

Abstract
The molecular heterogeneity of human cancer cells at the level of signaling protein activities remains poorly understood. Using a panel of 64 colorectal (CRC) cancer cell lines the activity status of the MAP kinases Erk1 and Erk2 was investigated. Erk1/2 activity varied greatly within the CRC cell line panel and was not detectably associated with the speed of cell growth in 10 CRC lines analyzed. As expected, mutations in K-Ras or B-Raf were often, albeit not always, linked to high Erk1/2 activity. The phosphorylation of several known Erk1/2 targets investigated did not generally reflect Erk1/2 activity in the 10 CRC lines analyzed. However, the reduction of Erk1/2 activity with MEK inhibitors generally abolished cell growth but only led to an increase of cellular p27Kip1 levels in CRC cells with high Erk1/2 activity levels. The results indicate that high Erk1/2 activation is utilized by some CRC lines to override the cell cycle brake p27Kip1, while others presumably rely on different mechanisms in order to inactivate this important cell cycle brake. Such detailed knowledge of the molecular diversity of cancer cell signaling mechanisms may eventually help to develop molecularly targeted, patient-specific therapeutic strategies and treatments.
Findings
The limited knowledge about the heterogeneity of cancers on the signaling protein activity level is a major obstacle for better, individualized cancer therapies with signal transduction-modulating drugs. It is now well feasible to comprehensively analyze mutations and mRNA expression changes in tumor biopsies and isolated tumor cells with high-throughput techniques. By contrast, in-depth biochemical analyses of signaling protein activities are currently all but impossible with patient biopsy material. However, important insight into the individual diversity of cancers can be gained by analyzing large panels of cancer cells from a specific tumor type [1-3].
Erk1 and 2 are multifunctional kinases which are employed in a very wide range of normal and pathological cell types, in many cases in order to regulate cell proliferation or differentiation [4-6]. However, these Erks also play, for example, a role in the trans-endothelial migration of some CRC cells [7] and can promote angiogenesis and invasion [8,9]. The most studied signaling cascade engaging Erk1/2 is the Ras - Raf - MEK - Erk pathway that is transmitting the signals of numerous cell surface receptors. In many tumors, including CRC, Erk activation is linked to mutations of Ras GTPases or the S/T kinase B-Raf [10,11]. By contrast, cancer-related mutations in MEK1/2 and Erk1/2 appear to be very rare, although different germline mutations in MEKs have been recently reported in human cardio-facio-cutaneous disorders [12].
In this study we have analyzed 64 different CRC cell lines for the activity status of Erk1 and 2 (for origins of cells see Additional file 1). The aim was to define how Erk1/2 activity varies in different CRC cells and what the functional consequences are, if any. Initially, total cell lysates were generated (detailed methods provided in Additional file 2) and analyzed by western blotting for Erk1/2 activation using a phosphoepitope-specific antibody. This clearly showed a striking heterogeneity in Erk1/2 phosphorylation on the Thr202/Tyr204 epitope, a well-established indicator of Erk1/2 kinase activity levels (Figure 1). Heterogeneity in the activation of Erk1 versus Erk2 was also observed. Aberrant migration of phospho-Erk1 was observed in one cell line (CoCM-1), but this was not investigated further, since many proteins in this cell line display an unexpected size (data not shown), arguing for a more general defect in the protein expression or processing machinery, which is independent of Erk1. To study the causes and functions of different Erk1/2 activity levels in CRC, 10 cell lines, 5 with high and 5 with low Erk1/2 phosphorylation, were selected for further analyses.
Figure 1.

Phosphorylation of Erk1/2 in 64 CRC cell lines on its key regulatory epitope. Equal amounts of total cell RIPA protein extracts were analyzed by western blotting with anti-pT202/pY204 (for human Erk1; corresponds to pT183/pY185 in Erk2), which is well established to reflect the activation state of Erk1/2. C10 extract from the same batch is included on each blot for standardization. Note that numerous proteins from CoCM-1 cells display aberrant gel migration for reasons unknown to us (T.K. and S.F., unpublished data).
Ras GTP-loading assays and data base searches http://www.sanger.ac.uk/genetics/CGP/CellLines indicated that 4 of 5 lines with high pErk1/2 contain a mutation in the KRAS gene (Figure 2). The fifth cell line, Colo 741, is mutated in BRAF (V600E). Interestingly, LS 174T cells show constitutively elevated Ras·GTP levels and harbour a KRAS(G12D) mutation but display low Erk1/2 activity. This is indicative of additional factors like, for example, protein phosphatases that can substantially affect Erk1/2 activity levels. Several other cell lines in the panel known to have mutations in the KRAS gene (e.g. Colo 320DM, SK-CO-1, SNU-C2B, SW403, SW620, SW837, SW1116) or BRAF (e.g. HT-29, LS411, RKO) also display low Erk activity; see also http://www.sanger.ac.uk/genetics/CGP/CellLines), further supporting a key role for additional modifiers in determining the activity of Erk1/2 within a subset of CRC cell lines.
