

Abstract
This commentary discusses the current use of arbitrary boundaries to distinguish the continuum of incipient (clonal cytopenias), oligoblastic (subacute), and polyblastic (acute) myelogenous leukemia. The historical path that led to the application of these boundaries is discussed, and simplification of the current diagnostic classification is proposed.
Keywords: Classification, Leukemic myeloblasts, Myelodysplasia, Myelogenous leukemia, Neoplasia, Refractory anemia, Clonal evolution
This commentary discusses the current use of arbitrary boundaries to distinguish the continuum of the clonal cytopenias and oligoblastic (subacute) and polyblastic (acute) myelogenous leukemia. Each is the result of the neoplastic transformation of a primitive multipotential hematopoietic cell and, in many cases, the result of clonal evolution to a more rapidly progressive myeloid neoplasm. This paper examines the historical path that led to the application of these boundaries and cites several recent studies showing that the boundaries in current use are not based on pathobiological findings and are arbitrary. Simplification of the current diagnostic classification of the (a) incipient (clonal cytopenias), (b) oligoblastic, and (c) polyblastic myelogenous leukemias is proposed. These classifications may be more consistent with general concepts of the diagnostic designations of neoplastic diseases as well as less idiosyncratic and more understandable for biomedical scientists, health care students and trainees, demographers, epidemiologists, hematological oncologists and pathologists, patients, science journalists, and other interested lay parties.
Experienced hematopathologists and hematologists have dispensed with the principles applied to cancer pathobiology, classification, and diagnosis during their periodic exercises in the classification of myeloid neoplasms on behalf of the World Health Organization (WHO) [1]. One of the most glaring, but not the only, deviation in pathobiological and diagnostic principles is the failure in their classification of the myelodysplastic syndromes (MDS) to adhere to the practice of diagnosing a cancer based on the presence of cancer cells in the tissue of interest. Cancers have varying degrees of cellular features of malignancy and progression at the time of diagnosis. These features are determined by assessing (a) the degree of dedifferentiation (aberrancy or deviation from normal cytomorphology) and tissue disorganization of the malignant cells (tumor grade) and (b) the anatomical distribution of the disease (tumor stage). The latter two features, grade and stage, aid in making prognostic approximations and determining therapeutic approaches. In any category of grade and stage, the diagnosis of the specific cancer is still made, based on its histopathological and cytoimmunochemical features. There is a notable and unfortunate exception to this rule: the disregard, over 40 years, of the diagnostic significance of leukemic myeloblasts in the diagnosis and classification of myelogenous leukemia. This may result, in part, from classical grade and stage designations being either inapplicable or of limited value when considering the myeloid leukemias.
The finding of malignant cells in the epithelial layer of the cervix is far less a threat to the patient than the presence of pancytopenia and 15% leukemic myeloblasts in her marrow, but we do not hesitate to call the former circumstance “carcinoma (in situ),” whereas we call the latter “anemia,” albeit modified by a preceding adjective and succeeding prepositional phrase. In hematologic parlance, one refers to the presence of lower proportions of leukemic blast cells in the marrow as “myelodysplasia” and, more specifically, as “refractory anemia” or “refractory anemia with excess (myelo)blasts,” or some more recently devised neologism [1]. Leukemic myeloblasts are sometimes phenocopies of normal myeloblasts by light microscopy, but they do not represent an excess of normal myeloblasts, and thus the term “excess (myelo)blasts” is without pathobiological foundation [2, 3].
The 20% Rule
What has led to the dysfunctional terminology “refractory anemia” when referring to what is (subacute, oligoblastic, smoldering, or pauciblastic) myelogenous leukemia? The designation “Les anémies réfractaires avec excés de myéloblastes dans la moelle” was first articulated by six French hematologists 43 years ago [4]. The choice of terms by these hematologists in 1970 was related to the early 20th century practice of calling unexplained anemias that did not respond to hematinics “refractory” and to a failure to consider the findings in pathobiological terms. Much earlier, in 1943, one French hematologist saw the matter of different manifestations and rates of progression of myelogenous leukemia insightfully. Paul Chevallier discussed the odo-leucoses with the intent of unifying them [5]. He chose the prefix “odo,” from the Greek δóς (“oudós,”) meaning “threshold,” to highlight that these disorders were on the threshold of polyblastic (acute) myelogenous leukemia. Chevallier proposed “leucoses” as the generic term for “leucémies” so that marked variations in the white cell count and other variable presenting features would not engender inappropriate terminology. It was a sage proposal, especially for that time, but, alas, it was ignored. The work was published in French; it had neither the advantage of an electronic journal database to provide worldwide awareness nor a mechanism to invoke its suggestion, such as hematology societies or international health organizations. The state of therapy for acute myelogenous leukemia (AML) was abysmal, and thoughts of classification were secondary. In addition, the world was at war. (A more detailed history of the recognition and description of the incipient and subacute myelogenous leukemias can be found elsewhere [2, 6].)
In 1975, to explore these matters further, a conference entitled “Hematopoietic Dysplasia (Preleukemic States),” hosted by Marcel Bessis and Jean Bernard, was held at the Institut de Pathologie Cellulaire on the campus of Hôpital de Bicêtre in Le Kremlin-Bicêtre, a suburb south of Paris, France [7]. Bessis and Bernard were elected to L'Académie des Sciences, largely as a result of their contributions to the hematological sciences. At this conference, Bernard Dreyfus, the lead physician who first used the designation “refractory anemia with excess myeloblasts” in his paper in 1970, presented his concept of this disorder in English, under the title, “Preleukemic States: Refractory Anemia With an Excess of Myeloblasts in the Bone Marrow (Smoldering Acute Leukemia)” [8]. His inclusion of the term “smoldering acute leukemia” indicated that he knew that this group of hematopoietic disorders was composed of myelogenous leukemias, that is, neoplasias and not dysplasias. The early use of the term “preleukemia” as a synonym for these neoplasms misled some basic scientists studying these disorders into believing they were studying the precursor to myelogenous leukemia, when the neoplastic transformation had already occurred. In this paper, Dreyfus gave a detailed description of 29 cases that displayed the typical clinical and cytomorphological findings of the subacute phase of myelogenous leukemia, and he described in some detail other series of patients and various nomenclature assigned to the patients' diagnoses. In the end, he begged the question and tried to make distinctions between myelogenous leukemia and refractory anemia with excess myeloblasts based on grounds that were weak at the time and indefensible today.
