

Abstract
A hallmark of hyperoxic acute lung injury is the influx of inflammatory cells to lung tissue and the production of proinflammatory cytokines, such as IL-1β; however, the mechanisms connecting hyperoxia and the inflammatory response to lung damage is not clear. The inflammasome protein complex activates caspase-1 to promote the processing and secretion of proinflammatory cytokines. We hypothesized that hyperoxia-induced K+ efflux activates the inflammasome via the purinergic P2X7 receptor to cause inflammation and hyperoxic acute lung injury. To test this hypothesis, we characterized the expression and activation of inflammasome components in primary murine alveolar macrophages exposed to hyperoxia (95% oxygen and 5% CO2) in vitro, and in alveolar macrophages isolated from mice exposed to hyperoxia (100% oxygen). Our results showed that hyperoxia increased K+ efflux, inflammasome formation, release of proinflammatory cytokines, and induction of caspase-1 and IL-1β cleavage both in vitro and in vivo. The P2X7 agonist ATP enhanced hyperoxia-induced inflammasome activation, whereas the P2X7 antagonist, oxidized ATP, inhibited hyperoxia induced inflammasome activation. In addition, when ATP was scavenged with apyrase, hyperoxia-induced inflammasome activation was significantly decreased. Furthermore, short hairpin RNA silencing of inflammasome components abrogated hyperoxia-induced secretion of proinflammatory cytokines in vitro. These results suggest that hyperoxia induces K+ efflux through the P2X7 receptor, leading to inflammasome activation and secretion of proinflammatory cytokines. These events would affect the permeability of the alveolar epithelium and ultimately lead to epithelial barrier dysfunction and cell death.
Oxygen therapy is one of the most widely used clinical interventions to counteract insufficient pulmonary oxygen delivery in patients with severe lung injury. However, prolonged exposure to hyperoxia leads to inflammation and acute lung injury (ALI) (1–3). While the molecular mechanisms that activate inflammation in response to inhaled oxygen are not well described, the proinflammatory cytokine IL-1β is known to be one of the most biologically active cytokines in the lungs of patients with ALI (4). IL-1β is produced by activated macrophages and epithelial cells and leads to the subsequent production of chemokines and inflammatory cytokines, such as TNF-α, IL-6, or matrix metalloproteinases (4). It has been recently reported that caspase-1–mediated processing of IL-1β is mediated by the inflammasome (5).
The NALP3 inflammasome is a multiprotein complex, which contains NALP3, the caspase recruitment domain-containing protein Cardinal, apoptosis-associated speck-like protein (ASC) and caspase-1 (6). The NALP3 inflammasome protein complex has been implicated in sensing reactive oxygen species (ROS) stress (7). NALP3-inflammasome assembly is activated by K+ efflux through the P2X7 receptor (8) by inducing agents such as extracellular ATP, hypotonic stress, or toxins associated with cell injury. NALP3-inflammasome activation leads to secretion of the proinflammatory cytokine IL-1β. The factors that are critical for the assembly and activation of the inflammasome and subsequent secretion of IL-1β remain unknown (9–11). The role of the inflammasome in hyperoxic lung injury is also presently unknown. This study focuses on the mechanisms of inflammasome activation by hyperoxia and the contributions of inflammasome activation to lung injury and alveolar epithelial permeability.
Materials and Methods
Cells and reagents
ATP and oxidized ATP (oxATP) were purchased from Sigma-Aldrich (St. Louis, MO). N-acetyl-l-cysteine (NAC) and apyrase were purchased from Calbiochem (La Jolla, CA). Abs for immunoblotting included anti–caspase-1, NALP3, ASC, proIL-1β, and IL-1β–17 (Cell Signaling Technology, Beverly, MA).
Exposure to hyperoxia
Mice, aged 4–6 wk, were placed in cages in an airtight chamber (50 × 50 × 30 cm) and exposed to 100% oxygen for 72 h. The oxygen concentration in the chamber was monitored with an oxygen analyzer (Vascular Technology, Chelmsford, MA) as described previously (1, 12–14).
Isolation of mouse lung macrophages
Alveolar macrophages were isolated from mice as described previously (15). Lungs were perfused at 37°C with buffer A (1% NaCl, 0.1% glucose, 25 mM HEPES, 5 mM KCl, 0.1 mg/ml streptomycin sulfate, 0.07 mg/ml penicillin G, 0.05 mg/ml EGTA, 5 mM Na2HPO4, 5 mM NaH2PO4, pH 7.4). The lungs were then lavaged five times at 37°C using buffer B (buffer A plus 5 mM MgSO4 and 3 mM CaCl2). The lung tissue was digested with 7–8 ml elastase (3 U/ml in buffer A) at 37°C for 15 min. This process was repeated twice. After chopping the lung tissue, the cell suspension was mixed with 100 μg/ml DNase I, incubated for 5 min at 37°C with gentle rotation, and filtered through 160-μm and 37-μm nylon mesh once, followed by filtering through 15-μm nylon mesh twice. The cells were then incubated in bacteriologic 100-mm polystyrene Petri dishes which were precoated with mouse IgG (1 mg mouse IgG per dish) at 37°C for 3 h. The macrophages were detached by scraping from the culture dish, centrifuged at 200 × g for 10 min, and resuspended with 10 ml buffer C (RPMI 1640 medium containing 25 mM HEPES and 1% FBS) at a concentration of 15 × 106 cells/ml. The activation of the NALP3 inflammasome is a two-step process requiring a priming step (signal 1), which can include a number of infectious and noninfectious stimuli, followed by an activation step (signal 2). Exposure of monocytes and macrophages to primary inflammatory stimuli, such as LPS, will stimulate proIL-1β production, but little secretion of IL-1β will occur in the absence of a secondary stimulus (16). LPS priming is a known activator of inflammasome complex and was used as the priming step for all in vitro experiments (17).
Bronchoalveolar lavage
Following anesthesia, a median sternotomy was performed. The trachea was dissected free from the underlying soft tissues, and a 0.6-mm tube was inserted into the trachea through a small incision. Bronchoalveolar lavage (BAL) was performed as described earlier (1, 14). The BAL was repeated three times in each mouse and the cell-free BAL fluid was stored at −70°C.
