

Abstract
There is growing evidence that impaired sensory-processing significantly contributes to the cognitive deficits found in schizophrenia. For example, the mismatch negativity (MMN) and P3a event-related potentials (ERPs), neurophysiological indices of sensory and cognitive function, are reduced in schizophrenia patients and may be used as biomarkers of the disease. In agreement with glutamatergic theories of schizophrenia, NMDA antagonists, such as ketamine, elicit many symptoms of schizophrenia when administered to normal subjects, including reductions in the MMN and the P3a. We sought to develop a nonhuman primate (NHP) model of schizophrenia based on NMDA-receptor blockade using subanesthetic administration of ketamine. This provided neurophysiological measures of sensory and cognitive function that were directly comparable to those recorded from humans. We first developed methods that allowed recording of ERPs from humans and rhesus macaques and found homologous MMN and P3a ERPs during an auditory oddball paradigm. We then investigated the effect of ketamine on these ERPs in macaques. As found in humans with schizophrenia, as well as in normal subjects given ketamine, we observed a significant decrease in amplitude of both ERPs. Our findings suggest the potential of a pharmacologically induced model of schizophrenia in NHPs that can pave the way for EEG-guided investigations into cellular mechanisms and therapies. Furthermore, given the established link between these ERPs, the glutamatergic system, and deficits in other neuropsychiatric disorders, our model can be used to investigate a wide range of pathologies.
Keywords: brain, psychiatry, neurology, monkey, medicine
Schizophrenia is a multifaceted disorder that may originate from neuronal pathology in multiple brain systems (1). Current theories suggest that some of the sensory and cognitive symptoms of schizophrenia may, at least partially, result from dysfunction of the glutamate neurotransmitter system (2). In support of this theory, it has been found that acute subanesthetic doses of the N-methyl-d-aspartate receptor (NMDAR) antagonist ketamine induces sensory and cognitive deficits akin to those experienced by schizophrenia patients, as well as decreases of the mismatch negativity (MMN) and P3 event-related potential (ERP) amplitudes (3).
The MMN is thought to reflect preattentive detection of a deviant stimulus (4), whereas the P3 is thought to reflect the redirection of attention to that deviant stimulus (5). In an oddball paradigm, responses to deviant (or “oddball”) stimuli occurring among a sequence of standard stimuli are measured. The MMN is obtained by subtracting the ERP to the standard stimulus from the ERP to the deviant stimulus, whereas the P3a is typically observed in the ERP to deviants.
Schizophrenia patients appear less able to detect and direct attention to novel stimuli than healthy controls (6). Consistent with this behavioral deficit, the amplitudes of both the MMN (7) and the P3 (8) have been found to be reduced in schizophrenia patients, leading to the proposals that reduced MMN is a marker of progressive pathology (7) and that reductions in both MMN and P3a are markers of vulnerability for this disorder (8, 9).
Given the homology of human and rhesus macaque brains (10), the development of a nonhuman primate (NHP) model of schizophrenia holds great potential for understanding the underlying cellular pathophysiologies and for exploring potential treatments. Of particular importance is the development of methods that allow comparison of neurophysiological correlates of sensory and cognitive functions in NHPs and humans. To this end, we developed a noninvasive electroencephalography (EEG) system that uses common recording hardware and analyses for the two species. Our system uses a noninvasive EEG cap in NHPs, with electrode density identical to that used in humans. Our approach allows for the calculation of topographic voltage maps and localization of activity generators in the NHP brain.
To determine the utility of our NHP EEG system, we recorded ERPs from humans (64-electrode array) (Fig. S1A) and NHPs (22-electrode array) (Fig. S1B) during a passive auditory intensity oddball paradigm. For both species, we established that ERPs had timing and topographic distributions consistent with previous reports, and source localization suggested homologous neural generators. Next, we investigated the effect of transient administration of subanesthetic doses of ketamine on these components in NHPs. These experiments revealed transient but selective reductions of MMN and P3a components, which mimicked those previously seen in human subjects similarly treated with NMDAR blockers. Most significantly, they also mimicked the chronic MMN and P3a reductions characteristic of schizophrenia.
Our findings, thus, support the utility of this NHP EEG system, used in conjunction with a ketamine-administration model of schizophrenia, to assay sensory and cognitive deficits. Our approach can, thus, be used to facilitate understanding of neural circuit dysfunctions characteristic of schizophrenia. Additionally, a wealth of previous evidence has shown a significant correlation between behavioral deficits and modulations of the MMN and P3a ERPs in a variety of neurological and neuropsychiatric pathologies (e.g., Alzheimer’s disease, dementia, Parkinson disease, affective disorders, and disorders of consciousness, etc.) (7, 11–13). Thus, our approach may also enable exploration, at neuronal and behavioral levels, of therapies targeted at this collection of pathologies.
Results
Comparison of MMN in Humans and Monkeys.
