly involved in the control of neuronal activity and synaptic plasticity as a neuromodulator [33–35]. These functions are described in several areas of the CNS, such as hippocampus [36], cortex [37], amygdala [38], cerebellum [39], and spinal cord [34], and are involved in different forms of plasticity [40, 41]. Although initial studies mainly focused on glutamatergic synapses, the effects of BDNF on GABAergic transmission have lately received increasing attention [40]. Interestingly, early work on these effects performed in the rat hippocampus yielded a number of conflicting results, unveiling a more complex picture than expected. Indeed, in juvenile rodents BDNF was found to favor a substantial depression of GABAergic transmission via either pre- or postsynaptic mechanisms [42, 43]; conversely, studies in immature neurons showed an overall potentiating effect [44, 45]. To explain such a discrepancy, it was hypothesized that the effect of BDNF onto GABAergic transmission in hippocampal neurons might be developmentally regulated in parallel with the switch in GABAergic transmission from excitatory to inhibitory [46]. Thus, BDNF depresses GABAergic transmission in mature neurons when GABA is inhibitory and potentiates it in immature neurons when GABA is depolarizing, favoring activity-dependent synapse formation which has been relayed to GABA-mediated Ca2+ entry in developing neurons [37, 44, 46]. The fact that the changes in BDNF effects on GABA-mediated transmission are coincident with the developmental switch in GABAergic current polarity raised the question of whether BDNF has an effect on KCC2 function and/or expression. This was indeed demonstrated by Rivera and colleagues [29, 30]. The authors provided evidence that, in hippocampal slices, BDNF rapidly downregulates KCC2 expression through the BDNF preferred receptor TrkB (tyrosine kinase B receptor), thus reducing neuronal Cl− extrusion capacity [29]. The effect required the activation of two downstream cascades involving src homology 2 domain containing transforming protein/FGF receptor substrate 2 (Shc/FRS-2) and phospholipase C γ- (PLCγ-) cAMP response element-binding protein signaling, respectively [30]. Interestingly, the activation of the Shc pathway alone was surprisingly found to promote the upregulation of KCC2, which might elegantly explain the opposite effects of BDNF across brain development based on the specific intracellular pathways involved [30]. One important point of this study is that the membrane level of KCC2 undergoes a fast turnover rate, and this turnover is accelerated by exogenous BDNF or by an increased neuronal activity during which BDNF is released [30]. A logical consequence is that such a fast regulation of KCC2 activity, which happens in few hours or less, is not compatible with the physiological time course required for altering gene expression, and a number of alternative mechanistic models have been proposed including protein phosphorylation, trafficking, and quaternary structure [47]. In particular, KCC2 activity and membrane localization seem to depend on the tyrosine phosphorylation level, and BDNF has been shown to promote KCC2 dephosphorylation, which in turn reduces surface protein expression [48]. Thus, KCC2 phosphorylation influences protein trafficking by either increasing endocytosis or reducing insertion [48]. Alternatively, KCC2 transport activity has been directly correlated with the capacity of the protein to form oligomers at the membrane level [49]. Thus, Cl− extrusion capacity is improved if KCC2 is organized in oligomers, and an increased oligomers/monomers ratio parallels the KCC2 upregulation during development [49]. Interestingly, KCC2 clustering is strongly reduced in the presence of a point mutation on the KCC2 tyrosine phosphorylation site, suggesting that phosphorylation and oligomerization might simply be different parts of the same process controlling the transporter activity [50]. Finally, KCC2 activity can also be rapidly affected by the activation of the Ca2+-dependent protease calpain [11], and these pathways may be under the control of BDNF/TrkB signaling [51].
Altogether, these findings provided clear evidence that Cl− homeostasis can be rapidly regulated by an extracellular signal, such as BDNF, thus inducing short- or long-term changes in neuronal activity that cannot be simply explained in terms of classical synaptic plasticity but rather as a novel form of “ionic plasticity” [16].
After the initial studies on the effects of BDNF on Cl− homeostasis in CA1 pyramidal neuron of the hippocampus [29, 30], similar mechanisms were subsequently observed in different regions across the CNS, including the spinal dorsal horn [27] and ventral horn [21], the ventral tegmental area [52], the cortex [53, 54], and the cerebellum [39]. These findings attracted attention to the fact that BDNF may play a pivotal role as a regulator of neuronal Cl− homeostasis in the brain and, by ricochet, of inhibition and hence neuronal network excitability.
3. Microglia Are a Central Source of BDNF
The expression of BDNF in synaptic vesicles and its synaptic release from different neuronal populations [34, 55] support the role of the neurotrophin in activity-dependent downregulation of KCC2 [30]. However, BDNF is not only expressed by neurons but is also found in astrocytes [56] and microglia [32]. Microglial BDNF was first shown in microglia cultures [57, 58] and soon confirmed in different regions of the CNS during the course of various neurological disorders, such as viral encephalitis [59], traumatic injury [60, 61], ischemia [62], multiple sclerosis [63], Parkinson's disease [64], neuropathic pain [27], and spasticity [21]. That microglia are a potential source of BDNF is a crucial point to predict the role of the neurotrophin in neurological disorders. Indeed, microglia primary function is to sense and react to alterations of the extracellular milieu with a protective and defensive role. In the presence of factors signaling potentially harmful, microglia undergo morphological and functional alterations collectively identified under the term of “microglia activation,” and, depending on the signaling pathways involved, this process may lead to secretion of specific messengers, including BDNF [65]. Once released, the neurotrophin in turn sculpts neuronal circuit excitability via the signaling cascade described above.