SDS-PAGE and Western blotting
Cells were harvested, washed twice with ice-cold PBS containing 1 mM sodium orthovanadate and 1 mM sodium fluoride, and lysed with ice-cold RIPA lysis buffer (PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 10 μg/ml leupeptin, 1 mM sodium orthovanadate). Cell lysates were clarified by centrifugation at 10,000 × g for 15 min, and supernatant protein concentrations were determined with a Bradford protein assay kit (Bio-Rad, Hercules, CA). Protein lysates were prepared for SDS-PAGE by adding an equal volume of 6× SDS-PAGE sample buffer (100 mM Tris-Cl, pH 6.8, 200 mM DTT, 4% SDS, 0.2% bromophenol blue, 20% glycerol) and heating the mixture at 100°C for 3 min. Twenty micrograms of protein was separated via SDS-PAGE and transferred onto a polyvinylidene difluoride membrane by electrophoresis (Immobilon P; Millipore, Bedford, MA). After blocking with TBS Tween buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween-20) containing 5% nonfat dry milk for 1 h at room temperature, the membranes were incubated with blocking solution containing the indicated Ab (diluted 1:1,000–1:2,000) overnight at 4°C. Membranes were washed and incubated with a suitable HRP conjugated secondary Ab (Jackson ImmunoResearch Laboratories, West Grove, PA). HRP activity was detected using an ECL kit according to the manufacturer’s instructions (Pierce, Rockford, IL) (12, 13).
Coimmunoprecipitation
Coimmunoprecipitation analysis was preformed as described previously (12, 13, 18). Macrophage lysates were collected with a buffer containing 2% CHAPS; 500 μg protein lysate was immunoprecipitated with 1 μg anti-NALP3, anti-ASC, or anti–caspase-1 Ab and 50 μl protein A/G conjugated agarose beads. The resulting immunoprecipitates were dissolved in SDS-PAGE sample buffer for electrophoresis and immunoblot analysis as described earlier (12). These immunoprecipitates were used to detect the presence of ASC or NALP3.
Quantification of ATP in cell culture supernatant
A bioluminescence assay with recombinant firefly luciferase and its substrate D-luciferin (Molecular Probes, Eugene, OR) was used to detect and quantify the ATP released into cell culture supernatants according to the manufacturer’s instructions (Pierce, Rockford, IL). The transmitted light was detected with a luminometer (Berthold Systems, Aliquippa, PA).
Intracellular K+ concentration
Macrophages were seeded at a density of 1 × 106 cells per well in 12-well tissue culture dishes and maintained for 24 h. The next day, the medium was removed at the indicated time points and the cells were washed once with PBS. Cells were lysed in 1 ml of 10% nitric acid. The extracellular (medium) and the intracellular K+ content was quantified using atomic absorbance spectroscopy (19).
YO-PRO-1 measurements by flow cytometry
YO-PRO-1(Molecular Probes, Carlsbad, CA) is a fluorescent nucleic acid dye, which exhibits significant fluorescence upon binding to RNA and DNA. YO-PRO-1 passes through P2X7 receptor channels of live cells (19), but not other members of the cyanine monomer family. The uptake of YO-PRO-1 was detected by flow cytometry by detection of the shift in fluorescence intensity.
Caspase-1 activity assay
Caspase-1 activity was analyzed using the fluorogenic substrate Ac-WEHD-AMC (Alexis Biochemicals, San Diego, CA) and a caspase 1 fluorometric assay kit with the manufacturer’s instructions (20).
Quantification of cytokines by ELISA
IL-1β, TNF-α, and IL-6 cytokines were analyzed in lung homogenates or extracellular culture media with a commercially available ELISA kit (Pierce, Rockford, IL) according to the manufacturer’s instructions.
Measurement of transepithelial albumin flux
Alveolar type II (ATII) cells were isolated as described earlier (21) and grown in 24-well transwell plates until the cellular monolayers reached confluence. THP-1 cells (1.5 × 105) were added to the appropriate ATII monolayers, cocultured for 3 h, and exposed to hyperoxia for 1 h. In some experiments, THP-1 cells were transfected with control short hairpin RNA (shRNA) or NALP3 shRNA and then added to monolayer of ATII cells. Transepithelial albumin flux across each monolayer was determined by the addition of 0.05 μCi [125I]-labeled human serum albumin to each upper compartment and incubated for 1 h, after which the contents from the lower compartment were collected and counted in a Wizard γ-counter (Perkin-Elmer, Waltham, MA) as described previously (4).
Statistical analysis
All the experiments were conducted in triplicate. When appropriate, data were expressed as mean ± SEM. Data sets were examined by one-way and two-way ANOVA tests, and individual group means were then compared using the Student unpaired t test.
Results
Effect of hyperoxia on ATP efflux by alveolar macrophages
ATP activates the NALP3 inflammasome through P2X7 receptor mediated K+ efflux (9, 19). To determine whether hyperoxia exposure induces ATP efflux, we exposed alveolar macrophages to hyperoxia (95% O2) and analyzed the extracellular ATP concentration (Fig. 1). The experiment was performed in the presence or absence of serum to control for the contribution of serum growth factors to ATP generation (medium without serum had no growth factors added). The extracellular ATP concentration 30 min after the initiation of hyperoxia exposure was 0.07 ± 0.01 nM/106 cells in 21% O2 and 15 ± 0.02 nM/106 cells in 95% O2 in the presence of serum (Fig. 1). Accumulation of ATP in the extracellular medium was maximal at 60 min, 25.03 ± 2.00 nM/106 cells in 95% O2 compared with 0.08 ± 0.01 nM/106 cells in 21% O2 (Fig. 1). After 60 min, the ATP concentration decreased (data not shown). This time frame is based on our previous finding that exposure of HUVECs to 60 min of oxidant stress (H2O2) lead to mitochondrial fragmentation (14).
FIGURE 1.
