

Significance
ERK activity contributes to AMPA-type glutamate receptor (AMPAR) membrane insertion, but until now, there was no clear understanding how this regulation could happen. We have identified the motif of ERK inhibiting PDE4 and controlling PKA output as a key step in the dopamine-induced membrane trafficking of GluA1. The significance of this regulatory motif is that it provides a point of integration for ERK activating signals to modulate neuronal excitability by tuning the trafficking of AMPAR.
Abstract
AMPA-type glutamate receptor (AMPAR) trafficking is essential for modulating synaptic transmission strength. Prior studies that have characterized signaling pathways underlying AMPAR trafficking have identified the cAMP/PKA-mediated phosphorylation of GluA1, an AMPAR subunit, as a key step in the membrane insertion of AMPAR. Inhibition of ERK impairs AMPAR membrane insertion, but the mechanism by which ERK exerts its effect is unknown. Dopamine, an activator of both PKA and ERK, induces AMPAR insertion, but the relationship between the two protein kinases in the process is not understood. We used a combination of computational modeling and live cell imaging to determine the relationship between ERK and PKA in AMPAR insertion. We developed a dynamical model to study the effects of phosphodiesterase 4 (PDE4), a cAMP phosphodiesterase that is phosphorylated and inhibited by ERK, on the membrane insertion of AMPAR. The model predicted that PKA could be a downstream effector of ERK in regulating AMPAR insertion. We experimentally tested the model predictions and found that dopamine-induced ERK phosphorylates and inhibits PDE4. This regulation results in increased cAMP levels and PKA-mediated phosphorylation of DARPP-32 and GluA1, leading to increased GluA1 trafficking to the membrane. These findings provide unique insight into an unanticipated network topology in which ERK uses PDE4 to regulate PKA output during dopamine signaling. The combination of dynamical models and experiments has helped us unravel the complex interactions between two protein kinase pathways in regulating a fundamental molecular process underlying synaptic plasticity.
The strength of synaptic transmission depends on the number of AMPA-type glutamate receptors (AMPARs) localized to the synaptic membrane. The regulated trafficking of AMPARs in and out of the postsynaptic membrane controls the number of synaptic AMPARs and is thought to underlie synaptic plasticity (1). AMPARs are composed of four subunits (GluA1–4), which assemble as homo- or hetero-tetramers to mediate excitatory transmissions in the brain. There are a number of intracellular pathways that regulate signal-initiated trafficking of GluA1-containing AMPARs. For instance, PKA and PKG, the cyclic nucleotide-activated kinases, phosphorylate GluA1 at S845 (2, 3). Phosphorylation of S845 is required for GluA1 synaptic insertion because mutation to A845 prevents GluA1 exocytosis (4).
Dopamine, a modulatory neurotransmitter that increases cAMP/PKA levels, promotes GluA1 phosphorylation at S845 and AMPAR insertion into the plasma membrane (3, 5, 6). Additional signaling pathways influence this process, but the role they play in dopamine-mediated AMPAR trafficking is not known. ERK, a downstream effector of dopamine, promotes AMPAR membrane insertion even though ERK does not directly phosphorylate GluA1 (7, 8). The objective of this study was to identify the mechanism by which ERK regulates dopamine-mediated GluA1 membrane insertion. Based on our observation that ERK inhibition decreases dopamine-mediated GluA1 phosphorylation at S845, we looked for ERK substrates that could affect cAMP levels. One possibility was that ERK phosphorylation and activation of cytosolic phospholipase A2 (cPLA2) could increase PKC activity, leading to activation of AC5, the main adenylyl cyclase in the striatum (9). Another substrate of ERK that could affect GluA1 trafficking is phosphodiesterase 4 (PDE4), a phosphodiesterase phosphorylated and inhibited by ERK (10). We tested for the involvement of both ERK substrates on GluA1 membrane insertion. We developed a computational model to explore the ERK regulation of dopamine-induced GluA1 membrane insertion. The model predictions were validated experimentally by monitoring dopamine-stimulated cAMP levels and GluA1 trafficking by live cell imaging in striatal primary neurons. The data presented here show that dopamine-activated ERK increases cAMP levels by phosphorylation and inhibition of PDE4 and results in the elevation of PKA mediated GluA1 phosphorylation and membrane insertion. Our approach allowed us to unravel the complex interaction between PKA and ERK pathways within the dopamine-signaling network.
Results
We examined the dopamine-dependent insertion of GluA1 by monitoring superecliptic pHluorin (SEP) N-terminally tagged GluA1 in primary striatal cultures. SEP is a pH-sensitive GFP variant used to monitor exocytosis in real time because its fluorescence is quenched when exposed to the acidic lumen of endocytic vesicles and recovers upon plasma membrane insertion (11, 12). Dopamine treatment led to a dose-dependent increase in GluA1 membrane insertion (Fig. 1A). Regardless of dopamine concentration, saturation of GluA1 membrane insertion was seen after addition of forskolin (FK) and 3-isobutyl-1-methylxanthine (IBMX) 30 min after dopamine stimulation. Dopamine receptors are classified as D1 or D2 type based on their coupling to heterotrimeric G proteins (13, 14). D2-type receptors (D2/D3/D4R) can activate adenylyl cyclase via interaction with the beta-gamma subunits of G protein i/o (15). D1R-type receptor (D1/D5R) agonists induced GluA1 phosphorylation and GluA1 membrane insertion (Fig. S1 A–C), whereas a D2R-type agonist did not stimulate GluA1 phosphorylation or GluA1 membrane insertion (Fig. S1 A and D). Hence, we did not consider any D2R-initiated signals in our study because the data suggest the predominant involvement of D1/D5R in GluA1 trafficking.