The synthesis and release of BDNF in microglia appear to be tightly associated with the purinergic receptor P2X4R [65–67]. Purinergic receptors are endogenously activated by ATP (Adenosine-5′-triphosphate), which is typically stored in the cytoplasm of neuronal and nonneuronal cells and released in the extracellular space following tissue damage [68]. Alternatively, ATP may be released by neurons [69] or astrocytes [70]. Microglia sense extracellular ATP trough different types of purinergic receptors [68], such as P2Y12Rs, which can detect tiny gradient of extracellular ATP and promote microglia migration [71, 72], or P2X7Rs, which trigger morphological changes in microglia from a resting to an activated state [73]. Microglial P2X4Rs, instead, do not appear to be involved in the morphological alterations leading to the activated phenotype, but rather their involvement is a functional consequence of microglia activation [65]. Indeed, P2X4Rs are normally expressed at negligible levels in resting microglia, and they need to be upregulated to promote BDNF synthesis and release [65]. Which external factors are involved in the upregulation of P2X4R in activated microglia is still a matter of debate. Chemokines released from injured neurons, such as CCL2 and CCL21, have been regarded as potential inductors of P2X4R expression [74, 75]. In particular, CCL21 application in vivo and in vitro strongly promoted P2X4R upregulation in spinal microglia [74]. Interestingly, in both CCL21 [74] and P2X4R [65] deficient mice microglia activation is not compromised, which implies a mechanistic separation between the morphological changes and the subsequent downstream effects. Also CCL2, which instead plays an important role in microglia activation after injury [76, 77], has been suggested to participate in the P2X4R upregulation process; however, CCL2 does not seem involved in de novo expression of the protein, but rather it has been suggested to promote P2X4R trafficking from intracellular stores to the cell membrane [75]. Finally, a few nonneuronal endogenous molecules have been also identified as potential inductors of P2X4R in microglia, namely, the proinflammatory cytokines INF-γ [78], the mast cell-derived tryptase activated PAR2 [79], and fibronectin, a component of the extracellular matrix [80, 81]. At the nuclear level, the interferon regulatory factor 8 (IRF8) has been recently proposed as a key transcription factor involved in the upregulation of P2X4Rs in activated microglia [82].
Once upregulated, P2X4Rs can efficiently respond to extracellular fluctuation in ATP concentration and initiates the intracellular cascade leading to BDNF synthesis and release. Being particularly highly Ca2+ permeable, P2X4 channels cause a significant Ca2+ influx and the downstream activation of Ca2+-dependent intracellular pathways, among which the phosphorylation of p38 MAP kinase, which is directly involved in the synthesis and release of BDNF [67]. In addition Ca2+ influx through P2X4Rs is also necessary to directly facilitate the release of BDNF by acting on the vesicle-releasing machinery, which is typically an NSF-attachment protein-(SNARE-) mediated exocytosis [67]. Alternative pathways (i.e., ERK1/2) have been also suggested to promote BDNF synthesis in cultured microglia [83, 84]; however, these hypotheses need to be properly confirmed in vivo.
4. The Special Case of Neuropathic Pain
Based on the findings outlined above, the following conclusions can be drawn: (1) various extracellular signals may activate microglia and upregulate P2X4Rs; (2) P2X4R activation triggers the release of BDNF from microglia; (3) BDNF-TrKB signaling alters KCC2 function leading to a reduced Cl− extrusion capacity which dampens GABAAR/GlyR mediated inhibition. Assuming that all these events happen in sequence, one should expect that microglia, under certain functional states, influence synaptic inhibition. This is indeed the case of neuropathic pain [66]. Nociceptive transmission is normally conveyed to higher centers through spinal nociceptive pathways. In the most simple configuration, this involves peripheral neurons located in the dorsal root ganglia, which contact second-order neurons in the spinal dorsal horn, and a spinal projection neurons which transmits the information to the thalamus. In the spinal dorsal horn, pain transmission is controlled by a network of local inhibitory interneurons which assure the separation of nociceptive sensory pathways from nonnociceptive sensory pathways by releasing GABA and Gly [85]. Indeed, a spinal administration of GABAAR or GlyR antagonists induces tactile allodynia [86, 87], a clinical condition in which innocuous stimuli are perceived as painful. Tactile allodynia is a classical symptom of neuropathic pain and indicates an erroneous encoding of low threshold stimuli through the nociceptive channel. Several causal events have been postulated to promote spinal disinhibition, including presynaptic mechanisms affecting the amount of transmitter released and intracellular pathways regulating postsynaptic GABA and glycine receptor function/expression [7]. In our laboratory, we found that altered Cl− homeostasis in the superficial spinal dorsal horn appears as a key mechanism underlying neuropathic pain symptoms [88] and that this alteration results from the release of BDNF from microglia [27]. Microglia had already been implicated in the pathogenesis of neuropathic pain [89–91], and in particular the upregulation of P2X4Rs in microglia was early identified as a crucial step in the central sensitization process [92]. P2X4Rs are in fact necessary for the development of mechanical allodynia after nerve injury and are required for the release of BDNF from microglia [65, 67]. BDNF in turn binds TrkB receptors in neurons of the superficial dorsal horn, thus compromising KCC2 function and altering Cl− homeostasis [27]. Blocking the microglia-to-neuron cascade at any level reverses established allodynia in neuropathic animals by restoring spinal inhibitory GABAergic/glycinergic transmission [27, 28]. Subsequent studies have provided additional evidence that this form of spinal disinhibition happens in a different model of pathological pain, such as spinal cord injury [93], diabetes-induced neuropathy [94], and orofacial pain [95]. Moreover, we have very recently shown that the pain hypersensitivity induced by morphine (better known as morphine-induced hyperalgesia) is mediated by the same P2X4Rs-BDNF-TrkB-KCC2 cascade, thus recapitulating the sequence of events described in neuropathic pain [28]. In the latter study, we used a transgenic mouse in which BDNF expression was genetically ablated in microglia only, and we showed that, without microglial BDNF, morphine hyperalgesia does not take place. The involvement of spinal microglia in this specific form of hypersensitivity is due to the expression of opioid receptors in microglia [84] whose activation promotes P2X4Rs [28]. In turn, morphine appears to act on microglia via a nonopioid receptor-dependent pathway to enable BDNF release upon P2X4Rs activation [28].
In conclusion, ten years of investigations on the spinal mechanisms of nociceptive transmission have provided compelling evidence that neuropathic pain critically depends on microglia-to-neuron signals which alter GABA/glycine-mediated inhibition.
5. Microglia-BDNF-KCC2 Signaling in the Pathogenesis of Multiple Neurological Conditions
The microglia-to-neuron communication discovered in the dorsal horn of the spinal cord can be virtually replicated in all those regions of the CNS where functional TrkB receptors are expressed and may play a role in the development of multiple central disorders [10]. Accumulating evidence in recent years supports this hypothesis.