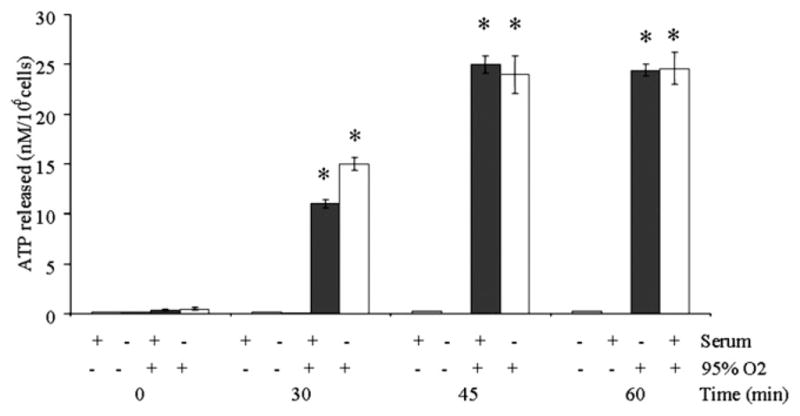
Hyperoxia exposure leads to release of ATP into extracellular media. Macrophages were isolated from C57BL/6 mice and exposed to hyperoxia (95% O2), and the ATP concentration was measured in the media. Experiments were conducted in the presence or absence of serum to control for the effects of growth factors. Results are presented as the mean ± SEM (*p < 0.05, compared with normoxic controls). Data shown represent one experiment (n = 4 percondition). Similar results wereobtainedin five separate experiments.
Hyperoxia induced K+ efflux
Recent reports suggest that oxidative stress increases K+ efflux by pancreatic islets (22). K+ efflux induces pore formation through stimulation of the P2X7 receptor, which is essential for caspase-1 activation and IL-1β processing. To determine whether hyperoxia decreases cytoplasmic [K+], we measured the intracellular K+ levels in macrophages in the presence or absence of hyperoxia exposure. The baseline cytoplasmic K+ levels in macrophages were 400 mmol per well in the absence of hyperoxia (Fig. 2A). Following exposure to hyperoxia, there was an 80% decrease in the level of cytoplasmic K+. To verify these results, we compared cytoplasmic K+ levels in macrophages stimulated with or without LPS followed by a short pulse with ATP as positive control. There was ~70% decrease in the intracellular K+ concentration after stimulation with ATP. The depletion of cytoplasmic K+ was detected within 20 min and was maintained for 60 min after ATP stimulation. In addition, the baseline extracellular K+ levels in media were 100 mmol per well in the absence of hyperoxia. We found that exposure to hyperoxia lead to a 3-fold increase in the level of K+ levels in the extracellular media (Fig. 2B).
FIGURE 2.
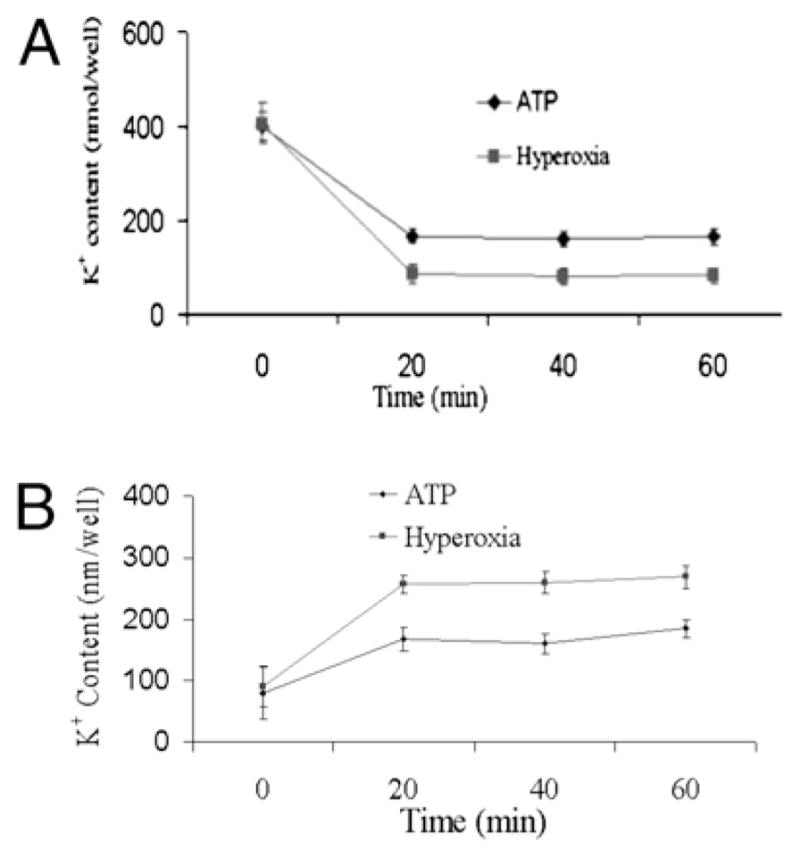
Hyperoxia exposure leads to K+ efflux. Macrophages were isolated from C57BL/6 mice, primed with LPS (100 ng/ml) for 1 h, exposed to hyperoxia (95% O2), and analyzed for intracellular (A) and extracellular (B) K+ levels by atomic absorption flame photometry. ATP was used as a positive control for K+ efflux
Caspase-1 was activated in alveolar macrophages exposed to hyperoxia
Recent reports suggest that caspase-1 is crucial for activation and processing of IL-1β by exogenous ATP (25). To evaluate caspase-1 activation, mice were exposed to hyperoxia (100% O2), and macrophages were isolated from the lung. Caspase-1 activity was determined by Western blot and ELISA. We found that exposure to hyperoxia (100% O2) for 72 h lead to enhanced caspase-1 cleavage (Fig. 3A) compared with normoxic control mice.
FIGURE 3.

Hyperoxia exposure leads to increased caspase-1 and IL-1 β activation. A, Caspase-1 cleavage and caspase-1 activity in isolated lung macrophages were measured by immunoblot and ELISA, respectively. B, Mice were exposed to hyperoxia (100% O2) for various time points, and macrophages isolated from whole lungs and analyzed for IL-1β cleavage. IL-1β levels were also analyzed in BAL fluid.
Hyperoxia exposure enhanced IL-1β processing in alveolar macrophages
Previous reports have demonstrated that large amounts of exogenous ATP induce IL-1β processing and release from macrophages the cell type that serves as a major source of inflammatory cytokines in lung injury and inflammation (24). Because hyperoxia induces ATP (25) and IL-1β (1) production, we investigated whether hyperoxia induced ATP generation has any effect on IL-1β processing and release by macrophages. The baseline levels of IL-1β in the BAL fluid of normoxic mice were near the limits of detection for our assays (Fig. 3B). In vivo, 3 d of hyperoxia are required for mice to develop severe lung injury and was the time point when changes in IL-1 and caspase-1 were measured (1, 2). The release of IL-1β from alveolar macrophages after 72 h of exposure to hyperoxia in vivo increased 3- to 4-fold over normoxic levels. IL-1β processing increased in a time-dependent manner and correlated with caspase-1 activation. These results suggest that hyperoxia enhanced IL-1β processing.
