

Significance
The study describes a previously unknown role for the cytosolic ubiquitin ligase Ubr1 in endoplasmic reticulum-associated protein degradation, thereby connecting the protein quality control processes of the endoplasmic reticulum and of the cytosol.
Keywords: protein quality control, stress response, heat shock
Abstract
Quality control and degradation of misfolded proteins are essential processes of all cells. The endoplasmic reticulum (ER) is the entry site of proteins into the secretory pathway in which protein folding occurs and terminally misfolded proteins are recognized and retrotranslocated across the ER membrane into the cytosol. Here, proteins undergo polyubiquitination by one of the membrane-embedded ubiquitin ligases, in yeast Hrd1/Der3 (HMG-CoA reductase degradation/degradation of the ER) and Doa10 (degradation of alpha), and are degraded by the proteasome. In this study, we identify cytosolic Ubr1 (E3 ubiquitin ligase, N-recognin) as an additional ubiquitin ligase that can participate in ER-associated protein degradation (ERAD) in yeast. We show that two polytopic ERAD substrates, mutated transporter of the mating type a pheromone, Ste6* (sterile), and cystic fibrosis transmembrane conductance regulator, undergo Ubr1-dependent degradation in the presence and absence of the canonical ER ubiquitin ligases. Whereas in the case of Ste6* Ubr1 is specifically required under stress conditions such as heat or ethanol or in the absence of the canonical ER ligases, efficient degradation of human cystic fibrosis transmembrane conductance regulator requires function of Ubr1 already in wild-type cells under standard growth conditions. Together with the Hsp70 (heat shock protein) chaperone Ssa1 (stress-seventy subfamily A) and the AAA-type ATPase Cdc48 (cell division cycle), Ubr1 directs the substrate to proteasomal degradation. These data unravel another layer of complexity in ERAD.
Constantly occurring statistic folding errors, as well as misfolding due to stress such as heat, heavy metal ions, or oxygen require a rigorous protein quality control system in all cellular compartments. Irreversibly misfolded proteins are degraded by a selective proteolysis machinery, the ubiquitin proteasome system. In humans, impairment of the protein quality control and elimination system contributes to several severe diseases, including Parkinson disease, Alzheimer’s disease, and Creutzfeldt–Jakob disease (1, 2), underscoring the importance of these quality control mechanisms. About one third of the cellular proteome consists of proteins passing the secretory pathway. Most of them are translocated into the endoplasmic reticulum (ER), where they are folded and permanently scanned for their functional structure. Only properly folded proteins are allowed to exit the ER and pass on to their site of action (3). Proteins that cannot fold properly are withdrawn from the secretory pathway, retrotranslocated across the ER membrane into the cytosol, polyubiquitinated and degraded by the 26S proteasome in a process termed ER-associated protein degradation (ERAD) (4).
The eukaryotic model organism Saccharomyces cerevisiae has been a driving force in the discovery and elucidation of ERAD (4–10). It has been known for years that two polytopic ligases located in the ER membrane, Hrd1/Der3 (HMG-CoA reductase degradation/degradation of the ER) and Doa10 (degradation of alpha), play a central role in ERAD in yeast, by directing ERAD substrates to proteasomal degradation (11–16). Previous studies on a variety of ERAD substrates revealed that, in most cases, the absence of one of the two canonical ER ubiquitin ligases does not lead to a complete block of degradation of the tested substrate. It was thought that this might be the result of a complementary effect of the remaining ligase, which takes over part of the ubiquitination activity of the missing ligase (15, 17, 18). However, even in the few cases in which the fate of a substrate was analyzed in strains missing both ligases, Hrd1/Der3 and Doa10, no complete cessation of degradation could be observed, indicating an additional unknown degradation route (17–19).
Here, we report that the cytosolic ubiquitin ligase Ubr1 functions as an additional E3 ligase in ERAD in yeast. We show that in the absence of the two canonical polytopic ER membrane ligases, Ubr1 can provide ubiquitin ligation activity for the ERAD substrate Ste6* (mutated Ste6; sterile). Application of heat or ethanol stress to cells unmasks the involvement of Ubr1 in Ste6* elimination also in the presence of Hrd1/Der3 and Doa10. Remarkably, the degradation of the ERAD substrate cystic fibrosis transmembrane conductance regulator (CFTR) depends on Ubr1 even in unstressed wild-type cells. Our findings indicate a previously unknown connection between cytosolic and membrane ligases in the ERAD process of yeast, which is required for the efficient removal of certain misfolded proteins of the ER membrane. The data also imply the existence of a retrotranslocation mechanism of misfolded ERAD substrates independent of the canonical ER ligases.
Results
Membrane-Bound Ste6* Is Degraded in a Proteasomal Manner in the Absence of Canonical ERAD Ligases.
