

Significance
The upper respiratory tract is colonized by numerous bacteria that are usually confined to this site in healthy individuals. However, in coinfection with a preceding viral infection, these bacteria can colonize the lung, leading to lethal secondary bacterial pneumonia. In this report, a frequent pathogen in coinfections with influenza virus, Haemophilus influenzae, was examined at the whole-genome level to comparatively analyze genes conferring bacterial fitness in mice previously infected with influenza A virus vs. in non-virally infected mice. We identified altered requirements for many bacterial virulence factors and physiological adaptations, providing insight into conditions in the lung that exert selection on coinfecting pathogens. Bacterial factors required for colonization during coinfection represent potential targets for antimicrobials to combat secondary bacterial pneumonia.
Keywords: transposon-insertion sequencing, functional genomics
Abstract
Bacterial coinfection represents a major cause of morbidity and mortality in epidemics of influenza A virus (IAV). The bacterium Haemophilus influenzae typically colonizes the human upper respiratory tract without causing disease, and yet in individuals infected with IAV, it can cause debilitating or lethal secondary pneumonia. Studies in murine models have detected immune components involved in susceptibility and pathology, and yet few studies have examined bacterial factors contributing to coinfection. We conducted genome-wide profiling of the H. influenzae genes that promote its fitness in a murine model of coinfection with IAV. Application of direct, high-throughput sequencing of transposon insertion sites revealed fitness phenotypes of a bank of H. influenzae mutants in viral coinfection in comparison with bacterial infection alone. One set of virulence genes was required in nonvirally infected mice but not in coinfection, consistent with a defect in anti-bacterial defenses during coinfection. Nevertheless, a core set of genes required in both in vivo conditions indicated that many bacterial countermeasures against host defenses remain critical for coinfection. The results also revealed a subset of genes required in coinfection but not in bacterial infection alone, including the iron-sulfur cluster regulator gene, iscR, which was required for oxidative stress resistance. Overexpression of the antioxidant protein Dps in the iscR mutant restored oxidative stress resistance and ability to colonize in coinfection. The results identify bacterial stress and metabolic adaptations required in an IAV coinfection model, revealing potential targets for treatment or prevention of secondary bacterial pneumonia after viral infection.
The bacterium Haemophilus influenzae is a Gram-negative inhabitant of the human upper respiratory tract and a common agent in sinusitis, otitis media, lung infections in cystic fibrosis, and exacerbations of chronic obstructive pulmonary disease (COPD). In the context of prior infection by influenza A virus (IAV), H. influenzae is associated with secondary bacterial pneumonia (1). Annually, influenza and related complications cause ∼36,000 deaths, over 200,000 hospitalizations in the United States, and ∼5 million cases of severe illness worldwide (2, 3). Uncomplicated IAV infection can progress to pneumonia; however, secondary bacterial infection combined with viral infection is commonly the major cause of excess morbidity and mortality during epidemics and pandemics. For example, the 1918 influenza pandemic killed an estimated 50 million people worldwide, and the majority of deaths have been attributed to bacterial secondary infections in which Streptococcus pneumoniae, H. influenzae, and Staphylococcus spp. represent the most common isolates (1). β-Lactam antibiotics are commonly used for treatment, and yet ∼30% of H. influenzae isolates are β-lactamase–positive (4–6). Because of increasing levels of bacterial antibiotic resistance, and the continued threat of global pandemics with potential emergence of new IAV subtypes, combined IAV and bacterial infection remains a significant public health concern.
In 1945, Francis and Vicente de Torregrosa demonstrated lethality of H. influenzae when introduced into the lungs of mice after infection with IAV (7). More recently, pathogenic mechanisms associated with the mouse lung model of lethal IAV coinfection with H. influenzae type b (Hib) were investigated, implicating innate immunity in disease progression (8). Coinfection did not influence viral titers and yet led to dramatically increased multiplication and persistence of bacteria. Viral enhancement of host susceptibility to bacterial infection has been examined in coinfection models with diverse bacteria, implicating modification of mucosal surfaces and dysfunctional immune responses that prevent bacterial containment including altered phagocytic capacity, defective TLR responses, and enhanced pro- and anti-inflammatory cytokine production, and decreased tolerance to tissue damage (9–14). In contrast, bacterial factors involved in coinfection have received less attention. There have been no systematic studies to identify such factors, and genes of H. influenzae involved in IAV coinfection have not been identified.
We investigated the hypothesis that H. influenzae possesses genes that promote its ability to survive host defenses and exploit conditions in the lung generated by coinfection with IAV. Using a genome-scale analytical approach, we simultaneously monitored fitness of thousands of transposon mutants in the murine lung model in the presence and absence of prior IAV infection. The results reveal a core set of bacterial genes required in both models, as well as genes required uniquely in one environment but not the other. Coinfection altered bacterial requirements for known virulence genes conferring not only immune evasion properties but also those encoding regulatory factors and physiological pathways. Therefore, genome-wide analysis of the fitness of bacterial mutants serves as a probe for conditions created during bacterial/viral coinfection of murine lung and identifies bacterial adaptations that specifically promote their multiplication in this pathogenic context.
Results
IAV Renders Mice Highly Susceptible to Coinfection with Nonencapsulated H. influenzae.
