

Significance
Outer membrane proteins (OMPs) are involved in important cellular activities in Gram-negative bacteria. Although the bepA (formerly yfgC) gene encoding a putative metalloprotease has been implicated in quality control of OMPs, its specific function remains unclear. This study reveals that BepA promotes assembly of LptD, an OMP involved in the transport of lipopolysaccharides, which undergoes intramolecular disulfide rearrangement during its biogenesis. BepA also promotes degradation of incorrectly folded LptD. BamA, another OMP involved in OMP assembly, is also degraded in a BepA-dependent manner in the absence of periplasmic chaperone SurA. BepA thus controls the quality of OMPs by promoting either the biogenesis or elimination of OMPs, depending on their folding state.
Keywords: extracytoplasmic function sigma factor, protein quality control, disulfide bond formation, peptidase M48, tetratricopeptide repeat (TPR) motif
Abstract
Gram-negative bacteria are equipped with quality-control systems for the outer membrane (OM) that sense and cope with defective biogenesis of its components. Accumulation of misfolded outer membrane proteins (OMPs) in Escherichia coli leads to activation of σE, an essential alternative σ factor that up-regulates transcription of multiple genes required to preserve OM structure and function. Disruption of bepA (formerly yfgC), a σE-regulated gene encoding a putative periplasmic metalloprotease, sensitizes cells to multiple drugs, suggesting that it may be involved in maintaining OM integrity. However, the specific function of BepA remains unclear. Here, we show that BepA enhances biogenesis of LptD, an essential OMP involved in OM transport and assembly of lipopolysaccharide, by promoting rearrangement of intramolecular disulfide bonds of LptD. In addition, BepA possesses protease activity and is responsible for the degradation of incorrectly folded LptD. In the absence of periplasmic chaperone SurA, BepA also promotes degradation of BamA, the central OMP subunit of the β-barrel assembly machinery (BAM) complex. Interestingly, defective oxidative folding of LptD caused by bepA disruption was partially suppressed by expression of protease-active site mutants of BepA, suggesting that BepA functions independently of its protease activity. We also show that BepA has genetic and physical interaction with components of the BAM complex. These findings raised the possibility that BepA maintains the integrity of OM both by promoting assembly of OMPs and by proteolytically eliminating OMPs when their correct assembly was compromised.
The cell envelope of Gram-negative bacteria is composed of two layers of biological membranes, the outer membrane (OM) and the inner (cytoplasmic) membrane (IM); the periplasmic space that separates these membranes contains a thin layer of peptidoglycans. Outer membrane proteins (OMPs) span the OM with amphipathic, antiparallel β-strands that form a barrel structure. OMPs are synthesized in the cytoplasm with a cleavable signal peptide at their N termini and are translocated across the IM through the Sec translocon (1). Signal peptide-processed OMPs are transported across the periplasmic space with the aid of periplasmic chaperones, such as SurA, Skp, and DegP, and are inserted into the OM through the function of the β-barrel assembly machinery (BAM) complex (2). The BAM complex is composed of five components: BamA, an essential OMP belonging to the Omp85 superfamily that includes the mitochondrial Sam50 and chloroplast Toc75 proteins, and four lipoprotein subunits (BamB to -E) (3–6). BamD is essential for the growth of Escherichia coli whereas the other three lipoprotein subunits are dispensable (5–7). BamA contains periplasmically exposed polypeptide transport-associated (POTRA) domains that bind the lipoprotein subunits of the BAM complex as well as substrate OMPs (8).
LptD, another essential OMP of Gram-negative bacteria, is involved in the assembly of lipopolysaccharide (LPS), a major OM constituent, in the OM (9). LPS is essential for the function of the OM as a permeability barrier. It is synthesized in the IM and targeted to the outer leaflet of the OM through the functions of seven Lpt proteins (LptA to -G). LptBFGC and LptDE constitute IM and OM complexes, respectively, and LptA connects these two complexes by shuttling or forming a bridge between the two membranes (10–14). While LptD is involved in flipping of LPS from the periplasmic side to the outer side of the OM (9), LptE is thought to contribute to LPS transport through promotion of LptD folding (15). However, a recent view by Kahne and colleagues suggested that LptE also directly contributes to LPS transport by serving as a “plug” for the luminal pore of LptD and by acting as a part of the OM LPS translocon together with LptD (16, 17). LptD has two intramolecular disulfide bonds and requires DsbA, a periplasmic oxidoreductase, for its oxidative folding (18, 19). Recently, Chng et al. reported that LptD is assembled into the OM via formation of a folding intermediate containing nonnative disulfide bonds that are finally isomerized to native disulfide bonds (17). Importantly, folding of the LptD β-barrel is rate-limiting for maturation of LptD and precedes disulfide isomerization, the step triggered by association with LptE. Analysis with a DsbA mutant that forms kinetically stable mixed-disulfide intermediates with substrates suggested that DsbA is involved in formation of both nonnative and native disulfide bonds in LptD (17). A previous study also suggested that DsbC participates in the oxidative folding of LptD (17, 20) although disruption of the dsbC gene had little effect on the stability of LptD (19).
Consistent with the vital roles of the cell envelope, E. coli is equipped with multiple envelope stress-response systems to sense and cope with disorders of OM constituents (21). Among them, the σE-dependent stress response system is the only essential one and is probably one of the most important for the maintenance of the OM structure and function. Imbalanced biogenesis and/or defective assembly of OMPs and LPS leads to cytoplasmic activation of σE, an extracytoplasmic function σ factor responsible for the transcription of a class of genes involved in biogenesis and quality control of the OM components, including those for subunits of the BAM complex and LPS biosynthetic enzymes (22–24).
To date, 114 genes have been identified as members of the E. coli σE regulon (24); however, their functions are not fully understood, making them a treasure trove for identification of novel regulators of OM integrity. The products of the σE-controlled genes include four periplasmic protease homologs, DegP, PtrA, YhiJ, and YfgC. DegP is a protease/chaperone that can switch between these activities, thereby acting in both the assembly and elimination of OMPs (25–27). In contrast, functions of the other three protease homologs remain largely unknown. Recently, genome-wide comprehensive analyses of the antibiotic sensitivity of an E. coli single-gene knockout library showed that disruption of the yfgC gene renders cells sensitive to multiple drugs (28–31). Moreover, profiling of the chemical sensitivities of ΔyfgC cells has suggested that this putative protease is involved in the biogenesis and/or quality control of OMPs (31). The amino acid sequence of YfgC indicates that it belongs to the peptidase M48 family and possesses a typical zinc metalloproteinase active site motif, H136EXXH. In addition, the C-terminal portion of YfgC contains four copies of the tetratricopeptide repeat (TPR) motif, a structural element involved in protein–protein interactions (32). Members of the M48 family include mitochondrial Oma1 (33), Ste24p (34), and E. coli HtpX (35). HtpX is a membrane-anchored protease with its protease-active site exposed to the cytoplasm and is thought to collaborate with the AAA+ protease FtsH to eliminate misfolded IM proteins (36). YfgC has recently been reported to interact with and contribute to proper localization of LoiP (YggG), an OM-associated protease homologous to YfgC (37). However, the proteolytic activity of YfgC has not been demonstrated, and its specific cellular function remains obscure.