Hyperoxia enhanced the interaction between inflammasome complexes
ASC interacts with caspase-1 to form an active inflammasome complex that is necessary for the complete processing of IL-1β (10, 18, 23). We hypothesized that hyperoxia would alter this interaction in alveolar macrophages isolated from mice exposed to 100% O2. To test this hypothesis, cell lysates from isolated alveolar macrophages were subjected to immunoprecipitation with anti-ASC Ab, and the immunoprecipitates were analyzed for the presence of caspase-1 and NALP3. When endogenous ASC was immunoprecipitated from macrophages that are not exposed to hyperoxia, endogenous NALP3 and caspase-1 were not associated with ASC-containing immune complexes. However, hyperoxia exposure stimulated the interaction between ASC and caspase-1, an essential component of the inflammasome. Hyperoxia also induced an interaction between ASC and NALP3 (Fig. 4). The assembly of the NALP3 and ASC complex is thought to induce IL-1β processing by caspase-1. These results suggest that hyperoxia-mediated IL-1β processing involves assembly of the components of the inflammasome complex.
FIGURE 4.
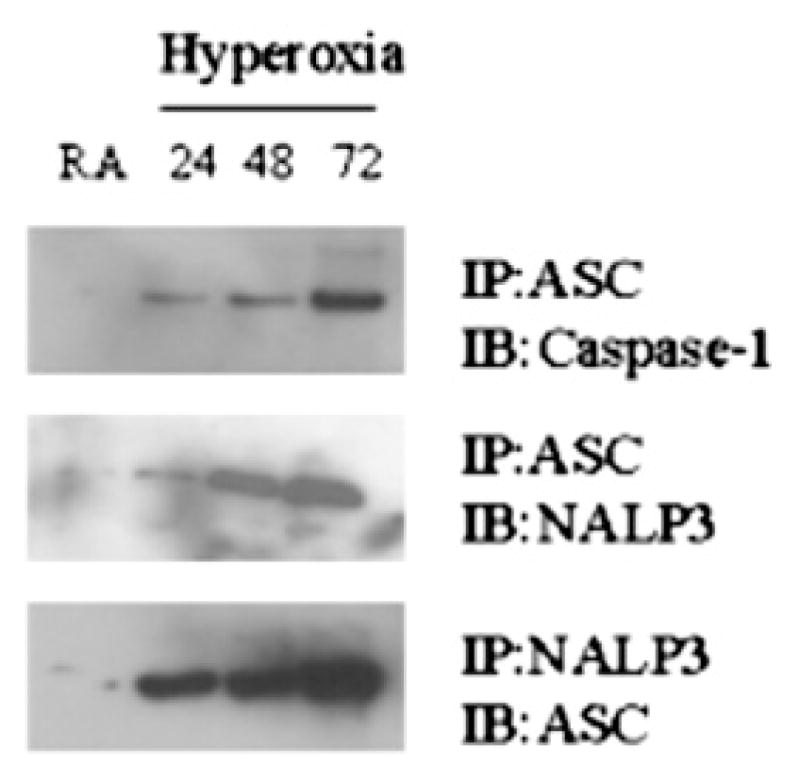
Hyperoxia enhances the interaction among inflammasome components. Mice were exposed to hyperoxia (100% O2) for various time points, and macrophages were isolated from whole lungs and analyzed for inflammasome complex formation by immunoprecipitation followed by Western blot analysis.
A P2X7 receptor antagonist inhibited the hyperoxia-induced interactions between inflammasome components
To determine whether the P2X7 receptor is required for hyperoxia-induced activation of the inflammasome, we treated alveolar macrophages with oxATP, a P2X7 antagonist, in the presence or absence of hyperoxia exposure. Alveolar macrophage lysates were subjected to immunoprecipitation using anti-ASC or NALP3 Ab and immunoblotted with anti-NALP3 or ASC Ab, respectively. Hyperoxia did not alter the expression level of NALP3 or ASC (Fig. 5A). However, hyperoxia enhanced the interaction between ASC and NALP3. OxATP inhibited the interaction between NALP3 and ASC. Because the P2X7 antagonist inhibited the inflammasome activation, it is clear that P2X7 plays an important role in hyperoxia induced inflammasome activation (Fig. 5B).
FIGURE 5.

P2X7 receptor antagonists inhibit the hyperoxia-induced interaction among inflammasome components. A, Macrophages were isolated from C57BL/6 mice, primed with LPS (100 ng/ml) for 1 h, exposed to hyperoxia (95% O2) for 1 h in the presence or absence of the P2X7 receptor antagonist oxATP, and immunoblotted for the inflammasome components NALP3, ASC and Caspase-1. B, Inflammasome complex formation was detected by immunoprecipitation followed by Western blot analysis.
Extracellular K+ inhibited hyperoxia mediated NALP3 inflammasome complex formation
Exposure of macrophages to high extracellular K+ concentrations (130 mM) prevents the lowering of cytoplasmic K+, IL-1β secretion, and caspase-1 activation that is induced by ATP in LPS-stimulated macrophages (26) and demonstrated not to be cytotoxic (27). To determine whether K+ efflux is required for hyperoxia-mediated NALP3 inflammasome activation, macrophages were exposed to ATP and hyperoxia in the presence or absence of extracellular K+. Cleavage and release of IL-1β and caspase-1 caused by either ATP or hyperoxia exposure was completely blocked when the extracellular concentration of K+ was increased to 130 mM (Fig. 6A). To determine whether this high K+ concentration inhibited inflammasome, ASC or NALP3 was immunoprecipitated from cells exposed to hyperoxia with or without 130 mM extracellular K+ (Fig. 6A). Exposure to hyperoxia or ATP induced NALP3 interaction with ASC, indicative of inflammasome complex formation. However, in the presence of high extracellular K+, this interaction was inhibited. Thus, physiologic extracellular concentrations of K+ blocked the recruitment of caspase-1 or NALP3 to ASC. The stimulation of P2X7 receptor with ATP causes pore formation and K+ efflux. Therefore, we determined whether the P2X7 receptor was required for inflammasome formation by treating the cells with the oxATP P2X7 receptor antagonist. P2X7 inhibition by oxATP completely blocked the hyperoxia-induced interaction of ASC with NALP3 inflammasome complex. The high levels of extracellular K+ inhibited hyperoxia-induced caspase-1 generation by macrophages (Fig. 6B). In addition, increasing extracellular potassium leads to a decrease in potassium efflux (Fig. 6C). These results suggest that hyperoxia induced K+ efflux and P2X7 receptor activation are required for inflammasome activation.