Bessis recognized that English was the lingua franca of science, so he expected the international participants to deliver and publish their papers in English. Accompanying the published papers were the discussions of each paper, an important part of the meeting and always published along with the articles. Initially, the papers from his conferences were published in English in the Nouvelle Revue Française d'Hématologie. The publishers informed Bessis that they would not continue to publish his symposia unless they were published in French; this was a period of the staunch defense of the French language from the incursion of English. In response, he started a new journal, Blood Cells, in 1975, for the principal purpose of publishing the papers and discussions from his symposia, held two to three times a year in Paris, in English. The first symposium published in Blood Cells was entitled, “Unclassifiable Leukemias,” and was published in April 1975. At that symposium, David Galton and John Dacie presented their influential paper on the classification of AML. The principal types were designated M0 through M6, and their discussion of the 238 cases on which their classification was based included their interpretation of the features of the seven morphological groups, and several subgroups, that determined their designations. Although others in attendance subsequently promoted it as the French-American-British classification, the ideas and the careful descriptions of the morphology that dictated the subclassifications were British, described by Galton and Dacie at the Royal Postgraduate Medical School in London, U.K. [9]. The French and American hematologists that marketed, and occasionally amplified, the classification were successful in that it was used for approximately 35 years until supplanted by the WHO classification in 2001. That classification reverted to the use of classical phenotypic, and a few prevalent genotypic, descriptive phrases.
The attendees at the symposium on hemopoietic dysplasias in September 1975 (published in June 1976) appeared to be misled by the innumerable variations in the expression of a neoplastic multipotential hematopoietic cell, resulting from the matrix of differentiation and maturation in each of eight blood cell lineages [10, 11]. They were so focused on the dysmorphia of cancer (not true dysplasia) in the red cell, granulocyte, and megakaryocyte-platelet lineages that they named the symposium, “Hematopoietic Dysplasias,” forever (apparently) assigning neoplastic diseases to the category of dysplastic diseases, an unfortunate pathobiological error. Recall that aplasia (hypoplasia), hyperplasia, metaplasia, dysplasia, and neoplasia are distinct pathological entities. Neoplasia receives its unique character by being the only tissue abnormality that is derived from the summation of somatic mutations and epigenetic modifications in a single tissue cell; the neoplasm is a clone, whereas true hematopoietic dysplasias may mimic the appearance of such a tissue change but they are polyclonal. They may result from an inherited germ-line gene mutation (e.g., congenital dyserythropoietic anemia) or, for example, from autoimmunity (e.g., pernicious anemia) as a consequence of the effects of a specific vitamin deficiency on blood cell morphology, which is truly dysplastic. In the late 19th and very early 20th centuries, the extreme marrow and blood cell dysplasia, pancytopenia, hypercellular marrow with a high marrow cell mitotic index, and patient fatality rate led some to consider pernicious anemia to be a neoplasm. It is a true MDS (polyclonal) without a propensity to progress to a hematopoietic neoplasm.
Bessis was a commanding, charming, erudite, and scholarly person with literary and artistic flare. He lived with his wife, Claude, in an elegant two-story apartment on the rue Saint Simon in Paris, just off the Boulevard Saint Germain. As a bibliophile, he had a large library with an extensive collection of books and antique microscopes, in the center of which was a carom billiard table to which he brought guests for a game or two after dinner. This game originated in France in the 18th century. The small apartment building itself was decorated with gargoyles, replicas of those on the face of the Château de Blois in the Loire valley. Bessis had a second home, a vacation home, in Honfleur, France, situated on the southern bank of the estuary of the Seine in lower Normandy. It is a town known for its extraordinarily picturesque and historic port and environs, various scenes of which were painted by Gustave Courbet, Eugène Boudin, and Claude Monet, among others, who formed the “école de Honfleur” early in the development of Impressionism. Bessis' atlas, Corpuscles, published in 1974 by Springer-Verlag, is a tribute to the beauty of shape and form as manifested by exquisite scanning electron micrographs of red cells. He was a respected blood and marrow cell morphologist and cell biologist, interested in structural and functional cellular aberrations, who pioneered the concept of studying single blood cells, highlighted in his text, Living Blood Cells and their Ultrastructure, published in 1972 [12]. He initiated these conferences by bringing together a small group of experimental hematologists from North America, Europe, Israel, and Japan, depending on the topic to be considered, which varied from the biophysical properties of blood cells to various clinical disorders of blood cells. He served as the host of the conferences, often sharing this duty with his mentor and colleague Bernard. On the occasion of this symposium on hematopoietic dysplasia, the discussion was focused on the dysmorphic features of this group of myeloid neoplasms and, although of diagnostic utility, resulted in overweighting the epiphenomena of the morphological dysmorphia of cancer when considering their essential nature.
One attendee of the conference saw the relationships among the syndromes being discussed in a more appropriate pathobiological context. Sven-Aage Killman, an insightful Danish hematologist from the Rigshospitalet in Copenhagen, Denmark, was invited to participate; he was a friend and colleague of Bessis, as were virtually all of the attendees. Killman wrote, “preleukemia per se does not exist but that preleukemic states with a rather high frequency sooner or later end in overt AML are actually true leukemias that, however, differentiate reasonably well. Another way of phrasing it is that preleukemic states are AMLs that present with partial and sometimes long-lasting remission, which only after months to years lose their differentiation ability and then are classified as AML” [13].