Fig. 1.
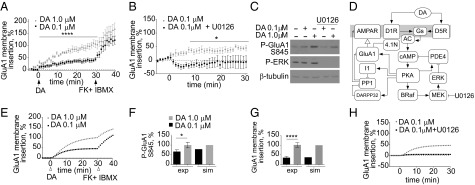
Dopamine-induced GluA1 membrane insertion depends on ERK activity. (A) Dopamine induces a dose-dependent increase in GluA1 membrane insertion. A concentration of 0.1 (black) or 1.0 (white) μM dopamine was added. ****P < 0.0001 (repeated-measures ANOVA Bonferroni post hoc; n = 9–11). At 30 min, 50 μM FK and 100 μM IBMX (FK + IBMX) were added. (B) ERK inhibition decreases 0.1-μM dopamine–GluA1 membrane insertion. A concentration of 0.1 μM dopamine and 10 μM U0126 (black) were added. *P = 0.013 (repeated-measures ANOVA Bonferroni post hoc; n = 6). (C) Inhibition of ERK decreases PKA phosphorylation of GluA1. Neuronal lysates were immunoblotted for phospho-GluA1 Ser-845 (PKA phosphorylation site) or phospho-ERK or β-tubulin. (D) Dopamine-signaling model of ERK regulation of GluA1 phosphorylation and membrane insertion. A diagram of the model topology is shown, depicting the dopamine signaling to GluA1 membrane trafficking. Filled arrows represent activation/production reactions. Plungers represent inhibition/degradation reactions. Open arrow represents membrane insertion. (E) Simulations of the time course dopamine-induced dose-dependent GluA1 membrane insertion. At t = 30, FK/IBMX were introduced. Compare with time course in A. (F) Comparison of experimental dopamine-induced GluA1 phosphorylation (exp) and simulation (sim) results after 30 min of 0.1- or 1.0-μM dopamine stimulation. *P = 0.0453 (t test; n = 5). (G) Comparison of experimental dopamine-induced GluA1 membrane insertion (exp) and simulation (sim) results at 30 min of 0.1- or 1.0-μM dopamine stimulation. *P = 0.0006 (Mann–Whitney test; n = 9–11). (H) Simulated time course of GluA1 membrane insertion after 0.1-μM dopamine stimulation in the presence of ERK (gray) or with no ERK activity (black).
Several computational models have explored the underlying signaling that controls AMPAR trafficking (16–20). Nakano et al. specifically explored the contribution of dopamine signaling to AMPAR trafficking in striatal neurons (18). Although the Nakano computational model contained the core signaling steps of dopamine-mediated AMPAR insertion, it did not agree with our experimental data. Specifically, the dopamine dose–response of GluA1 insertion and the FK/IBMX insertion saturation (Fig. S2) was not observed in the Nakano simulations, prompting us to search for additional modes of regulation. One possible missing regulation in the Nakano model is the ERK-mediated control of AMPAR trafficking because it is known that hippocampal glutamate-induced AMPAR trafficking is impaired when ERK is inhibited (21). We confirmed that in striatal neurons ERK inhibition reduced 0.1- and 1.0-μM dopamine-mediated GluA1 insertion (Fig. 1B and Fig. S3A). Also, ERK inhibition decreased D1R agonist-induced GluA1 insertion (Fig. S3B). We excluded the possibility of non-dopamine-mediated signaling affecting GluA1 trafficking because inhibiting ERK alone induced no changes in trafficking (Fig. S3C). Thus, these results show ERK as a regulator of dopamine-mediated GluA1 membrane insertion.
PKA phosphorylation of GluA1 at S845 is crucial for membrane insertion. We investigated whether ERK regulates GluA1 membrane insertion upstream or downstream of PKA phosphorylation of GluA1. Dopamine stimulation induced a dose-dependent increase in GluA1 phosphorylation and ERK activation (Fig. 1C and Fig. S3D), without changes to total GluA1 levels (Fig. S3 E and F). Inhibiting ERK with U1026 decreased both 0.1- and 1.0-μM dopamine-induced S845 phosphorylation (Fig. 1C and Fig. S3D), suggesting that ERK controls GluA1 trafficking by modulating the activity of PKA. Based on these data, we searched for ERK substrates that could enhance cAMP levels and increase GluA1 phosphorylation/membrane insertion. ERK can activate PKC via direct phosphorylation of cPLA2, leading to the production of arachidonic acid and the activation of PKC (22–24). PKC phosphorylates AC5 and enhances cAMP production (9). Thus, inhibiting ERK could potentially lead to a reduction in cAMP levels by decreasing the cPLA2–PKC-mediated activation of AC5. If the ERK–cPLA2–PKC loop is regulating cAMP production, cPLA2 inhibition should mimic the effect of ERK inhibition on GluA1 trafficking. However, we ruled out the involvement of the ERK–PKC loop in GluA1 trafficking because inhibiting cPLA2 had no effect on dopamine-induced or D1R agonist-induced GluA1 membrane insertion (Fig. S3 G and H).