The MMN is obtained by subtracting the ERP to the standard stimulus from the ERP to the deviant stimulus (see Materials and Methods). In humans, the auditory MMN is well documented as a fronto-central negative potential with a latency of 100–250 ms after the onset of stimulus presentation and has sources in auditory cortices and in the inferior frontal gyrus (14). Consistent with previous findings, our recordings revealed a human MMN occurring 56–188 ms after stimulus onset, with a peak amplitude of −1.83 μV at 104 ms [F(1,1259) = 97.12; P < 0.001; Fig. 1A; additional information is in Tables S1 and S2] and a broad central-scalp distribution [Fig. 1B, Upper; white arrow indicates the MMN (negative, blue) central-scalp distribution].
Fig. 1.
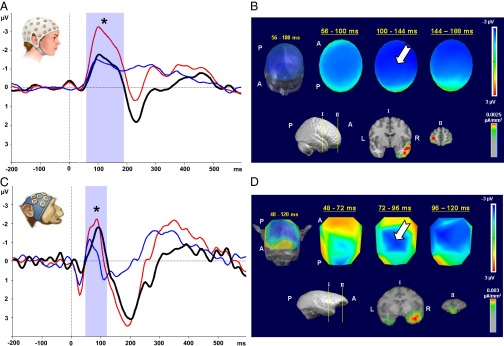
MMN in humans and NHPs. Left graphs show ERP plots of grand average from a central electrode (Cz) of five humans (A) and two NHP subjects (C). These graphs depict waveforms (averaged across low and high tones) from standard (blue line) and deviant (red line) conditions, as well as difference wave (black line). The blue shaded area identifies duration of the MMN [human: 56–190 m (peak amplitude, −1.83 μV at 104 ms; *P < 0.001); NHP: 48–120 ms (peak amplitude, −1.62 μV at 88 ms; *P < 0.001]. Human and monkey head icons identify species for results presented (they do not represent precise electrode placement or density). (B and D) Upper right images show scalp-voltage topographic maps, which reveal central negativity found in the difference wave for both species [human: time interval 56–188 ms (B); NHP: time interval 48–120 ms (D)] corresponding to the MMN [white arrow indicates MMN (negative, blue) central-scalp distribution]. Three-dimensional reconstruction of topographic maps [front-top view; Montreal Neurological Institute (MNI) human head template; rhesus macaque MRI] averaged over the entire time interval is shown at left. Three 2D top views, shown at right, represent snapshots along this time interval. Lower right images show source localization (LORETA inverse solution) for the entire time intervals corresponding to MMN in each species. (B) Three-dimensional reconstruction of template human brain (MNI) (side view) shown at left indicates location of MRI coronal sections depicted at right. Coronal sections illustrate locations of temporal [STG (I)] and frontal [inferior temporal gyrus (II)] areas identified as the main generators of this neurophysiological signal in humans. In D, the 3D reconstruction (NHP MRI; side view) shown at left indicates location of MRI coronal sections depicted at right. These coronal sections illustrate temporal [STG (I)] and frontal [RG (II)] areas identified as main generators of this neurophysiological signal in NHPs. A, anterior; L, left; P, posterior; R, right.
Unlike other previous studies that used epidural electrodes to establish MMNs in NHPs (Macaca fascicularis) (15, 16), we use high-density scalp electrodes, which enable scalp topographic voltage mapping and source localization. Javitt et al. reported that MMN in the macaque had a peak latency of ∼80 ms (15). We found NHP MMN 48–120 ms after stimulus onset, with a peak amplitude of −1.62 μV at 88 ms [F(1,409) = 11.17; P < 0.001; Fig. 1C; additional information is in Tables S1 and S2], and a central-scalp distribution [Fig. 1D, Upper; white arrow indicates the MMN (negative, blue) central-scalp distribution]. We have labeled this ERP as “mMMN” (i.e., monkey MMN).
Low-resolution brain electromagnetic tomography (LORETA) was used to estimate MMN generators. In both species, the superior temporal gyrus (STG) and frontal areas were estimated as primary neural generators (Fig. 1 B and D, lower images). For humans, the frontal generators included the inferior frontal gyrus (IFG) and the superior frontal gyrus (SFG). For macaques, the frontal generators included the rectus gyrus (RG) and the anterior cingulate gyrus (ACG). These data establish that comparable MMNs can be recorded with high-density scalp electrodes from both species. Our findings, moreover, provide functional evidence that the neural generators of those ERPs may be homologous in the two species.
Comparison of P3a in Humans and Monkeys.