Previous studies in yeast revealed that, in some cases, the absence of both canonical ER ubiquitin ligases, Hrd1/Der3 and Doa10, does not completely prevent degradation of a variety of ERAD substrates (17–19). Therefore, we hypothesized that an additional ERAD route exists that is independent of the canonical ligases and responsible for the residual degradation. To address this hypothesis, we selected the misfolded polytopic membrane protein Ste6* as a model substrate. Ste6* is a C-terminally truncated version of the transporter of the mating type a pheromone and is retained in the ER membrane as a result of the exposition of its misfolded domain to the cytosol (17, 20). In cells lacking the two canonical ubiquitin ligases, Ste6* is not fully stabilized but remains a target for degradation (17, 19). Within 90 min, over 50% of the initial amount of Ste6* is still degraded in a Δhrd1 Δdoa10 background (Fig. 1A).
Fig. 1.

Degradation of Ste6* in the absence of canonical ERAD ligases is proteasome dependent and facilitated by the cytosolic ubiquitin ligase Ubr1. (A–C) Cells of indicated strains expressing plasmid-encoded, HA-tagged Ste6* were subjected to PC analyses. Data reflect the mean of three to six independent experiments. Error bars indicate the SEM. (B) Cells lacking Hrd1/Der3, Doa10, and the multidrug transporter Pdr5 were treated with MG132 (○) to inhibit proteasomal degradation of Ste6* or equal amounts of solvent (■). (C) HRD1 DOA10 UBR1 triple-deleted cells were transformed with a plasmid-encoding, FLAG-tagged, inactive, RING-mutated (RM) Ubr1 (♦); with a FLAG-tagged wild-type Ubr1 (○); or with an empty vector (■).
As ERAD substrates usually end up in the proteasome for elimination (4–10, 21, 22), we tested whether the residual degradation of Ste6* in the absence of the two canonical ligases, Hrd1/Der3 and Doa10, remains proteasome dependent. Hence, we used MG132, a specific proteasomal inhibitor (23), which achieves optimum inhibitory activity in the absence of the multidrug-resistance transporter Pdr5 (pleiotropic drug resistance) (24). In a strain devoid of the two canonical ERAD ligases, as well as Pdr5, the degradation of Ste6* was strongly attenuated when cells were treated with MG132 (Fig. 1B). This indicates that elimination of Ste6* in Hrd1/Der3- and Doa10-deficient cells remains proteasome dependent and does not arise from other degradation systems. Because the assay in its final step was performed only with the membrane fraction separated from the cytosol after cell lysis with glass beads, the monitored degradation process refers only to Ste6* species that have been integrated properly into the ER membrane before their decomposition. These findings support the hypothesis of a yet unidentified ERAD route and propose the involvement of an additional ubiquitin ligase besides Hrd1/Der3 and Doa10 in ERAD. The ER membrane of yeast cells comprises only two membrane-embedded ligases, Hrd1/Der3 and Doa10, with no other anticipated candidates. Consequently, ubiquitination of Ste6* before proteasomal degradation would require either transport to a different cell compartment containing E3 ligases or recruitment of a cytosolic ligase to the ER membrane.
ERAD of Membrane-Bound Ste6* in the Absence of Canonical ER Ligases Is Executed by the Cytosolic Ubiquitin Ligase Ubr1.
Recently, the cytosolic RING (really interesting new gene) -type ubiquitin ligase Ubr1 was linked to ubiquitination and degradation of misfolded proteins in the cytosol (25–27). Therefore, we considered this ligase to ubiquitinate Ste6* in the absence of the canonical ERAD ligases. We compared the degradation rate of Ste6* in cells lacking Hrd1/Der3 and Doa10 and cells that also are devoid of Ubr1. Supplementary deletion of UBR1 resulted in nearly complete stabilization of Ste6* over 60 min of chase, indicating that Ubr1 can mediate ubiquitination and subsequent proteasomal degradation of membrane-bound Ste6* (Fig. 1A). Ubr1 carries conserved cysteine residues in its RING domain that are essential for its function as a ubiquitin ligase (28). Introduction of FLAG (hydrophilic 8-amino acid peptide with the sequence Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) -tagged, catalytically inactive, RING–cysteine-mutated Ubr1 into HRD1 DOA10 UBR1 triple-deleted cells could not complement Ubr1 function in degradation of Ste6*, whereas reintroduction of FLAG-tagged wild-type Ubr1 restored the function of genomic Ubr1 (Fig. 1C). Thus, the ligase activity of Ubr1 is indeed required for this newly identified ERAD route.
Several E2 Enzymes Contribute to Ubr1-Dependent ERAD.
