

Mutations in the gene-encoding β-catenin, CTNNB1, are highly prevalent in sporadic desmoid tumors and may predict the risk for recurrence. We sought to determine the prevalence of CTNNB1 mutations and to determine whether the CTNNB1 mutation status correlates with disease outcome.
Keywords: Desmoid tumor, Beta-catenin, Mutation status, CTNNB1, Fibromatosis
Learning Objectives
Describe the frequency of CTNNB1 mutations in sporadic desmoid tumors.
Summarize findings regarding CTNNB1 mutation status and disease outcome.
Abstract
Background.
Mutations in the gene-encoding β-catenin, CTNNB1, are highly prevalent in sporadic desmoid tumors and may predict the risk for recurrence. We sought to determine the prevalence of CTNNB1 mutations in a large cohort of sporadic desmoid tumors and to determine whether CTNNB1 mutation status correlates with disease outcome.
Methods.
Single-base extension genotyping of the CTNNB1 gene was performed on 145 sporadic, paraffin-embedded desmoid tumor specimens. Correlation of mutation status with outcome was performed on a subset of 115 patients who underwent macroscopically complete surgical resection.
Results.
CTNNB1 mutations were detected in 106 of 145 (73%) tumor specimens and in 86 of 115 (75%) specimens from patients who underwent curative-intent surgical resection, including discrete mutations in the following codons of CTNNB1 exon 3: T41A (46%), S45F (25%), S45P (1.7%), and S45C (0.9%). Desmoid tumors of the superficial trunk were significantly less likely to harbor CTNNB1 mutations than tumors located elsewhere, but none of the other examined clinicopathologic factors were found to be associated with CTNNB1 mutation status. At a median follow-up of 31 months, 5-year recurrence-free survival was slightly, although not statistically significantly, worse for patients with β-catenin-mutated tumors than for those with wild-type tumors (58% vs. 74%, respectively). The specific CTNNB1 codon mutation did not correlate with the risk for recurrence.
Conclusion.
CTNNB1 mutations are indeed common in sporadic desmoid tumors. However, our study did not detect a statistically significant difference in recurrence risk according to either the CTNNB1 mutation status or the specific CTNNB1 mutation.
Implications for Practice:
Mutations in the gene-encoding β-catenin, CTNNB1, are highly prevalent in sporadic desmoid tumors, yet whether mutation status and/or type predict outcome is not certain. In contrast to recently published studies from other groups, we did not detect a significant difference in recurrence risk according to either the CTNNB1 mutation status or the specific mutation. Accordingly, the impact of CTNNB1 mutation status in the treatment algorithm of these enigmatic tumors is uncertain at present. However, with the acquisition of further data, and as the efforts to target β-catenin as a therapy mature, knowledge of the CTNNB1 mutation status may eventually permit a much more rational, individualized treatment approach to desmoid tumors.
Introduction
Desmoid tumor, also known as aggressive fibromatosis, is a mesenchymal tumor that arises from fascial or deep musculoaponeurotic structures [1, 2]. Although desmoid tumors do not metastasize, they can be locally invasive and exhibit a propensity to recur despite aggressive surgical resection. Desmoid tumors may arise sporadically or in association with familial adenomatous polyposis (FAP), caused by a germ-line mutation in the adenomatous polyposis coli (APC) gene. Treatment consists of surgery, radiation therapy, and systemic therapies in various combinations. However, a consensus understanding of optimal treatment approaches and their outcomes is confounded by the rarity of desmoid tumors as well as by their unpredictable natural history, including the spontaneous regression of some desmoid tumors in the absence of treatment [3, 4].