IAV predisposes mice to H. influenzae coinfections in the lung, exhibiting a synergistic effect on multiplication, persistence, and virulence of Hib, a highly invasive encapsulated serotype (8). This effect is observed only when virus precedes Hib inoculation within a critical interval spanning the first 7 d between infections (8). Clinically prevalent nonencapsulated strains, termed nontypeable H. influenzae (NTHi), are primarily noninvasive and less virulent in animal models. To determine whether IAV coinfection would significantly enhance colonization and virulence of NTHi, we inoculated mice with IAV [H1N1 subtype influenza strain A/Puerto Rico/8/34 (PR8)], NTHi (strain NT127) (15), or IAV followed 5 d later with NTHi. Consistent with previous reports, mice that were not infected with virus readily cleared infections with NTHi at a dose of 108 colony-forming units (cfu), and clearance was nearly complete by day 4, with no bacteria recoverable from the mice by day 6 (Fig. S1A). In contrast, a low dose (100 cfu) of NTHi administered 5 d after infection with a nonlethal dose 200 times the tissue-culture ID50 (TCID50) of IAV (SI Methods) progressively multiplied, reaching a mean bacterial burden in the lung of 105 by day 4, at which time the coinfected mice died or exhibited signs of morbidity requiring euthanasia (Fig. S1B). Inoculation with virus alone (Fig. S2A) or with 108 heat-killed NTHi at 5 d post-IAV infection did not lead to morbidity (Fig. S2B), indicating that bacterial survival or multiplication is required for pathogenesis in the NTHi/IAV coinfection model. Therefore, despite its relative avirulence in healthy mice, NTHi gains a markedly enhanced ability to persist, rapidly multiply, and potentiate disease in IAV infected mice.
Genome-Wide Fitness Analysis of H. influenzae Mutants in the Murine IAV Coinfection Model in Comparison with Bacterial Infection Alone.
To identify the factors required by H. influenzae for colonization in this model, we applied a genome-scale approach, termed HITS (high-throughput insertion tracking by deep-sequencing), that monitors the relative role in fitness of each gene in the genome by enumerating each unique transposon mutant present in a large bank of mutants (∼75,000) pre- and postselection in the lung model (15). The relative abundance of mutants was quantified by direct, single-molecule sequencing of unique transposon-chromosome junctions using the Helicos sequencing platform (SI Methods) (16). The period examined was the first 24 h of coinfection, before mice exhibited signs of morbidity. In brief, five mice were inoculated with 200 TCID50 of IAV, followed 5 d later by infection with ∼105 transposon insertion mutants in H. influenzae generated with a minitransposon derived from the mariner Himar1 transposon. Concurrently, five nonvirally infected mice were inoculated with the mutant bank. At 24 h post-H. influenzae infection, lung bacterial load from single and coinfections was recovered and chromosomal DNA isolated for comparison with the inoculum via the HITS method.
The mariner minitransposon inserts specifically into TA dinucleotides in target DNA (17). In the input mutant bank, 64,132 (∼50%) of the 131,955 genomic TA target sites sustained unique mariner insertions, representing 5,271,416 unique sequencing reads with 4,282,783 in protein coding genes and 988,633 in RNA structural genes or intergenic regions. Genes essential in vitro cannot be analyzed and those required for optimal growth in vitro were excluded as virulence gene candidates as mutations in these genes are more likely to be pleiotropic (SI Methods). We excluded 297 putative essential genes sustaining no insertions, 164 genes with potential growth defects in vitro (based on an insertion density of <35% of available sites), duplicated genes, and 29 genes that were too small to analyze. Of the total 1,724 annotated genes, 1,194 remained that were considered nonessential in vitro and were further analyzed for their roles in bacterial fitness in single infection vs. in IAV coinfection.
Fitness of H. influenzae in single infection and in IAV coinfection was expressed as a survival index (s.i.) representing the ratio of insertions detected after infection (output) to those detected in the in vitro grown inoculum (input). As a threshold for detection of a survival difference in vivo vs. in vitro, a cutoff of s.i. ≤ 0.35 was chosen, and to control for variability, a t test was applied with a cutoff of P ≤ 0.005 (SI Methods). Genes with s.i. > 0.5 were designated as not required under a given in vivo condition. These thresholds resulted in 121 and 118 genes detected as required in H. influenzae single and coinfection, respectively, of which a common 85 were required in both in vivo conditions. Of 1,194 genes, 1,040 did not meet our criteria to be considered a candidate virulence gene in either single or coinfection. These categories are listed in Dataset S1 and displayed on a genome map along with total unique reads from the input mutant bank (Fig. S3). Fig. 1A shows the 24 and 18 genes that are considered uniquely required in either single or coinfection, respectively (also listed in Dataset S1). Representative of the HITS data pre- and postinfection, regions containing HI1305 (thiL) and HI0161 (gor) are shown (Fig. 1B) to illustrate classes of mutant phenotypes that were detected. The thiL gene encodes thiamine monophosphate (TMP) kinase, which catalyzes the conversion of TMP to thiamine diphosphate/pyrophosphate (TPP), the biologically active form of thiamine, and was needed in coinfection (s.i. = 0.07). The gor gene was needed in single infection (s.i. = 0.09) and encodes glutathione reductase, whose primary function is to provide antioxidant activity. Flanking thiL and gor are essential genes of riboflavin synthesis and glycerophospholipid metabolism [6,7-dimethyl-8-ribityllumazine synthase (ribH) and phosphatidylserine decarboxylase (psd)] with no insertions detected in their coding regions in the in vitro library. The nusB gene (transcription antiterminator) contains 52% TA insertion site saturation in vitro, and although it appears to be potentially required in vivo, it did not meet our criteria for statistical significance. HI1306 [phosphatidylglycerophosphatase A (pgpA)] and HI0159 represent genes with inferred growth defects in vitro (TA insertion site saturation, <35%). In contrast, HI1307 and HI0162 represent nonessential genes dispensable in both infections (s.i. > 0.5).
Fig. 1.