FIGURE 6.

Extracellular K+ inhibits inflammasome complex formation. Macrophages were isolated from C57BL/6 mice, primed with LPS (100 ng/ml) for 1 h, and exposed to hyperoxia (95% O2) for 1 h and oxATP in the presence or absence of 130 mM KCl. Lysates were prepared and (A) inflammasome assembly detected by immunoprecipitation and western blot analysis, and (B) caspase-1 activity detected by ELISA. *p < 0.05, compared with hyperoxia or treatment. C, Extracellular K+ levels were measured by atomic absorption flame photometry.
Reactive oxygen species were required for inflammasome activation
The mechanism by which K+ efflux induces inflammasome activation is not clear. Recent reports suggest that ROS-mediated caspase-1 activation is induced by ATP stimulation (6). To determine whether ROSs are required for the assembly and activation of the NALP3 inflammasome, macrophages were preincubated with the broad ROS inhibitor and antioxidant, NAC, followed by stimulation with hyperoxia. The NAC significantly inhibited IL-1β maturation in response to hyperoxia (Fig. 7), suggesting that in addition to K+, hyperoxia-induced ROSs are required for IL-1β processing. In addition, the NALP3 interaction with ASC was reversed in the presence of NAC, apocynin, or diphenylene iodonium (DPI; Fig. 7). These results suggest that hyperoxia-induced ROSs are necessary for IL-1β processing and inflammasome activation.
FIGURE 7.

Reactive oxygen species are required for K+ efflux and inflammasome activation. Macrophages were isolated from C57BL/6 mice and treated with NAC, apocynin, or DPI. Cells were exposed to hyperoxia (95% O2) for 1 h and then analyzed for inflammasome assembly by immunoprecipitation and Western blot analysis. ATP served as a positive control.
Hyperoxia induced cell permeabilization was inhibited by blocking the P2X7 receptor with oxATP
Stimulation of the P2X7 receptor with ATP leads to cell membrane pore formation and K+ efflux, which then triggers the maturation and secretion of IL-1β (16, 19). The formation of ATP-induced membrane P2X7 pores that allow permeabilization of molecules up to a molecular mass of 900 Da can be detected with the YO-PRO-1 dye (11). YO-PRO-1 is 629 Da and therefore able to enter the cell through the P2X7 pores, where it can be detected by the >100-fold increase in fluorescence intensity that is induced by its intercalation with RNA or DNA. Using flow cytometry to detect the fluorescence dye uptake by living cells, we observed a marked increase in the uptake of YO-PRO-1 by LPS-primed macrophages exposed to hyperoxia, compared with cells that are not exposed to hyperoxia (Fig. 8). To investigate whether the cell permeabilization visualized by YO-PRO-1 uptake can be attributed to the binding of ATP to P2X7, we added the irreversible P2X7 inhibitor oxATP before exposure of the cells to hyperoxia. The oxATP decreased YO-PRO-1 uptake, confirming that the observed cell permeabilization required ATP (Fig. 8). These results support the critical role of the P2X7 receptor in hyperoxia-induced pore formation.
FIGURE 8.
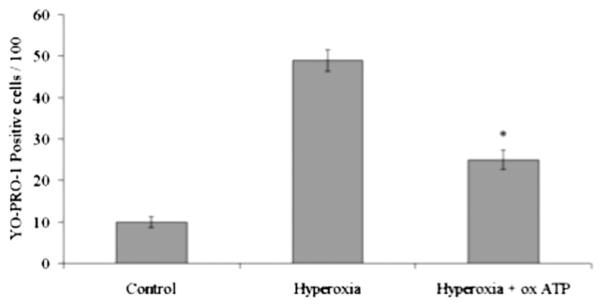
Hyperoxia-induced pore formation was inhibited by oxATP. Macrophages were isolated from C57BL/6 mice, primed with LPS (100 ng/ml) for 1 h, and exposed to hyperoxia (95% O2) for 1 h in the presence or absence of oxATP. Cells were analyzed for YO-PRO-1 uptake by flow cyotometry. Addition of oxATP resulted in a significant decrease in YO-PRO-1 uptake. *p < 0.01, compared with hyperoxia.
Apyrase inhibited hyperoxia-induced release of IL-1β
To test whether hyperoxia-induced autocrine ATP release is responsible for the activation of P2X7 and the release of IL-1β, we repeated our experiments in the presence of apyrase, an enzyme known to inhibit autocrine ATP signaling by hydrolyzing extra-cellular ATP to AMP (20). When added at a concentration of 20 U/ml, apyrase significantly inhibited hyperoxia-induced IL-1β release (Fig. 9). In addition, apyrase inhibited the release of IL-1β induced by exogenous ATP. These results suggest that hyperoxia-induced ATP is responsible for IL-1β release.
FIGURE 9.
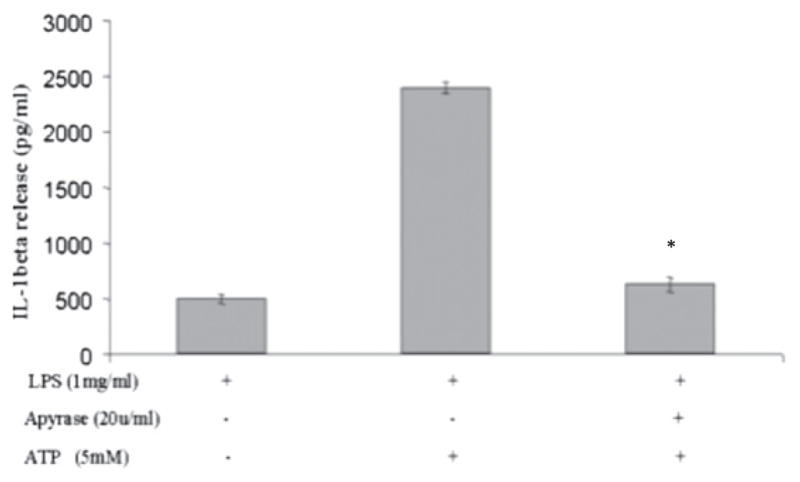
Hyperoxia-induced IL-1β release was inhibited by apyrase. Macrophages were isolated from C57BL/6 mice, primed with LPS (100 ng/ml) for 1 h, and exposed to hyperoxia (95% O2) for 1 h in the presence or absence of apyrase. IL-1β levels in the media were measured by ELISA. Addition of apyrase resulted in a significant decrease in IL-1β release. *p < 0.05, compared with ATP alone.