The occurrence of desmoid tumors in patients with FAP, along with cytogenetic studies of aggressive fibromatoses from patients without FAP that showed the frequent loss of a portion of the long arm of chromosome 5, suggested the potential role of APC mutations and β-catenin dysregulation in sporadic desmoid tumorigenesis [5]. APC controls the cytosolic levels of β-catenin, a cadherin-binding protein involved in cell-cell adhesion and a transcriptional activator of genes such as c-MYC that promote cellular proliferation and survival [6]. The APC protein complex marks β-catenin for degradation by the proteosome by sequential phosphorylation of four critical amino acids (serines 45, 37, and 33 and threonine 41), all encoded by exon 3 of the β-catenin gene [7, 8]. Most CTNNB1 mutations occur within this exon of the gene in the region encoding for the β-catenin protein sequence that contains the serine and threonine residues listed above. These mutations lead to stabilization of the β-catenin protein, translocation from the cytoplasm to the nucleus, and transcriptional activation of target genes [9]. Indeed, several recent studies have demonstrated that the vast majority of sporadic desmoid tumors harbor mutations in the CTNNB1 gene and not in the APC gene, and only three specific mutations in the CTNNB1 gene have been identified, one at threonine 41 (T41A) and two at serine 45 (S45F and S45P) [5, 10–14]. Furthermore, two recent studies have shown that CTNNB1-mutated desmoid tumors have a significantly worse recurrence-free survival (RFS) than wild-type tumors [15, 16]. However, Lazar et al. [16] found that the S45F mutation in particular predicts a higher risk for recurrence; whereas, Domont et al. [15] found that no specific mutant genotype predicts outcome. These discrepant results may be the result of differences in the patient cohorts studied, as Domont et al. only considered extra-abdominal tumors and included a high percentage of recurrent tumors, whereas Lazar et al. included intra-abdominal desmoid tumors and a higher percentage of primary tumors.
In this study, we sought to determine the prevalence of CTNNB1 mutations in a large cohort of sporadic desmoid tumors, including both primary and recurrent tumors and both intra-abdominal and extra-abdominal sites, and to determine whether CTNNB1 mutation status correlates with disease outcome.
Patients and Methods
Patients and Tumor Samples
A total of 160 formalin-fixed, paraffin-embedded desmoid tumor specimens collected between 1984 and 2009 were retrieved from the pathology archives of the Massachusetts General Hospital. All specimens were evaluated by an experienced soft tissue pathologist (G.P.N.), and a histologic diagnosis of desmoid tumor was confirmed in each case. Clinical data, including demographic, treatment, tumor, and outcome variables were collected and tabulated. To investigate the correlation between β-catenin mutation status and outcome, we included only those patients undergoing macroscopically complete surgical resection (R0 and R1 resections), and we excluded those patients lost to follow-up and for whom complete clinicopathologic data were lacking. This study was approved by the institutional review board of the Massachusetts General Hospital.
Patient age was defined as the age at initial presentation to Massachusetts General Hospital. This definition was used for patients with both primary and recurrent tumors, because the date of initial diagnosis was not always available for patients who presented with recurrent tumors. Tumor size was determined by pathology reports. Tumor site was categorized as intra-abdominal or extra-abdominal, with the latter further categorized as extremities, girdles (shoulder/axilla and hip/buttock/groin), head and neck, or superficial trunk. Microscopic margin status was retrieved from the final pathology reports and was considered positive if the tumor was identified at the inked margin of the pathologic specimen. Radiation therapy was delivered in an adjuvant fashion at the discretion of the surgeon and radiation oncologist when a higher risk for recurrence was predicted on clinical grounds. In general, patients were seen for follow-up after definitive treatment every three months for the first two years, then every 6 months until year five, and thereafter patients were either seen on a yearly basis or were considered cured, depending on the anticipated risk for recurrence (e.g., higher risk for extremity tumors and lower risk for abdominal wall tumors). Imaging studies were typically obtained with each visit or with every other visit and included an abdominopelvic computed tomography scan for intra-abdominal tumors and a magnetic resonance imaging scan for extremity and body wall desmoid tumors.