Toward the end of the conference, George Brecher from the University of California at San Francisco School of Medicine in San Francisco, California, Claude Sultan and Bernard Dreyfus from Hôpital Henri Mondor in Creteil, France, and John Senn from Sunnyside Hospital in Toronto, Canada, focused on “myeloid dysplasia,” “myelodysplasia,” or “myelodysplastic syndromes” as a designation for the conditions under discussion [14]. Others preferred the phrase “hemopoietic dysplasia” because of the ambiguity of the term “myeloid,” which can refer to marrow or to the granulopoietic cell series. The discussants either were not convinced by Killman's analysis or did not understand his important arguments. Their discussion also indicated that they did not understand the pathological designation “dysplasia” to which they were attracted because pathologists often considered it a preneoplastic condition in certain tissues, not recognizing or appropriately emphasizing that what they were calling “myelodysplasia” was not preneoplastic but was, in fact, a neoplasm (myeloneoplasia). The participants were more focused on classifying based on epiphenomena than on integrating the pathobiology of the myelogenous leukemias. This approach led to a long period of confusion among physicians, scientists, epidemiologists, and demographers. The U.S. National Cancer Institute did not recognize MDS as neoplasms in the Surveillance Epidemiology and End Results database until the 21st century, 75 years after their early description as “refractory anemias,” that is, those anemias not responding to iron or liver extract and without an another underlying associated disease that results in anemia [15].
Instead of referring to the syndromes as neoplasms, by using such terms as “clonal anemia,” “clonal bicytopenia,” or “clonal tricytopenia” in the absence of overt evidence of leukemic blast cells and “oligoblastic myelogenous leukemia” when leukemic blast cells were quantitatively evident in the marrow (greater than 2% myeloblasts by microscopy), they were called by pseudonyms, such as refractory anemia. The former approach would have been harmonious with cancer biology [2, 3]. In order to make a distinction between MDS and AML, an arbitrary upper boundary of 30% blast cells was chosen initially to separate refractory anemia with excess blasts (<30% leukemic blast cells) from AML (≥30% leukemic blast cells). That boundary had no apparent pathobiological rationale. Later, no substantial distinction was found in response to treatment or survival among patients diagnosed with AML and those designated refractory anemia presenting with 20%–29% leukemic blast cells in their marrow examination (so-called refractory anemia with excess blasts in transition). The boundary between refractory anemia and AML was decreased from 30% to ≥20% leukemic blast cells in the marrow at diagnosis. Like 30% blast cells, using 20% blast cells as a boundary at which to call the disease AML was both arbitrary and ambiguous because acute myelomonocytic leukemia, acute monocytic leukemia, acute promyelocytic leukemia, acute erythroid leukemia, and other rarer morphological variants of AML (e.g., acute eosinophilic or acute basophilic leukemia) may have <20% leukemic myeloblasts in the marrow.
The inconsistencies in classification are more apparent when one considers that chronic myelogenous leukemia is a myelogenous leukemia even though it may have <5% leukemic blast cells in the marrow and <1% in blood at the time of diagnosis. Some leukemias are so designated without any (or with a very slight) increase in marrow blast cells (e.g., chronic neutrophilic leukemia). The failure to consider cancer biology in the classification of clonal myeloid diseases seems resistant to modernization. In contrast, the physicians expert in lymphomas pathology, among the group designated this task for the WHO, have responded to a deeper understanding in pathobiology of lymphoma. They have moved from reticulum cell sarcoma (an imaginary cell type) to histiocytic lymphoma (an oxymoron, unless the cells were a true chimera of lymphocytes and myeloid [macrophage] cells) to diffuse large B-cell lymphoma (and its subsets) and have used other advanced concepts and techniques in the histopathological categorization of the lymphomas.
In an analysis of cases labeled as MDS with 10%–19% marrow blasts and cases of AML with 20%–29% blasts, no significant differences were found when hemoglobin concentration; white cell count; platelet count; cytogenetic risk category (normal karyotype, intermediate risk, or unfavorable risk categories); abnormalities of chromosome 5, 7, or complex karyotypes; or the frequency of eight prevalent mutations (e.g., NPM1, FLT3-ITD) were compared [16]. The probability of progression on multivariate analysis was related to the age of the patient at diagnosis and the cytogenetic risk stratification but not to the marrow blast percentage. Like a 30% boundary, no pathobiological justification was found for distinguishing refractory anemia with excess blasts (10%–19% blast cells) from AML (≥20% blast cells).
The term “refractory anemia” has shown remarkable staying power, perhaps for as long as a century as an unartful, misleading, and misunderstood diagnosis. One should teach students that refractory anemia and myelodysplasia are less advanced manifestations of myelogenous leukemia. This would introduce them to a unifying concept of the varied expression of neoplasia because of the marked variability in the penetrance of the many combinations of mutations and epigenetic modifications that dictate the phenotype and behavior of the clone. This effect is especially notable in the neoplastic transformation of a multipotential hematopoietic cell capable of differentiating into at least eight distinct lineages that can each mature, unpredictably, to different extents and thereby manifest morphological and immunocytological variability within each lineage. This complex matrix also can show wide degrees of biochemical and structural anarchy as well as innumerable cytologic patterns. No two are exactly alike. In essence, they are each varied manifestations of myelogenous leukemia, defined in this paper as the neoplastic transformation of a primitive multipotential hematopoietic cell, the deterministic mutations of which are multiple and variable and may have a range of penetrance. The concept of the spectrum of minimal to severe deviation tumors has a proud history in experimental oncology [17] and highlights the variable penetrance of oncogenic mutations in the same tissue and the impact of the further accumulation of cooperating mutations. Moreover, the appellation “refractory” is becoming a less accurate description because thalidomide derivatives, 5-azacytidine and decitabine, multidrug chemotherapy, and hematopoietic stem cell transplantation can treat refractory anemia in its various manifestations, with more to come, I anticipate.