In the spinal motor system, a TrkB-KCC2 interaction has been described in motoneurons following spinal cord injury [21]. Here, the reduced Cl− extrusion capacity due to the downregulation of KCC2 was associated with hyperreflexia and spasticity, a clinical condition burdening a large number of patients with spinal trauma [96]. The authors did not investigated the origin of BDNF in their model; however microglia are clearly involved in the pathophysiology resulting from spinal cord injury [97], and a role for microglial P2X4Rs has also been envisaged [98], suggesting a microglial BDNF link.
In the brain, alterations in Cl− homeostasis have been shown to underlay epilepsy in animals and humans [99]. Based on experiments in vitro on hippocampal slices [29, 30], TrkB-KCC2 signaling was proposed as the molecular mechanism underlying hyperexcitability in epilepsy [30]. In this model, however, KCC2 downregulation was shown to be activity dependent, thus implying a neuronal source of BDNF whose release is directly related to the level of network excitability. On the other hand, epilepsy has multiple etiologies and might develop in different brain areas. A role for microglia can be therefore predicted in those pathological conditions which imply a neuronal damage and the subsequent reorganization of synaptic function, as in the case of a traumatic event [100]. Indeed, a TrkB-dependent downregulation of KCC2 has also been described in traumatic brain injury [101], a condition in which neuronal death and inflammation clearly promote the activation of microglia [102]. Interestingly, in animal models of traumatic brain injury, microglia were found to express P2X4Rs and phosphorylated p38 [103, 104], which is known to be the main upstream signal for BDNF synthesis and release in microglia [67].
Finally, a BDNF-mediated impairment of Cl− homeostasis has been shown to underlie the central mechanisms of opiate dependence in the ventral tegmental area (VTA) [52]. Although the main source of BDNF remains here elusive, chronic exposure to opioids is known to activate microglia and to induce the synthesis of BDNF [28, 84], which, in turn, impairs Cl− homeostasis in central neurons [28]. In addition, it has recently been described that functional modifications in microglia are involved in mechanisms of opiate dependence in the nucleus accumbens where the early exposure to morphine in young rats was shown to influence drug-seeking behavior in adulthood increasing the risk of drug-induced reinstatement [105].
Taken together, these evidence indicate that BDNF-TrkB signaling drives disinhibition by targeting KCC2 function. Such an effect does not directly depend on the source of BDNF (neuron, astrocytes, or microglia) but rather on the intracellular pathways linking TrkB to KCC2 [29]. This is exemplified in immature neurons where BDNF-TrkB signaling, rather than causing KCC2 downregulation, stimulates the synthesis of KCC2 and favors the developmental switch of GABAergic transmission from excitatory to inhibitory [106]. In contrast, microglial BDNF has gained special attention as underlying neurological diseases in adult tissue, and this is mainly due to the specific role played by microglia in the CNS. Indeed, microglia-driven disinhibition via BDNF-TrkB signaling can be regarded as a peculiar consequence of microglial reaction to injury or to certain pharmacological treatments, potentially occurring in different areas of the CNS. The “pathological” consequences of such process are usually dramatic, leading for instance to an altered nociceptive behavior or to seizure. It remains enigmatic what the normal “physiological” meaning of the release of BDNF from microglia and the subsequent downregulation of KCC2 is. The primary role of microglia is in fact to react in response to a variety of external challenges supposedly with the aim of protecting neurons. In this context, the release of BDNF can be considered as a part of a neuroprotective strategy, being neurotrophins classically involved in neuronal survival process [107]. A neuroprotective and reparative role for microglial BDNF has indeed been postulated during the course of encephalitis [59], brain ischemia [62], and traumatic injury [60]. Interestingly, the posttraumatic loss of KCC2 in mature neurons induced by BDNF and the subsequent GABA-mediated depolarization was found necessary for neuronal survival of injured neurons, a mechanism which is strongly reminiscent of the trophic effect of excitatory GABA during CNS development [101]. In contrast, the central inflammatory reaction in the spinal dorsal horn following peripheral nerve injury appears to be substantially maladaptive and detrimental. Indeed, the activation of spinal microglia following nerve injury produces a release of BDNF onto spinal neurons which are not directly injured. The main effect of microglial activation in this case is thus the suppression of spinal inhibition and the activation of nociceptive pathways, leading to the clinical development of neuropathic pain [27]. Microglia therefore appear as an ambiguous actor, in some cases protective and in other cases having deleterious actions [108]. An accurate prediction of the balance between neuroprotection and neurotoxicity appears thus important to understand how microglia intervene in diseases to develop appropriate therapeutic strategies.
Yet, regardless of the positive or negative outcome of microglia action on neuronal survival and repair, the activation of the P2X4R-BDNF-TrkB-KCC2 cascade allows microglia to critically control network excitability and to unmask hidden neuronal circuits that are normally kept silent by the physiological Cl−-mediated inhibition (Figure 1) [109].
Figure 1.
