

Abstract
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death in the United States and the incidence is increasing as the population ages. Cigarette smoking is the primary risk factor; however, only a minority of smokers develop the disease. Inhalation of cigarette smoke introduces an abundance of free radicals into the lungs, causing oxidative stress and inflammation. We hypothesized that after the initial burst of oxidative stress associated with cigarette smoke exposure, a sustained source of endogenous free radical production is modulated by the antioxidant enzyme extracellular superoxide dismutase (ECSOD) and the superoxide-generating complex NADPH oxidase (NOX). Primary mouse macrophages exposed to cigarette smoke extract exhibited increased oxidative stress as indicated by fluorogenic dyes and isoprostane concentration, which was suppressed in the presence of both a superoxide dismutase mimetic and a NOX inhibitor. Similarly, primary macrophages isolated from ECSOD-overexpressing mice or NOX-deficient mice showed reduced oxidative stress in response to cigarette smoke treatment. In addition, both reduced glutathione and cytokines (MIP2 and IFNγ) were increased in bronchoalveolar lavage fluid of wild-type mice exposed to cigarette smoke but not in ECSOD-overexpressing or NOX-deficient mice. These data suggest that the mechanisms underlying the host defense against cigarette smoke-induced oxidative damage and subsequent development of COPD may include endogenous oxidases and antioxidant enzymes.
Keywords: Extracellular superoxide dismutase, NADPH oxidase, Smoke, Macrophages, ROS, Antioxidants, Oxidative stress, GSH, Cytokines, Lung, 4-HNE, BALF, COPD, Free radicals
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death in the United States and is characterized by a progressive decline in lung function that is often associated with emphysema or chronic bronchitis [1]. More than 90% of COPD patients have a history of smoking; however, the majority of smokers do not develop COPD [2]. This paradox suggests that many smokers exhibit host defenses that protect the lung from the more than 1015 oxidants per puff of cigarette smoke [3]. The mechanism for this protection is unclear but antioxidants, including superoxide dismutase (SOD) and glutathione, are candidate defense systems.
All eukaryotic cells contain antioxidant enzymes that function to offset oxidative stress. This conservation during evolution suggests that scavenging and eliminating reactive oxygen species (ROS) are essential to life. One of the major families of antioxidant enzymes is SOD [4]. The primary function of SOD is to dismutate superoxide (O−2) to oxygen (O2) and hydrogen peroxide (H2O2). In mammals there are three SOD enzymes: cytosolic SOD (CuZnSOD or SOD1), mitochondrial SOD (MnSOD or SOD2), and extracellular SOD (ECSOD or SOD3). ECSOD is thought to be the primary source of extracellular enzymatic antioxidant activity in the lung [5–7]. Deletion of the ECSOD gene (SOD3) leads to severe lung damage and enzyme dysfunction and has also been associated with pulmonary diseases such as lung cancer, pulmonary fibrosis, and adult respiratory distress syndrome [8–13]. Recently SOD3 single-nucleotide polymorphisms have been associated with altered lung function and COPD [14,15]. The mechanism by which ECSOD protects the lung from cigarette smoke is unknown but may be related to its ability to attenuate oxidative damage and to reduce lung inflammation [16,17].
Airway and lung inflammation is a hallmark of COPD and is more prominent in patients with severe disease [18,19]. Cigarette smoke elicits a well-documented initial inflammatory response in the lungs; however, in patients who develop COPD, the presence of inflammatory cells in the airways is maintained beyond the cessation of smoking [20–23]. The predominant inflammatory cells in the airways of smokers are macrophages, which generate ROS when activated [24]. Markers of ROS injury (oxidative footprints) are elevated in COPD patients [25,26]. Inflammatory cells generate oxidative stress by activation of NADPH oxidase. NOX and DUOX proteins comprise both membrane-bound and cytoplasmic components of the NADPH oxidase complex. Upon introduction of an inflammatory signal, the cytosolic components are recruited to the plasma membrane and undergo highly regulated assembly to form the active complex [27]. Activated NADPH oxidase (NOX) produces O−2, which reacts quickly to produce a burst of additional oxidants including H2O2 and the highly destructive hydroxyl radical (•OH). Because inflammation and oxidative stress are coupled through mechanisms including NOX activity and NF-κB signaling, it follows that inhibition of oxidative stress via antioxidant enzymes affects inflammation. In this study, we investigated whether ECSOD could attenuate cigarette smoke-induced macrophage activation using both in vitro and in vivo models. We also explored the role of NOX in smoke-induced free radical generation by inhibiting its function or knocking down expression.