The P3a emerges after the MMN and has a latency of 200–500 ms in humans (17). We investigated the P3a in the averaged response to low and high deviants (see Materials and Methods for details). In humans, the P3a lasted from 208–256 ms, with a peak amplitude of 0.72 μV at 228 ms (t = 37.53; P < 0.01; Fig. 2A; additional information is in Tables S3 and S4). In macaques, the P3a lasted 104–248 ms, with peak amplitude of 3.5 μV at 196 ms (t = 31.89; P < 0.01; Fig. 2C; additional information is in Tables S3 and S4). We have labeled this ERP as “mP3a” (i.e., monkey P3a). Both species presented a central-scalp distribution [Figs. 2B and 3D, upper images; white arrow indicates the P3a (positive, red) central-scalp distribution]. Source analysis, again, implicated the STG and frontal areas (IFG and SFG in humans and RG and ACG in NHPs) as the main neural generators (Fig. 2 B and D, lower images). Additional sources included dorsal parietal area, visual cortex, and cerebellum.
Fig. 2.
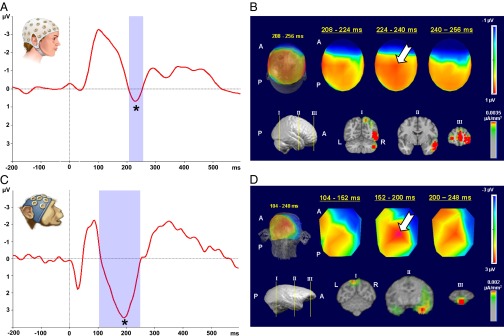
P3a ERP component in human and nonhuman primates. The left graphs show ERP plots of grand average from a central electrode (Cz) of five human subjects (A) and two NHP subjects (C). Depicted are waveforms (average of low and high tones) of the deviant (red line) condition. The blue shaded area identifies the duration of the P3a component [human: 208–256 ms (peak amplitude, 0.72 μV at 228 ms; *P < 0.01); NHP: 104–248 ms (peak amplitude, 3.5 μV at 196 ms; *P < 0.01)]. Upper right images show scalp-voltage topographic maps, which reveal maximal central positivity for P3a in both species [human: time interval, 208–256 ms (B); NHP: time interval, 104–248 ms (D); white arrow indicates P3a (positive, red) central-scalp distribution]. Three-dimensional reconstruction of topographic maps (back-top view; MNI human head template; NHP MRI) averaged over the entire time interval is shown at left. Three 2D top views, shown at right, represent snapshots along this time interval. Lower right images show source localization (LORETA inverse solution) for the entire time intervals corresponding to P3a ERP component in each species. (B) Three-dimensional reconstruction of template human brain (MNI) (side view) shown at left indicates location of MRI coronal sections depicted at right. These coronal sections illustrate dorsal parietal, visual cortex, and cerebellum (I), temporal [STG (II)], and frontal [IFG, SFG) (III)] areas identified as the main generators of this neurophysiological signal in humans. (D) Three-dimensional reconstruction (NHP MRI) (side view) shown at left indicates location of MRI coronal sections depicted at right. Coronal sections illustrate dorsal parietal (I), temporal [STG (II)], and frontal [RG and ACG (III)] areas identified as generators of this neurophysiological signal in NHPs. A, anterior; L, left; P, posterior; R, right.
Fig. 3.

Acute subanesthetic ketamine effect on the MMN in NHPs. (A) Scalp-voltage topographic maps (2D top view) illustrating MMN effect under three conditions (Materials and Methods): ketamine, saline, and 5 h postketamine for the time interval of maximum MMN amplitude (72–96 ms). White arrow indicates MMN (negative, blue) central-scalp distributions. (B) ERP plot of grand average for difference waves (MMN) from a central electrode (Cz) of two NHPs. Data are plotted separately for three conditions: ketamine, brown curve (60–116 ms; peak amplitude, −0.94 μV at 88 ms); saline, green curve (68–136 ms; peak amplitude, −2.79 μV at 84 ms); and 5 h postketamine, orange curve (60–128 ms; peak amplitude, −2.62 μV at 84 ms). Topographic maps and ERP plots reveal marked and highly significant reduction of MMN magnitude under ketamine, relative to saline (ketamine vs. saline: P < 0.001). The ketamine effect reversed after 5 h of recovery (ketamine vs. 5 h postketamine: P < 0.001). The MMN magnitude for saline does not differ from that seen following ketamine washout (5 h postketamine vs. saline: P > 0.05).
Effects of Acute Subanesthetic Ketamine on MMN and P3a in NHPs.
Building on our finding of comparable MMN and P3a ERPs in humans and macaques, and earlier ERP studies (3) that established support for a ketamine model of schizophrenia in healthy human subjects, we investigated the effects of ketamine in the MMN and P3a in the macaque. We used our auditory oddball paradigm under three conditions: (i) acute subanesthetic ketamine injection (1 mg/kg); (ii) saline control injection; and (iii) 5 h postketamine injection [after 5 h, ketamine levels are expected to be very low (18)]. Ketamine (brown line) led to a significant reduction of both MMN (Fig. 3) [ketamine vs. saline; F(1,290) = 43.98; P < 0.001; additional information is in Tables S1 and S2] and P3a (Fig. 4) [ketamine vs. saline; F(1,301) = 27.73; P < 0.001; additional information is in Tables S3 and S4] amplitudes compared with saline (green line). This reduction is apparent in topographic voltage maps [MMN in Fig. 3A and P3a in Fig. 4A; white arrow indicates MMN (negative, blue) and P3a (positive, red) central-scalp distributions, respectively] and in the waveforms (MMN in Fig. 3B and P3a in Fig. 4B).