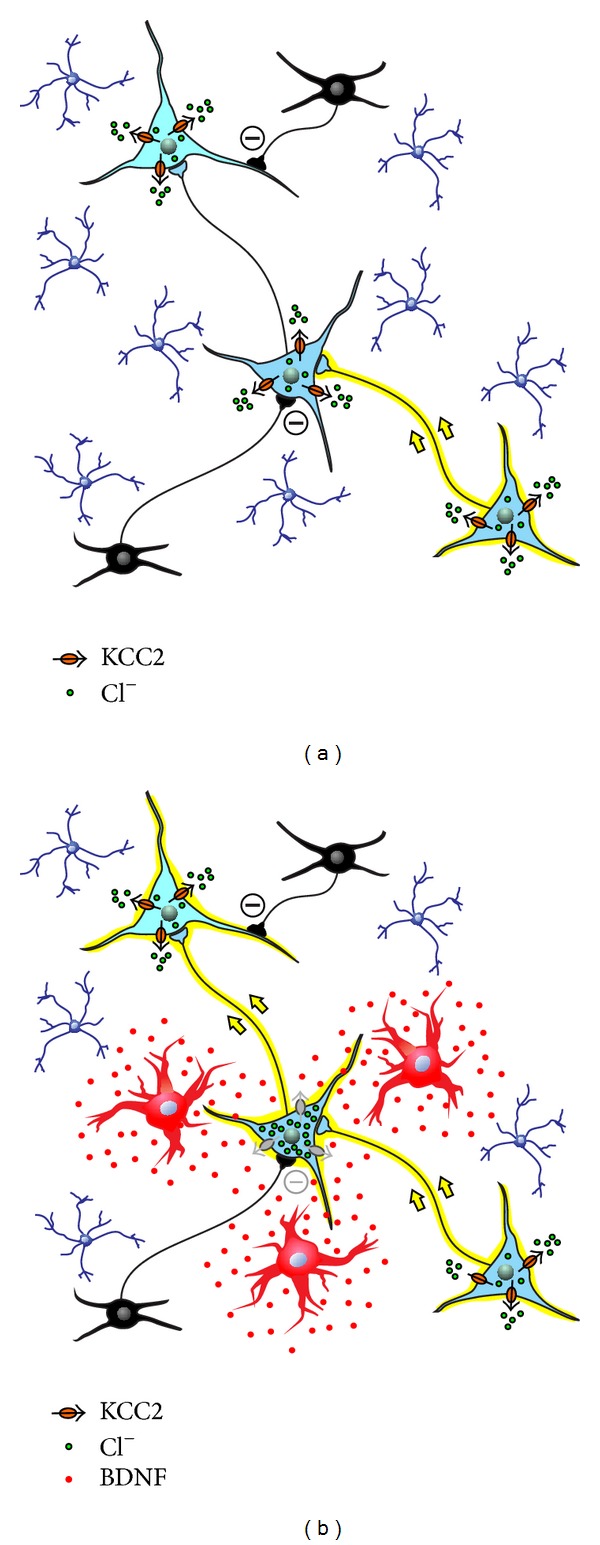
Microglia control neuronal network excitability via secretion of BDNF. The left panel illustrates a schematic neuronal network in the mature CNS under normal conditions: microglia (blue ramified cells) are in their resting state; small inhibitory interneurons release GABA or Gly to repress the flow of signals across the network; normal KCC2 activity extrudes Cl− (black arrows) to maintain the Cl− gradient, and, consequently, Cl− flows in through GABAAR/GlyR channels to inhibit activity. The right panel illustrates the same network after an external event has induced microglial activation (red cells) and the release of microglial BDNF: BDNF-TrkB signaling causes downregulation of KCC2; Cl− accumulates in neurons and the Cl− gradient collapses; GABAAR/GlyR-mediated inhibition is less effective in controlling neuronal firing, and previously silent neuronal pathways are unmasked (yellow arrows).
6. Future Directions
The signaling cascade described in this review represents a molecular substrate underlying the mechanism by which microglia target GABAergic/glycinergic neurotransmission. However, it is likely that BDNF released from microglia also challenge network excitability by mechanisms other than KCC2. In particular, BDNF-TrkB signaling also targets NMDA receptors [65, 110], and microglial BDNF has been suggested to underlie certain forms of pathological pain via the activation of spinal NMDA [111]. The outcome of both KCC2 downregulation and NMDA potentiation is an overall increase in network excitability. This raises the question of whether modulationsof KCC2 and NMDA functions are independent processes or are reciprocally connected. Several lines of evidence support the latter hypothesis [112, 113], and future investigations are encouraged to further explore such interactions in different neurological disorders. In addition to BDNF, microglia are known to directly or indirectly modulate synaptic transmission through the release of tens of other different molecules [114]. Most of past studies have differently focused on the effect of these molecules in the modulation of glutamatergic transmission. In this respect, a role has been described for cytokines [115], glycine [116], NMDA receptor agonists [117], adenosine [118], and ATP [119]. On the other hand, a growing body of studies reported that cytokines might also directly modulate GABAergic transmission [115]: interleukin 1β was found to depress GABA release in a model of autoimmune encephalitis [120] and to potentiate GABAergic transmission in CA1 [121] or in hypothalamic neurons [122]; both interleukin 6 and interleukin 1β were seen to reduce GABA- and Gly-mediated currents in the spinal dorsal horn [123]; tumor necrosis factor α was shown to promote GABAAR endocytosis in hippocampal neurons thus weakening the inhibitory synaptic strength [124]. In addition, microglia also produce lipophilic gaseous molecules, such as nitric oxide [125–127], and lipidic inflammatory mediators, such as prostaglandins [127, 128]. Interestingly, prostaglandin E2 directly suppresses glycine-mediated transmission in the spinal dorsal horn, a mechanism centrally involved in the development of inflammatory pain [129, 130]. Deeper insights into the role played by each of these messengers in normal and pathological conditions are required to improve our understanding of the role of microglia-to-neuron communication.
Yet, the effects reported in different studies for most of these microglia-derived molecules are often quite dissimilar and critically influenced by the experimental paradigms, drug concentrations, and neuronal populations considered [114]. In addition, the mechanisms leading to the release of specific molecules, as well as the molecular pathways activated in neurons, are still poorly understood, making it difficult to draw a coherent picture for their role in synaptic transmission. Conversely, the P2X4R-BDNF-TrkB-KCC2 cascade described here appears to connect altered extracellular conditions with microglia activation, neuronal excitability, and eventually the development of a pathological behavior. Collectively, these findings open new important therapeutic avenues for the control of neuropathic pain [25] and epilepsy [99]. Yet, many questions are left unanswered and need to be addressed to better delineate the range of applications for an effective microglia-targeted therapeutic strategy; in particular: which neurological disorders are associated with a microglia-driven loss of inhibition? In which brain areas? Do all microglia have the same potential to synthesize and release BDNF when exposed to a given extracellular challenge? Or, instead, are microglia a heterogeneous population with multiple phenotypes playing different roles in different CNS areas and in different pathological states?
Tackling the multiform universe of microglia-neuron interactions and understanding the underlying molecular pathways offer the opportunity to identify specific biomarkers for neurological disorders and potential targets for novel therapeutic approaches.
Acknowledgments
The authors are grateful to Mr. Sylvain Côté for his precious assistance in preparing the figure. The authors acknowledge the financial support from the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, the Fonds de recherche Québec-Santé, the Krembil Foundation, and the Regione Piemonte/University of Turin.