Next, we focused our attention on PDE4, an ERK substrate known to modulate cAMP levels. PDE4 is phosphorylated by ERK at the C terminus (Ser-579 in the PDE4D3 isoform). This phosphorylation reduces PDE basal activity by 80% (10). PDE4 can also be phosphorylated at the N terminus by PKA, enhancing its basal catalytic activity by 200% (25, 26). Given this complex regulation of PDE4, it is difficult to predict a priori the net effect of dopamine, an activator of both PKA and ERK, on PDE4 activity and cAMP levels. We developed a compartmental ordinary differential-equation-based model to test whether the inclusion of the ERK–PDE4–PKA loop could recapitulate the ERK-mediated regulation of GluA1 phosphorylation and membrane insertion seen in our experimental data. We used the Nakano model as a starting point of signal flow from D1R/D5R receptors to GluA1 membrane insertion. We added additional regulation and used rate constants reported in the biochemical literature. We constrained unknown parameters in an iterative fashion using input–output relationships of experiments described in the literature and performed in our laboratory for this study (SI Text and Dataset S1). Our model includes the dopamine-induced activation of PKA and ERK, the regulation of PDE4 by ERK and PKA, and the PKA-mediated insertion of GluA1. It also includes the PKA-mediated regulation of dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) and its inhibition of protein phosphatase 1 (PP1), the phosphatase that dephosphorylates GluA1 at S845. A diagram of all of the reactions in the model is shown in Fig. S4. A simplified diagram depicting the key components is presented as Fig. 1 D and E. In our simulations, we compared the phosphorylation and membrane insertion of GluA1 in response to dopamine with our experimental data (Fig. 1A). For ease of comparison, all simulated and experimental data were scaled to maximum values obtained with 1.0-μM dopamine treatment for 30 min. The simulation results and the experiments showed a similar dose-dependent increase in PKA-phosphorylated GluA1 and GluA1 membrane insertion after dopamine stimulation (Fig. 1 F and G). The simulation results also mimicked the experimental FK/IBMX-induced saturation of GluA1 membrane insertion. With the inclusion of the ERK–PDE4 regulation, the simulations also recapitulated key experimental data, chiefly that ERK enhances the dopamine-dependent trafficking of GluA1 to the membrane. Inhibiting ERK abolished GluA1 membrane insertion at 0.1 μM dopamine (Fig. 1H) and reduced membrane insertion at 1.0 μM dopamine (Fig. S5). These comparisons show that our model reasonably recapitulates the biochemical dynamics of the dopamine-signaling network controlling GluA1 membrane insertion. The full model is described in SI Description of Dopamine Signaling Model and has been deposited at the Virtual Cell web site, http://vcell.org.
ERK–PDE4 Loop Enhances Dopamine-Activated PKA Output.
Previous models of dopamine signaling have highlighted the importance of a positive feedback loop between PKA, PP1, and DARPP-32/I1 on AMPAR trafficking (18, 20). In this study, we propose a unique regulatory motif involving ERK-mediated regulation of PDE4, resulting in an increase in the activation of PKA and its downstream effectors. The inclusion of ERK-mediated regulation of PDE4 leads to a feedback (PKA–ERK–PDE4) motif nested within an inhibitory feedback loop (PKA–PDE4–cAMP) (Fig. 2A). The net balance of PKA and ERK activity on PDE4 determines cAMP and PKA levels. PKA activity induces ERK activation but also depends on the status of ERK activation over time. When the PKA–ERK–PDE4 feedback loop is disrupted, as when ERK is inhibited, ERK regulation of PDE4 activity is lost. Consequently, the PKA–PDE4–cAMP motif acts as a simple negative feedback loop to self-dampen cAMP levels and PKA output (Fig. 2A).
Fig. 2.

ERK inhibition decreases PKA output. (A) Model topology depicting a feedback (PKA–ERK–PDE4) motif nested within an inhibitory feedback loop (PKA–PDE4–cAMP). (B) Simulation of dopamine-induced cAMP percentage (Left) or membrane GluA1 percentage (Right) at 30 min. Three pertubations are shown: no ERK activity, addition of BDNF, or no PDE4 activity.
The predicted effects of abolishing ERK activity or PDE4 activity on downstream effectors, such as cAMP levels, PKA phosphorylation, and membrane insertion of GluA1 are shown in Fig. 2B and Fig. S6 A and B. Inhibiting ERK leads to decreases in cAMP levels, a decrease in GluA1 phosphorylation, and an ultimate decrease in GluA1 membrane insertion (Fig. 2B and Fig. S6 A and B). DARPP-32, an inhibitor of PP1, is another PKA target that is essential in the regulation of GluA1 dynamics. The model predicted that ERK inhibition would decrease PKA-mediated phosphorylation of DARPP-32 and thus reduce inhibition of PP1 by DARPP-32 (Fig. S6 A and B). BDNF, a known ERK-activating neurotrophin, has been implicated in synaptic plasticity. The model predicted that upon 0.1-μM dopamine treatment, BDNF would enhance both GluA1 phosphorylation and membrane insertion (Fig. 2B and Fig. S6 C and D). Inhibition of PDE4 enhanced cAMP levels, GluA1 phosphorylation, and membrane insertion at 0.1 μM dopamine (Fig. 2B and Fig. S6 E and F).
Although the influence of ERK-mediated inhibition of PDE4 on GluA1 trafficking was difficult to predict a priori, our computer simulations led to specific, mechanistic, testable hypotheses. Based on the simulations, we guided our experimental questions. If ERK-mediated inhibition of PDE4 is involved in dopamine-induced GluA1 trafficking, then: (i) BDNF should enhance 0.1-μM dopamine-mediated GluA1 phosphorylation as well as membrane insertion; also, inhibition of ERK should decrease phosphorylation of other PKA targets such as DARPP-32; (ii) dopamine should induce the ERK-phosphorylation of PDE4, and thus ERK inhibition should decrease cAMP levels; and, finally, (iii) inhibition of PDE4 should increase GluA1 phosphorylation and membrane insertion by increasing cAMP levels.