Fig. 4.

Acute subanesthetic ketamine effect on the P3a in NHPs. (A) Scalp-voltage topographic maps (2D top view) illustrating P3a component under three conditions: ketamine, saline, and 5 h postketamine for the time interval of maximum P3a amplitude (152–200 ms). The white arrow indicates P3a (positive, red) central-scalp distributions. (B) ERP plot of grand average for deviant condition from a central electrode (Cz) of two NHPs. Data are plotted separately for three conditions: ketamine, brown line (108–232 ms; peak amplitude, 1.55 μV at 168 ms); saline, green line (108–244 ms; peak amplitude, 3.04 μV at 200 ms); and 5 h postketamine, orange line (120–268 ms; peak amplitude, 2.78 μV at 192 ms). Topographic maps and ERP plots reveal marked and highly significant reduction of P3a magnitude under the ketamine, relative to saline (ketamine vs. saline: P < 0.001). The ketamine effect reversed after 5 h of recovery (ketamine vs. 5 h postketamine: P < 0.001). P3a magnitude for saline does not differ from that seen following ketamine washout (5 h postketamine vs. saline: P > 0.05). mP3a indicates monkey P3.
It has been reported previously that schizophrenia-like symptoms, such as impairments in task switching (19, 20), disappear relatively rapidly (<1 h) after ketamine administration. As an additional control, we, thus, examined MMN and P3a components 5 h after ketamine injection. The drug effects were no longer significant after this delay (orange line), as shown for the MMN in Fig. 3 and for the P3a in Fig. 4 [MMN ketamine vs. 5 h postketamine (F(1,403) = 58.48; P < 0.001); 5 h postketamine vs. saline (F(1,290) = 0.15; P > 0.05); P3a ketamine vs. 5 h postketamine (F(1,411) = 44.34; P < 0.001); 5 h postketamine vs. saline (F(1,301) = 0.06; P > 0.05); additional information is in Tables S1–S4].
Taken together, our findings demonstrate that the NMDAR antagonist ketamine significantly reduces the amplitude of the MMN and P3a ERP components in the macaque, as monitored by a high-density scalp EEG system. Our results parallel those seen in human ERP studies of the effects of ketamine and, thus, offer a NHP model to investigate potential therapies and cellular mechanisms that underlie deficits seen in schizophrenia patients and in healthy subjects administered ketamine.
Discussion
The Etiology of Schizophrenia: The Dopamine and Glutamate Hypotheses.
Over the past 50 y, a wide range of studies have given rise to two main neurotransmitter hypotheses regarding the pathophysiology of schizophrenia: the dopamine (DA) and glutamate hypotheses. Since the 1970s, the DA hypothesis of schizophrenia has provided the dominant framework for the understanding and treatment of schizophrenia (21). There are, however, numerous limitations to this framework including: (i) limited efficacy of DA antipsychotic drugs (which modulate DA levels) in treatment of negative symptoms and cognitive deficits (22); (ii) positive symptoms (for which DA antipsychotics are usually efficacious) persist in some cases despite aggressive treatment with DA antipsychotics (23); and (iii) lack of explanatory power for widespread sensory and cognitive deficits (24), including those indexed by disruptions of MMN and P3a (24).
The discovery of glutamate’s role in schizophrenia dates to the demonstration that the dissociative anesthetics phencyclidine (PCP) and ketamine can induce psychosis (25). This was followed by discovery of the “PCP receptor” (26) and later by the realization that both PCP and ketamine act by blocking the NMDAR channel (2). Since then, strong correlations between the action of NMDA antagonists and various stereotypical deficits observed in schizophrenia patients, including executive functioning, attention/vigilance, verbal fluency, and visual and verbal working memory (27), have been reported. The glutamate model reformulates how we think about psychosis and suggests a different set of targets for treatment than does the DA model. Whereas the DA model suggests a localized dysfunction reflecting the limited range of dopaminergic projections, glutamate is the main excitatory neurotransmitter of the brain and any dysfunction of that transmitter system would be expected to have widespread effects. This expectation is consistent with the sensory and cognitive effects seen with schizophrenia, which are similar to those induced by ketamine administration in normal subjects (3).
Nonetheless, no single pharmacologic approach can fully mimic the constellation of impairments present in heterogeneous disorders like schizophrenia, because perturbations of one transmitter system necessarily impact other systems (28). For example, it has been argued that the mimicry of schizophrenia symptoms by NMDAR antagonists may be attributable, in part, to secondary DA effects (29). The development of NHP models will aid in elucidating neurotransmitter interactions that underlie schizophrenia-related pathophysiology and development of therapeutics for this devastating disorder.