Single-Base Extension Mutational Analysis of CTNNB1 Codons 41 and 45
The histologic diagnosis of desmoid tumor was confirmed by a soft tissue pathologist (G.P.N.), and tumor-rich formalin-fixed paraffin-embedded blocks were identified for DNA extraction, as described previously [17]. Genotyping with a slightly modified protocol was then performed. Briefly, tumor DNA samples were analyzed for recurrent point mutations in CTNNB1 codons T41 and S45 using the single-base extension SNaPshot assay. CTNNB1 exon 3, which contains both target codons 41 and 45, was amplified in a single PCR reaction. After thermocycling 38 cycles, PCR were cleaned up by adding 2 μL of an exonuclease I (Exo) and shrimp alkaline phosphatase (SAP) mixture. The extension primer pool mixed with RR mix (2 μL) was added directly to the cleaned up PCR amplification product for single-base extension. The extension primers were designed to interrogate four bases corresponding to the two target codons, all in the reverse direction: CTNNB1 c.121A, c.122C, c.133T, and c.134C. The extension products were treated with SAP using the same procedure used in the Exo-SAP step and then analyzed with the ABI 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA http://www.appliedbiosystems.com). Detailed primer information is available upon request to the authors.
Statistical Methods
Binary and categoric comparisons were made using the chi-squared test. Medians were compared using the Kruskal-Wallis test. Time to local failure and RFS were calculated from the time of surgery at our institution to first local recurrence. Patients who did not have a local recurrence at or before their date of last contact or death were censored at this date. Local recurrence was defined as clinical or radiologic evidence of tumor regrowth at the primary site. RFS curves were estimated using Kaplan-Meier methodology, and comparisons were made using the log-rank test. Univariate and multivariate Cox proportional hazards modeling was used to estimate hazard ratios for RFS. Logistic regression modeling was used to determine factors associated with CTNBB1 mutations. Two-sided p values are provided. All statistical analyses were performed using SAS version 9.2 (SAS Institute, Inc., Cary, NC, http://www.sas.com).
Results
CTNNB1 Mutations are Common in Sporadic Desmoid Tumors