The ubiquitin-conjugating enzymes Ubc1, Ubc6, and Ubc7 have been shown to be the canonical E2s in the ERAD process. Depending on the E3 ligase involved in the ubiquitination process, there is a certain requirement for E2 enzymes. Hrd1/Der3 is thought to act mainly in concert with the E2s Ubc1 and Ubc7, whereas Doa10 mainly works together with Ubc6 and Ubc7 (14, 29, 30). Canonical ERAD of the substrate Ste6* depends on Doa10; therefore, Ubc6 and Ubc7 are the preferred ubiquitin-conjugating enzymes in this process (20). In Ubr1-dependent ERAD, the combined deletion of UBC6 and UBC7 or combined deletion of UBC1 and UBC7 showed a small but measurable decline of Ste6* degradation (Fig. 2 A and B). The ongoing degradation of Ste6* indicates that Ubr1 does not act primarily in concert with the canonical E2 enzymes of either of the well-established ERAD ligases in this process. In its function as the ligase of the N-end rule pathway, Ubr1 was shown to work prominently together with Ubc2 (31). Deletion of UBC2 led to significant, although not complete, stabilization of Ste6* in a mutant lacking the two canonical ubiquitin ligases Hrd1/Der3 and Doa10 (Fig. 2C). This finding indicates that Ubr1 also acts in its function as an ERAD ligase together with Ubc2. The remaining degradation in the absence of Ubc2 hints at yet another E2 enzyme involved in this degradation route. As a possible candidate, we tested Ubc4, which plays an important role in cytosolic ubiquitination events (ref. 32 and included references). However, we could not find any involvement of Ubc4 in Ubr1-dependent ERAD under these conditions (Fig. 2D). Obviously, with respect to its function in ERAD, several E2s can work together with Ubr1 and contribute to the degradation of Ste6*.
Fig. 2.
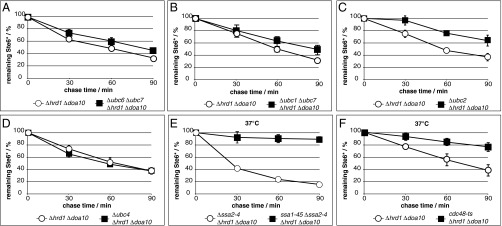
Ubr1-dependent ERAD is assisted by several E2 enzymes and relies on Ssa1 and Cdc48. (A–F) Cells of indicated strains expressing plasmid-encoded, HA-tagged Ste6* were subjected to PC analyses. Data reflect the mean of three to seven independent experiments. Error bars indicate the SEM. (E) Cells devoid of Ssa2, Ssa3, and Ssa4 but harboring wild-type Ssa1 (○) and cells carrying the temperature-sensitive ssa1-45 allele instead of wild-type SSA1 (■) were analyzed for Ubr1-dependent ERAD of Ste6*. (F) The function of wild-type Cdc48 (○) was impaired by use of the temperature-sensitive (ts) cdc48-T413R allele (■). (E and F) Cells were shifted to 37 °C 1 h before time point 0.
Ubr1 Interacts with Ste6*.
To accomplish ubiquitination, at least a transient interaction between the ligase and the substrate is required. Therefore, we analyzed the interaction between Ubr1 and Ste6* in cells devoid of the canonical ligases Hrd1 and Doa10. To exclude the unspecific binding of Ubr1 to solubilized membrane complexes or cytosolic proteins involved in ERAD, binding of an abundant cytosolic protein, phosphoglycerol kinase (PGK), was used as a negative control. Whereas Ubr1 and the established cytosolic ERAD segregase Cdc48 (cell devision cycle) coprecipitated with Ste6*, PGK did not (Fig. 3A). This indicates specific interactions among the substrate Ste6*, the ligase Ubr1, and Cdc48 and supports the initial hypothesis of an additional ERAD route dependent on the ligase Ubr1.
Fig. 3.
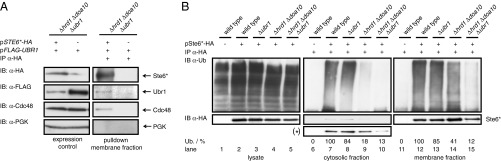
The cytosolic ligase Ubr1 interacts with the ERAD substrate Ste6* and sustains its ubiquitination in the absence of the canonical ER ligases. Cell lysates of indicated strains were separated via ultracentrifugation (100,000 × g) into cytosolic and membrane fractions and subsequently subjected to immunoprecipitation under conditions preserving (A) and preventing (B) interaction with noncovalent binding partners, respectively. The lower panels show 10× higher exposure time compared with the respective upper panels. (A) HRD1 DOA10 UBR1 triple-deleted cells were complemented with plasmid-encoded, fully functional FLAG-tagged Ubr1. Plasmid-encoded Ste6* HA was precipitated from solubilized proteins of the membrane fraction with the help of HA antibodies. (B) Ste6* HA was precipitated with the help of HA antibodies. Subsequent Western blot analyses and detection with Ub antibodies revealed the ubiquitination status of the substrate. IB, immunoblot; IP, immunoprecipitation; Ub./%, signal of detected ubiquitination normalized to background signals and the amount of precipitated Ste6* HA detected with HA antibodies.