The <5% Rule
Not only did hematologists conjure an arbitrary diagnostic boundary for the diagnosis of myelogenous leukemia—a marrow leukemic blast cell proportion ≥30% and then ≥20%—they also conjured an arbitrary boundary for the diagnosis of refractory anemia. The WHO committee on the classification of hematopoietic malignancies has continued the use of <5% marrow blast cells as representing refractory anemia (without excess blasts) at the time of diagnosis and ≥5% blast cells as representing a diagnosis of refractory anemia with excess blasts. In my experience, 3% or 4% myeloblasts in the marrow with hematologic lineage abnormalities, seen after infancy, represents myelogenous leukemia or a closely related neoplastic myeloid disease. The myeloblast proportion in marrow is a tightly regulated variable. In inflammatory states with striking neutrophilia, for example, the proportion of blast cells decreases in the marrow as a result of a marked expansion of the postblast myelocyte precursor population.
A large series of patients with MDS was found to have a difference in cumulative survival and risk of progression to polyblastic (acute) myelogenous leukemia if the diagnostic marrow contained 3%–4% blast cells compared with 0%–2% blast cells [18]. In this study, marrow blast percentage at diagnosis had a stepwise increased risk of shorter survival and progression to polyblastic myelogenous leukemia. Today, we use a quantitative variable (e.g., percent myeloblasts) to distinguish the various phenotypic manifestations of myelogenous leukemia. We are on the verge of reliably determining if 1% or 2% blasts in a marrow sample are leukemic blast cells at the time of diagnosis. In a clonal cytopenia, 1% or 2% blasts represents leukemic myeloblasts. They must be part of the clone in virtually all cases, unless the marrow is a mosaic of clonal and polyclonal hematopoiesis—a rare occurrence, especially in this age group. Multicolor flow cytometric analysis of the features of CD34+ cells in clonal cytopenias with low percentages of blast cells shows a very high frequency of aberrant antigen expression, indicating that the cells are abnormal (leukemic) blast cells. This approach to analysis has shown increasing reliability [19, 20].
From where did this boundary of <5% arise? In part, it is a reflection of digit preference. Using numbers other than those ending in 0 or 5 in making boundaries is egodystonic. We celebrate a 50th anniversary extravagantly but not the 57th, despite it being more impressive. We celebrated the “start” of the millennium on January 1, 2000, but the current millennium started on January 1, 2001. Unlike anniversaries, selecting 5% marrow myeloblasts as a boundary for the designation of refractory anemia with excess myeloblasts (myelogenous leukemia) has greater consequences.
C. Gordon Zubrod was a physician who had participated in the effort to develop and test malarial drugs to help decrease the large number of incapacities and deaths from malaria among allied troops in the Mediterranean and the Pacific theaters of operations during World War II. In 1954, he was recruited to the newly opened Clinical Center at the National Institutes of Health in Bethesda, Maryland, by James Shannon, a former colleague in the malaria program [21]. Shannon had become the scientific director of the National Heart Institute. The National Heart, Lung, and Blood Institute was founded as the National Heart Institute in 1948. Later, these additional disciplines were added (lung in 1969 and blood in 1976), sequentially, to the institute's name to reflect their growth as medical specialties and their enhanced research activities and, thus, the institute's broader mission. Shannon was subsequently appointed director of the National Institutes of Health in 1955.
Zubrod took on the task of spearheading the treatment of cancer. Some of the principles he used were derived from his experiences developing new drugs for malaria as part of the effort to protect troops in the Mediterranean and Asian theaters of war. During World War II, allied morbidity and mortality from malaria were enormous. He established a team of clinical and basic scientists to work on the problem and fostered the design of clinical trials to test new drug effectiveness. He focused on acute leukemia by setting up an acute leukemia task force. This choice was made because several drugs had appeared by the mid-1950s with cytocidal activity against leukemic cells, especially lymphoblastic leukemia, folic acid antagonists, amethopterin (methotrexate; the first antifol used was aminopterin to treat childhood acute lymphoblastic leukemia by Sidney Farber and his colleagues in 1947), and purine analogs (e.g., 6-mercaptopurine).
James Holland, a pioneer of anticancer drug development, was at the National Cancer Institute when Zubrod arrived. Holland had an interest in the treatment of acute leukemia. Holland soon left the institute to take up a position as the chair of the Department of Medicine A at Roswell Park Memorial Institute in Buffalo, New York. As an aside, he and his colleagues there combined 7 days of a continuous intravenous infusion of arabinosyl cytosine (cytarabine) and 3 days of intravenous daunorubicin for the treatment of AML in 1973. This combination has universally been referred to as the “7 + 3” regimen and is still the backbone of treatment for most cases of AML therapy 40 years later. The history of this development has been reviewed recently [22]. In addition, it was thought that measuring the effects of therapy might be easier following leukemia treatment by examining the blood and the marrow than by following the results of most solid tumor treatments, given the imaging tools of that time. There were also intriguing studies being conducted on the L1210 mouse leukemia model developed by Lloyd Law at the National Cancer Institute that provided early insights for optimizing drug dose schedules and, thereby, developing curative approaches to murine leukemia with chemotherapy. These schedules would be applied to human treatment.