Of the 160 desmoid tumor samples (from 160 patients) that had sufficient DNA extracted for CTNNB1 exon 3 gene sequencing, results were definitive in 149 (93%). After excluding 4 patients with a history of FAP (none with CTNNB1 mutations), 145 patients remained. A mutation in exon 3 of the CTNNB1 gene was identified in 106 (73%) of the 145 samples, and all mutations were found at codons 41 and 45. These were identified as c.121 A > G, or pThr-41-Ala (T41A), which was identified in 65 samples (61%); c.134 C > T, or pSer45Phe (S45F), identified in 35 tumor samples (33%); c.133 T > C, or pSer45Pro (S45P), found in 3 samples (2.8%); and c.134 C > G, or pSer45Cys (S45C), identified in 1 tumor (0.9%) sample. Mutations in both T41A and S45P were identified in the 2 (1.9%) remaining samples.
Patient Characteristics According to CTNNB1 Genotype
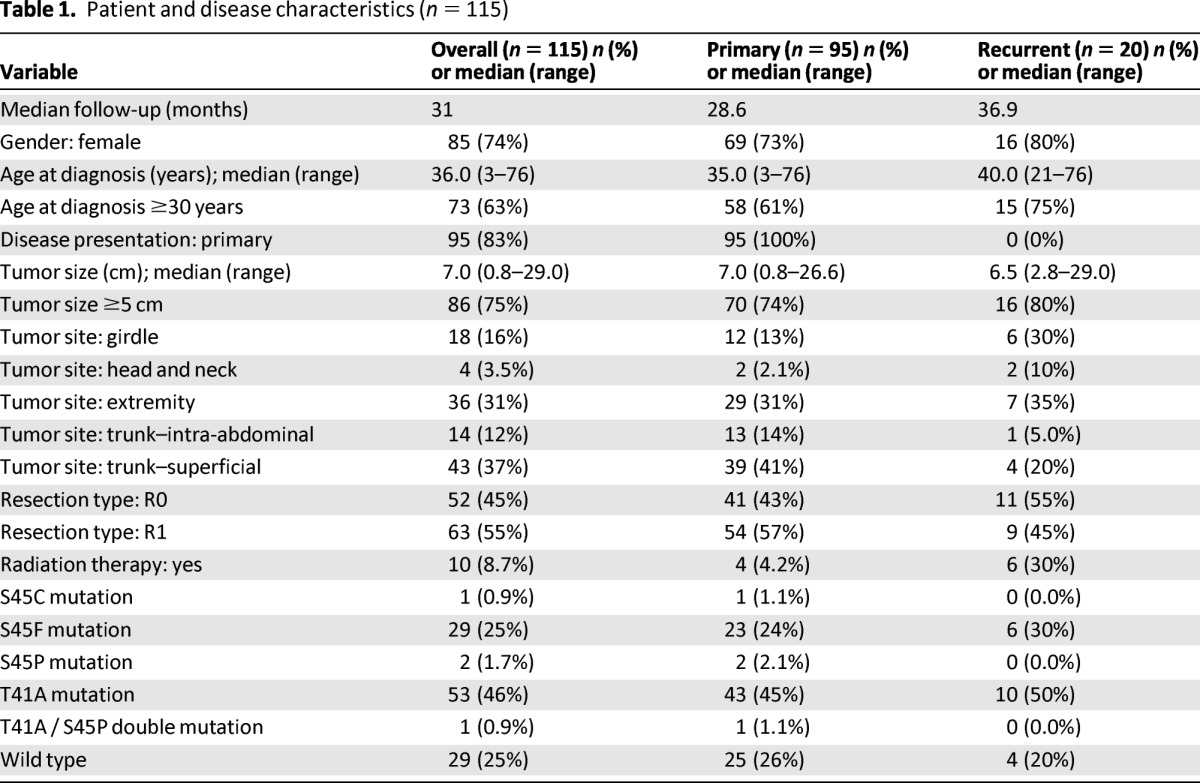
After excluding 30 patients who did not undergo macroscopically complete surgical resection or who had incomplete clinicopathologic and/or follow-up data, 115 patients remained and were included in the study (Table 1). Of these 115 patients, 85 (74%) were female and 30 (26%) were male. Median age was 36 years. Primary desmoid tumors were present in 95 patients (83%) at presentation, and 20 patients (17%) presented with recurrent tumors. Median tumor size was 7.0 cm (range, 0.8–29.0 cm). Surgical margins were microscopically negative in 52 cases and positive in 63 cases. Adjuvant radiation therapy was administered to 10 patients. The CTNNB1 mutation rate detected in this cohort (75%, or 86 of 115 cases) was similar to that identified for the entire series, as was the distribution of the specific mutations (T41A, 62%; S45F, 34%; S45P, 2.3%; S45C, 1.2%; and T41A and S45P double mutant, 1.2%). CTNNB1 mutation status was negatively associated with superficial trunk location (odds ratio = 0.31 [95% confidence interval, 0.13–0.73], p = .008), but it was not significantly associated with any other clinicopathologic factor examined, including age, sex, tumor site, presentation status, microscopic margin status, or receipt of radiation therapy. Tumor size, considered to be either a continuous variable or a binary variable (<5 cm or ≥5 cm), was also not significantly associated with CTNNB1 mutation status.
Table 1.