ERP Measures of Disrupted Sensory and Cognitive Function in Schizophrenia.
ERPs provide measures of all stages of sensory and cognitive processing and are well suited to identify deficits exhibited by schizophrenia patients (1). Of particular interest here are deficits in automatic change detection, reflected in the MMN, and deficits in attentional orienting, reflected in the P3. Abnormalities in these ERP components are consistently seen in schizophrenia patients and may be used as biological markers for the disease (1). Consistent with the glutamate hypothesis, previous studies report that administration of a subanesthetic dose of ketamine induces many of the sensory and cognitive impairments seen in patients with schizophrenia (3). Moreover, both MMN and P3 ERPs are reduced in healthy volunteers when exposed to acute ketamine administration, suggesting that this may be a useful model for schizophrenia.
As noted above, however, neurotransmitter systems do not work in isolation, and it would be surprising if other pharmacological agents did not also impact MMN and P3a ERPs. There is some evidence, for example, that nicotinic agents modulate the MMN (14). The emerging view, however, is that the most important and reliable modulation of the MMN is exerted through NMDARs (3, 30, 31). Furthermore, whereas dopaminergic antipsychotics, such as haloperidol, do not reliably affect the MMN, there is some evidence that they modulate the P300 (32), although this is still controversial (24). It is hoped that the NHP model offered here will help resolve some of these uncertainties.
MMN, P3a, and a Nonhuman Primate Model for Schizophrenia.
Animal models are necessary to gain an understanding of disease processes at a mechanistic level. NHP models are especially useful in the study of higher order sensory and cognitive deficits given the close relationship between humans and NHPs. There are several previous reports of MMN and “P3-like” components in a variety of primate species, including monkeys (16) and apes (33). For instance, Javitt et al. (15), using epidural electrodes, recorded an MMN-like component from cynomolgus monkeys.
Other prior studies reveal associations between physiological measures and behavioral deficits: (i) both humans (34) and monkeys exhibit schizophrenia-like deficits on task-switching (19) when treated with ketamine; and (ii) the amplitude reduction of MMN has been correlated with behavioral deficits present in schizophrenia patients (1, 7), and the reduction of both MMN and P3 has been associated with vulnerability for schizophrenia (8, 9). Here, to further explore these relationships and the suitability of the rhesus macaque as an animal model for schizophrenia, we studied the amplitude of MMN and P3a ERP responses in NHPs in relation to the administration of ketamine.
For this purpose, we have developed a high-density electrode cap that allows for recording of scalp EEG from NHPs. These caps, coupled with common experimental paradigms and analytical tools, allow for the recording of EEG signals that are directly comparable in NHP and human subjects. In particular, these methods allow for comparison of channel-specific responses (ERPs, frequency analysis, etc.) of full-scalp voltage maps and for source localization in NHPs and humans. This approach opens avenues for comparative studies designed to integrate findings made at the systems level in both species, with findings from the cellular level in NHPs.
In the current study, we have used this approach to compare human and NHP ERPs elicited in an auditory oddball paradigm and to examine feasibility of an NHP-ketamine model of schizophrenia. We found ERP components in NHPs that appear homologous to those found in humans. Moreover, the distributed neural architecture for MMN and P3a identified by source analysis is consistent with a recent report by Takahashi et al. (35) describing the use of an advanced version of LORETA source analysis (eLORETA) in large cohorts of nonpsychiatric subjects and schizophrenia patients.
We next examined the influence of acutely administered ketamine on ERP components in NHPs. We found decreases in the amplitudes of both MMN and P3a components, which are nearly identical to those seen in patients with schizophrenia and in normal volunteers given comparable subanesthetic doses of ketamine. These results are consistent with previous evidence that failures of glutamate neurotransmission underlie many of the symptoms of schizophrenia and that acute ketamine administration provides a good model of prodromal or acute incipient schizophrenia (3). Moreover, our findings support the validity of an NHP-ketamine model of schizophrenia.
Our results extend previous findings in several ways. Because our EEG NHP methods are the same as those used in our human work, we can directly compare NHP and human findings. These comparisons include dynamics, electrode identity, scalp distributions, and source localization. Moreover, because we use a high-density full-scalp cap, we have no requirement for a priori assumptions about optimal electrode placement, and we can detect unexpected components and source contributions. Our study opens the door to detailed studies of neural mechanisms of cognitive function, such as the predictive-coding model of the MMN (36).
Future directions may include the use of this system in NHPs to monitor pharmacological “treatment,” of ketamine-induced psychotomimesis, allowing for examination of changes in the distribution of electrical activity that accompany treatments and to identify potential sources. These sources can subsequently be targeted in “EEG-guided” investigation of neuronal signals at the cellular level. The same approach may also be extended to explore pathophysiology of other neuropsychiatric disorders.