The group's initial primary target was childhood acute lymphocytic leukemia. By the mid-1950s, they had three active drugs—methotrexate, glucocorticoids, and 6-mercatopurine—with vincristine soon to follow. Zubrod and his colleagues felt a definition of remission was required to quantify drug effects and to measure progress. Up to that time, patients were considered to improve if their blood leukemic blast cell count decreased with treatment, but there was no formal agreement on measuring the effects of drug therapy and no objective way to measure results at collaborating institutions. In August 1956, Holland, by then at Roswell Park Memorial Institute in Buffalo, Emil (Tom) Frei at the National Cancer Institute in Bethesda, and Joseph Burchenal at Memorial Hospital in New York City solicited opinions from several physicians who cared for patients with acute leukemia and presented a paper entitled, “Criteria for the Evaluation of Response to Therapy of Acute Leukemia,” at the International Society of Hematology meeting in Boston, Massachusetts. The three pioneering clinical scientists [21] defined a complete or “A1” remission as a post-treatment marrow that had <5% blast cells, a restoration of morphologically normal hematopoiesis, and near normal blood cell counts [23].
The concern about restricting remission to the normal marrow blast count (1 ± 0.4% standard deviation) was severalfold. Small numbers of reactive or large lymphocytes seen in the marrow after treatment, especially in the hypocellular marrow taken at the peak effect of chemotherapy, were very difficult to distinguish by light microscopy from a few percent residual leukemic blast cells. As important, the absence of effective methods of transfusing platelets made the risk of death from hemorrhage compelling with unduly prolonged intensive therapy. The antibiotics available to deal with infections during a period of severe neutropenia were very limited compared with today's arsenal. Consequently, one did not want to prolong the hypoplastic marrow phase after chemotherapy unless the need was unequivocal. Moreover, indwelling catheters, let alone small bore needles, had not been developed for infusions of blood, fluids, or medications. Unnecessary prolongation of treatment held great risk in the face of a marrow that was devoid of residual leukemic lymphoblast or myeloblast cells. Techniques for obtaining marrow biopsies were primitive and used infrequently, and a decision was made to consider the marrow devoid of residual leukemia if <5% of cells resembling lymphoblasts or myeloblasts were present. This breakpoint usually worked because inadequate therapeutic responses (refractory acute leukemia) often were accompanied by more evident residual blast cells in the marrow. This definition was useful principally in the management of childhood acute lymphoblastic leukemia because drugs had become available that could rid the marrow of blast cells. Later, when drugs became available that could place patients with AML in remission with some regularity, this 5% boundary continued to be applied to the treatment of AML without as strong a rationale. Again, it usually worked, fortuitously, because refractory leukemia usually had a higher residual blast cell count.
Importantly, in both types of acute leukemia, the <5% rule was intended to be used to evaluate the post-therapy marrow samples and had not been proposed as an interpretation of the significance of the marrow blast cell count at the time of diagnosis. Hence, the presence of 3% or 4% blast cells in the marrow at diagnosis or relapse (with blood cell lineage abnormalities (e.g., newly appearing anemia, neutropenia, thrombocytopenia) is another matter and usually indicates overt marrow leukemic hematopoiesis. Even 1% blast cells represents leukemic hematopoiesis in a clonal disorder, but in the absence of Chevallier's leucosis (see “The 20% Rule” section), calling a clonal cytopenia “leukemia” would be too dramatic for most physicians. The discussion with the patient should get very close to that explanation but should approximate the risk of progression based on several risk factors.
The Term “Acute” in the Classification of Myelogenous Leukemia
One of the disrupting variables in classification of the myelogenous leukemias is the implication of calling a disorder AML because this implies rapid progression and a very high risk of fatality, especially in the age group in which myelodysplasia is prevalent. When coined in the late 19th century, such terms were accurate. Acute myelogenous or acute lymphoblastic leukemia was fatal in weeks because there was no effective treatment. Although it is apparent that refractory anemia is a misnomer for a neoplasm but in most cases behaves in a more indolent fashion than AML, there is reluctance to use a term like “smoldering acute myelogenous leukemia,” as used parenthetically in Dreyfus' report in 1976. This issue led to institutionalized circumlocutions. The distinction in the prognosis implied by the usage of “acute” or “chronic” with regard to leukemia has been lost because of advances in therapy. Before the introduction of tyrosine kinase inhibitors, many patients with AML, especially in younger age groups, with favorable cytogenetics had a better prognosis than patients with BCR-ABL-positive chronic myelogenous leukemia. The significance of “acute” or “chronic” has waxed and waned with advances in management. Use of the terms “oligoblastic” and “polyblastic” myelogenous leukemia could help to mitigate the implications of “acute.”
Conclusion
The application of arbitrary boundaries or intervals is often provoked by the need to have harmonious comparisons in clinical trials. If multi-institutional participation is required, the comparability of cases is determined by certain criteria, such as leukemic blast percentages in marrow or blood, as for myelogenous leukemia. Unfortunately, such phenotypic criteria may lead to a misunderstanding of the pathophysiology of the disease because learning usually flows from academic centers. By using such criteria in the published studies of academic physicians, the arbitrary criteria come to be applied broadly to individual patients. In addition, the arbitrary boundaries used for multicenter comparability imply whether a patient with myelogenous leukemia should (a) receive ablative therapy (e.g., intensive chemotherapy and or hematopoietic stem cell transplantation), (b) receive something less than ablative therapy (e.g., gene-demethylating agents, which are in vogue but their method of action is uncertain), or (c) be observed for progression without specific therapy. Any of the three approaches may be used in a patient with myelogenous leukemia with a marrow leukemic myeloblast count of, for example, 4%, 14%, or 24%, depending on several other variables. In an individual case, the implications of arbitrary boundaries should be subjugated to reasoning on behalf of the patient.