References
- 1.Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nature Reviews Neurology. 2011;7(1):31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Viviani B, Gardoni F, Marinovich M. Cytokines and neuronal ion channels in health and disease. International Review of Neurobiology. 2007;82:247–263. doi: 10.1016/S0074-7742(07)82013-7. [DOI] [PubMed] [Google Scholar]
- 3.Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. Journal of Neuroscience. 2003;23(25):8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fiumelli H, Woodin MA. Role of activity-dependent regulation of neuronal chloride homeostasis in development. Current Opinion in Neurobiology. 2007;17(1):81–86. doi: 10.1016/j.conb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Harauzov A, Spolidoro M, DiCristo G, et al. Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. Journal of Neuroscience. 2010;30(1):361–371. doi: 10.1523/JNEUROSCI.2233-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kullmann DM, Moreau AW, Bakiri Y, Nicholson E. Plasticity of inhibition. Neuron. 2012;75(6):951–962. doi: 10.1016/j.neuron.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 7.Labrakakis C, Ferrini F, de Koninck Y. Inhibitory Synaptic Plasticity. New York, NY, USA: Springer; 2011. Mechanisms of plasticity of inhibition in chronic pain conditions; pp. 91–105. [Google Scholar]
- 8.Doyon N, Prescott SA, Castonguay A, Godin AG, Kröger H, de Koninck Y. Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis. PLoS Computational Biology. 2011;7(9) doi: 10.1371/journal.pcbi.1002149.e1002149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prescott SA, Sejnowski TJ, de Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Molecular Pain. 2006;2, article 32 doi: 10.1186/1744-8069-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Koninck Y. Altered chloride homeostasis in neurological disorders: a new target. Current Opinion in Pharmacology. 2007;7(1):93–99. doi: 10.1016/j.coph.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61(6):820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Price TJ, Cervero F, de Koninck Y. Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Current Topics in Medicinal Chemistry. 2005;5(6):547–555. doi: 10.2174/1568026054367629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain: a neuronal-specific isoform. Journal of Biological Chemistry. 1996;271(27):16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- 14.Rivera C, Voipio J, Payne JA, et al. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397(6716):251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 15.Cordero-Erausquin M, Coull JAM, Boudreau D, Rolland M, de Koninck Y. Differential maturation of GABA action and anion reversal potential in spinal lamina I neurons: impact of chloride extrusion capacity. Journal of Neuroscience. 2005;25(42):9613–9623. doi: 10.1523/JNEUROSCI.1488-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivera C, Voipio J, Kaila K. Two developmental switches in GABAergic signalling: the K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. Journal of Physiology. 2005;562, part 1:27–36. doi: 10.1113/jphysiol.2004.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-Ari Y, Gaiarsa J-L, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiological Reviews. 2007;87(4):1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- 18.Stil A, Jean-Xavier C, Liabeuf S, et al. Contribution of the potassium-chloride co-transporter KCC2 to the modulation of lumbar spinal networks in mice. European Journal of Neuroscience. 2011;33(7):1212–1222. doi: 10.1111/j.1460-9568.2010.07592.x. [DOI] [PubMed] [Google Scholar]
- 19.Chancey JH, Adlaf EW, Sapp MC, et al. GABA depolarization is required for experience-dependent synapse unsilencing in adult-born neurons. Journal of Neuroscience. 2013;33(15):6614–6622. doi: 10.1523/JNEUROSCI.0781-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge S, Pradhan DA, Ming G-L, Song H. GABA sets the tempo for activity-dependent adult neurogenesis. Trends in Neurosciences. 2007;30(1):1–8. doi: 10.1016/j.tins.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Boulenguez P, Liabeuf S, Bos R, et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nature Medicine. 2010;16(3):302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- 22.Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nature Neuroscience. 2009;12(4):438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- 23.Arion D, Lewis DA. Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Archives of General Psychiatry. 2011;68(1):21–31. doi: 10.1001/archgenpsychiatry.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyde TM, Lipska BK, Ali T, et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. Journal of Neuroscience. 2011;31(30):11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doyon N, Ferrini F, Gagnon M, de Koninck Y. Treating pathological pain: is KCC2 the key to the gate? Expert Review of Neurotherapeutics. 2013;13(5):469–471. doi: 10.1586/ern.13.40. [DOI] [PubMed] [Google Scholar]
- 26.Huberfeld G, Wittner L, Clemenceau S, et al. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. Journal of Neuroscience. 2007;27(37):9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coull JAM, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 28.Ferrini F, Trang T, Mattioli TA, et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl− homeostasis. Nature Neuroscience . 2013;16(2):183–192. doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivera C, Li H, Thomas-Crusells J, et al. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. Journal of Cell Biology. 2002;159(5):747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivera C, Voipio J, Thomas-Crusells J, et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. Journal of Neuroscience. 2004;24(19):4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Liu L-Y, Xu T-L. Reduced potassium-chloride co-transporter expression in spinal cord dorsal horn neurons contributes to inflammatory pain hypersensitivity in rats. Neuroscience. 2008;152(2):502–510. doi: 10.1016/j.neuroscience.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 32.Trang T, Beggs S, Salter MW. Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biology. 2011;7(1):99–108. doi: 10.1017/S1740925X12000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Annals of the New York Academy of Sciences. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merighi A, Salio C, Ghirri A, et al. BDNF as a pain modulator. Progress in Neurobiology. 2008;85(3):297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Santos AR, Comprido D, Duarte CB. Regulation of local translation at the synapse by BDNF. Progress in Neurobiology. 2010;92(4):505–516. doi: 10.1016/j.pneurobio.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381(6584):706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 37.Marty S, Da M, Berninger B. Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends in Neurosciences. 1997;20(5):198–202. doi: 10.1016/s0166-2236(96)01026-0. [DOI] [PubMed] [Google Scholar]