Inflammasome silencing inhibited transepithelial albumin flux in alveolar epithelial type II cells
To simulate in vivo hyperoxia-induced inflammation and apoptosis, we standardized a coculture model of THP-1 cells in combination with alveolar epithelial type II (ATII) cells. ATII cells were isolated and cultured as described previously (21). THP-1 cells were chosen because of their similarity to the activated mature alveolar macrophages present in BAL fluid and for the ease of transfection of these cells. We chose macrophages and epithelial cells because they are among the first lung sentinel cells to be exposed to hyperoxia (28). Frank et al. (24) and Gaca et al. (29) suggested that macrophages are the first line of defense for the alveolar epithelium, and macrophage depletion by clodronate protects from ALI. In this regard, the epithelial cell coculture with ATII and THP-1 cells provides a suitable model for the study of epithelial-macrophage interactions that occur in vivo. To determine whether the hyperoxia-activated inflammasome contributes to epithelial cell permeability, we silenced inflammasome expression in THP-1 cells and studied epithelial permeability in THP-1and AEC-II cocultures. When THP-1 cells were treated with NALP3 shRNA and added to AEC-II cells, hyperoxia-induced transepithelial permeability was significantly reduced across mouse ATII cell monolayers (Fig. 10A). shRNA silencing of NALP3 abrogated hyperoxia-induced secretion of proinflammatory cytokines in vitro (Fig. 10B). When THP-1 cells were treated with NALP3 shRNA, inflammasome complex formation was not detected by immunoprecipitation followed by Western blot analysis under hyperoxia (Fig. 10C).
FIGURE 10.
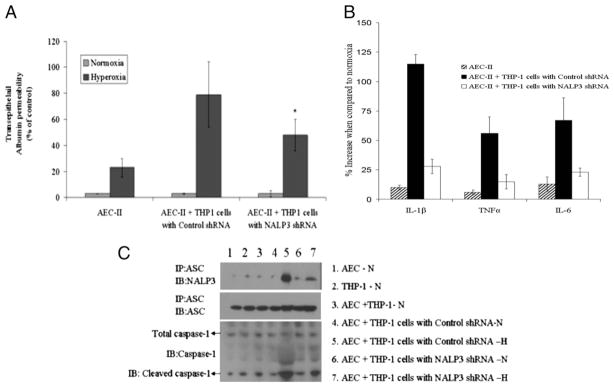
Silencing of NALP3 in THP1 cells abolishes protein permeability across mouse ATII cell monolayers in ATII and THP1 coculture models. A, ATII cells were grown in 24-well transwell plates until the cellular monolayers reached confluence. THP-1 cells were added to the appropriate ATII monolayers, cocultured for 6 h, and exposed to hyperoxic conditions for 1 h. In some experiments, THP-1 cells were transfected with control shRNA or NALP3 shRNA and then added to a monolayer of ATII cells. Transepithelial albumin flux across each monolayer was determined by the addition of [125I]-labeled human serum albumin to each upper compartment. The contents from the lower compartment were collected and counted in a Wizard γ-counter. *p < 0.05, relative to control shRNA. B, IL-1β, TNF-α and IL-6 proinflammatory cytokines were analyzed in extracellular media by ELISA. C, Inflammasome complex formation was detected by immunoprecipitation followed by Western blot analysis. H, hyperoxia; N, normoxia.
Discussion
The results presented in this study show that inflammasome formation and IL-1β processing is induced by hyperoxia in vivo and in vitro and is associated with increased alveolar epithelial protein permeability. Hyperoxia-stimulated K+ efflux, inflammasome formation, proinflammatory cytokine release, and marked induction of caspase-1 and IL-1β cleavage. The P2X7 agonist ATP enhanced hyperoxia-induced inflammasome activation, whereas the P2X7 antagonist, oxATP, inhibited hyperoxia-induced inflammasome activation. However, when ATP was scavenged with apyrase, hyperoxia-induced inflammasome activation was significantly decreased, indicating the possible involvement of the P2X7 receptor. Furthermore, shRNA silencing of inflammasome component expression abrogated hyperoxia-induced secretion of proinflammatory cytokines in vitro. These results suggest that hyperoxia induces K+ efflux through the P2X7 receptor and leads to inflammasome activation and secretion of proinflammatory cytokines.
Our studies showed that NALP3 inflammasome formation and IL-1β cleavage is induced by hyperoxia in vivo and in vitro, which was associated with increased alveolar epithelial protein permeability. Previous reports indicate that IL-1β is responsible for both alveolar epithelial and vascular endothelial permeability (4). Alveolar epithelial and vascular endothelial permeability is a key event in alveolar edema, and it is an extremely important component of lung injury. It is therefore possible that hyperoxia induces inflammasome activation followed by IL-1β cleavage that in turn contributes to edema. Our data support this hypothesis, and our results indicate that the inhibition of the inflammasome by using shRNA abrogates hyperoxia-induced secretion of IL-1β and ameliorates lung epithelial permeability. Thus, our results show that inflammasome activation is required for IL-1β cleavage and epithelial permeability in the response to hyperoxia. The findings reported in this study have potential clinical relevance. The transient blockade of the inflammasome can provide new causal therapies for pulmonary edema resulting from ALI, a substantial cause of morbidity and mortality in critically ill patients that is currently largely untreatable.
Inflammation is a major contributor to the pathogenesis of ALI (30). Recent studies suggest that inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice, and neutralizing Abs against the inflammasome successfully inhibit inflammasome activation in multiple models of CNS injury (31). Therefore, inhibition of the inflammasome in ALI is a novel therapeutic strategy to inhibit caspase-1 activation and thereby protect lung tissue from hyperoxia damage.