One of the difficulties with the aberrant terms and ambiguous boundaries applied to MDS and myelogenous leukemia is the impact they have in educating and training hematological oncologists and pathologists. These young physicians should be learning the pathobiology of the diseases they will be asked to diagnose, treat, and manage. I am concerned when I sit in clinicopathological conferences and hear senior participants indicate (a) that a patient with pancytopenia and 17% myeloblasts in their marrow has an “anemia” (even if modified by “refractory” or “excess blasts”) and not myelogenous leukemia, (b) that the marrow in treated patients with myelogenous leukemia containing 4% myeloblasts reflects a remission, or (c) that patients with trilineage hematopoietic abnormalities compatible with myeloid leukemia and 4% blasts have refractory anemia, when one can be virtually certain that the blast cells are part of the clone and thus leukemic.
With experience, some physicians learn to subvert these inappropriate designations and ambiguous boundaries and think more insightfully about the case, using various features of a patient's demography and disease characteristics to determine the management most appropriate for the circumstance. For some or perhaps many, the current classifications are both distracting and stultifying. Their use in hundreds of papers on the topics by experts in the field encourages many to use these boundaries with undue rigidity. This reaction is especially so among scientists in several fields, notably basic scientists, demographers, and epidemiologists, who are not intimately familiar with the clinical features of myelogenous leukemia or the pathobiology of the myelogenous leukemias, as well as among interested lay parties (e.g., patients, medical journalists, toxic tort lawyers). Moreover, one is struck by the increased frequency with which the coupled abbreviation “MDS/AML” is being used in the medical literature in recognition of the abbreviations representing the same pathobiological entity.
The use of the term “myelodysplasia” in some circumstances also provides too broad an umbrella when combining clonal cytopenia with oligoblastic myelogenous leukemia; “MDS” in the abbreviation MDS/AML usually refers to the latter, not the former. The broad use of the abbreviation MDS also limits the interpretation of studies of “MDS and sAML” [24]. In the latter terminology, sAML refers to AML secondary to myelodysplasia; however, unlike secondary AML resulting from DNA-damaging therapy, it is not really secondary because the neoplastic transformation to myelogenous leukemia had already occurred. It is more accurately designated as “clonally evolved” AML. Moreover, some studies do not define what state of the spectrum of myelodysplasia is being studied. If one is studying a patient who has had stable clonal anemia for several years and progresses to AML, those mutational changes may be quite different from those of someone who has had tricytopenia and 13% marrow myeloblasts and evolves to the arbitrary boundary of ≥20% myeloblasts. The latter patient had (oligoblastic) myelogenous leukemia at diagnosis, and that degree of clonal evolution is different and perhaps biologically less complex. It would be interesting to see what, if any, additional mutations are acquired in the latter case, assuming the apparent progression was not, on occasion, sampling error of the marrow biopsy.
Clonal evolution from a clonal cytopenia to polyblastic myelogenous leukemia had been established by clinical and laboratory observations, cytogenetic analysis, and the use of X chromosome-linked genes in informative women for approximately 50 years. Now genomic analysis has described the process of clonal evolution from MDS to AML in terms of a specific clone and its subclones and their deterministic and cooperating gene mutations [24, 25]. A more sophisticated prognostic classification should develop when we are in a position to identify the contents of the oncogenome in all patients with clonal cytopenias and oligoblastic and polyblastic myelogenous leukemia by using facile, advanced methods of sequencing or high-density microarrays; by storing, transmitting, and interpreting that information efficiently; and by showing relevance to disease progression (driver vs. cooperating vs. passenger mutations), including the effect of gene interactions [25, 26]. This goal is made difficult by the heterogeneity and interactions of mutations responsible for hematopoietic multipotential cell neoplastic transformations and for clonal evolution [25]. From the patient's standpoint, it will be most useful when the oncogenotype can determine the choice of specific, effective therapy. For now, it would be better to talk about the phenotypes as clonal mono-, bi-, or tricytopenia if the quantitative increase in (leukemic) blast cells is very low (≤2.0%) and oligoblastic myelogenous leukemia, in lieu of refractory anemia with excess blasts, or polyblastic (acute) myelogenous leukemia (Table 1).
Table 1.