In this study, we sought to determine the molecular functions of YfgC in maintaining the structure and function of the cell envelope. Our results suggest that YfgC exerts dual functions, promoting either the biogenesis or proteolytic elimination of OMPs, depending on the folding state of the OMPs. Therefore, we suggest that YfgC should be renamed as BepA (β-barrel assembly-enhancing protease).
Results
BepA Processes a Protease Activity That Is Important for Maintenance of the OM Integrity.
Although BepA carries a conserved zinc metalloprotease active site motif, H136EXXH, it is not known whether BepA possesses a protease activity. To verify whether BepA is indeed a protease, we purified C-terminally decahistidine (His10)-tagged wild-type BepA (BepAHis10) and its derivative with an amino acid substitution (E137Q) in the metalloprotease active site motif by metal affinity chromatography. Incubation of α-casein, a model substrate, with wild-type BepAHis10 gave rise to a small but significant amount of a band that migrated slightly faster than α-casein (Fig. 1A, open arrowhead). The N-terminal sequence of this band turned out to be FVAPF that matches the 24th to 28th residues of α-casein, indicating that it was generated by cleavage between F23 and F24. Generation of this fragment was inhibited when metal chelating reagent 1,10-phenanthroline or EDTA was included in the reaction mixture (Fig. 1B). Furthermore, incubation of α-casein with the protease active site motif mutant, BepA(E137Q)His10, did not produce such a fragment (Fig. 1A). Although the observed in vitro proteolytic activity of BepA is rather low for unknown reasons, the above results collectively demonstrate that BepA is indeed a metalloprotease.
Fig. 1.

Protease activity is required for BepA function. (A) Protease activity of BepA. α-casein (400 µg/mL) was incubated without (−) or with His10-tagged wild-type BepA (WT) or its E137Q derivative (E137Q) (200 µg/mL) at 37 °C for 0 or 24 h. Proteins were separated by SDS/PAGE and visualized by staining with Coomassie brilliant blue R-250. Proteolytic product of α-casein is indicated by an open arrowhead. Migration positions of molecular weight markers are shown on the left. (B) Effects of metal chelators on the BepA protease activity. Protease activity of C-terminally His10-tagged wild-type BepA was analyzed as in A except that the reaction mixture contained 50 μM ZnCl2 (Zn), 250 μM 1,10-Phenanthroline (PT), or 250 μM EDTA. (C) Erythromycin sensitivity of cells expressing BepA or its derivative. The minimum inhibitory concentration (MIC) of erythromycin for the ΔbepA or wild-type strain transformed with pUC18 vector, pUC-bepA, pUC-bepA(H136R), or pUC-bepA(E137Q) encoding wild-type, H136R mutant, or E137Q mutant BepA, respectively, at 30 °C is shown. (D) Cellular levels of BepA. BepA levels in the ΔbepA cells used in C were determined by immunoblotting with anti-BepA antiserum. Maltose-binding protein (MBP) was also detected by immunoblotting with anti-MBP antibodies and was used as a loading control.
Disruption of the bepA gene causes significantly elevated sensitivity to high-molecular mass antibiotics, such as erythromycin and vancomycin (28–31) (Fig. 1C), indicating that loss of BepA compromises the function of OM as a permeability barrier. We examined the complementation activities of two active site motif mutants, BepA(E137Q) and BepA(H136R), against the ΔbepA mutation. In contrast to wild-type BepA, these mutants failed to restore drug resistance to the ΔbepA mutant (Fig. 1C) although they accumulated at levels comparable with the wild-type protein (Fig. 1D). Importantly, these protease-dead mutants exhibited a dominant-negative phenotype, as their expression in the wild-type (bepA+) strain significantly increased sensitivity to erythromycin (Fig. 1C). It is not unexpected that proteases with a compromised proteolytic active site can exhibit a dominant-negative phenotype because they often retain other activities (e.g., substrate binding, oligomerization) and thus compete with the wild-type protein. These results suggest that protease activity of BepA is important for its function to maintain OM integrity. Indeed, as described in later sections (see Figs. 5 and 8), our results demonstrate that BepA is involved in degradation of misfolded OMPs.
Fig. 5.

BepA destabilizes LptD in LptE-limiting conditions. (A) LptE-depleted cells carrying bepA (bepA+) or its disruptant were pulse-labeled and analyzed as described in the legend to Fig. 4A. (B and C) The stability of LptD was assessed as in A using the ΔbepA (B) or wild-type (C) cells transformed with empty vector or either of the plasmids encoding C-terminally His10-tagged wild-type BepA or its H136R or E137Q derivative.
Fig. 8.

BepA-dependent degradation of BamA in the ΔsurA strain. Total cellular protein was prepared from wild-type or mutant cells lacking bepA and/or surA (lanes 1–4) or ΔsurA/ΔbepA cells harboring empty vector or either of the plasmids encoding C-terminally His10-tagged wild-type BepA or its H136R or E137Q derivative (lanes 5–8) and was subjected to SDS/PAGE followed by immunoblotting with anti-BamA or anti-BepA antisera. Putative BepA degradation products of BamA are indicated by asterisks.
Isolation of Multicopy Suppressors of the bepA-Null Mutation.
Despite apparent dysfunction of OM, disruption of bepA gene has little impact on the profiles of OMPs and heat modifiability of a major OMP, OmpA (Fig. S1). To gain insight into the role of BepA in the envelope function, we sought multicopy suppressors of the ΔbepA mutation. Multicopy suppressors provide clues to important functions of a deleted gene by identifying alternative routes to performing the key function disabled by the gene deletion. Although the ΔbepA strain was sensitive to erythromycin, the ΔbamE/ΔbepA strain showed greater sensitivity to multiple drugs than the bepA deletion alone (see Fig. 6 A and B). We thus used the double-mutant strain as the basis for the multicopy suppressor screen. For this screen, we used the ASKA clone library of 4,123 E. coli ORFs, individually cloned under the isopropyl-thio-β-D-galactopyranoside (IPTG)-inducible promoter of a chloramphenicol-resistant multicopy vector (38). Pools of ASKA clones were introduced into the ΔbamE/ΔbepA strain, and transformants were selected on agar plates containing 20 μg/mL chloramphenicol and 5 μg/mL erythromycin, with (100 μM) or without IPTG to induce the cloned genes. Both regimes were used because high expression can sometimes be toxic. Among the ASKA pools, we found 9 erythromycin-resistant transformants without IPTG and 248 transformants with IPTG. After verifying that drug resistance was linked to each plasmid (by retransformation with plasmids isolated from the first isolated drug-resistant bacteria), cloned genes were identified by DNA sequencing. We then tested whether these multicopy suppressors conferred resistance to other ΔbamE/ΔbepA-derived drug sensitivities (e.g., SDS and vancomycin) (Table S1) and retained the 14 clones that conferred resistance to all of the selection reagents. Of these, only the clone carrying lptE conferred erythromycin and vancomycin resistance to a level comparable with a plasmid carrying wild-type bepA, even in the absence of IPTG (Fig. 2A and Table S1). Furthermore, when lptE was under tight arabinose control, the erythromycin sensitivity of the ΔbepA strain was suppressed in an arabinose-dependent manner (Fig. 2B). These results established that overexpression of LptE compensated for the lack of BepA, at least with respect to the drug-sensitive phenotypes examined. LptE/D form the essential OM translocon that is required for export of LPS to the outer leaflet of the OM (9, 10, 16), and LptE assists the biogenesis and/or function of LptD (10, 15–17), suggesting that BepA may participate in the formation of functional LptD.