Experimental procedures
Genetically modified mice
ECSOD transgenic mice used in this study have a C57BL/6 genetic background and overexpress human ECSOD constitutively under a surfactant protein C (SPC) promoter. These mice were maintained at National Jewish Health (Denver, CO, USA). Additionally, ECSOD transgenic mice with a genetic background of C57BL/6 that over-express human ECSOD under a β-actin promoter were also maintained at National Jewish Health [28]. NADPH knockout (gp91phox−/−) mice were originally purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained as a breeding colony housed at National Jewish Health. All animals received care in accordance with the guidelines and approval of the Institutional Animal Care and Use Committee and were maintained on food and water ad libitum.
Isolation and maintenance of primary mouse macrophage cultures
Bone marrow was isolated from the femoral, tibial, and pelvic bones of mice by inserting a 25-gauge needle into the central cavity and expelling the bone marrow with a jet of bone marrow medium (Dulbecco’s modified Eagle’s medium (DMEM)+10% fetal bovine serum+10% L929 preconditioned medium as a source of CSF-1) into a sterile tube. Isolated cells were centrifuged at 1000 rpm for 10 min, resuspended into 10 ml of medium, and gently drawn in and out of a large-gauge needle (18 gauge) to break up any cell aggregates. Cells were plated on plastic tissue culture plates and incubated for 5–7 days at 37 °C before use as previously described [29].
Preparation of aqueous cigarette smoke extract (CSE)
CSE was prepared daily as previously described [30]. Briefly, commercial cigarettes (University of Kentucky, batch 3R4F) were smoked continuously over the course of 5 min. Constant flow was obtained at 2 L/min using an Accucal flowmeter (Gilmont Instruments, Barrington, IL, USA). Mainstream smoke was drawn through prewarmed (37 °C) DMEM for 5 min. Three cigarettes were used for every 30 ml of CSE solution. The medium was filter-sterilized and diluted in cell culture medium to 10%. Aliquots were frozen and stored at −20 °C so that all experiments reported here were conducted with CSE from the same preparation. Control solutions were made using an identical preparation except that the cigarettes were unlit.
In vivo cigarette smoke exposure
Mice (wild type, ECSOD-overexpressing, or NADPH oxidase) were exposed in TE-10z smoking chambers (Teague Enterprises, Davis, CA, USA). One machine holds 40 mice and goes through five cigarettes every 10 min. Mice were exposed to smoke for 2 weeks (Monday through Friday). On day 0 exposure of the mice to cigarette smoke for 5 h per day (~300 cigarettes) began. Control mice were placed in the same room, but not in the smoking chamber. At the conclusion of the smoke exposure, the mice were euthanized by isoflurane overdose and the lungs lavaged.
Mouse bronchoalveolar lavage (BAL)
BAL was performed five times with 1 ml of phosphate-buffered saline (PBS) with 0.1 mM EGTA (Sigma). The BAL returns were pooled from each sample and centrifuged at 1000 g for 10 min. The supernatant was removed and frozen at −80 °C and the cell pellet was resuspended in 1 ml of PBS for cell counting.
ROS-sensitive fluorimetric labeling
Dihydroethidium (DHE) reacts with ROS to produce a fluorescent oxidized product. 5-(And-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) is also a reliable fluorimetric marker for ROS in live cells (Invitrogen; Eugene, OR, USA). DHE (0.1 mM) or DCFDA (0.02 mM) was added to primary macrophage cells cultured on an eight-well chamber slide, incubated at 37 °C for 30 min, and then washed (2×) with warm Hanks’ buffered saline solution. Slides containing stained, live cells were placed on ice and imaged immediately. Images were scanned by a long-working-distance Marianas microscope with a 40× lens. Three images from each condition were captured. Slidebook software by Intelligent Imaging Innovations was used for image processing and analysis.