Proposed simplification of diagnostic classification of myelodysplastic syndromes
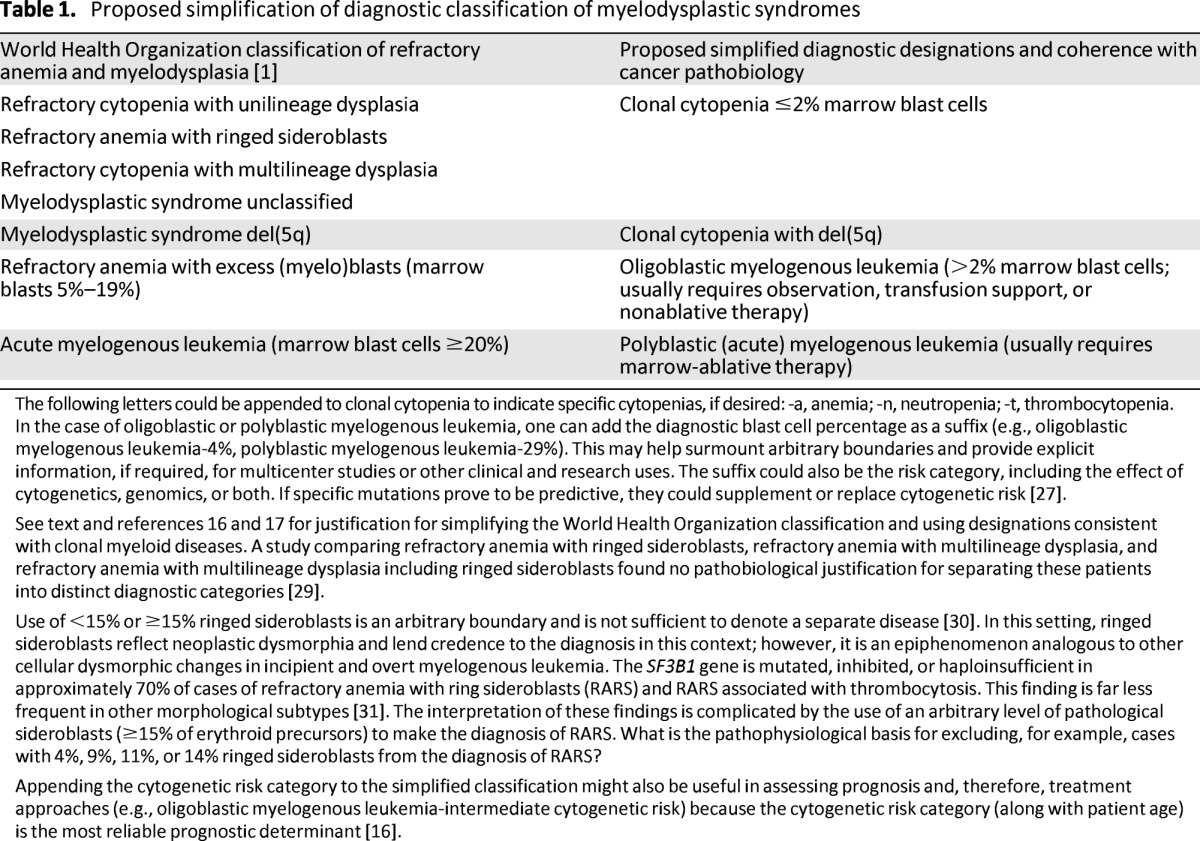
The following letters could be appended to clonal cytopenia to indicate specific cytopenias, if desired: -a, anemia; -n, neutropenia; -t, thrombocytopenia.
In the case of oligoblastic or polyblastic myelogenous leukemia, one can add the diagnostic blast cell percentage as a suffix (e.g., oligoblastic myelogenous leukemia-4%, polyblastic myelogenous leukemia-29%). This may help surmount arbitrary boundaries and provide explicit information, if required, for multicenter studies or other clinical and research uses. The suffix could also be the risk category, including the effect of cytogenetics, genomics, or both. If specific mutations prove to be predictive, they could supplement or replace cytogenetic risk [27].
See text and references 16 and 17 for justification for simplifying the World Health Organization classification and using designations consistent with clonal myeloid diseases. A study comparing refractory anemia with ringed sideroblasts, refractory anemia with multilineage dysplasia, and refractory anemia with multilineage dysplasia including ringed sideroblasts found no pathobiological justification for separating these patients into distinct diagnostic categories [29].
Use of <15% or ≥15% ringed sideroblasts is an arbitrary boundary and is not sufficient to denote a separate disease [30]. In this setting, ringed sideroblasts reflect neoplastic dysmorphia and lend credence to the diagnosis in this context; however, it is an epiphenomenon analogous to other cellular dysmorphic changes in incipient and overt myelogenous leukemia. The SF3B1 gene is mutated, inhibited, or haploinsufficient in approximately 70% of cases of refractory anemia with ring sideroblasts (RARS) and RARS associated with thrombocytosis. This finding is far less frequent in other morphological subtypes [31]. The interpretation of these findings is complicated by the use of an arbitrary level of pathological sideroblasts (≥15% of erythroid precursors) to make the diagnosis of RARS. What is the pathophysiological basis for excluding, for example, cases with 4%, 9%, 11%, or 14% ringed sideroblasts from the diagnosis of RARS?
Appending the cytogenetic risk category to the simplified classification might also be useful in assessing prognosis and, therefore, treatment approaches (e.g., oligoblastic myelogenous leukemia-intermediate cytogenetic risk) because the cytogenetic risk category (along with patient age) is the most reliable prognostic determinant [16].
Within these broader categories, further distinctions based on chromosomal risk category or oncogenetic changes could then be specified. The latter has been started in the current classification of polyblastic myelogenous leukemia with recurring translocations and in low risk clonal cytopenias, which have been shown to be have prognosis more precisely defined by examination of a prevalent set of gene mutations than by multivariable risk stratifications [27]. This approach or another similar method of communication would help bring hematology back into coherence with cancer biology and would make our nomenclature more understandable to scientists from other fields and to lay people.
The proposal shown in Table 1 is one suggestion, an approach we have proposed for many years [28]. The many experts in these disorders can improve the current classification if they can bring new ideas to the process and are open to considering the myelogenous leukemias in the broad framework of neoplasias, not dysplasias or refractory anemias.
Acknowledgments
I thank Jane L. Liesveld, M.D., Rochester, New York; Berelli Seshi, M.D., Los Angeles, California; and Jacob M. Rowe, M.D., Jerusalem, Israel, for reading the manuscript and making helpful suggestions. James F. Holland, M.D., New York, New York, vetted the section referring to his contribution to the definition of remission of acute myelogenous leukemia in 1956.
Disclosures
The author indicated no financial relationships.