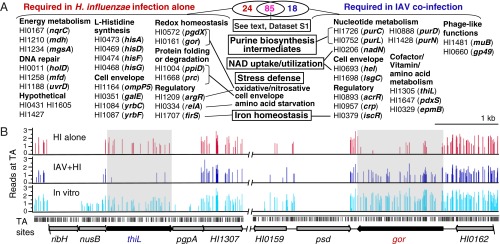
Genome-scale evaluation by HITS of the effect of IAV coinfection on lung colonization by H. influenzae mutants. (A) Coinfection alters survival requirements of H. influenzae in the lung. Candidate virulence genes with no observed effects on fitness in vitro were sorted into categories based on their roles in fitness detected in vivo. Genes required in both in vivo conditions are listed in Dataset S1 and discussed in the text. (B) Representative HITS data preinfection (in vitro) and postinfection with H. influenzae alone (HI alone) or IAV/H. influenzae coinfection (IAV+HI) for loci containing thiL and gor. Colored bars on the x axes designate sites of transposon insertions detected via sequencing and heights indicate relative abundance of insertions detected at each site (y axis: log10-transformed reads). Black bars below the x axis represent genomic TA dinucleotide positions.
A core set of 85 genes required in both models of infection included genes for biosynthesis of aromatic amino acids, asparagine, methionine, serine, and branched-chain amino acids. Also common were many of the genes (e.g., opsX, rfaF, orfH, galU) responsible for biosynthesis of carbohydrate structures of the inner and outer core of the lipooligosaccharide (LOS), a major H. influenzae virulence factor analogous to lipopolysaccharides of other species. We also note genes with functions in the following: cell shape (e.g., mreBCD, rodA), cell wall biogenesis (e.g., dacA, pbp2, mltC), and cell division (e.g., ftsN, zipA); DNA homologous recombination (e.g., recA, recB, recC, ruvB, xerC, xerD), mismatch/base excision repair (dam, xthA), and replication (rnhA); carbohydrate metabolism (nagB, glk, rpiA, talB); protein folding and stabilization (e.g., dsbA, dsbB); purine biosynthetic (e.g., purA, purB, guaA, guaB) and salvage (deoD, apaH) pathways; transport of divalent cations (yfeABCD), copper (copA), and zinc (znuA, znuB, znuC, zevB); regulatory functions (e.g., icc, sspA); and RNA processing (pnp, rnc). Many genes within the common core set were found in our previous HITS results for H. influenzae single infection in a similar murine lung model (15); however, in the current study, more members of each pathway were detected. This greater degree of pathway concordance potentially resulted from either the greater sequencing coverage attained in the current experiment or the use of single molecule sequencing which obviated the PCR amplification used in the previous study.
Coinfection changed the genes required in the lung compared with those required in H. influenzae single infection (Fig. 1A). The 24 genes required in single infection but not in coinfection included several in the pathway for histidine biosynthesis (hisG,D,A,F), the tricarboxylic acid cycle [malate dehydrogenase (mdh)] and electron transport chain [Na(+)-translocating NADH-quinone reductase subunit C (nqrC)] (18), and a sensor of ferrous iron (firS) (19). Stress defense was implicated including oxidative stress [glutathione-dependent peroxidase (pgdx) (20), glutathione reductase (gor)], nitrosative stress [arginine repressor (argR)] (21), amino acid starvation [GTP pyrophosphokinase (relA)] (22), cell envelope stress [peptidyl-prolyl cis-trans isomerase (ppiD), carboxyl-terminal protease (prc)] (23, 24), and stress imposed by accumulation of inhibitory phosphorylated intermediates [methylglyoxal synthase (mgsA)] (25). DNA excision and mismatch repair genes that may be protective against these stresses were implicated [DNA polymerase III subunit psi (holD), transcription-repair coupling factor (mfd), DNA-dependent helicase II (uvrD)]. Also included are genes affecting changes to the bacterial cell surface [outer-membrane adhesin (ompP5)] (26), yrbC and yrbF encoding ABC-transport functions associated with phospholipid trafficking affecting cell surface LOS arrangement (27) and galE (UDP-glucose 4-epimerase), which catalyzes the reversible interconversion of UDP-galactose and UDP-glucose in galactose metabolism and biosynthesis of the outer core of the LOS (28).
The 18 genes needed in coinfection but not in single infection include genes with functions in synthesis of purines (purC,D,L,N), vitamin B1 (thiL) and vitamin B6 [synthesis of pyridoxal 5′-phosphate (pdxS)], NAD uptake/utilization [nucleotide phosphatase lipoprotein E (hel), periplasmic NAD nucleotidase (nadN) (29, 30)], and transport of heme and antimicrobial peptides (sapA) (31, 32). We also note the requirement for the lsgC glycosyltransferase gene, which has a potential role in LOS chain extension (33); crp (encoding cAMP receptor), which regulates the response to carbon shortage; acrR, a repressor for a multidrug efflux pump (34); epmB (l-lysine 2,3-aminomutase), which functions in posttranslational modification of elongation factor P (35); and bacteriophage-derived proteins (DNA transposition protein MuB and Gp49) and iscR, a regulator controlling iron-sulfur (Fe-S) cluster formation. Of note, IscR has been implicated in oxidative-stress resistance (36). Because an alternative oxidative-stress resistance system encoded by pgdX was required only in single infection, the results suggest that H. influenzae uses alternative resistance mechanisms in the two models (Discussion).
Validation of Roles of Selected Bacterial Genes in Coinfected vs. Nonvirally Infected Mice.