Determination of glutathione (GSH) concentration
Cell lysate or BALF was mixed with an equal volume of 5% metaphosphoric acid. After a 10-min incubation on ice, samples were centrifuged at 14,000 g for 10 min and supernatants were stored at −80 °C before analysis. GSH was measured as previously described [31]. Briefly, GSH was incubated with 5,5′-dithiobis-2-nitrobenzoic acid to produce 2-nitro-5-thiobenzoic acid (TNB). In this reaction, GSH is oxidized to GSSG, which is then reconverted to GSH in the presence of glutathione reductase and NADPH. The rate of TNB formation was measured using a microplate reader at 412 nm.
Immunostaining for ECSOD
Wild-type and ECSOD-overexpressing mice were sacrificed, and their lungs were inflated with 4% paraformaldehyde at 20 cm of water pressure and then embedded in paraffin. Four-micrometer sections were rehydrated and boiled in 0.1 M sodium citrate, pH 6.0, for 10 min for antigen retrieval. The slides cooled for 20 min and were washed in 1× PBS for 5 min, twice. The slides were then incubated in 3% hydrogen peroxide for 5 min and washed in 1× PBS for 5 min, twice. A mouse-on-mouse peroxidase kit (M.O.M.) was used according to the manufacturer’s directions (Vector Laboratories, Burlingame, CA, USA). The sections were then incubated with 1/1000 mouse monoclonal anti-human ECSOD IgG (clone 4G11G6 available through StressMarq Biosciences, Victoria, BC, Canada) or 1/2000 IgG in blocking buffer for 30 min at room temperature. Vectastain ABC Elite and diaminobenzidine (Vector Laboratories) kits were used as described in the kit instructions. The sections were then stained for 1 min with hematoxylin and washed with distilled H2O. They were then dehydrated and mounted with coverslips.
ECSOD Western blots
Two million cells or 42 ml of medium was spun for 5 min (800 g). Cells were then placed in 100 μl of lysis buffer (50 mM Tris–HCl, 120 mM NaCl, 1% NP-40, 5 mM EDTA with one Roche Complete Mini Protease inhibitor tablet per 10 ml of buffer). Samples were boiled 5 min in 1:6 concentrated sample buffer with 10% β-mercaptoethanol to 100 μl of buffer. Experiments were done in duplicate. Fifty microliters of sample or Kaleidoscope Precision Plus protein standard (Bio-Rad, Hercules, CA, USA) was pipetted into each well and run on a 10% Tris–HCl gel (Bio-Rad) at 150 V for 1 h. Proteins were transferred to a PVDF membrane for 1 h at 100 V and blocked in 5% nonfat milk in TBS-T overnight at 4 °C. Primary antibody (rabbit anti-human ECSOD) was added at a dilution of 1:5000 in 5% nonfat milk/TBS-T. Blots were incubated overnight at 4 °C, washed three times for 5 min each in TBS-T, and then incubated 1 h in conjugated goat anti-rabbit secondary antibody (1:100,000) in 5% nonfat milk/TBS-T. The blot was washed three times for 5 min and developed using an ECL-Plus kit (GE Healthcare).
F8-isoprotanes measurement
Two million cells per well of a six-well plastic plate were exposed to 0.5 ml of control medium or 10% CSE (N=3 each) for 2 h. BHT (0.005%) was added to supernatant and then spun at low speed (209 g) to remove debris. Free 8-isoprostane was determined using an 8-isoprostane enzyme immunoassay (Cayman Chemical, Ann Arbor, MI, USA).
