

Significance
Ideal photovoltaic cells would be black, absorbing all of the Sun’s radiation, whereas Nature’s machinery for solar energy harvesting—photosynthesis—looks green. Organic semiconductor devices, based on molecular building blocks, lie conceptionally between the extremes of inorganic and photosynthetic light harvesting. How can organic solar cells appear almost black if they are based on molecular units? Using single-molecule spectroscopy, we identify the fundamental electronic building blocks of organic solar cells and reveal that discrete molecule-like transitions scatter over the entire visible spectrum. The fundamental molecular unit is narrowband, but disorder induces a continuum reminiscent of that characterizing highly ordered inorganic crystals.
Keywords: structure–property relations, conformational disorder, photophysics, organic solar cells
Abstract
The spectral breadth of conjugated polymers gives these materials a clear advantage over other molecular compounds for organic photovoltaic applications and is a key factor in recent efficiencies topping 10%. However, why do excitonic transitions, which are inherently narrow, lead to absorption over such a broad range of wavelengths in the first place? Using single-molecule spectroscopy, we address this fundamental question in a model material, poly(3-hexylthiophene). Narrow zero-phonon lines from single chromophores are found to scatter over 200 nm, an unprecedented inhomogeneous broadening that maps the ensemble. The giant red shift between solution and bulk films arises from energy transfer to the lowest-energy chromophores in collapsed polymer chains that adopt a highly ordered morphology. We propose that the extreme energetic disorder of chromophores is structural in origin. This structural disorder on the single-chromophore level may actually enable the high degree of polymer chain ordering found in bulk films: both structural order and disorder are crucial to materials physics in devices.
Despite half a century of research into organic photovoltaics (1), the promise of versatile paint-on solar-cell modules based on conjugated polymers has prompted a present flurry of activity in the field (2–5). A particular appeal of such excitonic solar cells is that very little material is needed to efficiently absorb light. This is due to the fact that the oscillator strength of primary photoexcitations, electron–hole pairs with binding energies far exceeding kT, is focused in the excitonic transition. The obvious downside is that this concentration also means excitonic transitions are inherently narrow and usually offer only mediocre spectral overlap with the broad solar spectrum. How then can excitonic solar cells be designed with appropriate spectral breadth? Merely introducing energetic disorder in the underlying excitonic material should lead to low-energy traps, impeding charge harvesting.
Although the optical and electronic properties of conjugated polymers are not perfectly suited to photovoltaics, their absorption spectra are surprisingly broad despite the excitonic nature of the transitions. Moreover, the diversity in functional characteristics revealed by varying processing conditions has fueled the quest to formulate robust structure–property relationships between the electronic and optical properties and the underlying polymer structure (6–12). Polythiophene derivatives have evolved into one of the chosen workhorse materials for solar-cell research (13, 14). Early spectroscopic studies established extraordinary solvatochromic and thermochromic characteristics, which were attributed to large conformational changes in response to the immediate environment (15–17). It is little surprise then that such diversity in device characteristics exists when using these materials (4). Poly(3-hexylthiophene) (P3HT) (structure shown in Fig. 1A) and related compounds were the first polymeric materials to exhibit signatures of 2D electronic delocalization (18) due to a high level of interchain ordering most clearly visualized in X-ray diffractometry (19). Despite this ordering, the optical absorption remains extremely broad, rendering the material favorable for photovoltaics. The striking dependence of P3HT ensemble optical properties on processing conditions has led to proposals that substantial dimerizing interchain interactions (20–23) could be involved. However, the signatures of excitonic dimerization, or H-aggregation, are not always straightforward to resolve conclusively (24). H-aggregation should lead to a reduction in radiative rate (25). Typically, upon aggregation of P3HT, a strong decrease in fluorescence quantum yield is observed, but this is accompanied by an increase in fluorescence rate (26) because nonradiative decay rates are also enhanced. It is not always trivial to distinguish an increase in nonradiative decay from deceleration of radiative decay, complicating a precise determination of transition oscillator strengths. For this reason, most of the focus to date has been on interpreting spectral shape and vibronic coupling (23). Although the origin of the magnitude of spectral shift between isolated and bulk-packed chains has been simply assigned to a “crystal shift” (23), a high degree of interchain ordering should also imply substantial planarization of the polymer with the associated increase in conjugation length (27)—which in turn decreases the strength of dipolar coupling that could lead to dimerization as the excited state becomes more delocalized (21).
Fig. 1.

Unraveling the spectral heterogeneity of P3HT using low-temperature single-molecule spectroscopy. (A) Comparison of P3HT PL spectra in dilute toluene solution (green) and in a drop-cast bulk film (red) at room temperature. Six representative low-temperature (4 K) single-molecule spectra (gray), consisting of a zero-phonon line and a vibronic progression, are shown as examples, spanning the spectral region from dilute solution to bulk film. (B) Normalized PL spectra of 115 single molecules, sorted by the peak energy of the 0–0 transition. The same 0–1 vibronic transition, shifted by 180 meV from the main peak, is seen for all spectra (Lower). (C) Correlation of 0–0 peak width with transition energy. The green circles represent averages over eight molecules, and the red line is a guide to the eye. (D) The intensity of the vibronic peak does not depend on chromophore transition energy.
Conclusively discriminating interchain from intrachain order is only possible by resorting to subensemble techniques such as single-molecule spectroscopy (28–35). Here, we demonstrate the feasibility of reconciling fundamental spectroscopy of P3HT, shown in this study to be the most heterogeneous polymeric material studied to date on the single-chromophore level, with the excitonic single-chromophore picture (36–38) established for the most ordered polymers such as polyfluorenes (39) (PF), ladder-type poly(para-phenylenes) (40, 41) (LPPP), polydiacetylene (42, 43) (PDA), and poly(phenylene-ethynylene) (28) (PPE). On the level of single chromophores, P3HT behaves like other prominent materials, such as poly(phenylene-vinylenes) (41, 44) (PPVs), with the exception of exhibiting giant variability in transition wavelengths spanning almost 200 nm. The dominant photophysics of the ensemble—the striking red shift between solution and film photoluminescence (PL)—is controlled by energy transfer to low-energy intrachain chromophores, in absence of the previously proposed “crystal shift” (23).