That some of the genes identified by genome-wide fitness analysis were required in coinfection or during infection with H. influenzae alone, but not in both models, implicated differences in bacterial virulence strategies in these environments. To verify this observation, selected genes were deleted in strains Rd (Fig. 2 A–C) or NT127 (Fig. 2D) by precise replacement of their coding regions with antibiotic resistance genes and the mutants were tested in both lung infection models via in vivo competition assays with LacZ+ reference strains. The iscR gene was evaluated as a member of the class of genes required only in coinfection. At 24 h after bacterial inoculation, the ratios of the coinoculated bacterial strains recovered from the lungs confirmed that the iscR mutant was attenuated in IAV coinfection (Figs. 2A and 3B) but had no defect during infection with H. influenzae alone (Figs. 2 B and E and 3B). To verify the role of a gene required only in single infection, we evaluated a galE mutant. As predicted by HITS analysis, galE was required for colonization of the lung in the absence of virus (Fig. 2B) but was nonessential in coinfection (Fig. 2 C and F).
Fig. 2.

Effect of IAV coinfection on lung colonization by iscR and galE mutants. Wild-type (WT) and H. influenzae mutants generated in Rd (A–C) or NT127 (D) strain backgrounds in competition with H. influenzae reference strains expressing lacZ in the corresponding wild-type strain background in the lungs of C57BL/6 mice 5 d following inoculation with IAV strain PR8 (+PR8) or without PR8 (-PR8). Bars represent mean ratios of colony-forming units of each strain recovered from the lungs relative to the LacZ+ strain. P values were from the unpaired t test (A, C, and D) and one-way ANOVA with Tukey’s multiple comparison test (B); n.s., not statistically significant. HITS data preinfection (in vitro), postinfection (HI alone), or after IAV/H. influenzae coinfection (IAV+HI) for iscR (E) and galE (F). Bars show sites of transposon insertions and heights indicate relative abundance of insertions detected at each site (y axis: log10-transformed read counts). Black bars below the x axis denote genomic TA dinucleotide positions.
Fig. 3.

Ectopic overexpression of Dps protects against sensitivity of H. influenzae iscR mutant to hydrogen peroxide and rescues survival in IAV coinfection. (A) H. influenzae Rd containing empty vector (parent), Rd overexpressing dps (Rd+dps), iscR mutant containing empty vector (∆iscR), iscR mutant with complementing copy of iscR in trans (iscR+iscR), and iscR mutant with dps overexpressed in trans (∆iscR+dps) were treated with 2 mM H2O2 for 30 min and plated to enumerate survivors on sBHI (SI Methods). Survival ratios are plotted as the percentage of colony-forming units obtained from H2O2 treated/samples receiving buffer alone (mock) from three independent cultures; error bars show SDs and fold differences are above the brackets. (B) The strains in A were coinoculated intranasally with an H. influenzae reference strain expressing lacZ into C57BL6 mice 5 d following infection with IAV strain PR8 (+PR8) or without PR8 (-PR8). Lungs were harvested at 24 h post-bacterial inoculation for determination of bacterial colony-forming units. Bars represent the mean ratio of colony-forming units of each designated strain relative to the LacZ+ reference strain. Log-transformed ratios were statistically significant (*) between the iscR mutant vs. the parent, complemented and dps overexpressing strains (P < 0.001; one-way ANOVA with Tukey’s multiple comparison test) for A and B.
Detection of a regulatory gene, iscR, specifically required in coinfection was of particular interest as it suggested a mechanism by which H. influenzae can sense and respond to conditions present during coinfection. Therefore, we verified that its role in coinfection is not strain-specific. Using a nonpolar iscR mutant of NTHi clinical isolate, NT127, we found that iscR was dispensable for colonization by H. influenzae NT127 infection alone but required for optimal survival in coinfection (Fig. 2D).
H. influenzae iscR Modulates Sensitivity to Oxidative Stress.
Pathological damage during infection of mice with IAV involves increased levels of oxidative stress in the lung (37–42). It is not known whether the oxidative stress experienced by host cells also affects coinfecting bacteria; however, an iscR homolog in at least one bacterial species, Pseudomonas aeruginosa, has been implicated in oxidative stress resistance (36). Thus, identification of a requirement for iscR in coinfection and not in single infection suggested a difference in exposure to oxidative stress or in the bacterium’s mode of combating such stress in the two environments. To evaluate whether the H. influenzae iscR mutant is hypersensitive to oxidative stress, we tested the effect of H2O2 on its viability. Because transcriptional regulation by IscR homologs is reportedly more pronounced under low-oxygen conditions (43), growth of the wild-type strain and the iscR mutant was analyzed in the absence and presence of 0.5 and 0.75 mM H2O2 following pregrowth of the strains under low and high oxygen conditions (Fig. S4). Under both conditions, wild-type and the iscR mutant showed a dose-dependent inhibition of growth by H2O2; however, the effect of the iscR mutation on H2O2 sensitivity was greater when cells were pregrown in low oxygen. In the absence of H2O2, both strains grew similarly under either condition. That H2O2 treatment resulted in a greater decrease in viability of the iscR mutant was verified in H2O2 sensitivity assays in which H. influenzae pregrown in low oxygen was incubated for 30 min in the absence and presence of 2 mM H2O2, followed by plating to enumerate survivors. The iscR mutant (∆iscR) showed a significant decrease in the number of survivors compared with its parent (∼100-fold) after H2O2 treatment, and complementation restored the survival phenotype (Fig. 3A).
IscR-Mediated Regulation of Iron-Sulfur Cluster Biogenesis Genes.