BDNF Enhances Dopamine-Induced GluA1 Phosphorylation and Membrane Insertion.
BDNF is one of the main activators of ERK independently of cAMP in the striatum. When the BDNF pathway was included in the model, it led to an increase in 0.1-μM dopamine-induced phosphorylation of GluA1 as well as membrane insertion (Fig. 2B). To further implicate ERK in dopamine-induced GluA1 membrane insertion, we tested whether BDNF could further increase dopamine-mediated GluA1 membrane insertion. Stimulation of 0.1 μM dopamine plus BDNF increased the phosphorylation of GluA1, but the effect was not statistically significant in cotreatments with 1 μM dopamine (Fig. 3 A and B and Fig. S7A). BDNF cotreatment with 0.1 μM dopamine (Fig. 3 C and D) or a D1R-type specific agonist (Fig. S7B) increased GluA1 membrane insertion, and, in accordance with the phosphorylation results, BDNF did not enhance GluA1 membrane insertion at 1.0 μM dopamine (Fig. S7 C and D). Application of BDNF alone was not able to enhance GluA1 membrane insertion (Fig. S7E). We concluded that ERK activity is solely responsible for the BDNF effect because inhibition of ERK abolishes the BDNF increase (Fig. S7F). The model also predicted that other PKA targets such as DARPP-32 should be regulated by ERK (Fig. S6 A and B). Indeed, we found that, upon ERK inhibition, PKA-mediated phosphorylation of DARPP-32 at Thr-34 decreased significantly at both dopamine concentrations tested (Fig. 4 A and B).
Fig. 3.

BDNF enhances dopamine–GluA1 phosphorylation and membrane insertion. (A) BDNF increases dopamine–GluA1 phosphorylation. BDNF was followed by 0.1 or 1.0 μM dopamine for 30 min. Cellular lysates were immunoblotted for phospho-GluA1 Ser-845, GluA1, phospho-ERK, and GAPDH. (B) Comparison of experimental BDNF enhancement of dopamine-induced GluA1 phosphorylation (exp) and simulation (sim) results after 30 min of 0.1-μM dopamine stimulation. *P = 0.0428 (t test; n = 5). (C) BDNF increases 0.1-μM dopamine–GluA1 membrane insertion. A concentration of 0.1 μM dopamine (gray) or 0.1 μM dopamine and BDNF (black) were added. *P = 0.0007 (repeated-measures ANOVA Bonferroni post hoc; n = 9). (D) Simulated time course of 0.1-μM dopamine-induced GluA1 membrane insertion (gray) or 0.1 μM dopamine and BDNF (black) stimulation.
Fig. 4.
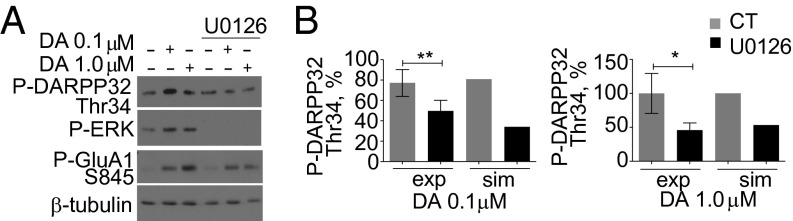
Inhibition of ERK decreases PKA phosphorylation of DARPP-32. (A) U0126 decreases dopamine-induced PKA phosphorylation of DARPP-32. Neurons were treated with or without U0126 followed by varying concentrations of dopamine for 30 min. Cellular lysates were immunoblotted for phospho–DARPP-32 Thr-34, phospho-GluA1 Ser-845, or phospho-ERK or β-tubulin. (B) Comparison of densitometry of U0126-mediated loss of dopamine-induced DARPP-32 phosphorylation (exp) and simulation (sim) results after 30 min of 0.1-μM (Left) and 1.0-μM (Right) dopamine stimulation. **P = 0.0051; *P = 0.0370 (t test; n = 9 for each concentration).
Dopamine Induces ERK Phosphorylation of PDE4, and ERK Activity Increases cAMP.
We next explored the dopamine-induced phosphorylation of PDE4 by ERK. PDE4 is phosphorylated and inhibited by growth-factor-activated ERK (10). We tested whether dopamine is able to induce the ERK-dependent phosphorylation of PDE4B or PDE4D and thus control cAMP levels. After dopamine treatment, PDE4B or PDE4D was immunoprecipitated and blotted by using a pan phospho-Ser/Thr ERK substrate antibody that recognizes any ERK-phosphorylated substrate. Dopamine treatment induced the ERK phosphorylation of PDE4B and PDE4D (Fig. 5A), which demonstrates the direct regulation of PDE4 activity by dopamine-activated ERK. A representative blot is found in Fig. S8A. We also tested whether ERK inhibition decreases cAMP levels in response to dopamine using the cAMP FRET reporter ICUE3 (27). Dopamine induced a dose-dependent increase in cAMP (Fig. S8B). Inhibiting ERK decreased dopamine-induced cAMP levels (Fig. 5B), and this deficit was reversed by adding the PDE4 inhibitor rolipram (Fig. 5C). Together these data confirm that activation of ERK by dopamine leads to inhibition of PDE4 and increases levels of cAMP.
Fig. 5.