Results
Comparison of Ensemble and Single-Chromophore Spectroscopy.
Nominally different conjugated polymer materials exhibit very similar spectral characteristics on the single-chromophore level (28). Because long polymer chains can contain many chromophores (40), it is not always straightforward to ensure that a single chromophore is identified within the polymer. Comparison with oligomeric model compounds, use of narrow-band excitation, and cryogenic cooling of the sample have, however, enabled a reproducible framework for the identification of single chromophores (40). We begin by comparing the ensemble PL spectra of P3HT in dilute toluene solution (10−4 mM) and in a drop-cast bulk film in Fig. 1A. Going from solution to the solid phase shifts the spectrum by over 100 nm (0.4 eV) to the red and modifies the spectral form. The bulk spectrum has been interpreted to show a shift of oscillator strength from the 0–0 to the 0–1 transition, assigned to H-aggregation (22), because bulk-phase P3HT exhibits a high level of structural ordering in electron microscopy (5), scanning-probe microscopy (45), and other elastic scattering techniques (11, 46, 47). The large spectral shift between solution and bulk has been assigned to the occurrence of a crystal shift in the H-aggregated chromophores embedded in an ordered environment of many other conjugated polymer chains (23). Such a large spectral shift between solution and film is not observed in other common conjugated polymers. Six representative single-chromophore spectra, recorded at 4 K from isolated chains directly deposited from toluene onto a SiO2 surface, are superimposed on the ensemble spectrum in gray. The single-chromophore spectra all have the same shape, a comparatively narrow asymmetric zero-phonon line with a low-energy acoustic phonon wing and a distinct vibronic band offset by 180 meV. The reduction of disorder broadening on the single-chromophore level facilitates determination of vibronic frequencies compared with the ensemble. The observed vibronics can be assigned to the ring C–C stretch (171 meV) and the symmetric C=C stretch modes (179 meV) (48). The single-chromophore spectra, obtained under excitation at 458 nm, span a spectral range of 195 nm (0.8 eV). In the red-most spectrum, excitation and emission are separated by 0.9 eV, and yet the same universal (41), narrow single-chromophore spectral shape is observed.
To highlight the universality of chromophores, we plot 115 spectra in Fig. 1B, sorted by 0–0 peak energy (see Fig. S1 for alternative forms of presenting the data). Including the vibronic progression, narrow spectral lines are found between 485 and 775 nm. The general spectral characteristics, in particular the energy of the dominant vibronic plotted beneath, remain unchanged regardless of peak energy. The distribution of peak positions is plotted in a histogram in Fig. S2 and appears to be trimodal (see the discussion in SI Text for possible origins of this distribution).
Interestingly, the 0–0 peak narrows slightly with decreasing energy, as plotted in Fig. 1C. Such spectral narrowing is seen in PF (28) and PDA (49), where lower-energy transitions correspond to improved chain ordering (the formation of the β-phase in PF). The opposite is seen in PPV (50), where chain bending induces a red shift and an increase in spectral jitter. Over the entire range of chromophore energies, the ratio between 0–0 and 0–1 PL peak intensities scatters substantially but does not vary systematically with transition energy (Fig. 1D). Strong variations in the PL intensity of the vibronic progression between single chromophores have been reported previously, even in nominally rigid materials such as LPPP (51). Such variations arise due to the slight distribution in ground-state molecular conformations, leading to differing structural relaxation energies and the resulting changes in the Franck–Condon progression, which become visible precisely because electronic and vibronic transitions are so narrow at low temperature. Examples of the interchromophoric scatter in 0–0/0–1 peak ratio are given in Fig. S3.
The single-chromophore PL spectra are identical in shape to that of different conjugated polymers (28) such as LPPP, PPV, PDA, PPE, and PF as illustrated in Fig. S1, and exhibit the common blinking and spectral diffusion (spectral jitter; Fig. S4). This similarity in spectral form strongly suggests that the narrow lines observed over such a broad spectral range arise from isolated chromophores (38). Because narrow single-chromophore zero-phonon lines are observed as far to the red as 680 nm, we conclude that the high-energy side of the bulk spectrum (Fig. 1A, red curve) is appropriately described by the presence of isolated rather than aggregated chromophores: the red shift of over 100 nm between solution and film can be mostly attributed to energy transfer from high-energy to low-energy chromophores. This conclusion, however, does not exclude the possibility of H-aggregate emission contributing to the red tail of the bulk PL spectrum. We stress that single-chromophore spectroscopy, by its very definition, probes only the emission from single chromophores; we cannot probe potential H-aggregate emission without resorting to well-defined dimer structures (52).
Difference in Polymer Chain Conformation Between Solution and Bulk Phase.