Human tissue lysate preparation
Normal donor lungs were obtained through the International Institute for the Advancement of Medicine (Jessup, PA, USA), and COPD lungs were obtained from the NHLBI Lung Tissue Research Consortium. Samples were weighed and 1 ml of ice-cold RIPA buffer (50 mM Hepes, pH 7.6; 1 mM EDTA; 0.7% deoxycholate; 1% NP-40; 0.5 M LiCl) containing a protease inhibitor cocktail (Roche, Basel, Switzerland) was added to approximately 300 g of tissue. Samples were then sonicated to homogenize the tissue, rotated at 4 °C for 30 min, and then centrifuged at 12,000 rpm for 20 min at 4 °C. Supernatants were collected and protein concentrations were determined using a BCA protein assay kit (Pierce, Rockford, IL, USA). Aliquots of each sample (~20 μg) were diluted in 1× sodium dodecyl sulfate–polyacrylamide gel electrophoresis sample buffer, boiled for 5 min, and electrophoresed through polyacrylamide gels. The resolved proteins were then transferred to PVDF membranes (Amersham Pharmacia Biotech) for Western blotting.
4-Hydroxynonenal (4-HNE) Western blotting
PVDF membranes were first blocked for 1 h at room temperature in 1% bovine serum albumin and 0.01% sodium azide diluted in PBS containing 0.1% Tween 20 (PBS-T). Primary antibody (4-HNE horseradish peroxidase (HRP)-conjugated; abcam 46542) was diluted in PBS-T and membranes were incubated overnight at 4 °C. Immunoreactive proteins were detected with enhanced chemiluminescence.
Results
Altered redox environment induced by cigarette smoke exposure in vitro, in vivo, and in human patients with COPD
To determine the effect of cigarette smoke exposure on redox environment, several markers of oxidative stress were assessed. First, fluorogenic dyes were employed to measure oxidative stress in primary mouse macrophages exposed to CSE (Fig. 1). Cultures were incubated with 10% CSE for 0, 30, 60, or 120 min and exposed to the oxidation-sensitive dyes DCFDA or DHE for 15 min, and then the mean fluorescence was measured. CSE exposure caused a marked, time-dependent increase in levels of oxidative stress as indicated by increased DCFDA fluorescence (Figs. 1A and B) as well as DHE fluorescence (Fig. 1C). Reduced glutathione (GSH) levels also provide an indication of redox environment within a cell. Therefore, GSH levels were measured using a colorimetric assay in primary mouse macrophages exposed to CSE. GSH levels were increased in response to prolonged (24 h) CSE exposure. This increase was both dose- and time-dependent (Fig. 1D).
Fig. 1.

Exposure to cigarette smoke alters redox environment in primary mouse macrophages. Primary bone marrow-derived mouse macrophages (BMMs) were treated with 0 or 10% CSE in medium without serum for 0, 30, 60, or 120 min (m). Cells were incubated in the dark with (A, B) 2 μM DCFDA or (C) 10 μM DHE for 15 min. Three representative images for each condition were obtained and mean fluorescence intensity was calculated. Images from one of three independent experiments are shown in (A) and include bright-field images (far left) to show representative confluency of the cell cultures. (D) BMMs were also treated with varying CSE concentrations (0, 2.5, 5, and 10%) at the following time points: 5 min, 4 h, 24 h. GSH concentration was measured using a colorimetric microplate assay and concentration values were normalized to protein. *p≤0.05, ***p≤0.001 compared to 0% CSE at the corresponding time point. Data represent means±SEM (N=3).
Cigarette smoke-induced recruitment of inflammatory cells was also assessed in mice exposed to cigarette smoke for 5 h/day, 5 days/week for 2 weeks (Fig. 2). Total cell counts in the BALF of mice exposed to 0, 25, or 100 μg/m3 total suspended particulates (TSP) were obtained. A TSP concentration of 100 μg/m3 induced a modest, but statistically significant (p≤0.05) increase in total cell count (Fig. 2A). In addition, the percentage of neutrophils in the BALF cell pellet was increased in mice exposed to cigarette smoke (Fig. 2B).
Fig. 2.