Dopamine-activated ERK increases cAMP levels by inhibition of PDE4B/D leading to regulation of GluA1 phosphorylation and membrane insertion. (A) Dopamine induces the ERK phosphorylation of PDE4B and PDE4D. After 30 min of 0.1- or 1.0-μM dopamine stimulation, PDE4B or PDE4D was immunoprecipitated and immunoblotted for a pan phospho-ERK substrate and PDE4B or PDE4D. Densitometry of immunoprecipitation is shown. (ANOVA post Tukey; n = 3–6; P = 0.0450). (B) ERK inhibition reduces 0.1-μM dopamine-induced cAMP levels. A concentration of 0.1 μM dopamine (gray) or 0.1 μM dopamine and 10 μM U0126 (black) was added. *P = 0.027 (repeated-measures ANOVA Bonferroni post hoc test; n = 5–10). (C) U0126 reduction of 0.1-μM dopamine-induced cAMP levels is reversed by rolipram coapplication. A concentration of 0.1 μM dopamine (gray), 0.1 μM dopamine and 10 μM U0126 (red), or 0.1 μM dopamine, 10 μM U0126, and 10 μM rolipram (black) was added. *P = 0.0182 (repeated-measures ANOVA Bonferroni post hoc test; n = 6–13). (D) Rolipram, an inhibitor of PDE4, increases dopamine phosphorylation of GluA1. Neurons were treated with 10 μM rolipram followed by 0.1- or 1.0-μM dopamine stimulation for 30 min. Cellular lysates were immunoblotted for phospho-GluA1 Ser-845 and GAPDH (loading control). (E) Rolipram enhances 0.1-μM dopamine-induced GluA1 phosphorylation. Comparison of experimental rolipram effect on 0.1-μM dopamine-induced GluA1 phosphorylation (exp) and simulation (sim) results is shown. *P = 0.0172 (t test; n = 5). (F) PDE4 inhibition increases 0.1-μM dopamine–GluA1 membrane insertion. A concentration of 0.1 μM dopamine (black) or 0.1 μM dopamine plus 10 μM rolipram (gray) was added. *P = 0.006 (repeated-measures ANOVA Bonferroni post hoc; n = 4–11). (G) Simulated rolipram enhancement of 0.1-μM dopamine-induced GluA1 membrane insertion. (H) Rolipram recovers U0126 effect on dopamine–GluA1 membrane insertion. A concentration of 0.1 μM dopamine, 10 μM rolipram, and 10 μM U0126 (gray) was added. A 0.1-μM dopamine–GluA1 membrane insertion experiment (black) from Fig. 1A and 0.1 μM dopamine with U0126–GluA1 membrane insertion experiment (red) from Fig. 1B are replotted along with Sp-cAMPS results (repeated-measures ANOVA Bonferroni post hoc; P = 0.0047; n = 13).
PDE4 Modulates Dopamine-Induced GluA1 Phosphorylation and Membrane Insertion.
The simulations predicted, following ablation of PDE4, an increase in the phosphorylation of GluA1 at 0.1 μM dopamine (Fig. 2B). Inhibition of PDE4 enhanced 0.1-μM dopamine-induced cAMP levels and GluA1 phosphorylation (Fig. S8C and Fig. 5 D and E). As predicted, PDE4 inhibition enhanced both 0.1-μM dopamine-induced and D1R agonist-induced GluA1 membrane insertion (Fig. 5 F and G and Fig. S9A). There was no significant effect on GluA1 membrane insertion at the higher dopamine concentration upon cotreatment with rolipram (Fig. S9B), despite the increase in phosphorylation of GluA1 (Fig. S9C). This result suggests that at high dopamine concentration, GluA1 membrane insertion is saturated. Inhibiting PDE4 alone had minimal effect on GluA1 membrane insertion (Fig. S9D). Rolipram and dopamine cotreatment rescued the ERK inhibition-induced deficit in GluA1 membrane insertion (Fig. 5H). This finding is further supported by the observation that addition of adenosine 3′,5′-cyclic monophosphorothioate, Sp isomer (Sp-cAMPS), a nonhydrolysable analog of cAMP and activator of PKA, also partially reversed the ERK inhibition deficit in 0.1-μM dopamine–GluA1 membrane insertion (Fig. S9E). This result suggests that when there is a lack of ERK activity, higher cAMP levels can counteract overactive PDE4.
ERK Regulation of PDE4 Affects PKA/PP1 Balance of GluA1 Membrane Insertion.