Why do different chromophores dominate ensemble solution and bulk film spectra (Fig. 1A)? The fundamental difference between the two cases is found in the underlying chain conformation, which can be controlled through the polarity of the immediate environment of the polymer (30): a “good” solvent, or matrix, will lead to optimal chain extension, driving the formation of well-solvated yet disordered spheres or globules. In contrast, a “poor” solvent promotes collapse of the polymer into toroidal or rod-like structures (29). Solvent quality for nonpolar organic compounds like conjugated polymers generally decreases with increasing polarity of the solvent. This effect can be replicated in the solid state for single chains of P3HT by embedding them in different polymer matrices. Fig. 2A compares dilute (10−4 mM, 100 times the concentration used in single-molecule experiments) solid solutions of P3HT in a virtually nonpolar Zeonex derivative (Zeonex 480) and in poly(methyl-methacrylate) (PMMA), which is more polar. The spectrum of P3HT embedded in Zeonex (green) closely resembles that of toluene solution, whereas the spectrum of P3HT embedded in PMMA (red) matches the peak and red tail of bulk P3HT. The mismatch in spectra at higher energy likely arises due to the presence of incompletely folded (i.e., blue-emitting) chains in PMMA. The effect of solvation is demonstrated in fluorescence micrographs of the films in Fig. 2B. The P3HT/Zeonex film appears uniform in emission, whereas the P3HT/PMMA film shows discrete bright spots. Both images are displayed on the same intensity scale. In the P3HT/PMMA film, the background is darker than in P3HT/Zeonex, but the spots are much brighter. The formation of bright spots in PMMA suggests that multiple chains can aggregate together. In the following, we demonstrate that even single isolated chains in PMMA collapse into ordered structures.
Fig. 2.

Replicating bulk and solution spectra in dilute matrix environments at room temperature. (A) The ensemble spectrum of chains dispersed in the inert Zeonex matrix (green) closely matches that of isolated chains in toluene (gray). In contrast, PMMA leads to chain aggregation: the spectrum of P3HT diluted in PMMA at the same concentration as in Zeonex (red) resembles the bulk film emission (gray). In both cases, the concentration of P3HT is 100 times higher than in single-molecule experiments. (B) The effect of matrix-induced phase separation is visible in fluorescence micrographs of the spin-coated films, plotted on the same intensity scale. In Zeonex, the film PL appears uniform, whereas in PMMA bright spots are seen corresponding to the formation of large multichain aggregates.
Fig. 3 reports measurements of the fluorescence modulation of single chains at room temperature under rotation of the polarization angle, θ, of the exciting laser within the sample plane. For a straight object, the overall transition dipole should lie along the axis of the π-orbitals, leading to a strong cosine-squared modulation of PL with laser polarization (29). Such a modulation in intensity is sketched in Fig. 3A. The depth of modulation, 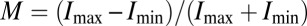 , provides information on the degree of order regarding the transition dipoles of an individual chain. Examples of measurements are shown in Fig. S5. Fig. 3B compares histograms for 738 single chains in Zeonex and 587 chains in PMMA. Whereas the PL excitation (i.e., absorption) of the molecules in Zeonex is mostly weakly polarized due to disorder, P3HT in PMMA (31) is predominantly ordered with M peaking around 0.8. Based on these M values, possible chain conformations are sketched at the top of Fig. 4.
, provides information on the degree of order regarding the transition dipoles of an individual chain. Examples of measurements are shown in Fig. S5. Fig. 3B compares histograms for 738 single chains in Zeonex and 587 chains in PMMA. Whereas the PL excitation (i.e., absorption) of the molecules in Zeonex is mostly weakly polarized due to disorder, P3HT in PMMA (31) is predominantly ordered with M peaking around 0.8. Based on these M values, possible chain conformations are sketched at the top of Fig. 4.
Fig. 3.

Shape dependence of single P3HT chains on matrix material at room temperature. (A) The modulation of PL intensity under rotation of the plane of polarization of the exciting laser is recorded. The modulation depth M provides a measure of chain extension and order: chains that mix well with the matrix (i.e., are well solvated) form random-coil structures that absorb any polarization of light. Poorly solvated chains, however, collapse, leading to ordered anisotropic rod-like structures. (B) Histogram of M values for single chains in Zeonex and PMMA. In Zeonex, isotropic structures are formed, whereas PMMA gives rise to highly anisotropic arrangements of the chains.
Fig. 4.

Excited-state properties of solvated (Zeonex) and collapsed (PMMA) single P3HT chains at room temperature. In solvated chains, the chromophores emit independently of each other (cartoon). In collapsed chains, energy transfer occurs to the lowest-energy chromophore. (A) Fluorescence intensity and linear dichroism D, determined by splitting the emission into two orthogonal polarization components as illustrated in the cartoon. In Zeonex, multistep blinking is observed. The emission is only weakly polarized (D ∼ 0), exhibiting slight fluctuations as different chromophores switch on and off. In the PMMA collapsed-chain configuration, the fluorescence intensity is constant and shows single-step blinking. The emission is strongly polarized (D ≠ 0) with no fluctuations. Representative single-chain PL spectra are shown in the Inset. (B) Photon statistics in emission, given by the cross-correlation of two photodetectors in the emission pathway. The molecules are excited by laser pulses (25-ns period), controlling photon arrival times τ. In Zeonex, multiple chromophores emit, leading to only a 20% dip in photodetector coincidence rates. In PMMA, a dip in coincidence rate by 80% is observed, because no more than one single photon is emitted for each laser pulse. Based on photon counts and background signal for the two cases, the maximum dip expected for a perfect single-photon or two-photon source is indicated by the dashed lines.
Chromophoric Emission at Room Temperature.
Intrachain conformation should have a dramatic impact on single-chain photophysics: in the random-coil structure, the chromophores will couple only weakly to each other, effectively emitting independently. In the ordered structures, energy transfer should occur between chromophores because interchromophoric distances are reduced. Two distinct experiments in Fig. 4 clearly demonstrate this interplay between conformation and photophysics. The Insets in Fig. 4A show two representative single-molecule spectra at room temperature, measured in Zeonex and PMMA, which closely resemble the ensemble (Fig. 2A) for the solvated and collapsed structures, respectively. Fig. 4A displays the PL intensity of a single chain as a function of time. The fluorescence beam is separated into two paths by a polarizing beam splitter and recorded with two separate photodiodes, allowing us to identify any change in orientation of the emissive transition dipole by quantifying the linear dichroism D as defined in the schematic. An ensemble of different dipole orientations will lead to D ∼ 0, as will a single dipole coincidentally oriented at 45° with respect to both detectors. The example P3HT chain in Zeonex is approximately five times brighter than in PMMA, but exhibits strong fluctuations and a gradual overall decrease in intensity (bleaching). The emission intensity does not drop completely to zero. In contrast, in PMMA, discrete blinking is observed. In Zeonex, the linear dichroism fluctuates around zero, exhibiting small jumps, which imply the involvement of multiple different chromophore emitters. In contrast, in the PMMA example, a high static D is found: either all dipoles are oriented along the same axis or only one single chromophore in the polymer is active at once. The first conclusion is inferred from the excitation polarization modulation in Fig. 3B. Below, we demonstrate that indeed only one chromophore emits at a time from the single chain.