Ubr1 Maintains Ubiquitination of Membrane-Bound Ste6* in the Absence of the Canonical ERAD Ligases.
Confirmation that ligase activity of Ubr1 is indeed required for substrate ubiquitination and not for other purposes came from ubiquitination assays. In wild-type cells, Ste6* was shown to be ubiquitinated at the membrane and thereafter extracted from the membrane in full length before its degradation by the proteasome (33). Therefore, after lysis of cells with glass beads, cytosolic and membrane fraction were separated via ultracentrifugation to distinguish between soluble and membrane-associated ubiquitinated species of Ste6*. Compared with the wild type, the ubiquitination status of Ste6* was reduced only slightly in the absence of Ubr1. Relative to the amount of precipitated Ste6*, the ratio of ubiquitinated species found in the cytosolic and membrane fraction remained the same upon deletion of UBR1 (Fig. 3B; compare lanes 7 and 12 as well as 8 and 13). This suggests that in wild-type and UBR1 deleted cells, the canonical ligases Hrd1/Der3 and Doa10 dictate ubiquitination (Fig. 3B) and the successive degradation kinetics of Ste6* (see Fig. 5A). When the two canonical ERAD ligases are absent, Ste6* remains ubiquitinated, although considerably reduced compared with the wild type. Most of the ubiquitinated Ste6* species in Δhrd1 Δdoa10 cells remain in the membrane fraction (Fig. 3B, lanes 9 and 14), indicating a lower extraction efficiency of ubiquitinated Ste6* species from the membrane in the absence of canonical ligases. When in addition to HRD1 and DOA10, UBR1 also is deleted, the ubiquitination of Ste6* is abolished to nearly background levels (Fig. 3B, lane 10 and 15). This proves that ubiquitination of Ste6* in cells lacking the canonical ligases Hrd1/Der3 and Doa10 is maintained by the action of the cytosolic ligase Ubr1 (Fig. 3B; compare lanes 14 and 15).
Fig. 5.
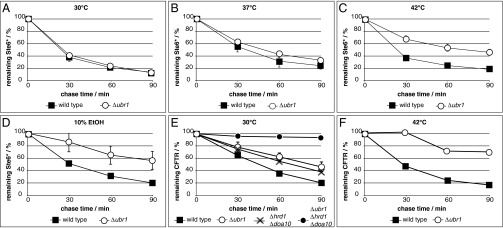
Ubr1 takes over ERAD of Ste6* under heat and ethanol stress, whereas CFTR becomes a target of Ubr1 already under standard conditions. Degradation of Ste6* or CFTR was analyzed in cells of indicated strains at 30 °C (A, D, and E), 37 °C (B), and 42 °C (C and F), and in the presence of 10% (vol/vol) ethanol (D), respectively. (A–E) Data represent the mean of three independent experiments, except for the following strains in E that represent the mean of two independent experiments and therefore do not include error bars: Δhrd1 Δdoa10 and Δubr1 Δhrd1 Δdoa10. Error bars indicate the SEM. (F) Data represent the mean of two independent experiments.
The Hsp70 Chaperone Ssa1 and the AAA-Type ATPase Cdc48 Are Required for Ubr1-Dependent ERAD of Ste6*.
The cytosolic Hsp70 (heat shock protein) chaperone Ssa1 (stress-seventy subfamily A) was implicated in proteasomal degradation of Ste6* in wild-type cells. In this process, Ssa1 is thought to facilitate the integration of the substrate into the Doa10 ligase complex before its ubiquitination (17, 33). Ssa1 also might mask hydrophobic regions of Ste6* after extraction from the ER membrane to prevent aggregation (33). We first tested whether Ssa1 also is required for Ubr1-dependent ERAD of Ste6* in cells deficient in the canonical ligases. For this experiment, we used cells devoid of the three Ssa Hsp70s, Ssa2, Ssa3, and Ssa4, as well as Hrd1/Der3 and Doa10 (Δssa2-4 Δhrd1 Δdoa10). To analyze Ssa1 involvement, a strain carrying a temperature-sensitive allele of SSA1, termed ssa1-45 (34), instead of the wild-type SSA1 allele was generated. This strain grows comparably to the wild type at 25 °C (Fig. S1), indicating there are no considerable metabolic changes in the mutant strain under permissive conditions. Degradation of Ste6* was abolished completely under restrictive conditions (37 °C) in ssa1-45 cells (Fig. 2E). This requirement of Ssa1 for the Ubr1-triggered degradation process may reflect the fact that Ssa1 promotes Ste6* recognition by the ligase Ubr1 as proposed for recognition of Ste6* by Doa10 (33).