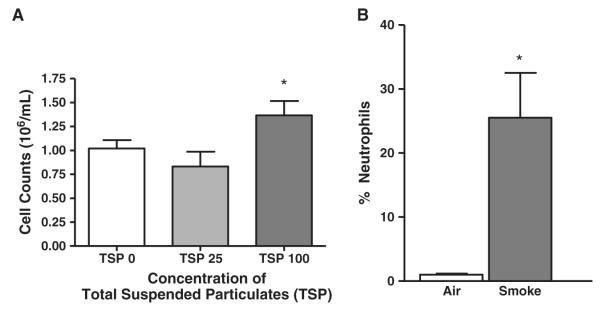
Mice exposed to cigarette smoke show increased recruitment of inflammatory cells. C57BL/6 wild-type mice were exposed to cigarette smoke in TE-10z smoking chambers for 5 h/day, 5 days/week for 2 weeks. The concentration of TSP was kept at approximately 0 (air control), 25 (low dose), or 100 μg/m3 (high dose). Mice were sacrificed and BALF was collected. (A) Total cell counts were obtained using a hemacytometer. (B) Cell pellets were collected from BALF and cytospin preparations were obtained. Total cell counts as well as differential cell counts were obtained from 10 images captured at random to determine the percentage of neutrophils for each condition (using an ImagePro algorithm). *p≤0.05 compared to air control condition. Data represent means±SEM (N=4 mice/group).
ECSOD attenuates cigarette smoke-induced oxidative stress in primary mouse macrophages
We used a SOD mimetic and ECSOD-overexpressing cells to investigate the protective role of the antioxidant in smoke-induced oxidative stress. Cultured mouse macrophages were treated for 60 min with the potent, small-molecule SOD mimetic MnTE-2PyP (30 μM), before incubation with 10% CSE for 60 min, and mean DCFDA fluorescence was assessed as a measure of oxidative stress. MnTE-2PyP significantly reduced the amount of measurable ROS induced by CSE in mouse macrophages (Fig. 3A). Additionally, the effect of ECSOD on smoke-induced oxidative stress was investigated by isolating primary bone marrow-derived macrophages from wild-type or ECSOD-overexpressing mice. Wild-type or transgenic cells were then exposed to 10% CSE for 60 min and mean DCFDA fluorescence was measured. Oxidative stress induced by CSE was markedly reduced in primary macrophages that overexpress ECSOD compared to those derived from wild-type mice (Figs. 3B and C).
Fig. 3.

Treatment with a SOD mimetic or overexpression of ECSOD blocks oxidative stress induced by cigarette smoke exposure in primary mouse macrophages. (A) Wild-type BMMs were treated with 30 μM MnTE-2Pyp (SOD mimetic) for 60 min before incubation with 10% CSE (where indicated) for an additional 60 min. (B and C) ECSOD-overexpressing (OE) BMMs were cultured and treated with 10% CSE for 0, 30, 60, or 120 min alongside BMMs isolated from wild-type mice. Three representative images for each condition were obtained and mean DCFDA fluorescence intensity was calculated. *p≤0.05, **p≤0.01 compared to 0% CSE at the corresponding time point. (D) 8-Isoprostane concentrations in BMMs that were exposed to 0 or 10% CSE for 2 h. ***p≤0.001 compared to air control for each condition (WT vs ECSOD). Data represent means±SEM (N=3).
Free 8-isoprostanes, a reliable oxidative stress marker, significantly increased in the medium after 2 h of exposure to 10% CSE; however, free 8-isoprostanes were not significantly different between wild-type and ECSOD-overexpressing BMMs (Fig. 3D).
ECSOD overexpression in these mice was controlled by an actin promoter, which allows for constitutive and ubiquitous expression of the enzyme in all cell types [28]. To verify that primary macrophages isolated from these mice did in fact show increased ECSOD expression compared to wild type, we performed immunostaining for ECSOD in lung tissue and Western blot analysis of medium from cultured BMMs. Intense staining for human ECSOD was observed in lung tissue sections of ECSOD-overexpressing (OE) mice. Positive staining was observed predominantly in macrophages and neutrophils from ECSOD-OE mice but was absent from the lungs of wild-type mice (Fig. 4A). Western blotting revealed expression of human ECSOD protein in medium from ECSOD-overexpressing BMMs versus wild-type cells. An approximately 32-kDa band was present in ECSOD-OE medium, but not in WT medium (Fig. 4B). These data are in concordance with previous work, which has found inflammatory cells to be a source of ECSOD [32].
Fig. 4.