The number of multiple chromophores involved in the emission can be quantified by photon statistics obtained using a cross-correlation between photodiodes detecting the fluorescence divided into two pathways with a (nonpolarizing) beam splitter (Fig. 4B) (53–55). Fluorescence is excited by a pulsed laser (488 nm) with a period of 25 ns. If only one photon is emitted by the molecule per laser pulse, it cannot be simultaneously picked up by both detectors. Therefore, for single emitters, the cross-correlation must drop to zero at zero delay τ between detector signals, a phenomenon known as photon antibunching. Because such cross-correlation analysis requires high photon counts, we average (53) over 80 and 30 single molecules for PMMA and Zeonex, respectively. In Zeonex, the cross-correlation signal at τ = 0 is nearly identical to that at other delays, implying that, on average, multiple chromophores on the polymer emit at once and do not couple efficiently. In PMMA, the cross-correlation at τ = 0 drops to 20% of coincidence photon counts compared with τ ≠ 0, implying predominant single-chromophore emission. Given the large number of chromophores on a chain, this phenomenon must result from energy transfer to the lowest-energy chromophore and concurrent singlet–singlet annihilation in the multichromophoric assembly (56).
Discussion
Cryogenic single-chain spectroscopy of P3HT reveals clear signatures of discrete intrachain chromophores. The diversity of spectral characteristics found in the ensemble when going from isolated to aggregated chains originates from these chromophores. Universal single-chromophore spectra, comparable in shape to those observed in many other materials, scatter over almost 200 nm, providing a measure of the level of interchromophoric inhomogeneous broadening of ∼0.8 eV, greatly exceeding prior estimates (23). In comparison, the corresponding disorder extracted from identical experiments for LPPP is 0.03 and 0.2 eV for PPV (41). Such a level of energetic disorder as found in P3HT is unprecedented in molecular emitters, and there is no immediately obvious explanation for this scatter based on conventional models of conjugation in P3HT (57).
Ensemble P3HT shows a large spectral shift upon going from dilute solution to the bulk film (Fig. 1A). Because the transition between solution and film probes such a broad spectral range, intermediate states have been generated by means of solvent-vapor annealing to optimize both absorption and charge transport in solar cells (3, 8). Despite this heterogeneity of ensemble P3HT, key elements of the bulk photophysics (22, 23) are consistent with the formation of discrete intrachain chromophores. We stress, however, that bulk spectral emission features extending beyond 700 nm may be associated with H-aggregation or charge-transfer phenomena (1), which would only arise in the bulk and cannot be probed readily by single-molecule techniques. Notable examples of spectral characteristics of bulk PL that we cannot probe here are the monotonic changes of spectral shape with temperature (23), and the evolution of the vibronic progression and temporal red shift in time-resolved PL (58). These features can both be modeled with the H-aggregate picture (23).
The same universal spectral features are observed independent of energetic separation between excitation and emission. Ultrafast dissipation of excitation energy in P3HT has previously been concluded from photon-echo spectroscopy and was attributed to efficient coupling to vibrations (59). However, the dissipation of 0.9 eV, the difference between excitation at 458 nm and emission at 680 nm, would require five quanta of the dominant vibration, which seems implausible. Because the emission intensity of all chromophores is comparable, we conclude that each chain most likely has very similar absorbing chromophores (presumably short conjugated units), which then populate the emissive chromophore by energy transfer. We note that when only one chromophore is present on a polymer chain, such as in β-phase PF, absorption and emission spectra exhibit the expected near-perfect mirror symmetry (39). Even though multiple chromophores must exist on single P3HT chains, it is reasonable to assume that each single chromophore identified in Fig. 1 by its emission also has a complementary specific mirror-symmetric absorbing feature (39). The coexistence of short and long chromophores within a single polymer chain, distinguished by their transition energy and vibronic coupling strength, has been demonstrated previously in polyindenofluorene using single-chain light-harvesting action spectroscopy (60).
Intuitively, one may be inclined to speculate that the most extended chromophores are also the lowest-energy units. However, slight bending of the backbone, chain torsion, and changes in bond alternation can mask a strict correlation between conjugation length and lowering of the optical gap (50). Semiempirical calculations have suggested that even substantial bending of the π-system in polythiophene need not necessarily disrupt conjugation (57). Given the high degree of ordering of single-chain P3HT seen in polarization fluorescence modulation (Fig. 3), it is tempting to surmise that, in these objects, the π-system is also the most extended (27). However, the fact that the vibronic progression does not depend systematically on transition wavelength is crucial (Fig. 1D). In PDA, for example, the most extended chains show a dramatic increase of 0–0 transition oscillator strength relative to the 0–1 peak at low temperature, because the extended π-system constitutes an effective linear J-aggregate (43). It is conceivable that exciton self-trapping limits excitonic coherence and thus the transfer of oscillator strength to the purely electronic (0–0) transition. Such an effect has been suggested for β-phase PF (39), which also shows substantial vibronic coupling in near-perfectly extended chains under cryogenic conditions, in contrast to PDA. It is also conceivable that the broad energetic distribution of zero-phonon lines (Fig. 1B) arises from an intrinsic Stark shift due to trapped charges (61). However, in this case a correlation should exist between increased red shift and increased line width (62), which is not observed; the line width appears to decrease with decreasing transition energy.