IscR, first discovered in E. coli, is an [2Fe-2S] iron sulfur-cluster–containing transcriptional repressor of the iscRSUA operon, which functions in Fe-S cluster assembly (44). IscR provides feedback in maintaining Fe-S homeostasis by repressing the isc operon when Fe-S cluster formation is not needed. When Fe-S cluster biogenesis is in demand, IscR loses its Fe-S cluster and becomes inactive as a repressor of the isc operon, leading to increased expression of iscRSUA (45). Repression of iscRSUA by IscR is a potential mechanism for its role in oxidative stress resistance, because accumulation of excess Fe-S clusters is detrimental in the presence of oxidants that can degrade the clusters with release of Fe2+, fueling Fenton chemistry to produce reactive oxygen species (ROS) (46).
To evaluate the role of IscR in regulation of Fe-S biogenesis genes in H. influenzae, genes of the isc locus similar to those of E. coli, including iscS, hscB, and hscA, were evaluated by reverse transcriptase–quantitative PCR in cells grown with low vs. high levels of culture aeration (SI Methods and Table S1). In addition, a gene of unknown function unique to H. influenzae and several closely related species, HI0374 and an adjacent upstream putative rRNA/tRNA methyltransferase homolog, HI0380, were also evaluated. Expression of these genes was increased in the iscR mutant (Table S1 and Fig. S5), consistent with a greater effect of IscR on H2O2 sensitivity after growth in low oxygen (Fig. 3A and Fig. S4). Complementation with iscR expressed in single copy from the xyl locus restored expression of each gene to levels similar to those in the parental strain (Fig. S5).
Rescue of Oxidative Stress Resistance in the iscR Mutant by Overexpression of Dps.
If susceptibility of the iscR mutant to oxidative stress results from excess mobilization and exposure of intracellular iron to reactive Fenton chemistry, then sequestration of iron would be expected to alleviate the bactericidal effect of H2O2. We evaluated this hypothesis by ectopic expression of the dps gene, which is not regulated by iscR in H. influenzae (Fig. S5). The ferritin-like Dps protein, first discovered in E. coli as a DNA-binding protein, protects DNA from hydrogen peroxide-mediated oxidative damage (47), and members of this family function in iron storage/detoxification (48). Fig. 3A shows that overexpression of dps in the iscR mutant (∆iscR+dps) rescued survival to a level slightly above that of the parent strain. Moreover, overexpression of dps in the parent strain (Rd+dps) enhanced its survival as well.
Dps Overexpression Rescues iscR Mutant in Coinfection.
Because iscR is likely to regulate numerous genes and physiological pathways, we addressed whether the ability of iscR to mitigate oxidative stress in particular can account for its role in the coinfection model. If IscR is required for control of additional colonization factors unrelated to anti-oxidant defense, we would predict that dps overexpression would fail to rescue the iscR mutant in coinfection, whereas if the major role of IscR in this model is to combat oxidative stress, then the level of restoration should mirror that observed in H2O2 sensitivity assays. Indeed, in vivo competition assays showed that overexpression of dps in the mutant (∆iscR+dps) restored colonization to a level even higher than that of the parent and complemented strains in IAV coinfection (Fig. 3B). Additionally, overexpression of dps in the parent strain (Rd+dps) also enhanced colonization levels over that of the parent. Consistent with a role for iscR in ROS resistance that is specific to IAV coinfection, all strains showed similar colonization levels in the absence of IAV (Fig. 3B).
Discussion
Infection with IAV provokes an extensive inflammatory response, leading to immune pathology, pathogen control, and associated metabolic changes in the lung. These changes promote rapid multiplication of bacteria in the lung, which can then lead to fatal pneumonia. Application of HITS to the coinfection model revealed the genome-wide profile of bacterial adaptations required in this environment. This information can be used to obtain insight into host responses or metabolic changes in the lung that are most important in altering selection pressures the bacterium encounters during pathogenesis.
HITS data indicate differential adaptations to nutrient limitation, immune effectors, and cellular stressors in the two models of infection. For example, genes of histidine biosynthesis were required in single infection but not in coinfection. Histidine may be available to bacteria in coinfection as IAV infection of cultured epithelial cells promotes release of several amino acids, including histidine (49). GalE is required for addition of galactose moieties to the outer core of the LOS, which inhibits deposition of complement and antimicrobial peptides. GalE was needed for single infection but nonessential in coinfection, suggesting that coinfection inactivates immune defenses that are normally antagonized by LOS-galactose structures. Although several genes involved in defense against various stress conditions were dispensable in coinfection, others were needed, including epmB, which participates in translating mRNAs involved in stress resistance and virulence in Salmonella enterica (50), and iscR, encoding a regulator of Fe-S cluster biogenesis.
We further investigated the mechanism by which IscR participates in coinfection. IscR of H. influenzae (Fig. S5 and Table S1) directs a similar regulatory pattern to that mediated by E. coli IscR at the isc locus (44). IscR of E. coli regulates additional genes including the sufABCDSE operon (51), a second Fe-S assembly system, and although H. influenzae does not possess a suf operon, IscR may control expression of other genes that have not been identified in H. influenzae. We show in this report that iscR of H. influenzae is required for resistance to oxidative stress (Fig. 3 and Fig. S4) and colonization in coinfection (Figs. 2 and 3) and that these phenotypes are restored in the iscR mutant by overexpression of Dps (Fig. 3). Because Dps enzymes function to protect against ROS and archetypal Dps family members sequester iron from Fenton chemistry while detoxifying hydroxyl radicals, the results implicate IscR’s function in defense against ROS, likely via maintaining Fe-S homeostasis, as its major contribution to coinfection. Thus, ROS, although damaging to host tissues, exerts selective pressure on bacteria in the coinfection model, requiring adaptation by IscR. A potential clinical implication of this result is that ROS may mediate some of the limited antibacterial defense remaining in the coinfected host. Approaches to antagonize ROS generation have been considered as therapies for IAV infection (40); therefore, it will be important to evaluate the effects of antioxidant-based therapy on severity of bacterial superinfection.