Patient and disease characteristics (n = 115)
CTNNB1 Mutation Status Does Not Significantly Correlate With Outcome
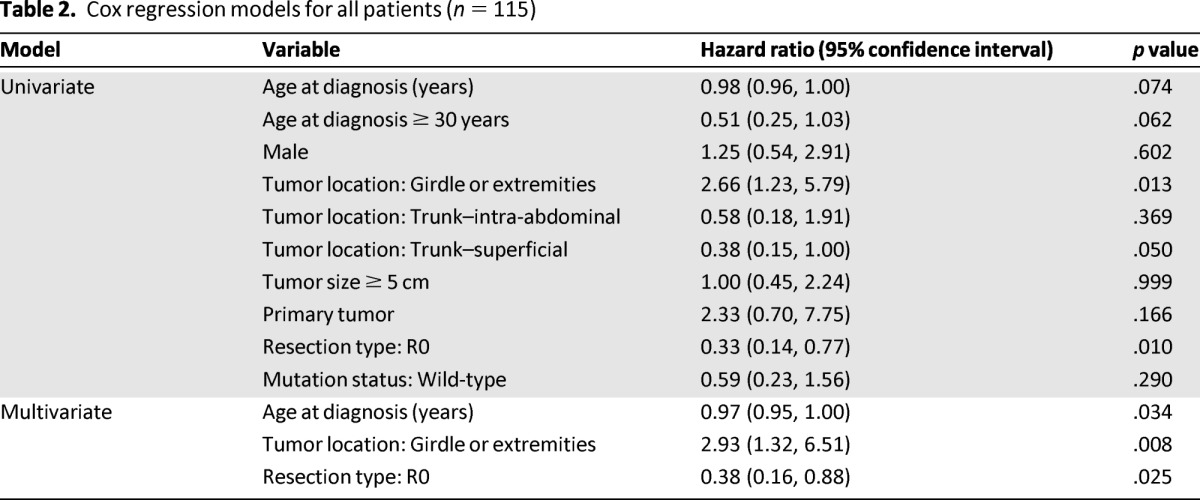
We sought to determine whether CTNNB1 mutation status correlated with clinical outcome; namely, RFS. There were a total of 31 local recurrences (27% of the cohort), 5 of which occurred in patients with wild-type tumors and 26 of which occurred in patients with CTNNB1-mutated tumors. Five-year RFS was slightly, although not significantly, worse in the CTNNB1-mutated tumors (58.0%) than in the wild-type tumors (73.6%) (p = .285) (Fig. 1). Five-year RFS did not differ significantly among the different CTNNB1 genotypes (Fig. 2). The estimated 5-year RFS rate was 59.8% (95% CI, 35.4%, 77.5%) for patients with the 45F mutation, 54.9% (35.6%, 70.5%) for patients with the 41A mutation, and 73.6% (46.7%, 88.4%) for patients with wild-type tumors. Because only four samples harbored S45P, S45C, or double mutations, these tumors were excluded from the analysis. In a multivariate Cox regression model that included all patients, girdle/extremity tumors were associated with a shorter time to local recurrence, whereas advanced age and R0 resection status were associated with longer RFS (Table 2). Given the small proportion of patients treated with adjuvant radiation and systemic therapies as well as the diversity and highly variable start and stop dates of the systemic agents, we could not legitimately analyze these data in a time-to-event statistical analysis.
Figure 1.

Recurrence-free survival for wild-type tumors versus CTNNB1-mutated tumors in all patients.
Abbreviation: RFS, recurrence-free survival.
Figure 2.

Recurrence-free survival by CTNNB1 genotype.
Abbreviation: RFS, recurrence-free survival.
Table 2.
Cox regression models for all patients (n = 115)
Because of the potential prognostic impact of presentation status (primary vs. recurrent) on outcome, we analyzed the CTNNB1 genotype in patients with primary tumors (n = 95) and found slightly higher 2-year rates of recurrence in patients with mutated tumors (RFS, 64%) compared with rates in patients with wild-type tumors (RFS, 77%); however, this failed to reach statistical significance (p = .128) (Fig. 3). Similarly, CTNNB1 mutation status failed to predict outcome for patients with recurrent desmoid tumors. Because of the potential prognostic impact of surgical margin status on outcome, we also analyzed the CTNNB1 genotype in patients who underwent R0 resection (n = 52), and we found the risk for recurrence to be similar in patients with CTNNB1-mutated tumors and in those with wild-type tumors (Fig. 4).
Figure 3.

Recurrence-free survival for CTNNB1-mutated tumors versus wild-type tumors in patients with primary tumors.
Abbreviation: RFS, recurrence-free survival.
Figure 4.

Recurrence-free survival for wild-type tumors versus CTNNB1-mutated tumors in patients undergoing R0 resection.
Abbreviation: RFS, recurrence-free survival.