The most red-emitting single chromophores (Fig. 1) constitute the monomolecular precursor states for the red species in bulk P3HT films. Emission in the bulk occurs preferentially from these species, populated by energy transfer provided the polymer chain is sufficiently collapsed and ordered, as is the case in PMMA matrices or in the bulk. In this regard, P3HT behaves like many other conjugated polymers, such as poly[2-methoxy-5-(2′-ethylhexyloxy)-p-phenylene vinylene] (MEH-PPV) (63, 64), with the fundamental difference that interchromophoric energetic disorder is at least twice as large. As more and more chains pack together, the quantum yield of fluorescence of the mesoscopic objects within the bulk film is reduced because the probability of exciton dissociation and charge formation increases with greater average exciton migration distances (24, 33). Although we have identified isolated chromophores that emit at wavelengths of ∼680 nm, a further red shift with respect to this purely chromophoric transition observed in bulk films may arise due to the previously described phenomenon of weak H-aggregation (22), due to a Stark effect resulting from the electric field emanating from local trapped charges (65), or from solid-state solvatochromic effects (1). Finally, we note that it is both the conformation of the overall chain that changes with polarity (matrix) as well as the energetic distribution of chromophores that are responsible for such broad spectral variability. Nanoscale conformation therefore also affects ensemble absorption, enabling the tuning of absorption spectrum by solvent-vapor annealing (3, 8).
The most obvious origin of the energetic spread in universal single-chromophore spectra lies in structural variations between single chromophores. We conclude that single chromophores are both flexible and can adopt a wide range of subtly varying conformations: structural and energetic disorder dominates on the single-chromophore level. In contrast, in bulk films, an extraordinary high degree of order can be reached (19). We propose that this unique feature of P3HT arises directly from the disorder on the single-chromophore level: as chains fold back on themselves or aggregate with other chains, there are always suitably shaped chromophores present so that a closely packed structure can be formed. It is conceivable that disorder on the single-chromophore level could actually breed order in the bulk.
P3HT constitutes one of the most heterogeneous material systems explored in the context of organic electronics, which, besides some of the impressive device characteristics (14), accounts for its continuing popularity in the field (13). Nevertheless, conventional incremental materials development risks being impeded by lack of a robust understanding of primary photoexcitations in these systems. Is the primary photophysics of polythiophenes dominated by intermolecular interactions and H-aggregate species, or does the material actually adhere to the established concepts (36, 38, 41) of intrachain chromophores as derived from a wide range of compounds? Our single-molecule experiments tend to favor the latter notion, while leaving room for features of H-aggregation or other bulk solvation effects in the red tail of the emission spectrum. In contrast to all polymeric materials studied previously, the energetic heterogeneity of chromophoric transitions spans almost 1 eV, i.e., much of the visible spectrum. Such a breadth of possible fundamental transition energies can only be accounted for by an extreme sensitivity of electronic structure to conformational variations, which explains the dramatic impact processing conditions have on bulk material properties (3, 8).
Methods
P3HT (regioregularity, 95.7%; weight average molecular weight, 65.2 kDa; polydispersity index, 2.2) was purchased from EMD Chemicals and used as received. PMMA (weight average molecular weight = 96.7 kDa; number average molecular weight, 47.7 kDa) and Zeonex (Zeonex 480) were obtained from Sigma-Aldrich and Zeon Europe, respectively. Low-temperature single-molecule spectroscopy was carried out in a home-built fluorescence microscope as described previously (28). The fluorescence was excited at 458 nm using a frequency-doubled Ti:sapphire femtosecond laser system (HarmoniXX, APE; and Chameleon Ultra II, Coherent). To minimize contamination of the weak fluorescence signal by background luminescence, single-chromophore spectra were recorded from samples deposited, without a polymer matrix, directly on top of SiO2-covered Si wafers. The wafers were mounted in a liquid-helium cold-finger cryostat at 4 K under vacuum. Concentration series were performed to ensure that the single-molecule density varied as expected with solution concentration. Due to the low photon count rates, all experiments were carried out in spectral imaging mode [i.e., resolving the fluorescence spectrum (28)] rather than under direct 2D imaging. This approach ensured that the observed fluorescence really did originate from single P3HT chains.
Room temperature fluorescence was recorded on a separate microscope setup based on an Olympus IX71. P3HT chains were embedded in a PMMA or Zeonex 480 host matrix according to a previously described procedure (65): (i) Borosilicate glass coverslips were cleaned in a 2% (vol/vol) Hellmanex III (Hellma Analytics) solution, followed by rinsing with MilliQ water. (ii) The glass coverslips were additionally bleached by a UV-ozone cleaner (Novascan; PSD Pro Series UV). (iii) The P3HT was diluted in toluene to single-molecule concentration (∼10−12 to 10−13 M) and mixed with a 6% PMMA or Zeonex 480/toluene solution. (iv) This solution was dynamically spin-coated in a nitrogen glove box at 2,000 rpm onto the glass coverslips, leading to a film thickness of ∼200–300 nm with an average P3HT chain density of ∼20 individual P3HT chains in a range of 50 × 50 µm2. The sample was incorporated into a home-built gas flow cell and purged with nitrogen to prevent bleaching by oxygen. Excitation was carried out by a fiber-coupled diode laser (PicoQuant; LDH-D-C-485) at 485 nm under continuous-wave excitation for wide-field fluorescence microscopy or under pulsed excitation with a repetition rate of 40 MHz for confocal fluorescence microscopy and time-correlated single-photon counting. The excitation light was passed through a clean-up filter (AHF Analysentechnik; z485/10), expanded, and focused (or collimated) via a lens system onto the back focal plane of a 1.35 N.A. oil immersion objective (Olympus; UPLSAPO 60XO) through the back port of the microscope and a dichroic mirror (AHF Analysentechnik; z488RDC) for wide-field or confocal excitation. For wide-field microscopy, the fluorescence signal was imaged on an EMCCD camera (Andor; iXon 897) after an additional magnification of 1.6× and after passing a fluorescence filter (AHF Analysentechnik; RS488LP), whereas, for confocal measurements, the fluorescence signal was split either by a polarizing beam splitter (Thorlabs; CM1-PBS251) into two orthogonal polarizing detection channels or by a 50/50 beam splitter and detected by two avalanche photodiodes (PicoQuant; τ-SPAD-20). The excitation intensities were set to ∼1.5 and 150 W/cm2 for wide-field and confocal excitation, respectively. The polarization rotation of the excitation beam, confocal detection, and photon statistics analysis were carried out as described previously (65). For the antibunching measurements, we averaged the fluorescence traces of 80 single chains in PMMA and 30 single chains in Zeonex, respectively (53). By considering the count rate, photodetector dark counts and the background intensity, we estimate the expected magnitude of the antibunching dip for a perfect single emitter as 10% and 2.5% for P3HT/PMMA and P3HT/Zeonex, respectively. These thresholds are indicated as dashed lines in Fig. 4B.