- 38.Mahan AL, Ressler KJ. Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends in Neurosciences. 2012;35(1):24–35. doi: 10.1016/j.tins.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Y, Wang JJ, Yung WH. Coupling between GABA-A receptor and chloride transporter underlies ionic plasticity in cerebellar purkinje neurons. Cerebellum. 2013;12(3):328–330. doi: 10.1007/s12311-013-0453-3. [DOI] [PubMed] [Google Scholar]
- 40.Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Experimental Brain Research. 2009;199(3-4):203–234. doi: 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- 41.Woo NH, Lu B. Intercellular Communication in the Nervous System, Malenka Rpp. London, UK: Academic Press; 2009. BDNF in synaptic plasticity and memory; pp. 590–598. [Google Scholar]
- 42.Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. Journal of Neurophysiology. 1998;80(6):3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka T, Saito H, Matsuki N. Inhibition of GABA(A) synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. Journal of Neuroscience. 1997;17(9):2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldelli P, Novara M, Carabelli V, Hernández-Guijo JM, Carbone E. BDNF up-regulates evoked GABAergic transmission in developing hippocampus by potentiating presynaptic N- and P/Q-type Ca2+ channels signalling. European Journal of Neuroscience. 2002;16(12):2297–2310. doi: 10.1046/j.1460-9568.2002.02313.x. [DOI] [PubMed] [Google Scholar]
- 45.Baldelli P, Hernandez-Guijo J-M, Carabelli V, Carbone E. Brain-derived neurotrophic factor enhances GABA release probability and nonuniform distribution of N- and P/Q-type channels on release sites of hippocampal inhibitory synapses. Journal of Neuroscience. 2005;25(13):3358–3368. doi: 10.1523/JNEUROSCI.4227-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizoguchi Y, Ishibashi H, Nabekura J. The action of BDNF on GABAA currents changes from potentiating to suppressing during maturation of rat hippocampal CA1 pyramidal neurons. Journal of Physiology. 2003;548, part 3:703–709. doi: 10.1113/jphysiol.2003.038935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivakine EA, Acton BA, Mahadevan V, et al. Neto2 is a KCC2 interacting protein required for neuronal Cl-regulation in hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(9):3561–3566. doi: 10.1073/pnas.1212907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wake H, Watanabe M, Moorhouse AJ, et al. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. Journal of Neuroscience. 2007;27(7):1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blaesse P, Guillemin I, Schindler J, et al. Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. Journal of Neuroscience. 2006;26(41):10407–10419. doi: 10.1523/JNEUROSCI.3257-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe M, Wake H, Moorhouse AJ, Nabekura J. Clustering of neuronal K+-Cl- cotransporters in lipid rafts by tyrosine phosphorylation. Journal of Biological Chemistry. 2009;284(41):27980–27988. doi: 10.1074/jbc.M109.043620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zadran S, Jourdi H, Rostamiani K, Qin G, Bi X, Baudry M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. Journal of Neuroscience. 2010;30(3):1086–1095. doi: 10.1523/JNEUROSCI.5120-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vargas-Perez H, Kee RT-A, Walton CH, et al. Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naïve rats. Science. 2009;324(5935):1732–1734. doi: 10.1126/science.1168501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fukuchi M, Nii T, Ishimaru N, et al. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neuroscience Research. 2009;65(1):35–43. doi: 10.1016/j.neures.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 54.Molinaro G, Battaglia G, Riozzi B, et al. Memantine treatment reduces the expression of the K+/Cl- cotransporter KCC2 in the hippocampus and cerebral cortex, and attenuates behavioural responses mediated by GABAA receptor activation in mice. Brain Research. 2009;1265:75–79. doi: 10.1016/j.brainres.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 55.Pezet S, Malcangio M, McMahon SB. BDNF: a neuromodulator in nociceptive pathways? Brain Research Reviews. 2002;40(1–3):240–249. doi: 10.1016/s0165-0173(02)00206-0. [DOI] [PubMed] [Google Scholar]
- 56.Parpura V, Zorec R. Gliotransmission: exocytotic release from astrocytes. Brain Research Reviews. 2010;63(1-2):83–92. doi: 10.1016/j.brainresrev.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. Journal of Neuroscience. 1996;16(8):2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miwa T, Furukawa S, Nakajima K, et al. Lipopolysaccharide enhances synthesis of brain-derived neurotrophic factor in cultured rat microglia. Journal of Neuroscience Research. 1997;50(6):1023–1029. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1023::AID-JNR13>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 59.Soontornniyomkij V, Wang G, Pittman CA, Wiley CA, Achim CL. Expression of brain-derived neurotrophic factor protein in activated microglia of human immunodeficiency virus type 1 encephalitis. Neuropathology and Applied Neurobiology. 1998;24(6):453–460. doi: 10.1046/j.1365-2990.1998.00134.x. [DOI] [PubMed] [Google Scholar]
- 60.Batchelor PE, Liberatore GT, Wong JYF, et al. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. Journal of Neuroscience. 1999;19(5):1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dougherty KD, Dreyfus CF, Black IB. Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiology of Disease. 2000;7(6):574–585. doi: 10.1006/nbdi.2000.0318. [DOI] [PubMed] [Google Scholar]
- 62.Lee T-H, Kato H, Chen S-T, Kogure K, Itoyama Y. Expression disparity of brain-derived neurotrophic factor immunoreactivity and mRNA in ischemic hippocampal neurons. NeuroReport. 2002;13(17):2271–2275. doi: 10.1097/00001756-200212030-00020. [DOI] [PubMed] [Google Scholar]
- 63.Stadelmann C, Kerschensteiner M, Misgeld T, Brück W, Hohlfeld R, Lassmann H. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain. 2002;125, part 1:75–85. doi: 10.1093/brain/awf015. [DOI] [PubMed] [Google Scholar]
- 64.Knott C, Stern G, Kingsbury A, Welcher AA, Wilkin GP. Elevated glial brain-derived neurotrophic factor in Parkinson’s diseased nigra. Parkinsonism and Related Disorders. 2002;8(5):329–341. doi: 10.1016/s1353-8020(02)00008-1. [DOI] [PubMed] [Google Scholar]
- 65.Ulmann L, Hatcher JP, Hughes JP, et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. Journal of Neuroscience. 2008;28(44):11263–11268. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beggs S, Trang T, Salter MW. P2X4R+ microglia drive neuropathic pain. Nature Neuroscience. 2012;15(8):1068–1073. doi: 10.1038/nn.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. Journal of Neuroscience. 2009;29(11):3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Inoue K. Microglial activation by purines and pyrimidines. Glia. 2002;40(2):156–163. doi: 10.1002/glia.10150. [DOI] [PubMed] [Google Scholar]