Coinfection modified H. influenzae’s requirements for an additional, interrelated set of physiological adaptations relevant to previously reported changes in the lung induced by IAV. The superoxide-producing catabolic enzyme xanthine oxidase (XO) has been identified as a major source of ROS in IAV infection (37, 40). Increased XO activity in IAV infection has been implicated in depleting adenosine and its derivatives, hypoxanthine and xanthine (37), purine precursors that can be used by H. influenzae (52). Because genes of de novo purine intermediate synthesis leading to inosine (purC, purL, purD, purN) were implicated in coinfection, it appears that purine scavenging is insufficient in this context, consistent with IAV induced XO activity.
In parallel with increased oxidant production, IAV infection leads to induction of host pathways for antioxidant defense including glutathione peroxidase (53) with concurrent depletion of glutathione (GSH) (54), a primary defense of mammalian cells against ROS. Depletion of GSH, which H. influenzae cannot synthesize and acquires from exogenous sources (55), may account for the decreased requirement for the glutathione-dependent peroxidase PgdX in coinfection, because limitation of its substrate would render this enzyme ineffective. In the absence of PgdX activity, which normally detoxifies ROS, H. influenzae may rely on alternative ROS resistance mechanisms, including repression of Fe-S cluster biogenesis by IscR. Potentially linked to GSH depletion, another important antioxidant that may be depleted is TPP, a cofactor for NADPH-generating pathways critical for maintaining cellular redox balance and levels of GSH (56). H. influenzae is not able to synthesize thiamine de novo (52) and, therefore, must acquire thiamine, TMP, or TPP from the host. TMP is converted to TPP in H. influenzae by ThiL (TMP kinase), which was required in coinfection, suggesting that exogenous sources of TPP, the biologically active form of thiamine, are limiting in this context. Although TPP levels during coinfection are not known, our results raise the possibility that limitation of TPP availability to H. influenzae in coinfection might be exploited therapeutically by targeting ThiL with inhibitory compounds. Such inhibitors may be specific to bacteria as TMP kinase activity is absent in mammalian cells, which directly convert thiamine to TPP by thiamine pyrophosphokinase.
In summary, IAV/H. influenzae coinfection alleviates requirements for numerous virulence factors and physiological adaptations required by H. influenzae in the normal murine lung. Nevertheless, many bacterial genes are required in coinfection, suggesting that host defenses remain that could potentially be amplified therapeutically. Moreover, NTHi factors required in coinfection are likely to represent targets for vaccines or antimicrobials effective in this pathogenic context and potentially in other predisposing conditions involving lung inflammation such as COPD. Conversely, approaches for controlling viral pathogenesis in the context of bacterial/viral coinfection should take into account their effects on antibacterial defenses, and understanding bacterial colonization factors will aid in defining such strategies.
Methods
Detailed description of the materials and experimental methods is provided in SI Methods, including strains, growth conditions, mouse lung model, transposon junction sequence analysis, expression analysis, and strain construction. Also included are methods used for manual revision of genome annotations. Experiments were conducted with approval from the Institutional Animal Care and Use Committees at the University of Pennsylvania School of Medicine and at the University of Massachusetts Medical School.
Supplementary Material
Acknowledgments
We thank Dr. David Lapointe for bioinformatic support, Dr. Jeffrey Gawronski for providing the transposon mutant library, and Dr. John Thompson for assistance with Helicos sequencing. This work was supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases (NIH/NIAID) Multiple Principal Investigator Grant R01AI095740 (to B.J.A. and H.S.), NIH Grant R56AI049437 (to B.J.A.), and NIH/NIAID Grant U19AI083022 (to H.S.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1311217110/-/DCSupplemental.