ECSOD expression is increased in BMMs isolated from ECSOD-overexpressing mice compared to wild type. (A) Immunostaining for transgene-specific ECSOD from lung tissue sections reveals a pattern of predominantly positive human ECSOD staining in the macrophages and neutrophils (arrows) of the lung in (image A) the ECSOD-overexpressing mice (SOD3 gene controlled by a β-actin promoter), with absent staining in (image B) the wild-type mice. (B) Western blot showing expression of human ECSOD protein in medium from cultured BMMs isolated from mice that overexpress human ECSOD (OE) versus wild-type (WT) cells. The arrow shows the approximately 32-kDa band present in OE medium, but not in WT medium.
NOX enzymes act as an intrinsic source of ROS in an in vitro model of cigarette smoke-induced oxidative stress
To elucidate the endogenous source of free radical production in smoke-induced oxidative stress, we investigated the role of NOX enzymes. NOX inhibitors, diphenyleneiodonium (DPI) and apocynin, were used to determine the role of NOX activity in CSE-induced oxidative stress. Primary mouse macrophages were incubated for 60 min with DPI (5 μM) or apocynin (1 μM) followed by 10% CSE for an additional 60 min. Both DPI and apocynin decreased the amount of measurable DCFDA fluorescence elicited by CSE, although apocynin was more effective at reducing oxidative stress (Fig. 5A). Additionally, bone marrow-derived macrophages isolated from wild-type or NOX-deficient mice were exposed to 10% CSE for 60 min and oxidative stress was measured subsequently by DCFDA indicator dye fluorescence. NOX knockout macrophages showed almost completely reduced oxidative stress when exposed to CSE compared to wild-type cells (Figs. 5B and C).
Fig. 5.

Inhibition of NOX with an inhibitor or genetic knockout reduces oxidative stress induced by CSE treatment in primary mouse macrophages. (A) Wild-type BMMs were treated with 5 μM DPI or 1 μM apocynin for 60 min before incubation with 10% CSE (where indicated) for an additional 60 min. (B and C) BMMs isolated from NOX knockout mice were cultured and treated with 10% CSE for 0, 30, 60, or 120 min alongside BMMs isolated from wild-type mice. Three representative images for each condition were obtained and mean DCFDA fluorescence intensity was calculated. *p≤0.05, **p≤0.01 compared to 0% CSE at the corresponding time point. Data represent means±SEM (N=3).
Endogenous enzymes (ECSOD and NOX) regulate oxidative stress induced by cigarette smoke exposure in mice
We measured cytokine and antioxidant levels in BALF from wild-type and transgenic mice to determine the role of ECSOD and NOX enzymes in an intact animal model of smoke exposure. Macrophage-inflammatory protein-2 (MIP2) was measured in the BALF of wild-type and transgenic mice exposed to cigarette smoke for 5 h/day, 5 days/week for 2 weeks. MIP2 was increased in BALF from wild-type mice exposed to smoke compared to air control. However, in ECSOD-overexpressing mice (SPC promoter) and in NOX knockout mice, MIP2 levels remained similar in mice exposed to smoke versus air control (Fig. 6A). Interferon-γ (IFNγ) is another proinflammatory cytokine that was measured in wild-type and transgenic mice exposed to cigarette smoke for 2 weeks. IFNγ was also increased in response to smoke exposure, although this finding was not significant in our model. Levels of IFNγ remained steady in ECSOD-over-expressing mice or NOX KO mice exposed to cigarette smoke (Fig. 6B). Tumor necrosis factor-α levels were also measured in wild-type and transgenic mice, although no significant differences were observed with this amount of cigarette smoke exposure (data not shown).
Fig. 6.

ECSOD and NOX play regulatory roles in cigarette smoke-induced lung damage in vivo. Wild-type, ECSOD-overexpressing (SOD3 gene controlled by a SPC promoter), or NOX knockout mice were exposed to cigarette smoke in TE-10z smoking chambers for 5 h/day, 5 days/week for 2 weeks. (A and B) BALF was collected and cytokines (MIP2 and IFNγ) were measured from the first 1 ml of lavage fluid collected. (C) GSH concentration was also measured in the first 1 ml of lavage fluid collected using a colorimetric microplate assay. Data represent means±SEM (N=4 mice/group).