Materials and Methods
For additional information, please see SI Materials and Methods.
Subjects.
Humans.
Five adult male subjects (20–36 y old) were used in this study. All subjects reported being free of neurological and psychiatric disorders and informed consent was obtained. All procedures were conducted in accordance with the Salk Institute Institutional Review Board (IRB).
Rhesus macaques (M. mulatta).
Two adult male monkeys (8–9 y old) were used in this study. All procedures and animal care were approved by the Salk Institute Animal Care and Use Committee, and the research was carried out in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Stimuli Presentation.
We used a passive auditory-intensity oddball paradigm [100-ms (10 ms rise/fall) pure sinusoidal tones (1,500 Hz)] to present tones of different intensities (low, 60 dB; high, 80 dB) to subjects in a sound-isolated, dimly lit room. Frequent (“standard”) and infrequent (“deviant”) stimuli were presented 80 and 20% of the time, respectively. Interstimulus interval was 700 ms. Twelve-hundred standard and 300 deviant stimuli were presented in each recording session. Both high-deviant (low-standard) and low-deviant (high-standard) conditions were used to allow comparison of responses to identical stimuli (low or high) in different contexts (standard or deviant). Stimulus presentation was controlled by Cogent 2000 (University College London Functional Imaging Laboratory and Institute of Cognitive Neuroscience MATLAB toolbox) using a personal computer. Tones were presented using a Yamaha RX 397 amplifier and a Sony SS-F7000P speaker for NHPs and an Advent AV570 speaker for humans. To minimize movement artifacts, human subjects were asked to maintain central fixation, and NHPs were trained to maintain central fixation. The fixation target was a red circle (1° in diameter) on a black background presented using a 21-inch Sony GDM-C520 CRT monitor at a 40-cm viewing distance.
EEG Data Collection/Recordings.
For both human and NHP subjects, EEG scalp recordings were acquired with the Vision Recorder software (Brain Products) using a BrainAmp MR amplifier (Brain Products). We used a 64-channel EEG cap BrainCap MR (Brain Products) with Ag/AgCl electrodes for human subject data collection and customized 22-channel EEG caps, also with Ag/AgCl electrodes, for NHPs. Collection of NHP EEG data required several additional steps (SI Materials and Methods). NHPs were restrained in the chair in a sphinx-like position with head protruding, stabilized, and facing forward.
EEG Data Analysis.
EEG data were analyzed using Analyzer 2.0 software (Brain Products). The analysis procedure included preprocessing (rereferencing the datasets, band-pass filtering, down-sampling, segmentation, etc.) before calculating ERPs for each condition. The same analyses were applied for humans and NHPs.
Identification of Human and NHP ERPs.
We first identified MMN and P3a components in humans and then searched for homologous components in NHPs before pharmacological manipulation. ERP components were identified using established criteria. MMN was defined as the difference wave obtained by subtracting ERPs for standard from ERPs for deviant stimuli. The P3a was identified and analyzed from deviant stimulus trials. We ascertained the timing, electrode location, voltage scalp distribution, and neural generators for these ERP components. A 40-ms time window was placed around the maximal amplitude in the average ERP waveforms of each species and was used to extract mean amplitude values per subject from single trials. These values were used for statistical analysis [MMN, two-way repeated-measures ANOVA (factor 1, standard vs. deviant; factor 2, high vs. low); P3a, t test of response to deviants] (STATISTICA data analysis software, 2007; StatSoft).
Ketamine and Saline Injections.
Using the same passive auditory intensity oddball paradigm EEG data were collected from two NHPs under three physiological conditions: (i) “ketamine” (injection of ketamine; 1 mg/kg); (ii) “saline” (injection of saline solution); and (iii) “5 h postketamine” (injection of ketamine; 1 mg/kg). All injections were i.m. Recording began 12 min after injection for ketamine and saline conditions and 5 h after injection for 5 h postketamine condition. All recording sessions lasted 18 min. NHPs showed no behavioral signs of ketamine effects (i.e., no signs of drowsiness and no differential behavior between ketamine and saline conditions). A 40-ms time window was established around the maximal amplitude in the average ERP (MMN and P3a) waveforms and was used to extract mean amplitude values per subject from single trials. These values were used for statistical analysis [MMN, three-way repeated-measures ANOVA (factor 1, physiological condition; factor 2, standard vs. deviant; factor 3, high vs. low tone); P3a two-way repeated-measures ANOVA (factor 1, physiological conditions; factor 2, high vs. low)] (STATISTICA data analysis software, 2007; StatSoft).
Topographic Voltage Maps and Source Analysis.
Topographic voltage-distribution maps for both human and NHP data were calculated in Cartool 3.43 (D. Brunet, Functional Brain Mapping Laboratory, Geneva, Switzerland) using previously acquired electrode-position files for the 64-channel human and 22-channel NHP caps. Estimation of intracranial generators for MMN and P3a was performed using Cartool 3.43 software with LORETA. Neural generators were estimated across two time intervals per species: human (56–188 ms and 208–256 ms) and NHP (48–120 ms and 104–248 ms) corresponding to the MMN and P3a components, respectively.