We hypothesized a central role for PDE4 and its regulation by ERK in GluA1 phosphorylation and trafficking dynamics. Both model and experiments suggest that ERK regulation of PDE4 is essential for GluA1 membrane insertion at 0.1 μM dopamine. However, at higher dopamine concentrations, BDNF and PDE4 inhibition do not enhance GluA1 membrane insertion. We examined the model to understand this dichotomy. Simulation of 0.1 μM dopamine showed that the majority of PDE4 is in the ERK-phosphorylated (PDE4–ERK) or the double-phosphorylated (PDE4–ERK–PKA) form, with the PKA-phosphorylated form (PDE4–PKA) representing only 15.6% of total PDE4 levels (Fig. 6A). Inhibiting ERK shifted the distribution of each phosphorylated PDE4 species such that PDE4–PKA is the predominant form of PDE4 (Fig. 6A) and resulted in a threefold increase in total PDE4 activity (Fig. 6B). The topology of the PKA–ERK–PDE4 loop provides a point of integration in allowing other extracellular signals, such as the BDNF, to signal through ERK and enhance PKA output independently of cAMP. In the simulations, BDNF treatment increased the ratio of both PDE4–ERK and PDE4–ERK–PKA to PDE4–PKA and PDE4 (Fig. 6A) and led to a decrease in total PDE4 activity (Fig. 6B). Similar effects are seen with simulated dopamine treatment at 1.0 μM (Fig. S10A). Because PKA activity also depends on the amount of active adenylyl cyclase, and that changes in a dopamine-dependent manner, we plotted the proportions of active AC and total PDE4 activity along with the proportion of active PKA values obtained at steady state. BDNF increased the ratio of AC to PDE activity and led to increases in PKA at 0.1 μM dopamine (Fig. 6B), but the effect was lost at higher dopamine concentrations, suggesting that PKA reaches maximum activation despite further increases in cAMP levels (Fig. S10A). This effect is further illustrated in Fig. 6 C and D, where we modulated levels of active ERK by uncoupling it from its upstream activators (Fig. 6C). We plotted the resulting steady-state levels of active PKA and its downstream output, namely, active PP1 and membrane GluA1 levels in response to different dopamine concentrations. At 0.1 μM dopamine, modulation of active ERK by reducing it to levels comparable with U0126 treatment or increasing it to levels seen with BDNF treatment has a drastic effect on PKA activity and output, beyond what is seen with dopamine treatment alone (black box, Fig. 6D). At 1 μM dopamine, increasing ERK activation beyond levels that occur with dopamine treatment alone does not further enhance PKA activation or output (Fig. 6D, black arrow). Results show that at 0.1 μM dopamine, inhibiting ERK decreases the ratio of active PKA to active PP1 ([PKA]/[PP1]), promoting the dephosphorylation of GluA1. Upon BDNF cotreatment, the balance between PKA and PP1 changes, and PKA phosphorylation is promoted (Fig. 6E). At 1.0 μM dopamine, the BDNF effect on [PKA]/[PP1] is negligible (Fig. S10B).
Fig. 6.

Model predicts that ERK regulates GluA1 phosphorylation and membrane insertion by modulating PKA/PP1 activity ratio. (A) Simulation results showing distribution of four different PDE4 states at 30 min after treatments of 0.1 μM dopamine, 0.1 μM dopamine and U0126, or 0.1 μM dopamine and BDNF. Nonphosphorylated PDE4 percentage (white), PKA-phosphorylated PDE4 percentage (black), ERK-phosphorylated PDE4 percentage (light gray), and ERK/PKA double-phosphorylated PDE4 percentage (dark gray) are shown. (B) Simulation results showing percent of total activities of AC, PDE4, and PKA at 30 min after treatments of 0.1 μM dopamine, 0.1 μM dopamine and U0126, or 0.1 μM dopamine and BDNF. Active AC percentage (black), PDE4 activity percentage (gray), and PKA activity percentage (white) are shown. (C) Model topology depicting uncoupled ERK. (D) Simulation of dopamine-induced active PKA (Left), active PP1 (Center), and GluA1 membrane insertion (Right) against varying concentrations of active ERK and dopamine. All values were scaled to maximum values obtained with 1.0 μM dopamine after 30 min color-coded to represent low (blue) and high (red) concentrations. Arrows indicate concentration of active ERK concentrations when ERK is inhibited (U0126 treatment) or BDNF is added. The box represents basal to dopamine-activated concentrations of active ERK. (E) Simulation results of the ratio of active PKA and PP1 at 30 min after treatments of 0.1 μM dopamine alone or with U0126 or BDNF.
Discussion
Existing models of GluA1 membrane trafficking do not account for experimental observations that ERK plays a key role in this process. ERK activity is required for AMPAR insertion because its inhibition prevents GluA1-mediated insertion in response to glutamate or dopamine stimulation (8, 21). Several studies have reported the essential contribution of ERK to synaptic plasticity. For example, the use of specific ERK inhibitors or the expression of dominant negative Ras occludes hippocampal long-term potentiation, a form of synaptic plasticity, and impairs learning and memory (7, 28–30). However, the ERK substrates that mediate synaptic plasticity events are still unknown.
Here we identify PDE4 as a key ERK substrate in GluA1 membrane insertion. We developed a dynamical computational model to examine our central hypothesis that dopamine-activated ERK inhibits PDE4, which results in increased cAMP/PKA levels and enhanced GluA1 phosphorylation at S845 and membrane insertion. We show that the ERK–PDE4 loop enhances dopamine-induced PKA output by preventing the PKA–PDE4 negative feedback loop from decreasing cAMP/PKA levels (Fig. 2A). When the ERK–PDE4 feedback loop is abolished, as in our U0126 cotreatment experiments, the PKA–PDE4 negative feedback loop maintains low PKA activity.
Several lines of evidence support our conclusion that ERK-mediated regulation of GluA1 membrane insertion occurs via PDE4 inhibition. First, ERK inhibition results in a decrease in phosphorylation of GluA1 by PKA (Fig. 1C). The U0126-mediated deficit in GluA1 membrane insertion is rescued by increasing cAMP via Sp-cAMPS or rolipram treatment (Fig. 5H and Fig. S9E). We also show that ERK inhibition decreases dopamine-induced cAMP levels (Fig. 5B), and this decrease is rescued by rolipram treatment (Fig. 5C). We demonstrate that BDNF, an activator of ERK, can enhance GluA1 phosphorylation and membrane insertion (Fig. 3 B and C). Finally, we show that ERK-mediated signaling leading to phosphorylation and that inactivation of PDE4 enhances GluA1 membrane insertion (Fig. 5A).