Figure 2.

Comparison of Erk activities with Ras·GTP loading and known mutations in KRAS and BRAF. Ten CRC lines selected for particularly high or low Erk1/2 activity from the panel in Figure 1 were grouped and analyzed again for Erk1/2 activation (top panel) and total Erk1/2 expression levels (second panel), as well as Ras activation (third panel) in total cell protein extracts. Ras activation is measured by Ras·GTP loading using GST-Raf-RBD(1-149) as activation-specific precipitation probe. The list at the bottom summarizes publicly available data on the mutational status of the KRAS and BRAF genes in these CRC cell lines www.sanger.ac.uk/genetics/CGP/CellLines. Cell lines written in red show high Erk1/2 activity, cell lines in black contain low Erk activity.
The total Erk1/2 levels are similar in all 10 cell lines. Unexpectedly, the apparent activity of MEK1/2, analyzed by western blotting with a pMEK1/2 (pS217/pS221) antibody, did not correlate well with Erk1/2 activity (data not shown). We are at present unable to provide a molecular explanation for this, but a potential reason could again be the impact of Erk phosphatases, such as those of the MKP family.
Comparing the growth rates in the 10 cell lines failed to show any correlation between Erk1/2 activity and proliferation speed (Figure 3), initially suggesting to us that relatively low levels of Erk1/2 activation may be sufficient to drive basal cell proliferation in most, if not all, CRC cells. The high levels of Erk activity observed in some CRC lines could thus be random fluctuations without functional consequences, or they may serve other functions.
Figure 3.
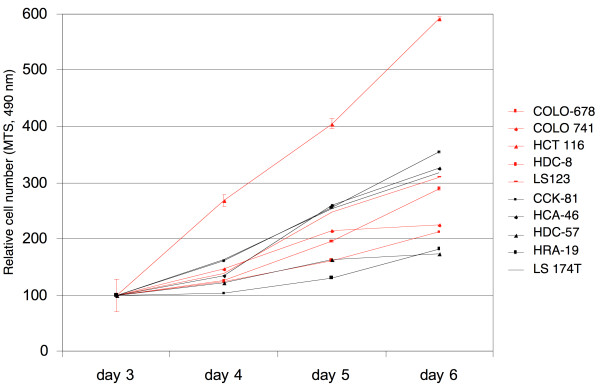
Growth of ten selected CRC cell lines with either high or low Erk activity analyzed by MTS assay. Cells were cultured in an excess of growth medium with 10% FBS which was replaced daily to avoid growth reductions due to medium depletion. Cell lines labeled in red contain high Erk1/2 activity, while cell lines shown in black possess low Erk activity. For clarity, representative error bars are only shown for the fast growing HCT 116 cells, but omitted for all other cells lines.
In order to address the latter possibility, selected Erk substrates and targets, including Elk1, Msk1, Myc and p90Rsk, were analyzed with phosphoepitope specific antibodies in the 10 CRC lines. It is not possible to perform a comprehensive analysis at this point, since more than 160 substrates and targets of Erk have been reported so far [13] and phospho-specific antibodies are not yet available for many of these targets. In all cases initially tested we failed to detect a perfect correlation between Erk activation and phospho-levels of potential target proteins (data not shown), suggesting to us that probably none of these proteins is an essential substrate for all CRC lines with high Erk1/2 activity. Clearly, this does not preclude a functionally important role of the phosphorylation of these proteins by Erk1/2 in individual cases.
p27Kip1 is an important cell cycle regulatory protein previously discussed as a direct or indirect target of Erk1/2, with phosphorylation leading to its proteasomal destruction [14-16]. Therefore, elevations of p27Kip1 phospho-levels may be difficult to monitor in steady state, but the inhibition of Erk1/2 may lead to a change in p27Kip1 abundance which would be expected to be easily detectable.
Indeed, inhibiting Erk activation through the potent MEK1/2 inhibitor U0126, led to an increased expression of p27Kip1 in all 5 CRC lines with high Erk activity and none of the lines with low Erk activity (Figure 4). HRA-19 cells, which have slightly higher Erk activity than the rest of the 'low Erk' cell lines display a subtle effect on p27Kip1 expression when treated with U0126.