- 69.Pankratov Y, Lalo U, Verkhratsky A, North RA. Vesicular release of ATP at central synapses. Pflugers Archiv European Journal of Physiology. 2006;452(5):589–597. doi: 10.1007/s00424-006-0061-x. [DOI] [PubMed] [Google Scholar]
- 70.Guthrie PB, Knappenberger J, Segal M, Bennett MVL, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. Journal of Neuroscience. 1999;19(2):520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nature Neuroscience. 2006;9(12):1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- 72.Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55(6):604–616. doi: 10.1002/glia.20489. [DOI] [PubMed] [Google Scholar]
- 73.Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. Journal of Neuroscience. 2009;29(12):3781–3791. doi: 10.1523/JNEUROSCI.5512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Biber K, Tsuda M, Tozaki-Saitoh H, et al. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. The EMBO Journal. 2011;30(9):1864–1873. doi: 10.1038/emboj.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toyomitsu E, Tsuda M, Yamashita T, Tozaki-Saitoh H, Tanaka Y, Inoue K. CCL2 promotes P2X4 receptor trafficking to the cell surface of microglia. Purinergic Signalling. 2012;8(2):301–310. doi: 10.1007/s11302-011-9288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abbadie C, Lindia JA, Cumiskey AM, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(13):7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang J, Xiang QS, Echeverry S, Mogil JS, de Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. Journal of Neuroscience. 2007;27(45):12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsuda M, Masuda T, Kitano J, Shimoyama H, Tozaki-Saitoh H, Inoue K. IFN-γ receptor signaling mediates spinal microglia activation driving neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(19):8032–8037. doi: 10.1073/pnas.0810420106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuan H, Zhu X, Zhou S, et al. Role of mast cell activation in inducing microglial cells to release neurotrophin. Journal of Neuroscience Research. 2010;88(6):1348–1354. doi: 10.1002/jnr.22304. [DOI] [PubMed] [Google Scholar]
- 80.Nasu-Tada K, Koizumi S, Tsuda M, Kunifusa E, Inoue K. Possible involvement of increase in spinal fibronectin following peripheral nerve injury in upregulation of microglial P2X4, a key molecule for mechanical allodynia. Glia. 2006;53(7):769–775. doi: 10.1002/glia.20339. [DOI] [PubMed] [Google Scholar]
- 81.Tsuda M, Toyomitsu E, Komatsu T, et al. Fibronectin/integrin system is involved in P2X4 receptor upregulation in the spinal cord and neuropathic pain after nerve injury. Glia. 2008;56(5):579–585. doi: 10.1002/glia.20641. [DOI] [PubMed] [Google Scholar]
- 82.Masuda T, Tsuda M, Yoshinaga R, et al. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Reports. 2012;1(4):334–340. doi: 10.1016/j.celrep.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miao J, Ding M, Zhang A, et al. Pleiotrophin promotes microglia proliferation and secretion of neurotrophic factors by activating extracellular signal-regulated kinase 1/2 pathway. Neuroscience Research. 2012;74(3-4):269–276. doi: 10.1016/j.neures.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 84.Takayama N, Ueda H. Morphine-induced chemotaxis and brain-derived neurotrophic factor expression in microglia. Journal of Neuroscience. 2005;25(2):430–435. doi: 10.1523/JNEUROSCI.3170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bardoni R, Takazawa T, Tong CK, et al. Pre- and postsynaptic inhibitory control in the spinal cord dorsal horn. Annals of the New York Academy of Sciences. 2013;1279:90–96. doi: 10.1111/nyas.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roberts LA, Beyer C, Komisaruk BR. Nociceptive responses to altered GABAergic activity at the spinal cord. Life Sciences. 1986;39(18):1667–1674. doi: 10.1016/0024-3205(86)90164-5. [DOI] [PubMed] [Google Scholar]
- 87.Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37(1):111–123. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- 88.Coull JAM, Boudreau D, Bachand K, et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424(6951):938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- 89.Aldskogius H. Regulation of microglia—potential new drug targets in the CNS. Expert Opinion on Therapeutic Targets. 2001;5(6):655–668. doi: 10.1517/14728222.5.6.655. [DOI] [PubMed] [Google Scholar]
- 90.Arruda JL, Sweitzer S, Rutkowski MD, Deleo JA. Intrathecal anti-IL-6 antibody and IgG attenuates peripheral nerve injury-induced mechanical allodynia in the rat: possible immune modulation in neuropathic pain. Brain Research. 2000;879(1-2):216–225. doi: 10.1016/s0006-8993(00)02807-9. [DOI] [PubMed] [Google Scholar]
- 91.Jin S-X, Zhuang Z-Y, Woolf CJ, Ji R-R. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. Journal of Neuroscience. 2003;23(10):4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsuda M, Shigemoto-Mogami Y, Koizumi S, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424(6950):778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 93.Lu Y, Zheng J, Xiong L, Zimmermann M, Yang J. Spinal cord injury-induced attenuation of GABAergic inhibition in spinal dorsal horn circuits is associated with down-regulation of the chloride transporter KCC2 in rat. Journal of Physiology. 2008;586, part 23:5701–5715. doi: 10.1113/jphysiol.2008.152348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jolivalt CG, Lee CA, Ramos KM, Calcutt NA. Allodynia and hyperalgesia in diabetic rats are mediated by GABA and depletion of spinal potassium-chloride co-transporters. Pain. 2008;140(1):48–57. doi: 10.1016/j.pain.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wei B, Kumada T, Furukawa T, et al. Pre- and post-synaptic switches of GABA actions associated with Cl- homeostatic changes are induced in the spinal nucleus of the trigeminal nerve in a rat model of trigeminal neuropathic pain. Neuroscience. 2013;228:334–348. doi: 10.1016/j.neuroscience.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 96.Malhotra S, Pandyan AD, Day CR, Jones PW, Hermens H. Spasticity, an impairment that is poorly defined and poorly measured. Clinical Rehabilitation. 2009;23(7):651–658. doi: 10.1177/0269215508101747. [DOI] [PubMed] [Google Scholar]