The ERK–PDE4 motif could signal in other receptor systems that induce the simultaneous activation of PKA and ERK, such as the beta-adrenergic receptor in the hippocampus (28, 31). This motif could also be a potential point of integration for Gs-coupled receptors and ERK activating agents such as growth factors, chemokines, and cytokines in many tissues and cell types. This type of integration could regulate PKA and potentially EPAC1–Rap1 output and thus play a role in a number of processes such as cytoskeletal reorganization, secretion, and metabolic control.
Phosphodiesterases, the main modulators of cellular cAMP levels, are key players in AMPAR trafficking, given the essential role of PKA-mediated phosphorylation of GluA1 in the membrane insertion of AMPARs. Here, we identified PDE4 as a regulator of GluA1 phosphorylation and membrane insertion. The PDE4 family is encoded by four genes—designated as PDE4A, PDE4B, PDE4C, and PDE4D—and each gene is expressed as multiple isoforms that are all sensitive to rolipram inhibition (32). However, only the PDE4B, PDE4C, and PDE4D isoforms are phosphorylated and regulated by ERK. We have yet to identify all of the PDE4 isoforms involved in dopamine-induced GluA1 trafficking, but based on our immunoprecipitation data, the long isoforms of PDE4B and PDE4D appear to be implicated because they are ERK phosphorylated following ERK signaling in a dopamine-dependent manner (Fig. 5A). Agents that increase AMPAR membrane insertion, such as rolipram, can potentially enhance synaptic strength and contribute to synaptic plasticity. Indeed, rolipram treatment facilitates long-term potentiation and memory formation (33). PDE4D knockout animals also show enhanced memory in spatial tasks (34). Furthermore, rolipram is able to rescue memory deficits induced by ERK inhibition (35).
Although our data show a clear role for PDE4 in GluA1 phosphorylation and membrane insertion, it is likely that there are other PDE families that have yet to be implicated in the regulation of GluA1 S845 phosphorylation and AMPAR trafficking. An additional player may be PDE10, which also regulates GluA1 S845 phosphorylation in vivo (36); however, the effect of PDE10 inhibition on AMPAR membrane insertion has yet to be explored. Our data suggest that PDE4 is not the only ERK substrate that modulates AMPAR trafficking. Sp-cAMPS cotreatment is only able to partially rescue the deficiency caused ERK inhibition, suggesting that non-PKA events may also be at play in the ERK regulation of AMPAR trafficking. BDNF and rolipram enhance 0.1-μM dopamine-mediated GluA1 insertion, but fail to increase 1.0-μM dopamine-mediated GluA1 insertion. Surprisingly, rolipram enhances the phosphorylation of S845 at both dopamine concentrations. The nature of this discrepancy remains unknown.
We show that ERK promotes the phosphorylation of Thr-34 on DARPP-32 by PKA. Previous studies have demonstrated a positive role for ERK activity in the metabotropic glutamate mGlu5 receptor and adenosine A2A receptor stimulation of DARPP-32 Thr-34 phosphorylation (37). Here we present a mechanism that describes how ERK can modulate PKA activity. Our data also suggest a mechanism for the regulation of PP1 activity by ERK signaling. The model predicts that the functional consequence of ERK-mediated regulation of DARPP-32 is enhanced inhibition of PP1, but that hypothesis has yet to be experimentally tested.
Our combined approach of computational modeling and experimental measurements of dopamine signaling and AMPAR trafficking uncovered a unique role for dopamine-activated ERK as a modulator of PKA output. Simulations guided the experiments to validate the role of the ERK–PDE4 loop in GluA1 trafficking. This approach could be useful in identifying novel therapeutic targets for dopamine pathologies such as Parkinson disease and addiction that result in dysregulation of the AMPAR response (38, 39).
Materials and Methods
A detailed description of all materials, methods, and the computational model can be found in SI Materials and Methods. Briefly, primary striatal neurons were dissected from embryonic day-18 embryos as described with modifications (40). Western blots and immunoprecipitation were performed as described with modifications (41).
Supplementary Material
Acknowledgments
We thank Drs. E. Sobie, R. Blitzer, R. Iyengar, and A. Boran for helpful comments on the manuscript; and Dr. J. Zhang for providing the cAMP reporter ICUE3 plasmid. This work was supported by National Institutes of Health Grants PSDGM071558 and 5R01DK087650.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The models used in this paper have been deposited at the Virtual Cell web site, vcell.org/vcell_models/published_models.html.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1311783110/-/DCSupplemental.