Supplementary Material
Acknowledgments
We thank D. Lidzey and G. Khalil for helpful discussions. J.M.L. is indebted to The David and Lucile Packard Foundation for providing a fellowship. A.T. acknowledges the Fonds der Chemischen Industrie for a Chemiefonds fellowship. D.V.B. acknowledges support from an Energy Frontier Research Center funded by the U.S. Department of Energy under Award Number DE-SC0001091. This work was partially funded by the European Research Council Starting Grant MolMesON (Grant 305020).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307760110/-/DCSupplemental.
References
- 1. Pope M, Swenberg CE (1999) Electronic Processes in Organic Crystals and Polymers (Oxford Univ Press, Oxford)
- 2.You J, et al. A polymer tandem solar cell with 10.6% power conversion efficiency. Nat Commun. 2013;4:1446. doi: 10.1038/ncomms2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campoy-Quiles M, et al. Morphology evolution via self-organization and lateral and vertical diffusion in polymer:fullerene solar cell blends. Nat Mater. 2008;7(2):158–164. doi: 10.1038/nmat2102. [DOI] [PubMed] [Google Scholar]
- 4.Kim Y, et al. A strong regioregularity effect in self-organizing conjugated polymer films and high-efficiency polythiophene: Fullerene solar cells. Nat Mater. 2006;5(3):197–203. [Google Scholar]
- 5.Yang XN, et al. Nanoscale morphology of high-performance polymer solar cells. Nano Lett. 2005;5(4):579–583. doi: 10.1021/nl048120i. [DOI] [PubMed] [Google Scholar]
- 6.Barnes MD, Baghar M. Optical probes of chain packing structure and exciton dynamics in polythiophene films, composites, and nanostructures. J Polym Sci B. 2012;50(15):1121–1129. [Google Scholar]
- 7.Erb T, et al. Correlation between structural and optical properties of composite polymer/fullerene films for organic solar cells. Adv Funct Mater. 2005;15(7):1193–1196. [Google Scholar]
- 8.Moulé AJ, Meerholz K. Controlling morphology in polymer-fullerene mixtures. Adv Mater. 2008;20(2):240–243. [Google Scholar]
- 9.Padinger F, Rittberger RS, Sariciftci NS. Effects of postproduction treatment on plastic solar cells. Adv Funct Mater. 2003;13(1):85–88. [Google Scholar]
- 10.Zen A, et al. Effect of molecular weight on the structure and crystallinity of poly(3-hexylthiophene) Macromolecules. 2006;39(6):2162–2171. [Google Scholar]
- 11.Salleo A, Kline RJ, DeLongchamp DM, Chabinyc ML. Microstructural characterization and charge transport in thin films of conjugated polymers. Adv Mater. 2010;22(34):3812–3838. doi: 10.1002/adma.200903712. [DOI] [PubMed] [Google Scholar]
- 12.McCulloch I, et al. Semiconducting thienothiophene copolymers: design, synthesis, morphology, and performance in thin-film organic transistors. Adv Mater. 2009;21(10–11):1091–1109. [Google Scholar]
- 13.Dang MT, Hirsch L, Wantz G. P3HT:PCBM, best seller in polymer photovoltaic research. Adv Mater. 2011;23(31):3597–3602. doi: 10.1002/adma.201100792. [DOI] [PubMed] [Google Scholar]
- 14.Zhao GJ, He YJ, Li YF. 6.5% efficiency of polymer solar cells based on poly(3-hexylthiophene) and indene-C(60) bisadduct by device optimization. Adv Mater. 2010;22(39):4355–4358. doi: 10.1002/adma.201001339. [DOI] [PubMed] [Google Scholar]
- 15.Roncali J. Conjugated poly(thiophenes)—synthesis, functionalization, and applications. Chem Rev. 1992;92(4):711–738. [Google Scholar]
- 16.McCullough RD. The chemistry of conducting polythiophenes. Adv Mater. 1998;10(2):93–97. [Google Scholar]
- 17.Perepichka IF, Perepichka DF, Meng H, Wudl F. Light-emitting polythiophenes. Adv Mater. 2005;17(19):2281–2305. [Google Scholar]
- 18.Osterbacka R, An CP, Jiang XM, Vardeny ZV. Two-dimensional electronic excitations in self-assembled conjugated polymer nanocrystals. Science. 2000;287(5454):839–842. doi: 10.1126/science.287.5454.839. [DOI] [PubMed] [Google Scholar]
- 19.Sirringhaus H, et al. Two-dimensional charge transport in self-organized, high-mobility conjugated polymers. Nature. 1999;401(6754):685–688. [Google Scholar]
- 20.Niles ET, et al. J-aggregate behavior in poly-3-hexylthiophene nanofibers. J Phys Chem Lett. 2012;3(2):259–263. [Google Scholar]
- 21.Yamagata H, Pochas CM, Spano FC. Designing J- and H-aggregates through wave function overlap engineering: Applications to poly(3-hexylthiophene) J Phys Chem B. 2012;116(49):14494–14503. doi: 10.1021/jp309407r. [DOI] [PubMed] [Google Scholar]
- 22.Clark J, Silva C, Friend RH, Spano FC. Role of intermolecular coupling in the photophysics of disordered organic semiconductors: Aggregate emission in regioregular polythiophene. Phys Rev Lett. 2007;98(20):206406. doi: 10.1103/PhysRevLett.98.206406. [DOI] [PubMed] [Google Scholar]