Discussion
In this study, we identified a high frequency (∼75%) of CTNNB1 mutations in a large cohort of sporadic desmoid tumors, including both primary and recurrent tumors at both intra-abdominal and extra-abdominal sites. This mutation rate is considerably higher than the rate of 39% to 54% reported in some of the earliest studies [11, 12, 14], but is quite consistent with that reported in three recent large series, all of which identified CTNNB1 mutations in 85% of their respective series of sporadic desmoid tumors [10, 15, 16]. Similarly, we also found that all of the mutations are clustered in exon 3 of the CTNNB1 gene at threonine 41 and serine 45, emphasizing the crucial role that these residues play in the Wnt signaling pathway. Although we identified a very limited CTNNB1 mutational spectrum in our series of desmoid tumors, including high percentages of T41A and S45F mutants, we also identified one patient with an S45C mutation and another patient with a T41A/S45P double mutation. To our knowledge, these two CTNNB1 mutations have not been described previously in desmoid tumors.
We also sought to determine whether CTNNB1 mutation status predicted disease outcome in a subset of 115 patients who underwent macroscopically complete surgical resection. We found that 5-year RFS was slightly, although not significantly, worse in mutated tumors (58%) compared with wild-type tumors (74%). This conclusion is true even when considering only primary tumors (i.e., not including recurrent tumors) and when considering only those tumors resected with negative margins (R0 resections). These results contrast those reported by Domont et al., who found a statistically significant difference in 5-year RFS between wild-type tumors and mutated tumors (75% vs. 43%; p = .02) [15]. It should be noted, however, that these authors included eight patients, all of whom had CTNNB1 mutations, who underwent R2 resections, and we reasonably suspect that all of them subsequently had recurrence, no doubt influencing the conclusions of the study. Nevertheless, our finding that no specific CTNNB1 mutation genotype (e.g., T41A or S45F) predicts outcome is in agreement with the findings reported by Domont et al., who found a statistically nonsignificant trend toward a higher risk for recurrence for S45F-mutated tumors. These data contradict the results of Lazar et al., who reported that the risk for recurrence in desmoid tumors was strongly associated with the S45F mutation [16].
These discrepancies may be because each of these studies included distinct populations of patients with desmoid tumors, included variable percentages of patients with primary versus recurrent tumors, included patients with intra-abdominal versus extra-abdominal tumors, and included patients with R0 versus R1 versus R2 resections. Furthermore, the therapies rendered to patients in these studies, including adjuvant radiation and medical therapies, are highly variable. Moreover, the recurrence rates reported in each of these three studies vary quite markedly. These significant differences in recurrence rates may be the result of any of a number of factors, including differences in tumor location, R0 resection rates, and patient age, and may mask the effect that CTNNB1 mutation status might have on risk for recurrence. For instance, in this study we report 5-year RFS rates of 58% and 73.6% for wild-type and mutated tumors, respectively. Domont et al. reported 5-year RFS rates of 43% and 75%, respectively, and Lazar et al. reported 5-year RFS rates of 23% for S45F-mutated tumors, 57% for T41A-mutated tumors, and 65% for wild-type tumors. Finally, our study is underpowered to show a 15%–20% difference in RFS according to mutation status. Specifically, this study has only 29% power to detect the 16% difference in 5-year RFS (58% vs. 74%) that we identified at a two-sided significance level of 5%. Thus, with greater patient numbers, perhaps by combining our population of patients with those from other centers, we may be able to demonstrate a statistically significant difference in RFS according to CTNNB1 mutation status.