References
- 1.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 2.Serulle Y, et al. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56(4):670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snyder GL, et al. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20(12):4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esteban JA, et al. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6(2):136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 5.Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25(32):7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mangiavacchi S, Wolf ME. D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J Neurochem. 2004;88(5):1261–1271. doi: 10.1046/j.1471-4159.2003.02248.x. [DOI] [PubMed] [Google Scholar]
- 7.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110(4):443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Wolf ME. Brain-derived neurotrophic factor rapidly increases AMPA receptor surface expression in rat nucleus accumbens. Eur J Neurosci. 2011;34(2):190–198. doi: 10.1111/j.1460-9568.2011.07754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawabe J, et al. Differential activation of adenylyl cyclase by protein kinase C isoenzymes. J Biol Chem. 1994;269(24):16554–16558. [PubMed] [Google Scholar]
- 10.Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999;18(4):893–903. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394(6689):192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 12.Shi SH, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284(5421):1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 13.Neve KA, Neve RL. The Dopamine Receptors. Totowa, NJ: Humana; 1997. [Google Scholar]
- 14.Civelli O, Bunzow JR, Grandy DK. Molecular diversity of the dopamine receptors. Annu Rev Pharmacol Toxicol. 1993;33:281–307. doi: 10.1146/annurev.pa.33.040193.001433. [DOI] [PubMed] [Google Scholar]
- 15.Yao L, et al. Betagamma dimers mediate synergy of dopamine D2 and adenosine A2 receptor-stimulated PKA signaling and regulate ethanol consumption. Cell. 2002;109(6):733–743. doi: 10.1016/s0092-8674(02)00763-8. [DOI] [PubMed] [Google Scholar]
- 16.Czöndör K, et al. Unified quantitative model of AMPA receptor trafficking at synapses. Proc Natl Acad Sci USA. 2012;109(9):3522–3527. doi: 10.1073/pnas.1109818109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayer A, Bhalla US. Molecular switches at the synapse emerge from receptor and kinase traffic. PLOS Comput Biol. 2005;1(2):137–154. doi: 10.1371/journal.pcbi.0010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakano T, Doi T, Yoshimoto J, Doya K. A kinetic model of dopamine- and calcium-dependent striatal synaptic plasticity. PLOS Comput Biol. 2010;6(2):e1000670. doi: 10.1371/journal.pcbi.1000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oliveira RF, Kim M, Blackwell KT. Subcellular location of PKA controls striatal plasticity: Stochastic simulations in spiny dendrites. PLOS Comput Biol. 2012;8(2):e1002383. doi: 10.1371/journal.pcbi.1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindskog M, Kim M, Wikström MA, Blackwell KT, Kotaleski JH. Transient calcium and dopamine increase PKA activity and DARPP-32 phosphorylation. PLOS Comput Biol. 2006;2(9):e119. doi: 10.1371/journal.pcbi.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson MA, Szatmari EM, Yasuda R. AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc Natl Acad Sci USA. 2010;107(36):15951–15956. doi: 10.1073/pnas.0913875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin LL, et al. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72(2):269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 23.Nemenoff RA, et al. Phosphorylation and activation of a high molecular weight form of phospholipase A2 by p42 microtubule-associated protein 2 kinase and protein kinase C. J Biol Chem. 1993;268(3):1960–1964. [PubMed] [Google Scholar]
- 24.Shinomura T, Asaoka Y, Oka M, Yoshida K, Nishizuka Y. Synergistic action of diacylglycerol and unsaturated fatty acid for protein kinase C activation: Its possible implications. Proc Natl Acad Sci USA. 1991;88(12):5149–5153. doi: 10.1073/pnas.88.12.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann R, Wilkinson IR, McCallum JF, Engels P, Houslay MD. cAMP-specific phosphodiesterase HSPDE4D3 mutants which mimic activation and changes in rolipram inhibition triggered by protein kinase A phosphorylation of Ser-54: Generation of a molecular model. Biochem J. 1998;333(Pt 1):139–149. doi: 10.1042/bj3330139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sette C, Conti M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J Biol Chem. 1996;271(28):16526–16534. doi: 10.1074/jbc.271.28.16526. [DOI] [PubMed] [Google Scholar]
- 27.Allen MD, et al. Reading dynamic kinase activity in living cells for high-throughput screening. ACS Chem Biol. 2006;1(6):371–376. doi: 10.1021/cb600202f. [DOI] [PubMed] [Google Scholar]
- 28.Winder DG, et al. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24(3):715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- 29.Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481(7379):71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- 30.Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5(3):173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 31.Neves SR, et al. Cell shape and negative links in regulatory motifs together control spatial information flow in signaling networks. Cell. 2008;133(4):666–680. doi: 10.1016/j.cell.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conti M, et al. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem. 2003;278(8):5493–5496. doi: 10.1074/jbc.R200029200. [DOI] [PubMed] [Google Scholar]
- 33.Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci USA. 1998;95(25):15020–15025. doi: 10.1073/pnas.95.25.15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li YF, et al. Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J Neurosci. 2011;31(1):172–183. doi: 10.1523/JNEUROSCI.5236-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang HT, et al. Inhibition of the phosphodiesterase 4 (PDE4) enzyme reverses memory deficits produced by infusion of the MEK inhibitor U0126 into the CA1 subregion of the rat hippocampus. Neuropsychopharmacology. 2004;29(8):1432–1439. doi: 10.1038/sj.npp.1300440. [DOI] [PubMed] [Google Scholar]
- 36.Nishi A, et al. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci. 2008;28(42):10460–10471. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishi A, et al. Metabotropic mGlu5 receptors regulate adenosine A2A receptor signaling. Proc Natl Acad Sci USA. 2003;100(3):1322–1327. doi: 10.1073/pnas.0237126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt MV, et al. Individual stress vulnerability is predicted by short-term memory and AMPA receptor subunit ratio in the hippocampus. J Neurosci. 2010;30(50):16949–16958. doi: 10.1523/JNEUROSCI.4668-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vialou V, et al. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci. 2010;13(6):745–752. doi: 10.1038/nn.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wenderski WC, Neves SR. Modeling of spatial intracellular signaling events in neurons. Methods Enzymol. 2012;505:105–124. doi: 10.1016/B978-0-12-388448-0.00014-0. [DOI] [PubMed] [Google Scholar]
- 41.Hwangpo TA, et al. G Protein-regulated inducer of neurite outgrowth (GRIN) modulates Sprouty protein repression of mitogen-activated protein kinase (MAPK) activation by growth factor stimulation. J Biol Chem. 2012;287(17):13674–13685. doi: 10.1074/jbc.M111.320705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.