References
- 1.International Agency for Research on Cancer. Myelodysplastic syndromes. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Geneva, Switzerland: World Health Organization; 2008. pp. 87–107. [Google Scholar]
- 2.Lichtman MA. Myelodysplasia or myeloneoplasia: Thoughts on the nosology of clonal myeloid diseases. Blood Cells Mol Dis. 2000;26:572–581. doi: 10.1006/bcmd.2000.0335. [DOI] [PubMed] [Google Scholar]
- 3.Lichtman MA. Language and the clonal myeloid diseases. Blood. 2002;99:725–726. doi: 10.1182/blood.v99.2.725. [DOI] [PubMed] [Google Scholar]
- 4.Dreyfus B, Rochant H, Sultan B, et al. Les anémies réfractaires avec excés de myéloblastes dans la moelle. Etude de 11 observations. Presse Med. 1970;78:359. [PubMed] [Google Scholar]
- 5.Chevallier P. Sur la terminologie des leucosis et les affections-frontiéres: Les odoleucoses. Sang. 1943;15:587–593. [Google Scholar]
- 6.Lichtman MA, Spivak JL. Commentary. In: Lichtman MA, Spivak JL, Boxer LA, et al., editors. Hematology: Landmark Papers of the 20th Century. Waltham, MA: Academic Press; 2000. pp. 285–286. [Google Scholar]
- 7.Bernard J, Bessis M. Hematopoietic dysplasias (preleukemic states) Blood Cells. 1976;2:5–364. [Google Scholar]
- 8.Dreyfus B. Preleukemic States. I. Definition and classification. II. Refractory anemia with an excess of myeloblasts in the bone marrow (smoldering acute leukemia) Blood Cells. 1976;2:33–55. [PubMed] [Google Scholar]
- 9.Galton DAG, Dacie JV. Classification of the acute leukaemias. Blood Cells. 1975;1:17–24. [Google Scholar]
- 10.Lichtman MA. The stem cell in the pathogenesis and treatment of myelogenous leukemia: A perspective. Leukemia. 2001;15:1489–1494. doi: 10.1038/sj.leu.2402247. [DOI] [PubMed] [Google Scholar]
- 11.Lichtman MA. The classification and clinical manifestations of the clonal myeloid diseases. In: Kaushansky K, Lichtman MA, Beutler E, et al., editors. Williams Hematology. 8th ed. New York, NY: McGraw-Hill; 2010. pp. 1211–1222. [Google Scholar]
- 12.Bessis M. Living Blood Cells and their Ultrastructure. Berlin, Germany: Springer-Verlag; 1973. [Google Scholar]
- 13.Killman S-A. Preleukemia. Does it exist? Blood Cells. 1976;2:81–105. doi: 10.1007/978-3-642-66312-3_6. [DOI] [PubMed] [Google Scholar]
- 14.Bessis M, Bernard J. Section IV. General discussion. I. Is preleukemic states an adequate designation? Blood Cells. 1976;2:347–351. [Google Scholar]
- 15.Rhoads CP, Barker WH. Refractory anemia: Analysis of 100 cases. JAMA. 1938;110:794–796. [Google Scholar]
- 16.Bacher U, Kern W, Alpermann T, et al. Prognosis in patients with MDS or AML and bone marrow blasts between 10% and 30% is not associated with blast counts but depends on cytogenetic and molecular genetic characteristics. Leukemia. 2011;25:1361–1364. doi: 10.1038/leu.2011.80. [DOI] [PubMed] [Google Scholar]
- 17.Lichtman MA. Understanding the mutational evolution of clonal cytopenias and oligoblastic myelogenous leukemia (“myelodysplasia”) Leukemia. 2013 Jul 11; doi: 10.1038/leu.2013.209. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 18.Germing U, Kündgen A. Prognostic scoring systems in MDS. Leuk Res. 2012;36:1463–1469. doi: 10.1016/j.leukres.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Ogata K, Kishikawa Y, Satoh C, et al. Diagnosticapplication of flow cytometric characteristics of CD34+ cells in low-grade myelodysplastic syndromes. Blood. 2006;108:1037–1044. doi: 10.1182/blood-2005-12-4916. [DOI] [PubMed] [Google Scholar]
- 20.Wood BL. Myeloid malignancies: Myelodysplastic syndromes, myeloproliferative disorders, and acute myeloid leukemia. Clin Lab Med. 2007;27:551–575i. doi: 10.1016/j.cll.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Laszlo J. The Cure of Childhood Leukemia: Into the Age of Miracles. New Brunswick, NJ: Rutgers University Press; 1995. [Google Scholar]
- 22.Lichtman MA. A historical perspective on the development of the cytarabine (7 days) and daunorubicin (3 days) treatment regimen for acute myelogenous leukemia: 2013 the 40th anniversary of 7 + 3. Blood Cells Mol Dis. 2013;50:119–130. doi: 10.1016/j.bcmd.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Holland JF, Frei ET, Burchenal JH. Criteria for the evaluation of response to therapy of acute leukemia. Proceedings of the International Congress: International Society of Hematolology, Boston (August 27, 1956); New York, NY: Grune & Stratton; 1958. pp. 213–214. [Google Scholar]
- 24.Walter MJ, Shen D, Shao J, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–1282. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter MJ, Shen D, Ding L, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscape. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bejar R, Stevenson KE, Caughey BA, et al. Val-idation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30:3376–3382. doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leisveld JL, Lichtman MA. Myelodysplastic syndromes (clonal cytopenias and oligoblastic myelogenous leukemia) In: Kaushansky K, Lichtman MA, Beutler E, et al., editors. Williams Hematology. 8th ed. New York, NY: McGraw-Hill; 2010. pp. 1249–1276. [Google Scholar]
- 29.Bacher U, Kern W, Alpermann T, et al. Prognoses of MDS subtypes RARS, RCMD and RCMD-RS are comparable but cytogenetics separates a subgroup with inferior clinical course. Leuk Res. 2012;36:826–831. doi: 10.1016/j.leukres.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Malcovati L, Porta MG, Pascutto C, et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: A basis for clinical decision making. J Clin Oncol. 2005;23:7594–7603. doi: 10.1200/JCO.2005.01.7038. [DOI] [PubMed] [Google Scholar]
- 31.Visconte V, Rogers HJ, Singh J, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012;120:3173–3186. doi: 10.1182/blood-2012-05-430876. [DOI] [PMC free article] [PubMed] [Google Scholar]