Given that there is significant controversy about the true prognostic impact of CTNNB1 mutation status in desmoid tumors, one might reasonably question what role, if any, this factor plays in the treatment algorithm of these complicated tumors. Certainly, genotyping of CTNNB1 exon 3 is useful as a diagnostic test to distinguish a desmoid tumor from an otherwise pathologically confusing entity such as postsurgical scar. However, it is not clear what impact CTNNB1 mutation status might have on the current “watch and wait” posture that many surgeons now take in the treatment of desmoid tumors in light of at least two recent studies supporting the safety of this more conservative approach [3, 18]. These studies confirm that up to 50% of patients with extra-abdominal desmoid tumors will exhibit spontaneous tumor growth arrest after initial progression, and that many if not most of these patients will not require any treatment whatsoever. Clearly, because up to 85% of sporadic desmoid tumors harbor a CTNNB1 exon 3 mutation, a large proportion of these tumors exhibiting growth arrest are in fact mutated tumors. This finding suggests that β-catenin mutation status, rather than trumping all other clinicopathologic factors, is just one of the many clinical, pathologic, and molecular factors to consider in the management of these tumors. In addition, there is no shortage of controversy when it comes to the prognostic impact of margin status, adjuvant radiation therapy, and other factors on desmoid tumor outcome.
Our practice at Massachusetts General Hospital is to perform CTNNB1 exon 3 genotyping on every desmoid tumor we biopsy, and we store these data in our clinical outcomes database. A large proportion of these patients, especially those patients who are asymptomatic or who have tumors in anatomically challenging locations, are simply followed expectantly on no therapy at all or on a medical therapy of low toxicity, such as sulindac. Another subset undergo surgical resection and/or radiation therapy, but these decisions are currently made on clinical grounds alone (i.e., degree of symptoms, tumor growth rate, estimated morbidity of the therapy in question), with little if any consideration given to the CTNNB1 mutation status. However, the knowledge that a given desmoid tumor harbors a CTNNB1 mutation, regardless of the type, and thus has elevated levels of nuclear β-catenin, may prove useful as the efforts to target β-catenin as a therapy mature. With the acquisition of further data, especially those garnered through a prospective trial, we hope to acquire a more refined understanding of the molecular determinants of outcome for these tumors to permit a more rational, individualized treatment approach.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Acknowledgments
This work was funded in part by the Federal Share of program income earned by Massachusetts General Hospital on C06 CA059267, Proton Therapy Research and Treatment Center.
Author Contributions
Conception/Design: John T. Mullen, Thomas DeLaney, Andrew E. Rosenberg, Wendy Kobayashi, Jackie Szymonifka, Yen-Lin Chen, David C. Harmon, Edwin Choy, Sam S. Yoon, Kevin A. Raskin, Francis J. Hornicek, Gunnlauger P. Nielsen
Provision of study material or patients: John T. Mullen, Thomas DeLaney, Andrew E. Rosenberg, Long Le, Gunnlauger P. Nielsen
Collection and/or assembly of data: John T. Mullen, Andrew E. Rosenberg, Long Le, John Iafrate, Wendy Kobayashi, Gunnlauger P. Nielsen
Data analysis and interpretation: John T. Mullen, Thomas DeLaney, Andrew E. Rosenberg, Long Le, Wendy Kobayashi, Jackie Szymonifka, Beow Y. Yeap, David C. Harmon, Edwin Choy, Sam S. Yoon, Kevin A. Raskin, Francis J. Hornicek, Gunnlauger P. Nielsen
Manuscript writing: John T. Mullen, Jackie Szymonifka, Beow Y. Yeap, David C. Harmon, Edwin Choy, Sam S. Yoon, Francis J. Hornicek, Gunnlauger P. Nielsen
Final approval of manuscript: John T. Mullen, Thomas DeLaney, Andrew E. Rosenberg, Long Le, John Iafrate, Wendy Kobayashi, Jackie Szymonifka, Beow Y. Yeap, Yen-Lin Chen, David C. Harmon, Edwin Choy, Sam S. Yoon, Kevin A. Raskin, Francis J. Hornicek, Gunnlauger P. Nielsen
Disclosures
Thomas DeLaney: Amgen Giant Cell Tumor Global Advisory Board (C/A); GlaxoSmithKline (OI); Long Le: ArcherDx (E, OI); Partners Healthcare (IP); John Iafrate: SNaPshot technology licensing (IP); Edwin Choy: Amgen (C/A, RF); Pharmacyclics (RF); Novartis (RF); Francis Hornicek: Stryker Corporation (C/A); BioMed Valley Discoveries (C/A); NIH (RF); Chordoma Foundation (RF); Sarcoma Foundation (RF). The other authors indicated no financial relationships.