Supplementary Material
Acknowledgments
We thank Steven Hillyard, Antigona Martinez, and Marla Zinni for valuable contributions to design and data analysis; Thomas Liu and Valur Olafsson for assistance in EEG setup; and Dinh Diep and Aaron Cortez for assistance in animal training and care. Additionally, we thank Denis Brunet for assistance with developing NHP inverse solutions. Stimulus presentation for this experiment was conducted using the Cogent 2000 and Cogent graphics software (MATLAB toolbox), developed by teams at the Wellcome Department of Imaging Neuroscience and University College London. Cartool software (http://brainmapping.unige.ch/cartool) was programmed by Denis Brunet (Functional Brain Mapping Laboratory) and supported by the Center for Biomedical Imaging of Geneva and Lausanne.
Footnotes
The authors declare no conflict of interest.
See Commentary on page 15175.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312264110/-/DCSupplemental.
References
- 1.Rissling AJ, Light GA. Neurophysiological measures of sensory registration, stimulus discrimination, and selection in schizophrenia patients. Curr Top Behav Neurosci. 2010;4:283–309. doi: 10.1007/7854_2010_59. [DOI] [PubMed] [Google Scholar]
- 2.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 3.Umbricht D, et al. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: Implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57(12):1139–1147. doi: 10.1001/archpsyc.57.12.1139. [DOI] [PubMed] [Google Scholar]
- 4.Garrido MI, Kilner JM, Kiebel SJ, Friston KJ. Evoked brain responses are generated by feedback loops. Proc Natl Acad Sci USA. 2007;104(52):20961–20966. doi: 10.1073/pnas.0706274105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutton S, Braren M, Zubin J, John ER. Evoked-potential correlates of stimulus uncertainty. Science. 1965;150(3700):1187–1188. doi: 10.1126/science.150.3700.1187. [DOI] [PubMed] [Google Scholar]
- 6.Baribeau-Braun J, Picton TW, Gosselin JY. Schizophrenia: A neurophysiological evaluation of abnormal information processing. Science. 1983;219(4586):874–876. doi: 10.1126/science.6823555. [DOI] [PubMed] [Google Scholar]
- 7.Wynn JK, Sugar C, Horan WP, Kern R, Green MF. Mismatch negativity, social cognition, and functioning in schizophrenia patients. Biol Psychiatry. 2010;67(10):940–947. doi: 10.1016/j.biopsych.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Stelt O, Belger A. Application of electroencephalography to the study of cognitive and brain functions in schizophrenia. Schizophr Bull. 2007;33(4):955–970. doi: 10.1093/schbul/sbm016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jahshan C, et al. Cross-diagnostic comparison of duration mismatch negativity and P3a in bipolar disorder and schizophrenia. Bipolar Disord. 2012;14(3):239–248. doi: 10.1111/j.1399-5618.2012.01008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sereno MI, Tootell RBH. From monkeys to humans: What do we now know about brain homologies? Curr Opin Neurobiol. 2005;15(2):135–144. doi: 10.1016/j.conb.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Näätänen R, et al. The mismatch negativity: An index of cognitive decline in neuropsychiatric and neurological diseases and in ageing. Brain. 2011;134(Pt 12):3435–3453. doi: 10.1093/brain/awr064. [DOI] [PubMed] [Google Scholar]
- 12.Pekkonen E, Jousmäki V, Reinikainen K, Partanen J. Automatic auditory discrimination is impaired in Parkinson’s disease. Electroencephalogr Clin Neurophysiol. 1995;95(1):47–52. doi: 10.1016/0013-4694(94)00304-4. [DOI] [PubMed] [Google Scholar]
- 13.Vecchio F, Määttä S. The use of auditory event-related potentials in Alzheimer’s disease diagnosis. Int J Alzheimers Dis. 2011;2011:653173. doi: 10.4061/2011/653173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garrido MI, Kilner JM, Stephan KE, Friston KJ. The mismatch negativity: A review of underlying mechanisms. Clin Neurophysiol. 2009;120(3):453–463. doi: 10.1016/j.clinph.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Javitt DC, Schroeder CE, Steinschneider M, Arezzo JC, Vaughan HG., Jr Demonstration of mismatch negativity in the monkey. Electroencephalogr Clin Neurophysiol. 1992;83(1):87–90. doi: 10.1016/0013-4694(92)90137-7. [DOI] [PubMed] [Google Scholar]
- 16.Woodman GF. In: The Oxford Handbook of Event-Related Potential Components. Kappenman ES, Luck SJ, editors. Oxford University Press, New York; 2011. [Google Scholar]