The AAA-type ATPase Cdc48 is thought to be the machine that pulls substrates out and away from the membrane (35). In the absence of the canonical ER ligases, Ste6* still interacts with Cdc48 (Fig. 3A and Fig. S2). To test Cdc48 involvement in the ERAD route dependent on Ubr1, we used a strain carrying a temperature-sensitive allele of Cdc48 [cdc48-T413R called cdc48-ts (36)]. At 25 °C, the mutant strain grows comparably to the wild type (Fig. S1), indicating a rather undisturbed cell metabolism under permissive conditions. As illustrated in Fig. 2F, degradation of Ste6* slows considerably in the cdc48-ts mutant cells under restrictive conditions of 37 °C, which reflects the participation of Cdc48 in Ubr1-dependent ERAD of Ste6*.
Ubr1-Dependent ERAD Requires a Cytosolic Domain on the Substrate.
Because Ste6* contains a misfolded domain exposed to the cytosol (17, 18, 20), recognition of this substrate by the cytosolic ligase Ubr1 seems plausible. Therefore, we tested whether in the absence of canonical ligases, ERAD substrates that carry their misfolded domain within the ER lumen also would be degraded via the Ubr1-dependent ERAD route. Two topologically distinct versions of the ERAD substrate mutated carboxypeptidase Y (CPY*) were analyzed for Ubr1-dependent degradation: ER luminal, soluble CPY* (4, 37) and membrane-tethered CT* consisting of a CPY* moiety fused to the last transmembrane domain of Pdr5 (38). Although both substrates still were partially degraded, CPY* and CT* did not show Ubr1-dependent degradation in the absence of Hrd1/Der3 and Doa10 (Fig. 4).
Fig. 4.

The ERAD substrates CPY* and CT*, lacking cytosolic domains, are not targeted to ERAD by Ubr1. Cells of indicated strains expressing genomically encoded CPY* (A) or plasmid-encoded CT* (B) were subjected to PC analyses to analyze the degradation of the respective CPY* derivative. Data represent the mean of three independent experiments. Error bars indicate the SEM.
Ubr1 Is Present at the ER Membrane of Wild-Type Cells and Becomes a Ligase for ERAD of Ste6* Under Heat and Ethanol Stress.
After analyzing the interaction between Ste6* and the ligase Ubr1 in a Δhrd1 Δdoa10 background (Fig. 3A), we were interested in determining whether Ubr1 is recruited to membrane-embedded Ste6* in wild-type cells. Indeed, Ubr1 seems to be in complex with Ste6* in wild-type cells harboring the membrane-embedded ligases Hrd1/Der3 and Doa10 (Fig. S3). This indicates that under certain conditions, Ubr1 might play a role in ERAD even in the presence of the two canonical ligases. To investigate this hypothesis, we tested the role of Ubr1 in ERAD in the presence of tunicamycin and DTT, known to be stimuli for ER stress. Neither stress condition triggered Ubr1 participation in ERAD of Ste6* in the presence of Hrd1/Der3 and Doa10 (Figs. S4 and S5). Because the cytoplasmic heat shock-inducible Hsp70 chaperone Ssa1 was shown to be involved in Ubr1-triggered degradation of Ste6* (Fig. 2E) and heat shock induction can influence ERAD (39, 40), we speculated that a function of Ubr1 in ERAD of Ste6* in wild-type cells could be unmasked under heat shock conditions. No alteration of Ste6* degradation was visible at the standard temperature of 30 °C in cells solely deleted in UBR1 (Fig. 5A). However, during moderate heat shock (37 °C) of UBR1-deleted cells, a small delay in degradation of Ste6* became visible (Fig. 5B). A further increase in the temperature to 42 °C showed a significant delay of Ste6* elimination in UBR1-deleted cells and revealed an important role of Ubr1 in the degradation process of Ste6* under this stress condition (Fig. 5C and Fig. S5). Clearly, Ubr1 is required for efficient ERAD of Ste6* under heat stress. This indicates that Hrd1/Der3 and Doa10 are the dominant ligases that mediate the ubiquitination process of this misfolded protein under the standard temperature of 30 °C (Fig. 5A). However, their function increasingly is taken over by the ligase Ubr1 under heat stress (Fig. 5 B and C). Exposure of cells to ethanol, known to induce a heat-like stress response (41, 42), led to the same result: Ubr1 took over functions of the canonical ligases in degradation of Ste6* (Fig. 5D and Fig. S6).
Human CFTR Expressed in Yeast Is a Target of Ubr1.