IL-1β cleavage is preceded by the activation of caspase-1 (6, 7, 32). Caspase-1 is an important mediator of both apoptosis and inflammation (33, 34). Our results show that hyperoxia exposure activates caspase-1 both in vivo and in vitro. The caspase-1 activators ATP and nigericin act through a mechanism that involves P2X7 stimulation and K+ efflux (9, 19, 26, 27). Our results also indicate that casapse-1 activation and inflammasome complex formation are impaired in the presence of P2X7 antagonist or extracellular K+ (130 mM; Fig. 6A), which suggests that P2X7 activation and K+ efflux are necessary steps in inflammasome activation. These results lead to the model that in normoxic conditions, and when the extracellular [K+] concentration is normal, caspase-1 activation is inhibited. In the early response to hyperoxia, potassium channels on the plasma membrane open, resulting in potassium efflux. This efflux acts as an alarm signal that induces inflammasome complex formation, caspase-1 activation, and the release of cytokines without killing cells. Therefore, cytokine processing rather than death prevails. At sufficiently low [K+] levels, ASC binds caspase-1 directly and forms the pyroptosome, causing full caspase-1 activation, cell death, and a burst of cytokine release (34). Inhibition of the inflammasome complex and caspase-1 activation at an early stage can limit these proinflammatory processes that are the key to the pathogenesis of ALI.
Hyperoxia induces ATP stimulation, but this ATP stimulation alone cannot induce IL-1β release. NALP3 inflammasome activation occurs in response to various states of cell stress, including LPS, peptidoglycan, nucleic acids, and certain crystals such as uric acid. NALP3 activation occurs when the cells are also exposed to ATP, which triggers the opening of the nonselective cation channel of the purinergic P2X7 receptor. Hence, LPS priming is used in this in vitro model. LPS priming is a necessary step to induce sufficient intracellular proIL-1β substrate for the ATP-induced caspase-1 activation (35). ATP has been shown to activate NALP3 dependent caspase-1 cleavage, stimulate secretion of IL-1β, stimulate P2X7 receptor-mediated K+ efflux, and thereby reduce intracellular K levels+ (9, 19, 20, 27). In our study, hyperoxia exposure lead to ATP release, which further stimulated P2X7 receptor-mediated K+ efflux and inflammasome complex activation. oxATP was able to inhibit the hyperoxia-induced inflammasome activation. However, we believe that because ATP is a known agonist for P2X7, the effect on P2X7 channels and K+ efflux that we observed in our study could be the cumulative effect of hyperoxia and ATP. One plausible mechanism is that hyperoxia and ROS lead to cellular damage and release of ATP leading to inflammasome complex formation. Because oxATP inhibits hyperoxia-induced inflammasome complex formation this would support this pathway. In addition, we found that ROSs are required for NALP3 activation. Recent data also suggest a role for ROS in activating caspase-1 in response to ATP treatment and in the NALP3 inflammasome (7). To further support a role for ROS in inflammasome activation, alveolar macrophages were treated with the ROS inhibitors NAC, apocynin, or DPI. IL-1β production was impaired in the presence of all three ROS inhibitors (Fig. 7), suggesting that ROS production is a necessary step in inflammasome activation. Because ATP itself is an inducer of ROS production, there is the possibility that ROS generation can also have occurred through ATP rather than hyperoxia alone in our study. A limitation of the current study is the difficulty in quantifying the relative contributions of ROS generation via hyperoxia or ATP. However, the signaling mechanism linking hyperoxia induced ROS to inflammasome activation remains unknown. Apoptosis signal regulating kinase-1 (ASK-1) can play a downstream role in ROS-induced purinergic P2X7 receptor-mediated apoptosis (36). Recent reports also suggest that ROS-detoxifying proteins might be part of this mechanism, such as thioredoxin, an inhibitor of ASK-1 that inhibits NALP3 inflammasome-mediated IL-1β secretion (27). We recently reported that increased ASK-1 expression leads to hyperoxic ALI (HALI) associated cell death (12). However, the mechanism leading to ASK-1–induced cell death in HALI is not clear. Additional investigation is necessary to clarify the link between K+ efflux, ROS generation, and ASK-1 activation in HALI-induced inflammasome activation.
We believe that the early stage of HALI involves the initiation of NALP3 complex formation and low levels of caspase-1 activation, as demonstrated by low caspase-1 levels after the first 24 h of hyperoxia exposure. In this model, the intermediate stage of HALI is characterized by enhanced NALP3 complex formation that occurs after 48 h of hyperoxia exposure. During the final stages of HALI, which occur after 72 h of hyperoxia exposure, the NALP3 complex is highly active and leads to the full activation of caspase-1 and a burst of cytokine release. The cytokine burst leads to cytokine-dependent inflammation and apoptosis. This can be particularly relevant in ALI, in which inflammatory processes increase from the early stages to the late stages of ALI. The cytokine burst also causes epithelial permeability, epithelial barrier dysfunction, and cell death. From a clinical perspective, the inhibition of K+ efflux and P2X7, or the use of pharmacologic approaches that disrupt inflammasome formation, can provide a strategy for the protection of lung tissue from cellular injury during acute or chronic illness associated with ALI syndromes.
Acknowledgments
This work was supported by National Institutes of Health Grants RO1HL074859 (to A.B.W) and T32-HL007874 (to N.K.) and American Heart Association National Scientist Development Grant 09SDG2260957 (to N.K.).