References
- 1.Wang XY, et al. Influenza and bacterial pathogen coinfections in the 20th century. Interdiscip Perspect Infect Dis. 2011;2011:146376. doi: 10.1155/2011/146376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson WW, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA. 2003;289(2):179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 3.Thompson WW, et al. Influenza-associated hospitalizations in the United States. JAMA. 2004;292(11):1333–1340. doi: 10.1001/jama.292.11.1333. [DOI] [PubMed] [Google Scholar]
- 4.Block SL, et al. Increasing bacterial resistance in pediatric acute conjunctivitis (1997-1998) Antimicrob Agents Chemother. 2000;44(6):1650–1654. doi: 10.1128/aac.44.6.1650-1654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein JO. Role of nontypeable Haemophilus influenzae in pediatric respiratory tract infections. Pediatr Infect Dis J. 1997;16(2) Suppl:S5–S8. doi: 10.1097/00006454-199702001-00002. [DOI] [PubMed] [Google Scholar]
- 6.Klein JO. Management of acute otitis media in an era of increasing antibiotic resistance. Int J Pediatr Otorhinolaryngol. 1999;49(Suppl 1):S15–S17. doi: 10.1016/s0165-5876(99)00125-1. [DOI] [PubMed] [Google Scholar]
- 7.Francis T, Jr, Vicente Torregrosa MV. Combined infection of mice with H. influenzae and influenza virus by the intranasal route. J Infect Dis. 1945;76(1):70–77. [Google Scholar]
- 8.Lee LN, et al. A mouse model of lethal synergism between influenza virus and Haemophilus influenzae. Am J Pathol. 2010;176(2):800–811. doi: 10.2353/ajpath.2010.090596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith MW, Schmidt JE, Rehg JE, Orihuela CJ, McCullers JA. Induction of pro- and anti-inflammatory molecules in a mouse model of pneumococcal pneumonia after influenza. Comp Med. 2007;57(1):82–89. [PMC free article] [PubMed] [Google Scholar]
- 10.Didierlaurent A, et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205(2):323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. 2008;14(5):558–564. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- 12.McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. 2006;74(12):6707–6721. doi: 10.1128/IAI.00789-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19(3):571–582. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamieson AM, et al. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340(6137):1230–1234. doi: 10.1126/science.1233632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gawronski JD, Wong SM, Giannoukos G, Ward DV, Akerley BJ. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci USA. 2009;106(38):16422–16427. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson JF, et al. Single-step capture and sequencing of natural DNA for detection of BRCA1 mutations. Genome Res. 2012;22(2):340–345. doi: 10.1101/gr.122192.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubin EJ, et al. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc Natl Acad Sci USA. 1999;96(4):1645–1650. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashi M, Nakayama Y, Unemoto T. Existence of Na+-translocating NADH-quinone reductase in Haemophilus influenzae. FEBS Lett. 1996;381(3):174–176. doi: 10.1016/0014-5793(96)00114-7. [DOI] [PubMed] [Google Scholar]
- 19.Steele KH, O’Connor LH, Burpo N, Kohler K, Johnston JW. Characterization of a ferrous iron-responsive two-component system in nontypeable Haemophilus influenzae. J Bacteriol. 2012;194(22):6162–6173. doi: 10.1128/JB.01465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pauwels F, Vergauwen B, Van Beeumen JJ. Physiological characterization of Haemophilus influenzae Rd deficient in its glutathione-dependent peroxidase PGdx. J Biol Chem. 2004;279(13):12163–12170. doi: 10.1074/jbc.M312037200. [DOI] [PubMed] [Google Scholar]
- 21.Hyduke DR, Jarboe LR, Tran LM, Chou KJ, Liao JC. Integrated network analysis identifies nitric oxide response networks and dihydroxyacid dehydratase as a crucial target in Escherichia coli. Proc Natl Acad Sci USA. 2007;104(20):8484–8489. doi: 10.1073/pnas.0610888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haseltine WA, Block R. Synthesis of guanosine tetra- and pentaphosphate requires the presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes. Proc Natl Acad Sci USA. 1973;70(5):1564–1568. doi: 10.1073/pnas.70.5.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowley G, Spector M, Kormanec J, Roberts M. Pushing the envelope: Extracytoplasmic stress responses in bacterial pathogens. Nat Rev Microbiol. 2006;4(5):383–394. doi: 10.1038/nrmicro1394. [DOI] [PubMed] [Google Scholar]
- 24.Bordes P, et al. Insights into the extracytoplasmic stress response of Xanthomonas campestris pv. campestris: Role and regulation of sigmaE-dependent activity. J Bacteriol. 2011;193(1):246–264. doi: 10.1128/JB.00884-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferguson GP, Tötemeyer S, MacLean MJ, Booth IR. Methylglyoxal production in bacteria: Suicide or survival? Arch Microbiol. 1998;170(4):209–218. doi: 10.1007/s002030050635. [DOI] [PubMed] [Google Scholar]
- 26.Bookwalter JE, et al. A carcinoembryonic antigen-related cell adhesion molecule 1 homologue plays a pivotal role in nontypeable Haemophilus influenzae colonization of the chinchilla nasopharynx via the outer membrane protein P5-homologous adhesin. Infect Immun. 2008;76(1):48–55. doi: 10.1128/IAI.00980-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura S, et al. Molecular basis of increased serum resistance among pulmonary isolates of non-typeable Haemophilus influenzae. PLoS Pathog. 2011;7(1):e1001247. doi: 10.1371/journal.ppat.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maskell DJ, Szabo MJ, Deadman ME, Moxon ER. The gal locus from Haemophilus influenzae: Cloning, sequencing and the use of gal mutants to study lipopolysaccharide. Mol Microbiol. 1992;6(20):3051–3063. doi: 10.1111/j.1365-2958.1992.tb01763.x. [DOI] [PubMed] [Google Scholar]
- 29.Reidl J, et al. NADP and NAD utilization in Haemophilus influenzae. Mol Microbiol. 2000;35(6):1573–1581. doi: 10.1046/j.1365-2958.2000.01829.x. [DOI] [PubMed] [Google Scholar]