In our effort to identify other substrates of Ubr1-dependent ERAD, we tested human CFTR expressed in yeast. Folding of this mammalian protein is highly sophisticated and rarely complete, even in its natural environment. This renders about 70% of CFTR to proteasomal degradation via ERAD in human cells (43, 44). In yeast, CFTR remains largely unfolded and is degraded completely by ERAD (18, 45). When we compared degradation rates of HA-tagged CFTR in the presence of the canonical ER ligases, we found already considerable stabilization of the protein in UBR1 single-deleted cells (Fig. 5E). Stabilization was comparable to the rate found in HRD1 DOA10 double-deleted cells (Fig. 5E). Under heat shock conditions, stabilization of CFTR is nearly complete in the absence of Ubr1 alone (Fig. 5F). Not surprisingly, the absence of all three ligases—Hrd1/Der3, Doa10, and Ubr1—led to complete stabilization of CFTR (Fig. 5E). These findings reveal an important role of Ubr1 in the ERAD process of CFTR in yeast and, in general, indicate involvement of Ubr1 in ERAD of specific substrates, even in the presence of the canonical ER ligases.
Discussion
In this study, we uncovered a previously unknown ERAD route in yeast that depends on the cytosolic N-end rule ubiquitin ligase Ubr1. Whereas specific substrates, such as human CFTR expressed in yeast, seem generally to undergo Ubr1-dependent degradation (Fig. 5E), others (Ste6*) are only targets under specific stress conditions, such as exposure to heat or ethanol (Fig. 5 A–D). Because the ER stress inducers tunicamycin and DTT do not switch on the Ubr1 function in ERAD of Ste6*, heat- and ethanol-induced Ubr1-dependent ERAD seems to be part of a specific stress response and not a general response to random stress stimuli.
The finding that Ubr1 can trigger degradation of CFTR and Ste6* in the presence and absence of the canonical ER ligases implies that further natural ERAD substrates might exist that are not recognized prominently by Hrd1/Der3 and Doa10 but instead are preferentially polyubiquitinated and targeted to proteasomal degradation by Ubr1. It also is likely that heat stress and exposure to a high concentration of ethanol generally lead to a high load of misfolded proteins, which no longer can be handled solely by the canonical ER ligases, making recruitment of Ubr1 necessary to eliminate the misfolded proteins efficiently. In addition, heat and ethanol may alter membrane fluidity massively and thereby largely impair the function of the canonical membrane-embedded ligases (41, 42). This scenario might be mimicked in cells with a Δhrd1 Δdoa10 background and may be the reason Ste6* becomes a target of Ubr1 in these cells (Fig. 1A). The fact that CFTR already is a target of Ubr1 under normal growth conditions in the presence of the canonical ER ligases might be explained by the massive misfolding of CFTR domains in the cytoplasm of yeast. In contrast, Ste6* lacks only part of its C terminus and most probably contains an otherwise rather intact structure that cannot be recognized easily by Ubr1 without further misfolding upon heat or ethanol treatment.
Because absence of the E2 enzyme Ubc2 does not cause complete stabilization of Ste6* in Δhrd1 Δdoa10 cells (Fig. 2C), additional E2 enzymes seem to be involved in the ubiquitination process of Ubr1-dependent ERAD. Although absence of Ubc4 does not seem to affect degradation of Ste6* (Fig. 2D), Ubc4 cannot be excluded from this task, because it exhibits high redundancy with its family member Ubc5 (32), and additional mutant analyses are necessary to clarify its possible participation in Ubr1-dependent ERAD.
Further studies are required to elucidate the targeting mechanism of Ubr1: Does it recognize the misfolded domains of the substrate with one of its known substrate-binding pockets; is it guided to the membrane bound substrate by the Hsp70 Ssa1; or is Ubr1 recruited to the ER membrane by a yet unknown factor embedded in the ER membrane?
Importantly, not every ERAD substrate becomes a target of Ubr1 when the canonical ERAD ligases Hrd1/Der3 and Doa10 are missing. Residual degradation of the ERAD substrates CPY* and CT* remains unaffected when UBR1 also is deleted (Fig. 4 A and B). Our findings on CT* (Fig. 4B) raise the question whether other cytosolic ligases are involved in the degradation of certain ERAD substrates or whether these substrates are passed on to the vacuole and degraded there. However, in the case of Ste6* and CFTR, the involvement of other cytosolic ligases is unlikely, because the substrates are fully stabilized in the absence of the canonical ERAD ligases and Ubr1. Based on the few substrates tested so far, one might propose that only substrates carrying a cytosolic domain can become targets of Ubr1-triggered ERAD.
Concerning retrotranslocation of these substrates from the ER to the cytoplasm, the cell seems to exhibit great plasticity. The previously proposed function of the ER membrane-embedded ligases Hrd1/Der3 and Doa10 as possible channel components for substrate retrotranslocation (9, 16, 46) cannot hold true for Ubr1-triggered ERAD of the tested substrates in the absence of the canonical ligases. The mechanism underlying the retrotranslocation of proteins from the ER to the cytoplasm in the absence of the canonical ligases using the Ubr1-dependent ERAD route will be a challenging goal in the future.