- 17.Polich J. Updating P300: An integrative theory of P3a and P3b. Clin Neurophysiol. 2007;118(10):2128–2148. doi: 10.1016/j.clinph.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wieber J, Gugler R, Hengstmann JH, Dengler HJ. Pharmacokinetics of ketamine in man. Anaesthesist. 1975;24(6):260–263. [PubMed] [Google Scholar]
- 19.Stoet G, Snyder LH. Effects of the NMDA antagonist ketamine on task-switching performance: Evidence for specific impairments of executive control. Neuropsychopharmacology. 2006;31(8):1675–1681. doi: 10.1038/sj.npp.1300930. [DOI] [PubMed] [Google Scholar]
- 20.Taffe MA, Davis SA, Gutierrez T, Gold LH. Ketamine impairs multiple cognitive domains in rhesus monkeys. Drug Alcohol Depend. 2002;68(2):175–187. doi: 10.1016/s0376-8716(02)00194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meltzer HY, Stahl SM. The dopamine hypothesis of schizophrenia: A review. Schizophr Bull. 1976;2(1):19–76. doi: 10.1093/schbul/2.1.19. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg TE, Weinberger DR. Effects of neuroleptic medications on the cognition of patients with schizophrenia: A review of recent studies. J Clin Psychiatry. 1996;57(Suppl 9):62–65. [PubMed] [Google Scholar]
- 23.Foussias G, Remington G. Antipsychotics and schizophrenia: From efficacy and effectiveness to clinical decision-making. Can J Psychiatry. 2010;55(3):117–125. doi: 10.1177/070674371005500302. [DOI] [PubMed] [Google Scholar]
- 24.Korostenskaja M, Kähkönen S. What do ERPs and ERFs reveal about the effect of antipsychotic treatment on cognition in schizophrenia? Curr Pharm Des. 2009;15(22):2573–2593. doi: 10.2174/138161209788957474. [DOI] [PubMed] [Google Scholar]
- 25.Luby ED, Gottlieb JS, Cohen BD, Rosenbaum G, Domino EF. Model psychoses and schizophrenia. Am J Psychiatry. 1962;119:61–67. doi: 10.1176/ajp.119.1.61. [DOI] [PubMed] [Google Scholar]
- 26.Zukin SR, Zukin RS. Specific [3H]phencyclidine binding in rat central nervous system. Proc Natl Acad Sci USA. 1979;76(10):5372–5376. doi: 10.1073/pnas.76.10.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: The final common pathway on the road to schizophrenia? Brain Res Bull. 2010;83(3-4):108–121. doi: 10.1016/j.brainresbull.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swerdlow NR. Are we studying and treating schizophrenia correctly? Schizophr Res. 2011;130(1-3):1–10. doi: 10.1016/j.schres.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seeman P, Guan H-C. Phencyclidine and glutamate agonist LY379268 stimulate dopamine D2High receptors: D2 basis for schizophrenia. Synapse. 2008;62(11):819–828. doi: 10.1002/syn.20561. [DOI] [PubMed] [Google Scholar]
- 30.Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: Implications for schizophrenia. Proc Natl Acad Sci USA. 1996;93(21):11962–11967. doi: 10.1073/pnas.93.21.11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Umbricht D, Koller R, Vollenweider FX, Schmid L. Mismatch negativity predicts psychotic experiences induced by NMDA receptor antagonist in healthy volunteers. Biol Psychiatry. 2002;51(5):400–406. doi: 10.1016/s0006-3223(01)01242-2. [DOI] [PubMed] [Google Scholar]
- 32.Kähkönen S, et al. Dopamine modulates involuntary attention shifting and reorienting: An electromagnetic study. Clin Neurophysiol. 2002;113(12):1894–1902. doi: 10.1016/s1388-2457(02)00305-x. [DOI] [PubMed] [Google Scholar]
- 33.Ueno A, et al. Auditory ERPs to stimulus deviance in an awake chimpanzee (Pan troglodytes): Towards hominid cognitive neurosciences. PLoS ONE. 2008;3(1):e1442. doi: 10.1371/journal.pone.0001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wylie GR, Clark EA, Butler PD, Javitt DC. Schizophrenia patients show task switching deficits consistent with N-methyl-d-aspartate system dysfunction but not global executive deficits: Implications for pathophysiology of executive dysfunction in schizophrenia. Schizophr Bull. 2010;36(3):585–594. doi: 10.1093/schbul/sbn119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi H, et al. Neural substrates of normal and impaired preattentive sensory discrimination in large cohorts of nonpsychiatric subjects and schizophrenia patients as indexed by MMN and P3a change detection responses. Neuroimage. 2012;66C:594–603. doi: 10.1016/j.neuroimage.2012.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wacongne C, Changeux J-P, Dehaene S. A neuronal model of predictive coding accounting for the mismatch negativity. J Neurosci. 2012;32(11):3665–3678. doi: 10.1523/JNEUROSCI.5003-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.