Fig. 6.
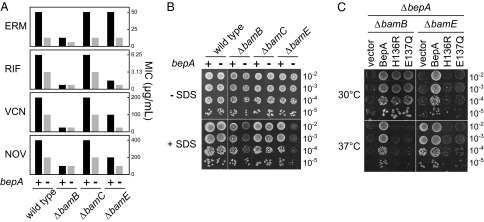
bamB/bepA and bamE/bepA double disruptants show elevated drug sensitivity. (A) MICs of erythromycin (ERM), rifampicin (RIF), vancomycin (VCN), and novobiocin (NOV) at 30 °C for strains lacking bepA, bamB, bamC, and bamE individually or in combinations as indicated. (B) Overnight cultures of strains lacking bepA, bamB, bamC, and bamE individually or in combinations as indicated were serially diluted with saline, spotted onto L agar with or without 0.5% SDS, and incubated at 30 °C for 24 h. (C) bamB/bepA and bamE/bepA double-knockout strains were transformed with a plasmid encoding wild-type BepA or its H136R or E137Q derivative and were grown on L agar containing 0.5% SDS for 24 h at 30 °C or 37 °C.
Fig. 2.

Overexpression of LptE suppresses the drug sensitivity of the ΔbepA strain. (A) Erythromycin sensitivity of the ΔbepA strain transformed with an ASKA clone carrying lptE. pCA24NΔNot was used as a vector control. For comparison, the MIC of the ΔbepA strain transformed with pTH-bepA-his10 encoding wild-type BepAHis10 is shown. (B) LptE was overexpressed from pMAN-lptE carrying lptE under the control of the araBAD promoter. Expression of LptE was confirmed by SDS/PAGE of total cellular proteins followed by anti-LptE immunoblotting.
LptD with Nonnative Disulfide Bonds Accumulates in the Absence of BepA.
LptE promotes the proper oxidation of LptD and stabilizes LptD protein (19, 39). It seemed thus possible that lack of BepA, like that of LptE, destabilizes LptD. Also, considering that the drug sensitivity of the ΔbepA strain was suppressed by overproduction of LptE, it might be also possible that the bepA disruption might cause decrease in the LptE level. However, these possibilities were ruled out, as the levels of LptD and LptE were little affected by the presence or absence of BepA (Fig. 3A, +ME). We next considered whether BepA affected disulfide bond formation in LptD. Mature LptD has two intramolecular disulfide bonds between nonconsecutive pairs of Cys residues (C31–C724 and C173–C725; mature LptD with correct disulfide bonds referred to here as LptDNC) (19). As reported previously (19), correctly disulfide-bonded LptDNC migrates more slowly than the fully reduced form (Fig. 3A, −ME, left lane). Importantly, the ΔbepA strain had an additional band (LptDC) that migrated more rapidly than the fully reduced band (Fig. 3A, −ME, right lane).
Fig. 3.

Defective oxidative folding of LptD in the absence of BepA. (A) Immunoblotting of LptD was carried out after reducing (+ME) or nonreducing (−ME) SDS/PAGE of total cellular proteins. Wild-type (+) or ΔbepA (−) cells were grown overnight at 30 °C. Anti-LptE immunoblotting was also carried out after reducing SDS/PAGE. The positions of reduced LptD (LptDRED), properly oxidized LptD (LptDNC), rapidly migrating LptD (LptDC), LptE, and molecular weight markers are indicated. (B) Schematic representation of disulfide bonds in LptDNC and LptDC. Asterisk indicates a nonspecific band.
To unambiguously establish that different disulfide bond connectivity was responsible for generation of LptDC, we examined the migration patterns of C-terminally hexahistidine (His6)-tagged LptD proteins (LptDHis6) having various combinations of their four Cys residues changed to Ser, thereby restricting which disulfide bonds were able to form. LptD(CCSS)His6, which formed only the sequential C31–C173 disulfide bond, gave only the rapidly migrating species, even in the wild-type (bepA+) strain (Fig. S2A). In contrast, LptD(SSCC)His6, which formed only the sequential C724–C725 disulfide bond, did not produce this rapidly migrating species. As reported (17), the C724–C725 disulfide bond can actually be formed, as this species was nonreactive with the sulfhydryl-reactive alkylating reagent maleimide PEG2-biotin, unless it had been reduced with DTT before the alkylating reaction (Fig. S2B). All species that could form the C31–C173 bond (CCCC, CCSC, and CCCS) promoted LptDC formation in the absence of BepA (Fig. S2A). We thus concluded that LptDC had nonnative disulfide bonds, at least between C31 and C173 and possibly between C724 and C725 (Fig. 3B). Subcellular fractionation revealed that, like LptDNC, LptDC was associated with the OM (Fig. S2 C and D).
BepA Promotes Formation of the Proper Intramolecular Disulfide Bonds in LptD.
It has been recently reported that LptD is assembled into the OM via formation of an intermediate containing nonnative disulfide bonds linked between consecutive Cys residues that are isomerized to native disulfide bonds, the step triggered by assembly with LptE (17). Our results suggest that LptDC corresponds to the folding intermediate with consecutive disulfide bonds and that the absence of BepA causes its accumulation. To determine whether LptDC is indeed an on-pathway species, we examined the kinetics of formation of LptDNC and LptDC by pulse-labeling cells with [35S]-methionine for 1 min, followed by chase with excess nonlabeled methionine. In wild-type cells, almost all labeled LptD molecules were present as LptDC after a 1-min chase and were then converted to LptDNC in a process in which an initial rapid (t1/2 ∼10 min) phase was followed by a slower conversion that took ∼40 min for completion (Fig. 4A). Like in wild-type cells, LptD was mostly detected as LptDC after a 1-min chase in ΔbepA cells and was converted to LptDNC upon chase. However, this conversion took a much longer time (t1/2 > 80 min) (Fig. 4A) in ΔbepA cells. Conversely, the rate of LptDC to LptDNC conversion was restored by overproduction of BepA in ΔbepA cells (Fig. 4B). The conversion was slightly but significantly accelerated by overproduction of BepA, even in wild-type cells (Fig. S3). These results indicate that BepA promotes disulfide isomerization of LptD and that the process promoted by BepA is rate-limiting for LptD maturation. Importantly, overexpression of LptE in ΔbepA cells promoted both the extent and rate of the conversion of LptD to approximately those of wild-type cells (Fig. S4), suggesting that the suppression of ΔbepA by LptE overexpression resulted mainly from restoration of LptDNC formation.
Fig. 4.

BepA facilitates disulfide isomerization in LptD. (A) Wild-type and ΔbepA cells were labeled with [35S]-methionine for 1 min and chased for the indicated durations at 30 °C. Acid-precipitated proteins were subjected to immunoprecipitation with anti-LptD antiserum and were analyzed by nonreducing (−ME) or reducing (+ME) SDS/PAGE followed by phosphorimaging. Migration positions of molecular weight markers are shown on the left. (B) Oxidative folding of LptD was monitored as in A using ΔbepA cells transformed with empty vector or either of the plasmids encoding C-terminally His10-tagged wild-type BepA or its H136R or E137Q derivative. LptDNC (%) in Right was calculated by dividing the band intensity of LptDNC by the sum of those of LptDC and LptDNC.