Abbreviations used in this paper
- ALI
acute lung injury
- ASC
apoptosis-associated speck-like protein
- ASK-1
apoptosis signal regulating kinase-1
- ATII
alveolar type II
- BAL
bronchoalveolar lavage
- DPI
diphenylene iodonium
- H
hyperoxia
- HALI
hyperoxic acute lung injury
- N
normoxia
- NAC
N-acetyl-l-cysteine
- oxATP
oxidized ATP
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Waxman AB, Einarsson O, Seres T, Knickelbein RG, Warshaw JB, Johnston R, Homer RJ, Elias JA. Targeted lung expression of interleukin-11 enhances murine tolerance of 100% oxygen and diminishes hyperoxia-induced DNA fragmentation. J Clin Invest. 1998;101:1970–1982. doi: 10.1172/JCI1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward NS, Waxman AB, Homer RJ, Mantell LL, Einarsson O, Du Y, Elias JA. Interleukin-6-induced protection in hyperoxic acute lung injury. Am J Respir Cell Mol Biol. 2000;22:535–542. doi: 10.1165/ajrcmb.22.5.3808. [DOI] [PubMed] [Google Scholar]
- 3.He CH, Waxman AB, Lee CG, Link H, Rabach ME, Ma B, Chen Q, Zhu Z, Zhong M, Nakayama K, et al. Bcl-2-related protein A1 is an endogenous and cytokine-stimulated mediator of cytoprotection in hyperoxic acute lung injury. J Clin Invest. 2005;115:1039–1048. doi: 10.1172/JCI23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, Sheppard D, Violette SM, Weinreb PH, Horan GS, et al. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res. 2008;102:804–812. doi: 10.1161/CIRCRESAHA.107.161067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 6.Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126:659–662. doi: 10.1016/j.cell.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117:3786–3799. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Virgilio F. Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci. 2007;28:465–472. doi: 10.1016/j.tips.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Sutterwala FS, Ogura Y, Flavell RA. The inflammasome in pathogen recognition and inflammation. J Leukoc Biol. 2007;82:259–264. doi: 10.1189/jlb.1206755. [DOI] [PubMed] [Google Scholar]
- 11.Drenth JP, van der Meer JW. The inflammasome—a linebacker of innate defense. N Engl J Med. 2006;355:730–732. doi: 10.1056/NEJMcibr063500. [DOI] [PubMed] [Google Scholar]
- 12.Kolliputi N, Waxman AB. IL-6 cytoprotection in hyperoxic acute lung injury occurs via suppressor of cytokine signaling-1-induced apoptosis signal-regulating kinase-1 degradation. Am J Respir Cell Mol Biol. 2009;40:314–324. doi: 10.1165/rcmb.2007-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolliputi N, Waxman AB. IL-6 cytoprotection in hyperoxic acute lung injury occurs via PI3K/Akt-mediated Bax phosphorylation. Am J Physiol Lung Cell Mol Physiol. 2009;297:L6–L16. doi: 10.1152/ajplung.90381.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waxman AB, Kolliputi N. IL-6 protects against hyperoxia-induced mitochondrial damage via Bcl-2-induced Bak interactions with mitofusins. Am J Respir Cell Mol Biol. 2009;41:385–396. doi: 10.1165/rcmb.2008-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavnikova N, Prokhorova S, Helyar L, Laskin DL. Isolation and partial characterization of subpopulations of alveolar macrophages, granulocytes, and highly enriched interstitial macrophages from rat lung. Am J Respir Cell Mol Biol. 1993;8:384–392. doi: 10.1165/ajrcmb/8.4.384. [DOI] [PubMed] [Google Scholar]
- 16.Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286:C1100–C1108. doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- 17.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galán JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 19.Franchi L, Kanneganti TD, Dubyak GR, Núñez G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem. 2007;282:18810–18818. doi: 10.1074/jbc.M610762200. [DOI] [PubMed] [Google Scholar]
- 20.Elssner A, Duncan M, Gavrilin M, Wewers MD. A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 beta processing and release. J Immunol. 2004;172:4987–4994. doi: 10.4049/jimmunol.172.8.4987. [DOI] [PubMed] [Google Scholar]
- 21.Barker GF, Manzo ND, Cotich KL, Shone RK, Waxman AB. DNA damage induced by hyperoxia: quantitation and correlation with lung injury. Am J Respir Cell Mol Biol. 2006;35:277–288. doi: 10.1165/rcmb.2005-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hennige AM, Lembert N, Wahl MA, Ammon HP. Oxidative stress increases potassium efflux from pancreatic islets by depletion of intracellular calcium stores. Free Radic Res. 2000;33:507–516. doi: 10.1080/10715760000301051. [DOI] [PubMed] [Google Scholar]
- 23.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 24.Frank JA, Wray CM, McAuley DF, Schwendener R, Matthay MA. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1191–L1198. doi: 10.1152/ajplung.00055.2006. [DOI] [PubMed] [Google Scholar]
- 25.Ahmad KA, Iskandar KB, Hirpara JL, Clement MV, Pervaiz S. Hydrogen peroxide-mediated cytosolic acidification is a signal for mitochondrial translocation of Bax during drug-induced apoptosis of tumor cells. Cancer Res. 2004;64:7867–7878. doi: 10.1158/0008-5472.CAN-04-0648. [DOI] [PubMed] [Google Scholar]
- 26.Petrilli V, Papin S, Tschopp J. The inflammasome. Curr Biol. 2005;15:R581. doi: 10.1016/j.cub.2005.07.049. [DOI] [PubMed] [Google Scholar]
- 27.Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 28.Hjort MR, Brenyo AJ, Finkelstein JN, Frampton MW, LoMonaco MB, Stewart JC, Johnston CJ, D’Angio CT. Alveolar epithelial cell-macrophage interactions affect oxygen-stimulated interleukin-8 release. Inflammation. 2003;27:137–145. doi: 10.1023/a:1023817811850. [DOI] [PubMed] [Google Scholar]
- 29.Gaca JG, Palestrant D, Lukes DJ, Olausson M, Parker W, Davis RD., Jr Prevention of acute lung injury in swine: depletion of pulmonary intravascular macrophages using liposomal clodronate. J Surg Res. 2003;112:19–25. doi: 10.1016/s0022-4804(03)00142-2. [DOI] [PubMed] [Google Scholar]
- 30.Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14:523–535. doi: 10.1016/s1359-6101(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 31.Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab. 2009;29:534–544. doi: 10.1038/jcbfm.2008.143. [DOI] [PubMed] [Google Scholar]
- 32.Lister MF, Sharkey J, Sawatzky DA, Hodgkiss JP, Davidson DJ, Rossi AG, Finlayson K. The role of the purinergic P2X7 receptor in inflammation. J Inflamm (Lond) 2007;4:5. doi: 10.1186/1476-9255-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gottlieb R. ICE-ing the heart. Circ Res. 2005;96:1036–1038. doi: 10.1161/01.RES.0000169466.84402.c1. [DOI] [PubMed] [Google Scholar]
- 34.Saleh M, Green DR. Caspase-1 inflammasomes: choosing between death and taxis. Cell Death Differ. 2007;14:1559–1560. doi: 10.1038/sj.cdd.4402203. [DOI] [PubMed] [Google Scholar]
- 35.Wewers MD, Sarkar A. P2X(7) receptor and macrophage function. Purinergic Signal. 2009;5:189–195. doi: 10.1007/s11302-009-9131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noguchi T, Ishii K, Fukutomi H, Naguro I, Matsuzawa A, Takeda K, Ichijo H. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J Biol Chem. 2008;283:7657–7665. doi: 10.1074/jbc.M708402200. [DOI] [PubMed] [Google Scholar]