Figure 4.

Inhibition of Erk1/2 activation increases expression of p27Kip1 in CRC cells with high basal Erk1/2 activity. Cells were treated with increasing doses of the MEK1/2 inhibitor U0126 or the compound solvent DMSO at the highest concentration used (m) for 48 hours and then harvested and analyzed by western blotting. Note that exposure times of blots for individual cell lines are adjusted to obtain signals of useful strengths. A Erk expression and activity levels upon U0126 treatment of 5 cell lines with high basal Erk activity (left panels) or low basal activity (right panels). B Effect of U0126 treatment of CRC cells on the expression levels of the cell cycle regulator p27Kip1.
The simplest explanation for these findings is that a subset of CRC cells utilizes the strong activation of Erk1/2 to down-regulate p27Kip1 expression. This may be mediated by targeting the Thr187 residue on p27Kip1, which is linked to its ubiquitinylation and degradation [14-16].
Despite these interesting results, we can, of course, by no means exclude the possibility that high Erk1/2 activity is also required to phosphorylate other key substrates in this subset of 'high Erk' CRC cells.
Erk1 and Erk2 boast a plethora of known substrates [13] and very likely more remain to be detected. In many cases, the subcellular distribution and expression levels of both, Erk kinases and their potential substrates, could determine which substrates are actually becoming phosphorylated in a specific cell or disease context. What is a critical Erk substrate in each individual cancer may thus depend greatly on the specific genetic composition of that particular tumor.
Nevertheless, from our study it would appear that down-regulation of p27Kip1 expression is at least a common, if not ubiquitous occurrence in CRC cells with high Erk1/2 activity. Similar findings have been reported in other tumor types [17]. In some genetic contexts, however, p27Kip1 may not need to be inactivated for tumors to develop, and could possibly even take on oncogenic functions according to recent results [18,19].
Interestingly, the absolute expression levels of p27Kip1 do not correlate with Erk activity levels (Additional file 3). Several reasons could explain this finding. Firstly, p27Kip1 may be differentially localized in different CRC cells, allowing in some cases only a portion of p27Kip1 to act as a cell cycle break that requires counter-action by high Erk activity. Secondly, the individual variability of the overall genetic composition of each tumor cells could lead to distinct levels of p27Kip1 being tolerated before a prominent effect on the cell cycle machinery is elicited.
It is presently unclear whether p27Kip1 is commonly a direct Erk target in CRC cells with high Erk activity, although phosphorylation of p27Kip1 by Erk1/2 in CRC has been suggested and complex formation of endogenous p27Kip1 and Erk1/2 was detected in LoVo cells by co-immunoprecipitation [20]. Indirect mechanisms of p27Kip1 regulation by Erk have also been reported [21].
p27Kip1 downregulation through high Erk activity levels is only one molecular route to eliminate the normal function of this important cell cycle regulator [22,23]. Many CRC cells of the analyzed panel with low Erk1/2 activity have presumably found alternative means to accomplish this task. This does, of course, not imply that those cells do not require Erk activity; in fact, as discussed above, inhibiting Erks close allies, MEK1/2, clearly reduces Erk activity and inhibits proliferation in these cells (data not shown).
We also observed that U0126 treatment of CRC cells led to marked morphology changes in some of the cell lines (see Additional file 4) further supporting multiple functional roles of MEK1/2 and, presumably, Erk1/2.
The results reported here imply that different Erk signal intensities are used in CRC cells to accomplish distinct molecular tasks, an observation that was also made in a previous study analyzing the activity of Src family kinases in this cell line panel [3]. Understanding the importance of different signal strengths in individual cancers is not only of interest to learn more about these still poorly grasped diseases, it may eventually also impact on the therapeutic use of signal transduction-modulating drugs. In the case of p27Kip1, which is not usually mutated in cancers, the inhibition of its proteasomal degradation could become a useful therapeutic option for some CRC patients, including those with constitutively high Erk activity.
List of Abbreviations
CRC: colorectal cancer; Erk: Extracellular signal-regulated kinase; MEK: MAP kinase (MAPK) or Erk kinase; U0126: a MEK inhibitor compound.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TK designed and conducted most experiments, analyzed data and wrote the manuscript. TR co-supervised TK, analyzed data and wrote the manuscript. SF conceived the project, designed and conducted experiments, analyzed data and wrote the manuscript. All authors read and approved the final version of the manuscript.
Supplementary Material
Origin of CRC cell lines.