We found that ectopic expression of the H136R or E137Q mutant of BepA in ΔbepA cells also promoted LptDC to LptDNC conversion, albeit less effectively than wild-type BepA (Fig. 4B). In contrast, these mutants slightly retarded this conversion process when expressed in wild-type (bepA+) cells (Fig. S3), which was consistent with their dominant-negative properties. These results indicated that BepA could promote oxidative folding of LptD independently of its protease activity, although the intact protease motif is still required for its full functioning.
Disulfide isomerization of periplasmic and OM proteins is mainly catalyzed by DsbC (18), but that of LptD proceeds even in the absence of DsbC (17) (Fig. S5A). Additional disruption of the dsbG gene, encoding possible periplasmic disulfide isomerase, had essentially no effect (Fig. S5A). Therefore, DsbC and DsbG may be dispensable for disulfide exchange in the process of LptD biogenesis. The oxidoreductase DsbA is required for the oxidative folding of LptD (17, 18). In the absence of DsbA, several bands, including both LptDC and LptDNC, were generated after a 1-min chase (Fig. S5B). When treated with a reducing reagent, they were converted to LptDRED (Fig. S5B), indicating that they were LptD species with different oxidative states. These results suggested that, in the absence of DsbA, both consecutive and nonconsecutive disulfide bonds were formed stochastically.
BepA Degrades LptD When LptD Fails to Form the OM LPS Translocon.
In agreement with the recent reports by Chng et al. (17), we found that the LptDC to LptDNC conversion was almost completely blocked in LptE-depleted cells (Fig. 5A). In addition, the pulse–chase experiment revealed that LptDC was significantly destabilized in this condition (Fig. 5A). Strikingly, LptDC became stable, even under LptE-limiting conditions, when bepA was disrupted (Fig. 5A), and the LptDC-stabilizing effect of ΔbepA was suppressed by expression of wild-type but not protease-dead BepA protein from a plasmid (Fig. 5B). Furthermore, ectopic expression of wild-type BepA led to increased destabilization of LptDC in LptE-depleted cells carrying chromosomal bepA+ whereas that of protease-dead BepA mutants dominantly stabilized LptDC (Fig. 5C). These results strongly suggest that BepA functions as a protease in vivo to degrade LptD that has failed to interact with LptE to form the functional LPS translocon.
Genetic Interaction of bepA with bamB and bamE.
The ΔbepA strain exhibits elevated drug sensitivity (28–31) (Fig. 1C), a phenotype reminiscent of deletion of some of the nonessential members (bamB, bamC, and bamE) of the BAM complex (31). The BAM complex, composed of one β-barreled subunit (BamA) and four lipoprotein subunits (BamB to -E), facilitates insertion of β-barrel proteins into the OM. The similarities in the antibiotic sensitivity phenotypes of these proteins suggested that BepA may have some functional interaction with the Bam components. Consistent with this idea, ΔbamB/ΔbepA or ΔbamE/ΔbepA double mutants displayed elevated sensitivity toward multiple antibiotics (Fig. 6A). These double mutants also exhibited significantly higher SDS sensitivity; they grew much more slowly than the wild-type cells in the presence of 0.5% SDS, although the viability of cells seemed almost the same (Fig. 6B). None of the single mutations conferred these phenotypes (Fig. 6B). These results show that bepA genetically interacts with bamB and bamE. Interestingly, the protease-dead BepA mutant proteins restored the SDS resistance of the ΔbamB/ΔbepA strain at 30 °C, supporting the above notion that BepA plays a nonproteolytic role (Fig. 6C) whereas they failed to restore the SDS sensitivity of the ΔbamE/ΔbepA strain. Moreover, these mutant proteins dominantly sensitized the ΔbamE/ΔbepA cells to SDS at 37 °C, underscoring the fact that the residual activity of protease-defective proteins could be deleterious to the cell (Fig. 6C).
BepA Is Localized in Close Proximity to the BAM Complex.
We then investigated possible physical interaction with envelope components, such as the BAM complex. The SOSUI program (40) predicted BepA to be a periplasmic protein with an N-terminal 27 amino acid-long signal peptide. As expected, upon cell fractionation, BepA was predominantly recovered in the soluble fraction (Fig. S6). However, a significant amount of BepA was also present in the membrane fraction (Fig. S6). As an unbiased approach to identify putative BepA binding partners, we used chemical cross-linking. Wild-type cells with a plasmid encoding C-terminally His10-tagged BepA (BepAHis10) or its H136R or E137Q derivatives were converted to spheroplasts and treated with bis(sulfosuccinimidyl) suberate (BS3), a membrane-impermeable and amine-reactive cross-linker, and analyzed by SDS/PAGE and anti-BepA immunoblotting (Fig. 7A). BepAHis10 gave rise to several cross-linked products (Fig. 7A), which were even more obvious with BepA(H136R)His10 and BepA(E137Q)His10. Enhanced cross-linking with protease-dead mutants may be attributable to stalling reactions caused by impaired protease activity. Metal affinity chromatography followed by liquid chromatography-mass spectrometry analysis of the strongest cross-linked band of BepA(E137Q)His10 (∼170 kDa) identified BamA, the central subunit of the BAM complex (Fig. S7). Immunoblotting with anti-BamA antibodies confirmed BamA–BepA cross-linking (Fig. 7B). As expected, BamA–BepA adducts were almost exclusively recovered in the membrane fraction (Fig. 7B). To examine the possible interaction of BepA with other subunits of the BAM complex, we compared the patterns of BS3-mediated adduct production in the presence or absence of the nonessential lipoprotein subunits, i.e., BamB, BamC, or BamE. In addition to the BamA-BepA(E137Q) adduct detected in all strains (Fig. 7 C and D), a cross-linked product of ∼110 kDa, reactive with both anti-BepA and anti-BamC antisera, was detected in wild-type, ΔbamB, and ΔbamE strains, but not in the ΔbamC strain (Fig. 7 C and F). Additionally, an ∼85-kDa band reactive with both anti-BepA and anti-BamD antisera was detected in the ΔbamC strain (Fig. 7 C and G). No cross-link between BepA and BamB was observed (Fig. 7E).
Fig. 7.