Section editors: Laurence Baker: DSMB for Morphotek, CytRx; Jaap Verweij: None
Reviewer “A”: None
Reviewer “B”: None
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Alman BA, Pajerski ME, Diaz-Cano S, et al. Aggressive fibromatosis (desmoid tumor) is a monoclonal disorder. Diagn Mol Pathol. 1997;6:98–101. doi: 10.1097/00019606-199704000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Li M, Cordon-Cardo C, Gerald WL, et al. Desmoid fibromatosis is a clonal process. Hum Pathol. 1996;27:939–943. doi: 10.1016/s0046-8177(96)90221-x. [DOI] [PubMed] [Google Scholar]
- 3.Bonvalot S, Eldweny H, Haddad V, et al. Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol. 2008;34:462–468. doi: 10.1016/j.ejso.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Salas S, Dufresne A, Bui B, et al. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: A wait-and-see policy according to tumor presentation. J Clin Oncol. 2011;29:3553–3558. doi: 10.1200/JCO.2010.33.5489. [DOI] [PubMed] [Google Scholar]
- 5.Alman BA, Li C, Pajerski ME, et al. Increased beta-catenin protein and somatic APC mutations in sporadic aggressive fibromatoses (desmoid tumors) Am J Pathol. 1997;151:329–334. [PMC free article] [PubMed] [Google Scholar]
- 6.Munemitsu S, Albert I, Souza B, et al. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orford K, Crockett C, Jensen JP, et al. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem. 1997;272:24735–24738. doi: 10.1074/jbc.272.40.24735. [DOI] [PubMed] [Google Scholar]
- 8.Rubinfeld B, Albert I, Porfiri E, et al. Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res. 1997;57:4624–4630. [PubMed] [Google Scholar]
- 9.Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9:15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 10.Amary MF, Pauwels P, Meulemans E, et al. Detection of beta-catenin mutations in paraffin-embedded sporadic desmoid-type fibromatosis by mutation-specific restriction enzyme digestion (MSRED): An ancillary diagnostic tool. Am J Surg Pathol. 2007;31:1299–1309. doi: 10.1097/PAS.0b013e31802f581a. [DOI] [PubMed] [Google Scholar]
- 11.Miyoshi Y, Iwao K, Nawa G, et al. Frequent mutations in the beta-catenin gene in desmoid tumors from patients without familial adenomatous polyposis. Oncol Res. 1998;10:591–594. [PubMed] [Google Scholar]
- 12.Saito T, Oda Y, Kawaguchi K, et al. Possible association between higher beta-catenin mRNA expression and mutated beta-catenin in sporadic desmoid tumors: Real-time semiquantitative assay by TaqMan polymerase chain reaction. Lab Invest. 2002;82:97–103. doi: 10.1038/labinvest.3780399. [DOI] [PubMed] [Google Scholar]
- 13.Shitoh K, Konishi F, Iijima T, et al. A novel case of a sporadic desmoid tumour with mutation of the beta catenin gene. J Clin Pathol. 1999;52:695–696. doi: 10.1136/jcp.52.9.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tejpar S, Nollet F, Li C, et al. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor) Oncogene. 1999;18:6615–6620. doi: 10.1038/sj.onc.1203041. [DOI] [PubMed] [Google Scholar]
- 15.Domont J, Salas S, Lacroix L, et al. High frequency of beta-catenin heterozygous mutations in extra-abdominal fibromatosis: A potential molecular tool for disease management. Br J Cancer. 2010;102:1032–1036. doi: 10.1038/sj.bjc.6605557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazar AJ, Tuvin D, Hajibashi S, et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173:1518–1527. doi: 10.2353/ajpath.2008.080475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: A clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2:146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fiore M, Rimareix F, Mariani L, et al. Desmoid-type fibromatosis: A front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol. 2009;16:2587–2593. doi: 10.1245/s10434-009-0586-2. [DOI] [PubMed] [Google Scholar]