Methods description.
Expression levels of p27Kip1 in 10 CRC cell lines with high or low Erk activity.
Effect of the MEK1/2 U0126 on the morphology of HCT 116 cells.
Contributor Information
Theresia R Kress, Email: theresia.kress@biozentrum.uni-wuerzburg.de.
Thomas Raabe, Email: thomas.raabe@uni-wuerzburg.de.
Stephan M Feller, Email: stephan.feller@imm.ox.ac.uk.
Acknowledgements
We are grateful to Walter Bodmer and many other colleagues for providing cell lines, and to Cancer Research UK, Heads Up and Oxford University for financial support. TK was kindly supported by the Bayerische Forschungsstiftung (Munich, Germany).
References
- Aleksic T, Feller SM. Gamma-secretase inhibition combined with platinum compounds enhances cell death in a large subset of colorectal cancer cells. Cell Commun Signal. 2008;8:8. doi: 10.1186/1478-811X-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A. et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;8:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Emaduddin M, Bicknell DC, Bodmer WF, Feller SM. Cell growth, global phosphotyrosine elevation, and c-Met phosphorylation through Src family kinases in colorectal cancer cells. Proc Natl Acad Sci USA. 2008;8:2358–2362. doi: 10.1073/pnas.0712176105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;8:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Jivan A, Shao C, Duan L, Goad D, Zaganjor E, Osborne J, McGlynn K, Stippec S, Earnest S. et al. The roles of MAPKs in disease. Cell Res. 2008;8:436–442. doi: 10.1038/cr.2008.37. [DOI] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;8:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Tremblay PL, Auger FA, Huot J. Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene. 2006;8:6563–6573. doi: 10.1038/sj.onc.1209664. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Oliva K, Wang Y, Quinn M, Rice G. Downregulation of urokinase plasminogen activator receptor expression inhibits Erk signalling with concomitant suppression of invasiveness due to loss of uPAR-beta1 integrin complex in colon cancer cells. Br J Cancer. 2003;8:374–384. doi: 10.1038/sj.bjc.6601098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;8:322–327. doi: 10.1016/S1470-2045(05)70168-6. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;8:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Hubbard SR. Oncogenic mutations in B-Raf: some losses yield gains. Cell. 2004;8:764–766. doi: 10.1016/S0092-8674(04)00256-9. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;8:1287–1290. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. pp. 21–44. [DOI] [PubMed]
- Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;8:321–324. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;8:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Gitig DM, Koff A. Cell-free degradation of p27(kip1), a G1 cyclin-dependent kinase inhibitor, is dependent on CDK2 activity and the proteasome. Mol Cell Biol. 1999;8:1190–1201. doi: 10.1128/mcb.19.2.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of RAF-->MEK-->ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1. Cancer Res. 2005;8:4870–4880. doi: 10.1158/0008-5472.CAN-04-2848. [DOI] [PubMed] [Google Scholar]
- Besson A, Hwang HC, Cicero S, Donovan SL, Gurian-West M, Johnson D, Clurman BE, Dyer MA, Roberts JM. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev. 2007;8:1731–1746. doi: 10.1101/gad.1556607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickeleit I, Zender S, Kossatz U, Malek NP. p27kip1: a target for tumor therapies? Cell Div. 2007;8:13. doi: 10.1186/1747-1028-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang C, Wu Y, Liu X, Cao X. Ca2+/Calmodulin-dependent Protein Kinase II Promotes Cell Cycle Progression by Directly Activating MEK1 and Subsequently Modulating p27 Phosphorylation. J Biol Chem. 2009;8:3021–3027. doi: 10.1074/jbc.M805483200. [DOI] [PubMed] [Google Scholar]
- Hu Y, Wang X, Zeng L, Cai DY, Sabapathy K, Goff SP, Firpo EJ, Li B. ERK phosphorylates p66shcA on Ser36 and subsequently regulates p27kip1 expression via the Akt-FOXO3a pathway: implication of p27kip1 in cell response to oxidative stress. Mol Biol Cell. 2005;8:3705–3718. doi: 10.1091/mbc.E05-04-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blain SW, Scher HI, Cordon-Cardo C, Koff A. p27 as a target for cancer therapeutics. Cancer Cell. 2003;8:111–115. doi: 10.1016/S1535-6108(03)00026-6. [DOI] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Origin of CRC cell lines.
Methods description.
Expression levels of p27Kip1 in 10 CRC cell lines with high or low Erk activity.
Effect of the MEK1/2 U0126 on the morphology of HCT 116 cells.