BepA interacts with the BAM complex. (A–G) BS3-mediated cross-linking. (A and B) Wild-type cells transformed with the pUC18 vector (−) or its derivative encoding the C-terminally His10-tagged wild-type BepA (WT) or its H136R or E137Q derivative were subjected to cross-linking with BS3 as described in Materials and Methods. Membrane (M) and periplasmic (P) fractions were analyzed to SDS/PAGE followed by immunoblotting with anti-BepA (A) or anti-BamA (B) antisera. The cross-linked product of BepA and BamA is indicated (× BamA; also indicated by asterisks). (C–G) The ΔbepA strain (WT) or the ΔbepA strain also having deletions of bamB (ΔB), bamC (ΔC), or bamE (ΔE) were transformed with empty vector (–) or a plasmid encoding tag-free BepA(E137Q) and were subjected to BS3 cross-linking. Membrane fractions were analyzed by WIDE RANGE PAGE (Nacalai Tesque) (7.5% acrylamide), except for D, in which normal SDS/PAGE was used, followed by immunoblotting with anti-BepA (C), anti-BamA (D), anti-BamB (E), anti-BamC (F), or anti-BamD (G) antisera. (H) Photo–cross-linking of BepA and BamA. The ΔbepA cells transformed with pEVOL-pBpF (aminoacyl-tRNA synthetase/suppressor tRNA) and the plasmid encoding a BepA derivative with an amber codon at position 428 were grown with or without pBPA and were UV-irradiated for 10 min. Acid-precipitated proteins were analyzed by SDS/PAGE and immunoblotting with anti-BepA and anti-BamA antisera. The cross-linked product between BepA(Q428pBPA) and BamA is indicated. (I) Pull-down assay. Wild-type strain (WT) transformed with empty vector or the ΔbamB strain transformed with a plasmid encoding C-terminally His6-tagged BamB (BamBHis6) was cotransformed with a plasmid encoding BepA(E137Q). Membrane proteins were solubilized and subjected to a pull-down assay using His6 tag of BamB. Solubilized membrane (input) and affinity-purified proteins (eluate, a fivefold equivalent for Bam component immunoblotting and a 50-fold equivalent for BepA and OmpA immunoblotting) were analyzed by SDS/PAGE and immunoblotting.
As an alternative approach to probe possible interaction, we carried out site-directed in vivo cross-linking experiments. The C-terminal portion of BepA contains four copies of the TPR motif, which is widely involved in protein–protein interactions. We expected that the BepA TPR motifs would participate in interaction with other proteins and substituted an amber codon for the Q428 codon in the fourth TPR motif of BepA, to allow suppressor tRNA-mediated introduction of a nonnatural, photo-reactive amino acid, p-benzoylphenylalanine (pBPA), at this position (41). Cells expressing BepA(Q428pBPA) were UV-irradiated and analyzed by SDS/PAGE and immunoblotting. We found that a band of about 135 kDa that was reactive with both anti-BepA and anti-BamA antibodies was yielded in UV-irradiation and pBPA-dependent manners (Fig. 7H), indicating that it represents a BamA-BepA(Q428pBPA) adduct.
The BAM complex is rather stable, enabling purification of the entire BAM complex using a polyhisitidine tag attached to a BAM component (6, 42). To examine whether formation of the cross-linked adducts reflects direct interaction between BepA and the BAM complex, BepA(E137Q) was coexpressed with C-terminally His6-tagged BamB (BamBHis6) in the ΔbamB strain. Metal affinity chromatography followed by SDS/PAGE and immunoblotting revealed that, along with other Bam components, a portion of BepA(E137Q) was coisolated with BamBHis6 (Fig. 7I). Taken together, these results strongly suggest that BepA directly interact with the BAM complex possibly via its TPR domain. Its membrane localization could be at least partly resulted from this interaction.
BepA Cleaves BamA in Cells Lacking SurA.
The above finding that BepA interacts with BamA prompted us to examine the possibility that BamA becomes a proteolytic substrate of BepA. In the wild-type background, however, we detected no difference in the profiles of anti-BamA–reactive bands between wild-type and ΔbepA strains (Fig. 8, Upper, lanes 1 and 2). We next compared the profiles of anti-BamA–reactive bands in the ΔsurA background. SurA is thought to play a primary role in the biogenesis of OMPs, and disruption of the surA gene causes massive decreases in the levels of OMPs and increased permeability to a wide variety of drugs (43). Absence of SurA does not affect β-barrel folding of BamA in cells grown in rich media (44). However, we detected several anti-BamA–reactive bands that were smaller than intact BamA in ΔsurA cells when the cells were grown in minimal media. These bands were hardly detected in ΔsurA/ΔbepA double mutants (Fig. 8, Upper, lanes 3 and 4) but regenerated upon ectopic expression of wild-type but not protease-dead BepA proteins (Fig. 8, Upper, lanes 5–8) in the same strain background, suggesting that they are BamA degradation products. Degradation of other OMPs such as LamB and OmpA was not observed (Fig. S8 A and B) under similar conditions. Loss of SurA could induce the σE stress response and cause up-regulation of BepA. However, BamA degradation observed in the ΔsurA strain was not the direct result of increased synthesis of BepA because overexpression of BepA in the surA+ strain did not cause BamA degradation (Fig. S8C). It is unclear why BamA degradation products were not detected in cells grown in rich media, but they may be further degraded by other proteases in that case. Collectively, these results suggest that proper assembly of BamA is compromised in the absence of SurA and that BepA degrades BamA under such a condition. Alternatively, as BamA has been suggested to assume different conformations during its functional cycle (45, 46), the absence of SurA might cause accumulation of the BepA-susceptible form of BamA.
We noticed that C-terminally His10-tagged wild-type BepA migrated faster than its H136R or E137Q derivative and migrated slightly slower than chromosomally encoded BepA upon SDS/PAGE (Fig. 8, Lower). It seems likely that BepAHis10 underwent self-cleavage within its C-terminally attached tag sequence, which further supports our result that BepA possesses proteolytic activity.
Discussion
In this study, we investigated the cellular functions of BepA, a σE-regulated, periplasmic protease homolog implicated in the maintenance of OM integrity. With a purified preparation, we demonstrated that BepA indeed possesses a proteolytic activity. Our genetic and biochemical analyses suggest that BepA acts in the assembly/degradation of two essential OMPs, LptD and BamA, the central components of OM translocons for LPS and OMPs, respectively. Importantly, we showed that BepA can promote disulfide isomerization of LptD independently of its protease activity and degradation of LptD and BamA when their proper assembly is compromised. Our results suggest that BepA acts as a protease/chaperone that contributes to OM quality control by directly and/or indirectly affecting biogenesis of both OMPs and LPS.
BepA Promotes Maturation of LptD.
LptD is converted into the mature form with nonconsecutive disulfide bonds (LptDNC) via an intermediate with nonnative, consecutive disulfide bonds (LptDC) (17). Although Chng et al. identified this intermediate using epitope-tagged LptD expressed from a plasmid (17), we showed that chromosomally encoded LptD also follows this assembly pathway. A detailed study by Kadokura and Beckwith suggested that, whereas disulfide bonds of proteins are formed vectorially from the N terminus to the C terminus in the periplasm when the protein is cotranslationally translocated across the IM, posttranslationally translocated proteins have more relaxed restrictions for the order of disulfide bond formation (47). Although LptD is presumably exported across the membrane posttranslationally, disulfide bonds are exclusively formed in the N-to-C order. The finding that the precursor form of LptD, observed after 1-min chase, migrated more rapidly in nonreducing SDS/PAGE than in reducing gels (Fig. 4A) supports that at least the disulfide bond between C31 and C173 can be formed before completion of polypeptide translocation. The physical distance between the two Cys pairs (separated by 550 amino acids) may contribute to the exclusive formation of LptDC, even in a posttranslational translocation pathway. DsbA is involved in the oxidative folding pathway of LptD (17–19), and disulfide bonds of LptD are formed stochastically in the absence of DsbA. Oxidants in the media or molecular oxygen may contribute to oxidizing some populations of LptDRED directly to LptDNC, as previously suggested (19), which may be why DsbA is nonessential even though it appears to have critical roles in the biogenesis of essential LptD.