This study adds another layer of complexity to the known mechanisms of ERAD and underscores that previously accepted unidirectional pathways of ubiquitin-triggered degradation of proteins can intersect in a cell: the cytosolic ubiquitin ligase Ubr1 known to be involved in the quality control of cytosolic N-end rule substrates (47, 48), as well as misfolded cytoplasmic proteins (25–27), now can be connected to ERAD (this study). On the other hand, the ERAD ligase Doa10, known mainly to facilitate degradation of ERAD substrates with a misfolded cytosolic domain, also ubiquitinates cytosolic N-acetylated substrates or substrates containing specific C-terminal appendages and targets them for degradation (49–51). Our findings on Ubr1 participation in ERAD open opportunities for future studies to elucidate the molecular interplay of ubiquitin ligases.
Materials and Methods
Detailed information about the experimental procedures, strains, and plasmids used in this study may be found in SI Materials and Methods and in Tables S1 and S2, respectively. In short, experiments have been carried out as follows.
Ubiquitination Assay.
Lysates of cells harvested in logarithmic phase were separated into cytosolic and membrane fractions. Ste6* HA was precipitated under denaturing conditions with the help of HA antibodies in the presence of N-ethylmaleimide to prevent deubiquitination. After Western blotting, ubiquitinated Ste6* species were detected with ubiquitin antibodies.
Pulse Chase Analysis.
For pulse chase (PC) analysis of Ste6* HA and CFTR HA, samples of radiolabeled cells were taken at indicated time points after addition of chase media. After cell lysis with glass beads, membranes were separated from the cytosol. Solubilized membrane proteins were subjected to immunoprecipitation with HA antibodies. After SDS/PAGE, radiolabeled material of the respective substrate was quantified. PC analysis of CPY* was described previously (52) and carried out for CT* accordingly.
Immunoprecipitation Experiments.
Immunoprecipitation experiments were described earlier (19). In brief, cells in logarithmic phase were lysed with glass beads. The lysate was separated into cytosolic and membrane fractions via ultracentrifugation. Immunoprecipitation with indicated antibodies of cytosolic proteins or solubilized membrane proteins was done under conditions preserving protein complexes. Coprecipitated proteins were detected on Western blot using the indicated antibodies.
Supplementary Material
Acknowledgments
We thank Jeffrey Brodsky, Susan Michaelis, Thomas Sommer, and Alexander Varshavsky for providing antibodies and plasmids and Goran Kungulovski for critical reading of the manuscript. Ingo Amm, Nicole Berner, Melanie Dochow, Konrad Otte, and Janina Preissler helped with the generation of strains. This work was supported by the Deutsche Forschungsgemeinschaft (Bonn).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304928110/-/DCSupplemental.
References
- 1.Dobson CM. Protein folding and misfolding. Nature. 2003;426(6968):884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 3.van Anken E, Braakman I. Versatility of the endoplasmic reticulum protein folding factory. Crit Rev Biochem Mol Biol. 2005;40(4):191–228. doi: 10.1080/10409230591008161. [DOI] [PubMed] [Google Scholar]
- 4.Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273(5282):1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- 5.Vembar SS, Brodsky JL. One step at a time: Endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9(12):944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sommer T, Wolf DH. Endoplasmic reticulum degradation: Reverse protein flow of no return. FASEB J. 1997;11(14):1227–1233. doi: 10.1096/fasebj.11.14.9409541. [DOI] [PubMed] [Google Scholar]
- 7.Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: An unconventional route to a familiar fate. Proc Natl Acad Sci USA. 1996;93(24):13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kostova Z, Wolf DH. For whom the bell tolls: Protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003;22(10):2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hampton RY, Sommer T. Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol. 2012;24(4):460–466. doi: 10.1016/j.ceb.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 10.Plemper RK, Wolf DH. Retrograde protein translocation: ERADication of secretory proteins in health and disease. Trends Biochem Sci. 1999;24(7):266–270. doi: 10.1016/s0968-0004(99)01420-6. [DOI] [PubMed] [Google Scholar]
- 11.Deak PM, Wolf DH. Membrane topology and function of Der3/Hrd1p as a ubiquitin-protein ligase (E3) involved in endoplasmic reticulum degradation. J Biol Chem. 2001;276(14):10663–10669. doi: 10.1074/jbc.M008608200. [DOI] [PubMed] [Google Scholar]
- 12.Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9(1):209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7(12):2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3(1):24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- 15.Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001;15(20):2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kreft SG, Wang L, Hochstrasser M. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI) J Biol Chem. 2006;281(8):4646–4653. doi: 10.1074/jbc.M512215200. [DOI] [PubMed] [Google Scholar]
- 17.Huyer G, et al. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279(37):38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- 18.Gnann A, Riordan JR, Wolf DH. Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol Biol Cell. 2004;15(9):4125–4135. doi: 10.1091/mbc.E04-01-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stolz A, Schweizer RS, Schäfer A, Wolf DH. Dfm1 forms distinct complexes with Cdc48 and the ER ubiquitin ligases and is required for ERAD. Traffic. 2010;11(10):1363–1369. doi: 10.1111/j.1600-0854.2010.01093.x. [DOI] [PubMed] [Google Scholar]