- 23.Spano FC, Clark J, Silva C, Friend RH. Determining exciton coherence from the photoluminescence spectral line shape in poly(3-hexylthiophene) thin films. J Chem Phys. 2009;130(7):074904. doi: 10.1063/1.3076079. [DOI] [PubMed] [Google Scholar]
- 24.Ruseckas A, et al. Luminescence quenching by inter-chain aggregates in substituted polythiophenes. J Photochem Photobiol A. 2001;144(1):3–12. [Google Scholar]
- 25.Chaudhuri D, et al. Enhancing long-range exciton guiding in molecular nanowires by H-aggregation lifetime engineering. Nano Lett. 2011;11(2):488–492. doi: 10.1021/nl1033039. [DOI] [PubMed] [Google Scholar]
- 26.Cook S, Furube A, Katoh R. Analysis of the excited states of regioregular polythiophene P3HT. Energy Environ Sci. 2008;1(2):294–299. [Google Scholar]
- 27.Paquin F, et al. Charge separation in semicrystalline polymeric semiconductors by photoexcitation: Is the mechanism intrinsic or extrinsic? Phys Rev Lett. 2011;106(19):197401. doi: 10.1103/PhysRevLett.106.197401. [DOI] [PubMed] [Google Scholar]
- 28.Lupton JM. Single-molecule spectroscopy for plastic electronics: Materials analysis from the bottom-up. Adv Mater. 2010;22(15):1689–1721. doi: 10.1002/adma.200902306. [DOI] [PubMed] [Google Scholar]
- 29.Hu DH, et al. Collapse of stiff conjugated polymers with chemical defects into ordered, cylindrical conformations. Nature. 2000;405(6790):1030–1033. doi: 10.1038/35016520. [DOI] [PubMed] [Google Scholar]
- 30.Huser T, Yan M, Rothberg LJ. Single chain spectroscopy of conformational dependence of conjugated polymer photophysics. Proc Natl Acad Sci USA. 2000;97(21):11187–11191. doi: 10.1073/pnas.97.21.11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adachi T, et al. Regioregularity and single polythiophene chain conformation. J Phys Chem Lett. 2011;2(12):1400–1404. [Google Scholar]
- 32.Adachi T, et al. Conformational effect on energy transfer in single polythiophene chains. J Phys Chem B. 2012;116(32):9866–9872. doi: 10.1021/jp306674t. [DOI] [PubMed] [Google Scholar]
- 33.Scheblykin IG, Yartsev A, Pullerits T, Gulbinas V, Sundström V. Excited state and charge photogeneration dynamics in conjugated polymers. J Phys Chem B. 2007;111(23):6303–6321. doi: 10.1021/jp068864f. [DOI] [PubMed] [Google Scholar]
- 34.Sugimoto T, Habuchi S, Ogino K, Vacha M. Conformation-related exciton localization and charge-pair formation in polythiophenes: Ensemble and single-molecule study. J Phys Chem B. 2009;113(36):12220–12226. doi: 10.1021/jp9060945. [DOI] [PubMed] [Google Scholar]
- 35.Khalil GE, et al. Spectroscopy and single-molecule emission of a fluorene-terthiophene oligomer. J Phys Chem B. 2011;115(42):12028–12035. doi: 10.1021/jp206368g. [DOI] [PubMed] [Google Scholar]
- 36.Bässler H, Schweitzer B. Site-selective fluorescence spectroscopy of conjugated polymers and oligomers. Acc Chem Res. 1999;32(2):173–182. [Google Scholar]
- 37.Rossi G, Chance RR, Silbey R. Conformational disorder in conjugated polymers. J Chem Phys. 1989;90(12):7594–7601. [Google Scholar]
- 38.Shand ML, Chance RR, Lepostollec M, Schott M. Raman photoselection and conjugation-length dispersion in conjugated polymer-solutions. Phys Rev B. 1982;25(7):4431–4436. [Google Scholar]
- 39.Da Como E, Borys NJ, Strohriegl P, Walter MJ, Lupton JM. Formation of a defect-free π-electron system in single β-phase polyfluorene chains. J Am Chem Soc. 2011;133(11):3690–3692. doi: 10.1021/ja109342t. [DOI] [PubMed] [Google Scholar]
- 40.Schindler F, et al. Counting chromophores in conjugated polymers. Angew Chem Int Ed Engl. 2005;44(10):1520–1525. doi: 10.1002/anie.200461784. [DOI] [PubMed] [Google Scholar]
- 41.Schindler F, Lupton JM, Feldmann J, Scherf U. A universal picture of chromophores in pi-conjugated polymers derived from single-molecule spectroscopy. Proc Natl Acad Sci USA. 2004;101(41):14695–14700. doi: 10.1073/pnas.0403325101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guillet T, et al. Emission of a single conjugated polymer chain isolated in its single crystal monomer matrix. Phys Rev Lett. 2001;87(8):087401. doi: 10.1103/PhysRevLett.87.087401. [DOI] [PubMed] [Google Scholar]
- 43.Lecuiller R, et al. Fluorescence yield and lifetime of isolated polydiacetylene chains: Evidence for a one-dimensional exciton band in a conjugated polymer. Phys Rev B. 2002;66(12):125205. [Google Scholar]