We screened for multicopy suppressors against the drug sensitivity phenotype of the ΔbepA strain and identified the lptE gene that encodes a protein involved in the function and/or folding of LptD (15). This result suggests that BepA is also involved in LptD biogenesis. In agreement with this notion, we found that disruption of bepA delayed the rearrangement of LptD disulfide bonds, causing accumulation of LptDC, which was suppressed by overexpression of LptE. Disulfide bond rearrangement in LptD is triggered by its association with LptE (17). Our finding of suppression by LptE overproduction of defective LptDC-to-LptDNC conversion due to loss of BepA suggests that BepA promotes LptD/E association. Through the isolation and analyses of mutations in bamA that allele-specifically suppress an lptE mutation, it was proposed that LptE interacts with LptD that is being assembled by the BAM complex (15). BepA may directly assist efficient interaction between LptD and LptE at the BAM complex or act at some other step during LptD assembly to eventually facilitate formation of a productive LptD/E complex (Fig. 9, Upper).
Fig. 9.

A model for BepA function in the assembly/degradation of the OM LPS translocon. (Upper) BepA promotes disulfide rearrangement of LptDC that is triggered by association of LptDC with LptE. BepA may directly assist LptD-LptE interaction or act indirectly to facilitate formation of the productive LptD-LptE complex. Square brackets represent disulfide bonds (Lower). In the absence of LptE, BepA acts to proteolytically eliminate accumulated LptDC.
Possible Dual Functions of BepA as a Chaperone and Protease.
Here, we purified BepA and showed that it has a low but significant protease activity against α-casein. Its protease activity was inhibited by metal chelating reagents or amino acid substitution in the putative protease active-site motif (HEXXH), suggesting that BepA is a metalloprotease as deduced from its sequence homology to the M48 family of zinc metalloproteinases. Protease-dead BepA mutants are defective in complementation of the ΔbepA mutation and exert dominant-negative effects when expressed in wild-type (bepA+) cells. Degradation of LptDC in LptE-depleted cells and BamA in ΔsurA cells was dependent on BepA with an intact protease active-site motif. Moreover, overexpression of wild-type BepA accelerated degradation of LptDC in LptE-depleted cells whereas protease-dead BepA mutants dominantly stabilized LptDC under the same conditions. Our observations collectively suggest that BepA directly proteolyzes OMPs such as LptD and BamA in vivo and that protease activity is important for its function in maintaining OM integrity (Fig. 9, Lower). Although the observed in vitro protease activity was quite modest, it might be improved by, for instance, optimization of the assay conditions and/or addition of unidentified partner proteins.
Several lines of evidence suggest that BepA also possesses a protease activity-independent function. We found that the SDS sensitivity of the bamB/bepA double disruptant was complemented by protease-dead mutants. Disulfide rearrangement of LptD was also promoted by these protease-dead BepA derivatives. Furthermore, overexpression of BepA in wild-type cells facilitated the rate of LptDC to LptDNC conversion (Fig. S3) without affecting the stability of LptD. It is thus conceivable that BepA can facilitate formation of the proper conformation of LptD independently of its protease activity. These characteristics of BepA are reminiscent of DegP, another σE-regulated protease that reversibly switches between a protease and a chaperone in a temperature- and substrate-dependent manner (25, 27). Our results suggest that BepA is able to function in two ways depending on the folding state of a substrate OMP: as a chaperone promoting the biogenesis of a substrate OMP or as a protease degrading it when it fails to assemble correctly into the OM.
It should be noted that the rate of LptDC to LptDNC conversion was not fully restored by the expression of protease-dead BepA mutants in the ΔbepA strain; the protease-dead mutants effectively recovered the initial rate of LptDC to LptDNC conversion, but the final yield of LptDNC in protease-dead BepA-expressing cells was much lower than that in the strain expressing wild-type BepA. Although the exact reason for the above observations is unclear, a fraction of LptDC may elude the protease-dead mutants having reduced or altered chaperone-like activity and enter an off-target pathway after a certain period to yield a dead-end product that resists the action of BepA. The protease activity of BepA may also be required for its maximum activity to remove a portion of LptDC that would misfold, even in the presence of LptE, and competitively inhibit the function of BepA.
BepA Is Involved in Maintenance of OM Integrity.
bepA mutants exhibit hypersensitivity to a variety of hydrophobic and high-molecular-mass drugs. Recent large-scale chemical genomics combined with quantitative fitness measurements have suggested that the function of the bepA gene is highly correlated with those of genes involved in OMP and LPS biosynthesis (30, 31). Consistent with the above suggestion, our in vivo observation that additional disruption of the bamB or bamE gene synergistically elevated the SDS and antibiotic sensitivity of the ΔbepA strain suggested that BepA has genetic interactions with these BAM components and thus is involved in normal OM assembly. Direct interaction between BepA and the BAM complex was suggested from our biochemical results demonstrating that BepA can be pulled down with the BAM complex and cross-linked with BamA, BamC, and BamD. Site-specific in vivo photo–cross-linking experiments suggested that the TPR domain of BepA is involved in the interaction with BamA. These observations raise the possibility that BepA acts in cooperation with the BAM complex.
Suppression of the antibiotic hypersensitivity of the ΔbepA strain by overexpression of LptE, which would specifically improve the folding/assembly of LptD, suggests that the antibiotic hypersensitivity of this strain is mainly attributable to folding defects in LptD. Inefficient assembly of LptD would result in delay of LPS translocation across the OM, which causes phospholipids to flip from the inner to the outer leaflet of the OM (48). Impaired lipid asymmetry would then abolish the barrier function of OM against drugs. However, BepA may play a more general role in the biogenesis/quality control of OMPs. Synthetic lethal or sick phenotypes are often observed following disruption of two genes that have overlapping or compensatory functions. Indeed, we found that simultaneous disruption of bepA together with either bamB or bamE synthetically sensitized cells to SDS. In addition, BepA can interact with the BAM complex. The genetic and physical interactions between BepA and the BAM complex raise the possibility that BepA promotes the correct assembly of not only LptD but also other OMPs. The surA/bepA double disruptant was recently reported to exhibit a temperature-sensitive growth phenotype (49). Lack of SurA would produce misfolded BamA that could be normally eliminated by BepA, and the growth defects of the surA/bepA double disruptant may result from dysfunction of the BAM complex to assemble OMPs. Because lack of BepA does not cause a drastic change in β-barrel folding of OMPs that can be assessed by the heat-modifiability assay, it is not easy to assay maturation for most OMPs other than LptD, maturation of which can be assayed by examining the disulfide rearrangement. Improvement of experimental techniques to monitor folding/assembly states of OMPs would be required to test this possibility.
Materials and Methods
Bacterial Strains, Plasmids, and Media.
The bacterial strains, plasmids, and primers used in this study are listed in Tables S2–S4, respectively. Their constructions are described in SI Materials and Methods. E. coli K12 strains AD16 (50), MC4100 (51), and AM604 (10) were used as wild-type strains. AM604 and GC187 (ParalptE) were a generous gift from Thomas J. Silhavy (Princeton University, Princeton, NJ). SM4106 (ΔbamB::tet) was a generous gift from Shin-ichi Matsuyama (Rikkyo University, Tokyo, Japan). KRX (Promega) and TOP10 (Invitrogen) were used as hosts for protein expression and routine cloning procedures, respectively. The Keio collection (52), the ASKA library (38), and plasmid pCP20 were provided by the National BioResource Project-E. coli (National Institute of Genetics, Mishima, Japan). pEVOL-pBpF was purchased from Addgene. Cells were grown in L medium (containing 10 g/L bactotryptone, 5 g/L yeast extract, and 5 g/L NaCl; the pH was adjusted to 7.2 by NaOH) or M9 medium (53) (with omission of CaCl2), supplemented with ampicillin and chloramphenicol at 50 and 20 μg/mL, respectively, when appropriate.