- 20.Loayza D, Tam A, Schmidt WK, Michaelis S. Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9(10):2767–2784. doi: 10.1091/mbc.9.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40(2):238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Stolz A, Wolf DH. Endoplasmic reticulum associated protein degradation: A chaperone assisted journey to hell. Biochim Biophys Acta. 2010;1803(6):694–705. doi: 10.1016/j.bbamcr.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Lee DH, Goldberg AL. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998;8(10):397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 24.Fleming JA, et al. Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc Natl Acad Sci USA. 2002;99(3):1461–1466. doi: 10.1073/pnas.032516399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisele F, Wolf DH. Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett. 2008;582(30):4143–4146. doi: 10.1016/j.febslet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 26.Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA. 2010;107(3):1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nillegoda NB, et al. Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol Biol Cell. 2010;21(13):2102–2116. doi: 10.1091/mbc.E10-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y, Varshavsky A. The E2-E3 interaction in the N-end rule pathway: The RING-H2 finger of E3 is required for the synthesis of multiubiquitin chain. EMBO J. 1999;18(23):6832–6844. doi: 10.1093/emboj/18.23.6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MAT alpha 2 repressor. Cell. 1993;74(2):357–369. doi: 10.1016/0092-8674(93)90426-q. [DOI] [PubMed] [Google Scholar]
- 30.Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2(7):379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- 31.Madura K, Dohmen RJ, Varshavsky A. N-recognin/Ubc2 interactions in the N-end rule pathway. J Biol Chem. 1993;268(16):12046–12054. [PubMed] [Google Scholar]
- 32.Stoll KE, Brzovic PS, Davis TN, Klevit RE. The essential Ubc4/Ubc5 function in yeast is HECT E3-dependent, and RING E3-dependent pathways require only monoubiquitin transfer by Ubc4. J Biol Chem. 2011;286(17):15165–15170. doi: 10.1074/jbc.M110.203968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132(1):101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16(8):4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolf DH, Stolz A. The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim Biophys Acta. 2012;1823(1):117–124. doi: 10.1016/j.bbamcr.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Barbin L, Eisele F, Santt O, Wolf DH. The Cdc48-Ufd1-Npl4 complex is central in ubiquitin-proteasome triggered catabolite degradation of fructose-1,6-bisphosphatase. Biochem Biophys Res Commun. 2010;394(2):335–341. doi: 10.1016/j.bbrc.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 37.Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15(4):753–763. [PMC free article] [PubMed] [Google Scholar]
- 38.Taxis C, et al. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem. 2003;278(38):35903–35913. doi: 10.1074/jbc.M301080200. [DOI] [PubMed] [Google Scholar]
- 39.Han S, Liu Y, Chang A. Cytoplasmic Hsp70 promotes ubiquitination for endoplasmic reticulum-associated degradation of a misfolded mutant of the yeast plasma membrane ATPase, PMA1. J Biol Chem. 2007;282(36):26140–26149. doi: 10.1074/jbc.M701969200. [DOI] [PubMed] [Google Scholar]
- 40.Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24(12):5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piper PW. The heat shock and ethanol stress responses of yeast exhibit extensive similarity and functional overlap. FEMS Microbiol Lett. 1995;134(2-3):121–127. doi: 10.1111/j.1574-6968.1995.tb07925.x. [DOI] [PubMed] [Google Scholar]
- 42.Morano KA, Grant CM, Moye-Rowley WS. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics. 2012;190(4):1157–1195. doi: 10.1534/genetics.111.128033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269(41):25710–25718. [PubMed] [Google Scholar]
- 44.Jensen TJ, et al. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83(1):129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, et al. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell. 2001;12(5):1303–1314. doi: 10.1091/mbc.12.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143(4):579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia Z, et al. Substrate-binding sites of UBR1, the ubiquitin ligase of the N-end rule pathway. J Biol Chem. 2008;283(35):24011–24028. doi: 10.1074/jbc.M802583200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varshavsky A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011;20(8):1298–1345. doi: 10.1002/pro.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Metzger MB, Maurer MJ, Dancy BM, Michaelis S. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J Biol Chem. 2008;283(47):32302–32316. doi: 10.1074/jbc.M806424200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science. 2010;327(5968):973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25(3):533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benitez EM, Stolz A, Wolf DH. Yos9, a control protein for misfolded glycosylated and non-glycosylated proteins in ERAD. FEBS Lett. 2011;585(19):3015–3019. doi: 10.1016/j.febslet.2011.08.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.