- 97.Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. Journal of Neuroscience. 2006;26(16):4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu WH, Wang CY, Chen PS, et al. Valproic acid attenuates microgliosis in injured spinal cord and purinergic P2X4 receptor expression in activated microglia. Journal of Neuroscience Research. 2013;91(5):694–705. doi: 10.1002/jnr.23200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Miles R, Blaesse P, Huberfeld G, et al. Chloride homeostasis and GABA signaling in temporal lobe epilepsy. In: Noebeles JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies [Internet] 4th edition. Bethesda, Md, USA: National Center for Biotechnology Information (US); 2012. [PubMed] [Google Scholar]
- 100.Bernard C. Alterations in synaptic function in epilepsy. In: Noebeles JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies [Internet] 4th edition. Bethesda, Md, USA: National Center for Biotechnology Information (US); 2012. [PubMed] [Google Scholar]
- 101.Shulga A, Thomas-Crusells J, Sigl T, et al. Posttraumatic GABAA-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. Journal of Neuroscience. 2008;28(27):6996–7005. doi: 10.1523/JNEUROSCI.5268-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Experimental Neurology. 2013;244:11–21. doi: 10.1016/j.expneurol.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 103.Bachstetter AD, Rowe RK, Kaneko M, et al. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. Journal of Neuroscience. 2013;33(14):6143–6153. doi: 10.1523/JNEUROSCI.5399-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Z, Artelt M, Burnet M, Trautmann K, Schluesener HJ. Lesional accumulation of P2X4 receptor+ monocytes following experimental traumatic brain injury. Experimental Neurology. 2006;197(1):252–257. doi: 10.1016/j.expneurol.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 105.Schwarz JM, Bilbo SD. Adolescent morphine exposure affects long-term microglial function and later-life relapse liability in a model of addiction. Journal of Neuroscience. 2013;33(3):961–971. doi: 10.1523/JNEUROSCI.2516-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ludwig A, Uvarov P, Soni S, Thomas-Crusells J, Airaksinen MS, Rivera C. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. Journal of Neuroscience. 2011;31(2):644–649. doi: 10.1523/JNEUROSCI.2006-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lipsky RH, Marini AM. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Annals of the New York Academy of Sciences. 2007;1122:130–143. doi: 10.1196/annals.1403.009. [DOI] [PubMed] [Google Scholar]
- 108.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339(6116):156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Keller AF, Beggs S, Salter MW, de Koninck Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Molecular Pain. 2007;3, article 27 doi: 10.1186/1744-8069-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Crozier RA, Bi C, Han YR, Plummer MR. BDNF modulation of NMDA receptors is activity dependent. Journal of Neurophysiology. 2008;100(6):3264–3274. doi: 10.1152/jn.90418.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang L-N, Yang J-P, Ji F-H, et al. Brain-derived neurotrophic factor modulates N-methyl-D-aspartate receptor activation in a rat model of cancer-induced bone pain. Journal of Neuroscience Research. 2012;90(6):1249–1260. doi: 10.1002/jnr.22815. [DOI] [PubMed] [Google Scholar]
- 112.Lee HHC, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nature Neuroscience. 2011;14(6):736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. Journal of Neuroscience. 2012;32(33):11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bechade C, Cantaut-Belarif Y, Bessis A. Microglial control of neuronal activity. Frontiers in Cellular Neuroscience. 2013;7, article 32 doi: 10.3389/fncel.2013.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Frontiers in Neuroendocrinology. 2012;33(1):116–125. doi: 10.1016/j.yfrne.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hayashi Y, Ishibashi H, Hashimoto K, Nakanishi H. Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. Glia. 2006;53(6):660–668. doi: 10.1002/glia.20322. [DOI] [PubMed] [Google Scholar]
- 117.Steiner J, Walter M, Gos T, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? Journal of Neuroinflammation. 2011;8, article 94 doi: 10.1186/1742-2094-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Piccinin S, di Angelantonio S, Piccioni A, et al. CX3CL1-induced modulation at CA1 synapses reveals multiple mechanisms of EPSC modulation involving adenosine receptor subtypes. Journal of Neuroimmunology. 2010;224(1-2):85–92. doi: 10.1016/j.jneuroim.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 119.Pascual O, Achour SB, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(4):E197–E205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mandolesi G, Grasselli G, Musella A, et al. GABAergic signaling and connectivity on Purkinje cells are impaired in experimental autoimmune encephalomyelitis. Neurobiology of Disease. 2012;46(2):414–424. doi: 10.1016/j.nbd.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 121.Bellinger FP, Madamba S, Siggins GR. Interleukin 1β inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Research. 1993;628(1-2):227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- 122.Tabarean IV, Korn H, Bartfai T. Interleukin-1β induces hyperpolarization and modulates synaptic inhibition in preoptic and anterior hypothalamic neurons. Neuroscience. 2006;141(4):1685–1695. doi: 10.1016/j.neuroscience.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 123.Kawasaki Y, Zhang L, Cheng J-K, Ji R-R. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. Journal of Neuroscience. 2008;28(20):5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α . Journal of Neuroscience. 2005;25(12):3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen- activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. Journal of Neuroscience. 1998;18(5):1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Galea E, Feinstein DL, Reis DJ. Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10945–10949. doi: 10.1073/pnas.89.22.10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Matsui T, Svensson CI, Hirata Y, Mizobata K, Hua X-Y, Yaksh TL. Release of prostaglandin E2 and nitric oxide from spinal microglia is dependent on activation of p38 mitogen-activated protein kinase. Anesthesia and Analgesia. 2010;111(2):554–560. doi: 10.1213/ANE.0b013e3181e3a2a2. [DOI] [PubMed] [Google Scholar]
- 128.Saito O, Svensson CI, Buczynski MW, et al. Spinal glial TLR4-mediated nociception and production of prostaglandin E2 and TNF. British Journal of Pharmacology. 2010;160(7):1754–1764. doi: 10.1111/j.1476-5381.2010.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ahmadi S, Lippross S, Neuhuber WL, Zeilhofer HU. PGE2 selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nature Neuroscience. 2002;5(1):34–40. doi: 10.1038/nn778. [DOI] [PubMed] [Google Scholar]
- 130.Harvey RJ, Depner UB, Wässle H, et al. GlyR α3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science. 2004;304(5672):884–887. doi: 10.1126/science.1094925. [DOI] [PubMed] [Google Scholar]