Selection of Multicopy Suppressors.
A total of 4,123 clones from the ASKA library were divided into 112 groups such that each group contained up to 48 clones. Mixtures of plasmids were prepared from each group and used to transform SN537 (ΔbepA ΔbamE). Transformants were selected at 30 °C on L agar containing 20 μg/mL chloramphenicol and 0 or 5 μg/mL erythromycin in the presence or absence of 100 μM IPTG. Each group yielded 0–14 erythromycin/chloramphenicol-resistant colonies whereas 100–300 transformants were obtained on plates containing chloramphenicol but not erythromycin. Plasmids were purified from erythromycin/chloramphenicol-resistant colonies and reintroduced into SN537 to check whether erythromycin resistance was linked to each plasmid. The verified plasmid clones were sequenced to identify the cloned genes.
Pulse–Chase and Immunoprecipitation Experiments.
AD16, GC187, and their derivatives were grown to an early log phase in M9 medium supplemented with 19 amino acids other than methionine, 2 μg/mL thiamine, and 0.2% glucose at 30 or 37 °C. Cells carrying a pTH18cr derivative were grown similarly, but in the presence of 0.2% maltose and 1 mM IPTG instead of glucose. Cells were labeled with 370 kBq/mL [35S]-methionine (American Radiolabeled Chemicals) for 1 min at 30 or 37 °C, and chase was initiated by the addition of 0.04% (final conc.) nonradioactive methionine. Aliquots were withdrawn after 1, 2, 5, 10, 20, 40, 60, and 80 min, and proteins were precipitated with trichloroacetic acid. Protein precipitates were dissolved in 50 mM Tris⋅HCl (pH 8.1) containing 1% SDS and 1 mM EDTA, boiled for 5 min, and diluted 20-fold with 50 mM Tris⋅HCl (pH 8.1) containing 150 mM NaCl, 2% (wt/vol) Triton X-100, and 0.1 mM EDTA. After removal of insoluble materials by centrifugation at 20,000 × g for 5 min, the supernatant was subjected to immunoprecipitation with anti-LptD antiserum and Dynabeads Protein A (Invitrogen). Immunoprecipitated proteins were eluted from beads by boiling for 5 min in SDS/PAGE sample buffer with or without 1% 2-mercaptoethanol and were analyzed by SDS/PAGE and phosphorimaging with BAS-1500 (Fujifilm). Band intensities were quantified with MultiGauge software (Fujifilm).
BS3-Mediated Cross-Linking.
MC4100 derivatives transformed with plasmids that encode BepA derivatives were grown to a late log phase at 30 or 37 °C in L medium. Cells were harvested, washed once with buffer A [20 mM sodium-phosphate buffer (pH 7.2) containing 300 mM sucrose], and resuspended in the same buffer. After addition of 40 μg/mL lysozyme, cells were converted to spheroplasts by incubation at 4 °C in buffer A containing 1 mM EDTA in the presence or absence of 1 mM BS3 (Thermo Fisher Scientific) for 30 min. Then, samples were separated into periplasmic and spheroplast fractions by centrifugation at 20,000 × g for 2 min. Spheroplasts were suspended in 50 mM Tris⋅HCl (pH 7.5) and disrupted by sonication. After removal of unbroken cells by centrifugation at 10,000 × g for 2 min, membranes were collected by ultracentrifugation at 100,000 × g for 30 min. Proteins in the periplasmic and membrane fractions were precipitated by the addition of 10% (wt/vol, final conc.) trichloroacetic acid and were subjected to SDS/PAGE and immunoblotting.
pBPA-Mediated Photo–Cross-Linking.
SN56 transformed with pUC-BepA(Q428Amber) and pEVOL-pBpF was grown at 30 °C to a late log phase in L medium supplemented with 0.2% arabinose and 1 mM IPTG in the presence or absence of 1 mM pBPA (Bachem). Cells were chilled on ice for 10 min and UV-irradiated at 365 nm on a Petri dish for 10 min by using a B-100AP UV lamp (UV Products) at a distance of 4 cm. Cells were then harvested, suspended in 20 mM Tris⋅HCl (pH 7.5) and disrupted by sonication. After removal of unbroken cells by centrifugation at 10,000 × g for 2 min, membranes were collected by ultracentrifugation at 100,000 × g for 30 min. Proteins in the membrane fractions were precipitated by trichloroacetic acid and subjected to SDS/PAGE followed by immunoblotting.
Supplementary Material
Acknowledgments
We thank Carol Gross and Monica Guo for critical reading and editing of the manuscript and helpful comments, Hiroyuki Mori for stimulating discussion, and Nassos Typas for helpful advice. We also thank Kunihito Yoshikaie, Michiyo Sano, Yasuko Abe, and Tokuya Hattori for technical support. We are grateful to Shin-ichi Matsuyama and Tom Silhavy for bacterial strains and antisera, Hajime Tokuda for antiserum and laboratory resources, and National BioResource Project E. coli for bacterial strains and plasmids. This work was supported by Japan Society for the Promotion of Science KAKENHI Grants 24370054 (to Y.A.) and 24570152 (to S.-i.N.) and research grants from the Institute for Fermentation, Osaka (to Y.A.) and from the Takeda Science Foundation (to S.-i.N.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312012110/-/DCSupplemental.