- 30.Kemmer G, et al. NadN and e (P4) are essential for utilization of NAD and nicotinamide mononucleotide but not nicotinamide riboside in Haemophilus influenzae. J Bacteriol. 2001;183(13):3974–3981. doi: 10.1128/JB.183.13.3974-3981.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mason KM, Raffel FK, Ray WC, Bakaletz LO. Heme utilization by nontypeable Haemophilus influenzae is essential and dependent on Sap transporter function. J Bacteriol. 2011;193(10):2527–2535. doi: 10.1128/JB.01313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shelton CL, Raffel FK, Beatty WL, Johnson SM, Mason KM. Sap transporter mediated import and subsequent degradation of antimicrobial peptides in Haemophilus. PLoS Pathog. 2011;7(11):e1002360. doi: 10.1371/journal.ppat.1002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johansen EB, Szoka FC, Zaleski A, Apicella MA, Gibson BW. Utilizing the O-antigen lipopolysaccharide biosynthesis pathway in Escherichia coli to interrogate the substrate specificities of exogenous glycosyltransferase genes in a combinatorial approach. Glycobiology. 2010;20(6):763–774. doi: 10.1093/glycob/cwq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaczmarek FS, et al. Genetic and molecular characterization of beta-lactamase-negative ampicillin-resistant Haemophilus influenzae with unusually high resistance to ampicillin. Antimicrob Agents Chemother. 2004;48(5):1630–1639. doi: 10.1128/AAC.48.5.1630-1639.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JH, et al. Post-translational modification by β-lysylation is required for activity of Escherichia coli elongation factor P (EF-P) J Biol Chem. 2012;287(4):2579–2590. doi: 10.1074/jbc.M111.309633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim SH, Lee BY, Lau GW, Cho YH. IscR modulates catalase A (KatA) activity, peroxide resistance and full virulence of Pseudomonas aeruginosa PA14. J Microbiol Biotechnol. 2009;19(12):1520–1526. doi: 10.4014/jmb.0906.06028. [DOI] [PubMed] [Google Scholar]
- 37.Akaike T, et al. Dependence on O2- generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J Clin Invest. 1990;85(3):739–745. doi: 10.1172/JCI114499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suliman HB, Ryan LK, Bishop L, Folz RJ. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am J Physiol Lung Cell Mol Physiol. 2001;280(1):L69–L78. doi: 10.1152/ajplung.2001.280.1.L69. [DOI] [PubMed] [Google Scholar]
- 39.Buffinton GD, Christen S, Peterhans E, Stocker R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic Res Commun. 1992;16(2):99–110. doi: 10.3109/10715769209049163. [DOI] [PubMed] [Google Scholar]
- 40.Oda T, et al. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science. 1989;244(4907):974–976. doi: 10.1126/science.2543070. [DOI] [PubMed] [Google Scholar]
- 41.Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur J Immunol. 2006;36(6):1364–1373. doi: 10.1002/eji.200635977. [DOI] [PubMed] [Google Scholar]
- 42.Peterhans E, Grob M, Bürge T, Zanoni R. Virus-induced formation of reactive oxygen intermediates in phagocytic cells. Free Radic Res Commun. 1987;3(1-5):39–46. doi: 10.3109/10715768709069768. [DOI] [PubMed] [Google Scholar]
- 43.Giel JL, Rodionov D, Liu M, Blattner FR, Kiley PJ. IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol Microbiol. 2006;60(4):1058–1075. doi: 10.1111/j.1365-2958.2006.05160.x. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz CJ, et al. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc Natl Acad Sci USA. 2001;98(26):14895–14900. doi: 10.1073/pnas.251550898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giel JL, et al. Regulation of iron-sulphur cluster homeostasis through transcriptional control of the Isc pathway by [2Fe-2S]-IscR in Escherichia coli. Mol Microbiol. 2013;87(3):478–492. doi: 10.1111/mmi.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imlay JA. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol. 2006;59(4):1073–1082. doi: 10.1111/j.1365-2958.2006.05028.x. [DOI] [PubMed] [Google Scholar]
- 47.Almirón M, Link AJ, Furlong D, Kolter R. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 1992;6(12B):2646–2654. doi: 10.1101/gad.6.12b.2646. [DOI] [PubMed] [Google Scholar]
- 48.Chiancone E, Ceci P, Ilari A, Ribacchi F, Stefanini S. Iron and proteins for iron storage and detoxification. Biometals. 2004;17(3):197–202. doi: 10.1023/b:biom.0000027692.24395.76. [DOI] [PubMed] [Google Scholar]
- 49.Ritter JB, Wahl AS, Freund S, Genzel Y, Reichl U. Metabolic effects of influenza virus infection in cultured animal cells: Intra- and extracellular metabolite profiling. BMC Syst Biol. 2010;4:61. doi: 10.1186/1752-0509-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Navarre WW, et al. PoxA, yjeK, and elongation factor P coordinately modulate virulence and drug resistance in Salmonella enterica. Mol Cell. 2010;39(2):209–221. doi: 10.1016/j.molcel.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeo WS, Lee JH, Lee KC, Roe JH. IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol Microbiol. 2006;61(1):206–218. doi: 10.1111/j.1365-2958.2006.05220.x. [DOI] [PubMed] [Google Scholar]
- 52.Holt LB. The growth-factor requirements of Haemopiius influenzae. J Gen Microbiol. 1962;27:317–322. doi: 10.1099/00221287-27-2-317. [DOI] [PubMed] [Google Scholar]
- 53.Choi AM, Knobil K, Otterbein SL, Eastman DA, Jacoby DB. Oxidant stress responses in influenza virus pneumonia: Gene expression and transcription factor activation. Am J Physiol. 1996;271(3 Pt 1):L383–L391. doi: 10.1152/ajplung.1996.271.3.L383. [DOI] [PubMed] [Google Scholar]
- 54.Hennet T, Peterhans E, Stocker R. Alterations in antioxidant defences in lung and liver of mice infected with influenza A virus. J Gen Virol. 1992;73(Pt 1):39–46. doi: 10.1099/0022-1317-73-1-39. [DOI] [PubMed] [Google Scholar]
- 55.Vergauwen B, Pauwels F, Vaneechoutte M, Van Beeumen JJ. Exogenous glutathione completes the defense against oxidative stress in Haemophilus influenzae. J Bacteriol. 2003;185(5):1572–1581. doi: 10.1128/JB.185.5.1572-1581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Depeint F, Bruce WR, Shangari N, Mehta R, O’Brien PJ. Mitochondrial function and toxicity: Role of the B vitamin family on mitochondrial energy metabolism. Chem Biol Interact. 2006;163(1-2):94–112. doi: 10.1016/j.cbi.2006.04.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.