- 44.Feist FA, Basché T. Fluorescence excitation and emission spectroscopy on single MEH-PPV chains at low temperature. J Phys Chem B. 2008;112(32):9700–9708. doi: 10.1021/jp802585m. [DOI] [PubMed] [Google Scholar]
- 45.Mena-Osteritz E, et al. Two-dimensional crystals of poly(3-alkylthiophene)s: Direct visualization of polymer folds in submolecular resolution. Angew Chem Int Ed. 2000;39(15):2680–2684. doi: 10.1002/1521-3773(20000804)39:15<2679::aid-anie2679>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 46.Collins BA, et al. Polarized X-ray scattering reveals non-crystalline orientational ordering in organic films. Nat Mater. 2012;11(6):536–543. doi: 10.1038/nmat3310. [DOI] [PubMed] [Google Scholar]
- 47.Kline RJ, McGehee MD, Toney MF. Highly oriented crystals at the buried interface in polythiophene thin-film transistors. Nat Mater. 2006;5(3):222–228. [Google Scholar]
- 48.Brown PJ, et al. Effect of interchain interactions on the absorption and emission of poly(3-hexylthiophene) Phys Rev B. 2003;67(6):064203. [Google Scholar]
- 49.Schott M. The colors of polydiacetylenes: A commentary. J Phys Chem B. 2006;110(32):15864–15868. doi: 10.1021/jp0638437. [DOI] [PubMed] [Google Scholar]
- 50.Becker K, et al. How chromophore shape determines the spectroscopy of phenylene-vinylenes: Origin of spectral broadening in the absence of aggregation. J Phys Chem B. 2008;112(16):4859–4864. doi: 10.1021/jp800870p. [DOI] [PubMed] [Google Scholar]
- 51.Müller JG, Anni M, Scherf U, Lupton JM, Feldmann J. Vibrational fluorescence spectroscopy of single conjugated polymer molecules. Phys Rev B. 2004;70(3):035205. [Google Scholar]
- 52.Liu S, et al. Coherent and incoherent interactions between cofacial π-conjugated oligomer dimers in macrocycle templates. J Phys Chem B. 2013;117(16):4197–4203. doi: 10.1021/jp301903u. [DOI] [PubMed] [Google Scholar]
- 53.Hollars CW, Lane SM, Huser T. Controlled non-classical photon emission from single conjugated polymer molecules. Chem Phys Lett. 2003;370(3–4):393–398. [Google Scholar]
- 54.Lee TH, et al. Oriented semiconducting polymer nanostructures as on-demand room-temperature single-photon sources. Appl Phys Lett. 2004;85(1):100–102. [Google Scholar]
- 55.De Schryver FC, et al. Energy dissipation in multichromophoric single dendrimers. Acc Chem Res. 2005;38(7):514–522. doi: 10.1021/ar040126r. [DOI] [PubMed] [Google Scholar]
- 56.Hofkens J, et al. Revealing competitive Forster-type resonance energy-transfer pathways in single bichromophoric molecules. Proc Natl Acad Sci USA. 2003;100(23):13146–13151. doi: 10.1073/pnas.2235805100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beenken WJD, Pullerits T. Spectroscopic units in conjugated polymers: A quantum chemically founded concept? J Phys Chem B. 2004;108(20):6164–6169. doi: 10.1021/jp037332l. [DOI] [PubMed] [Google Scholar]
- 58.Banerji N, Cowan S, Vauthey E, Heeger AJ. Ultrafast relaxation of the poly(3-hexylthiophene) emission spectrum. J Phys Chem C. 2011;115(19):9726–9739. [Google Scholar]
- 59.Wells NP, Blank DA. Correlated exciton relaxation in poly(3-hexylthiophene) Phys Rev Lett. 2008;100(8):086403. doi: 10.1103/PhysRevLett.100.086403. [DOI] [PubMed] [Google Scholar]
- 60.Walter MJ, Lupton JM. Unraveling the inhomogeneously broadened absorption spectrum of conjugated polymers by single-molecule light-harvesting action spectroscopy. Phys Rev Lett. 2009;103(16):167401. doi: 10.1103/PhysRevLett.103.167401. [DOI] [PubMed] [Google Scholar]
- 61.Schindler F, Lupton JM, Müller J, Feldmann J, Scherf U. How single conjugated polymer molecules respond to electric fields. Nat Mater. 2006;5(2):141–146. doi: 10.1038/nmat1549. [DOI] [PubMed] [Google Scholar]
- 62.Müller J, et al. Monitoring surface charge migration in the spectral dynamics of single CdSe/CdS nanodot/nanorod heterostructures. Phys Rev B. 2005;72(20):205339. [Google Scholar]
- 63.Bolinger JC, et al. Conformation and energy transfer in single conjugated polymers. Acc Chem Res. 2012;45(11):1992–2001. doi: 10.1021/ar300012k. [DOI] [PubMed] [Google Scholar]
- 64.Schwartz BJ. Conjugated polymers as molecular materials: How chain conformation and film morphology influence energy transfer and interchain interactions. Annu Rev Phys Chem. 2003;54:141–172. doi: 10.1146/annurev.physchem.54.011002.103811. [DOI] [PubMed] [Google Scholar]
- 65.Stangl T, et al. Temporal switching of homo-FRET pathways in single-chromophore dimer models of π-conjugated polymers. J Am Chem Soc. 2013;135(1):78–81. doi: 10.1021/ja3108643. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.