References
- 1.Pugsley AP. The complete general secretory pathway in gram-negative bacteria. Microbiol Rev. 1993;57(1):50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ricci DP, Silhavy TJ. The Bam machine: A molecular cooper. Biochim Biophys Acta. 2012;1818(4):1067–1084. doi: 10.1016/j.bbamem.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science. 2003;299(5604):262–265. doi: 10.1126/science.1078973. [DOI] [PubMed] [Google Scholar]
- 4.Ruiz N, Falcone B, Kahne D, Silhavy TJ. Chemical conditionality: A genetic strategy to probe organelle assembly. Cell. 2005;121(2):307–317. doi: 10.1016/j.cell.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 5.Wu T, et al. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell. 2005;121(2):235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Sklar JG, et al. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc Natl Acad Sci USA. 2007;104(15):6400–6405. doi: 10.1073/pnas.0701579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malinverni JC, et al. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol. 2006;61(1):151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- 8.Kim S, et al. Structure and function of an essential component of the outer membrane protein assembly machine. Science. 2007;317(5840):961–964. doi: 10.1126/science.1143993. [DOI] [PubMed] [Google Scholar]
- 9.Bos MP, Tefsen B, Geurtsen J, Tommassen J. Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc Natl Acad Sci USA. 2004;101(25):9417–9422. doi: 10.1073/pnas.0402340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu T, et al. (2006) Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc Natl Acad Sci USA 103(31):11754–11759. [DOI] [PMC free article] [PubMed]
- 11.Sperandeo P, et al. Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J Bacteriol. 2007;189(1):244–253. doi: 10.1128/JB.01126-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sperandeo P, et al. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J Bacteriol. 2008;190(13):4460–4469. doi: 10.1128/JB.00270-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruiz N, Gronenberg LS, Kahne D, Silhavy TJ. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc Natl Acad Sci USA. 2008;105(14):5537–5542. doi: 10.1073/pnas.0801196105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narita S, Tokuda H. Biochemical characterization of an ABC transporter LptBFGC complex required for the outer membrane sorting of lipopolysaccharides. FEBS Lett. 2009;583(13):2160–2164. doi: 10.1016/j.febslet.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 15.Chimalakonda G, et al. Lipoprotein LptE is required for the assembly of LptD by the beta-barrel assembly machine in the outer membrane of Escherichia coli. Proc Natl Acad Sci USA. 2011;108(6):2492–2497. doi: 10.1073/pnas.1019089108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freinkman E, Chng SS, Kahne D. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc Natl Acad Sci USA. 2011;108(6):2486–2491. doi: 10.1073/pnas.1015617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chng SS, et al. Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science. 2012;337(6102):1665–1668. doi: 10.1126/science.1227215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. Snapshots of DsbA in action: Detection of proteins in the process of oxidative folding. Science. 2004;303(5657):534–537. doi: 10.1126/science.1091724. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz N, Chng SS, Hiniker A, Kahne D, Silhavy TJ. Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc Natl Acad Sci USA. 2010;107(27):12245–12250. doi: 10.1073/pnas.1007319107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denoncin K, Vertommen D, Paek E, Collet JF. The protein-disulfide isomerase DsbC cooperates with SurA and DsbA in the assembly of the essential β-barrel protein LptD. J Biol Chem. 2010;285(38):29425–29433. doi: 10.1074/jbc.M110.119321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rowley G, Spector M, Kormanec J, Roberts M. Pushing the envelope: Extracytoplasmic stress responses in bacterial pathogens. Nat Rev Microbiol. 2006;4(5):383–394. doi: 10.1038/nrmicro1394. [DOI] [PubMed] [Google Scholar]
- 22.Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell. 2003;113(1):61–71. doi: 10.1016/s0092-8674(03)00203-4. [DOI] [PubMed] [Google Scholar]
- 23.Lima S, Guo MS, Chaba R, Gross CA, Sauer RT. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science. 2013;340(6134):837–841. doi: 10.1126/science.1235358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bury-Moné S, et al. Global analysis of extracytoplasmic stress signaling in Escherichia coli. PLoS Genet. 2009;5(9):e1000651. doi: 10.1371/journal.pgen.1000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spiess C, Beil A, Ehrmann M. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell. 1999;97(3):339–347. doi: 10.1016/s0092-8674(00)80743-6. [DOI] [PubMed] [Google Scholar]
- 26.Misra R, CastilloKeller M, Deng M. Overexpression of protease-deficient DegP(S210A) rescues the lethal phenotype of Escherichia coli OmpF assembly mutants in a degP background. J Bacteriol. 2000;182(17):4882–4888. doi: 10.1128/jb.182.17.4882-4888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krojer T, et al. Structural basis for the regulated protease and chaperone function of DegP. Nature. 2008;453(7197):885–890. doi: 10.1038/nature07004. [DOI] [PubMed] [Google Scholar]
- 28.Girgis HS, Hottes AK, Tavazoie S. Genetic architecture of intrinsic antibiotic susceptibility. PLoS ONE. 2009;4(5):e5629. doi: 10.1371/journal.pone.0005629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu A, et al. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob Agents Chemother. 2010;54(4):1393–1403. doi: 10.1128/AAC.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nichols RJ, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144(1):143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh E, et al. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell. 2011;147(6):1295–1308. doi: 10.1016/j.cell.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allan RK, Ratajczak T. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones. 2011;16(4):353–367. doi: 10.1007/s12192-010-0248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehses S, et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187(7):1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujimura-Kamada K, Nouvet FJ, Michaelis S. A novel membrane-associated metalloprotease, Ste24p, is required for the first step of NH2-terminal processing of the yeast a-factor precursor. J Cell Biol. 1997;136(2):271–285. doi: 10.1083/jcb.136.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakoh M, Ito K, Akiyama Y. Proteolytic activity of HtpX, a membrane-bound and stress-controlled protease from Escherichia coli. J Biol Chem. 2005;280(39):33305–33310. doi: 10.1074/jbc.M506180200. [DOI] [PubMed] [Google Scholar]
- 36.Akiyama Y. Quality control of cytoplasmic membrane proteins in Escherichia coli. J Biochem. 2009;146(4):449–454. doi: 10.1093/jb/mvp071. [DOI] [PubMed] [Google Scholar]
- 37.Lütticke C, et al. E. coli LoiP (YggG), a metalloprotease hydrolyzing Phe-Phe bonds. Mol Biosyst. 2012;8(6):1775–1782. doi: 10.1039/c2mb05506f. [DOI] [PubMed] [Google Scholar]
- 38.Kitagawa M, et al. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): Unique resources for biological research. DNA Res. 2005;12(5):291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 39.Chng SS, Ruiz N, Chimalakonda G, Silhavy TJ, Kahne D. Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc Natl Acad Sci USA. 2010;107(12):5363–5368. doi: 10.1073/pnas.0912872107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14(4):378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 41.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2010;395(2):361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 42.Hagan CL, Kim S, Kahne D. Reconstitution of outer membrane protein assembly from purified components. Science. 2010;328(5980):890–892. doi: 10.1126/science.1188919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sklar JG, Wu T, Kahne D, Silhavy TJ. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 2007;21(19):2473–2484. doi: 10.1101/gad.1581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tellez R, Jr, Misra R. Substitutions in the BamA β-barrel domain overcome the conditional lethal phenotype of a ΔbamB ΔbamE strain of Escherichia coli. J Bacteriol. 2012;194(2):317–324. doi: 10.1128/JB.06192-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rigel NW, Schwalm J, Ricci DP, Silhavy TJ. BamE modulates the Escherichia coli beta-barrel assembly machine component BamA. J Bacteriol. 2012;194(5):1002–1008. doi: 10.1128/JB.06426-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rigel NW, Ricci DP, Silhavy TJ (2013) Conformation-specific labeling of BamA and suppressor analysis suggest a cyclic mechanism for β-barrel assembly in Escherichia coli. Proc Natl Acad Sci USA 110(13): 5151–5156. [DOI] [PMC free article] [PubMed]
- 47. Kadokura H, Beckwith J (2009) Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138(6):1164–1173. [DOI] [PMC free article] [PubMed]
- 48.Ruiz N, Kahne D, Silhavy TJ. Transport of lipopolysaccharide across the cell envelope: The long road of discovery. Nat Rev Microbiol. 2009;7(9):677–683. doi: 10.1038/nrmicro2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weski J, Ehrmann M. Genetic analysis of 15 protein folding factors and proteases of the Escherichia coli cell envelope. J Bacteriol. 2012;194(12):3225–3233. doi: 10.1128/JB.00221-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kihara A, Akiyama Y, Ito K. FtsH is required for proteolytic elimination of uncomplexed forms of SecY, an essential protein translocase subunit. Proc Natl Acad Sci USA. 1995;92(10):4532–4536. doi: 10.1073/pnas.92.10.4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casadaban MJ. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;104(3):541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 52. Baba T, et al. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol 2:2006.0008. [DOI] [PMC free article] [PubMed]
- 53.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 1972. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.