Levels of reduced GSH were increased in BALF of wild-type mice exposed to cigarette smoke compared to wild-type mice exposed to air. Interestingly, mice that overexpress ECSOD and, therefore, more efficiently scavenge excess O−2, did not show the compensatory increase in reduced GSH levels that was observed in wild-type mice. Similarly, mice that are deficient for NOX and, therefore, show reduced production of O−2, also showed significant attenuation of this compensatory antioxidant mechanism (Fig. 6C).
Oxidative damage to lung tissue from human patients with COPD
The effects of smoke exposure and disease state on redox environment were also measured in lysates from lung tissue obtained from human subjects to determine the relevance of oxidative stress in COPD. Lipid peroxidation, a marker of oxidative damage, was measured with a 4-HNE-specific antibody. Increased 4-HNE reactivity was observed in patients with moderate or severe COPD as opposed to those with mild COPD or without a diagnosis of disease (Fig. 7).
Fig. 7.
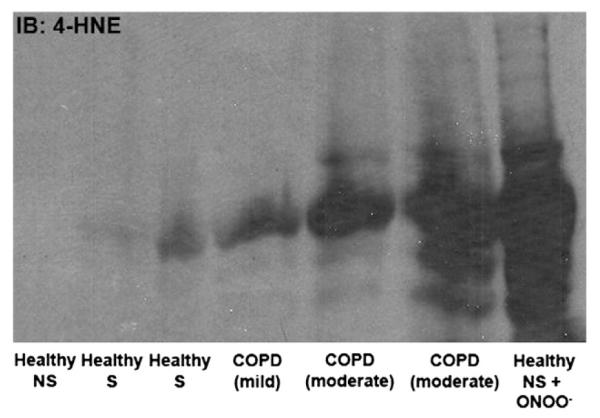
Markers of oxidative stress are correlated with severity of COPD in human subjects. Lysates were prepared from human lung lysate. Western blots were performed and blotted with a 4-HNE antibody conjugated to HRP. NS, nonsmoker; S, smoker.
Discussion
COPD is a devastating and increasingly prevalent disease that is closely associated with oxidative stress induced by chronic exposure to environmental stressors, especially cigarette smoke. Although tobacco smoke is highly concentrated in oxidants, not all individuals exposed to this hazard develop lung disease. Therefore, studying the endogenous enzymes that regulate cigarette smoke-induced oxidative stress may provide important clues to understand why some people are more susceptible to development of COPD than others. We hypothesize here that ECSOD may function to protect individuals from cigarette smoke exposure. This theory is substantiated by the observation that individuals with a particular ECSOD polymorphism (R213G) are 50% less likely to be diagnosed with COPD than control patients [14]. In the past 2 years there have been five publications implicating SOD3 polymorphisms as risk factors for lung disease [14,15,33–35]. There are several potential mechanisms that have been suggested to explain how ECSOD might protect individuals from diseases such as COPD. For example, ECSOD has been shown to modulate lung inflammation by preventing oxidative fragmentation of the extracellular matrix of the lung [17,36–38]. We were therefore interested to investigate the role of ECSOD in both inflammation and oxidative stress induced by cigarette smoke. NOX enzymes are also potential endogenous candidates for smoke-induced oxidative stress [39]. We therefore investigated the potential role of these enzymes in cigarette smoke exposure.
In this study, we show that exposure to cigarette smoke induces oxidative stress as measured by the fluorogenic indicator dyes DCFDA and DHE (Figs. 1A–C). Although these dyes have been used extensively to detect H2O2 and O−2, respectively, there is evidence to suggest that other ROS are capable of oxidizing the compounds, resulting in a fluorescent signal [40–42]. Therefore, we are careful to report the increased fluorescence observed here as a marker of global, cellular oxidative stress rather than an indication of the explicit species of free radical. Because ECSOD and NOX enzymes regulate O−2 specifically, and we show here that manipulation of these enzymes modulates oxidative stress, we hypothesize that O−2, in particular, is responsible for cigarette smoke-induced oxidative stress and inflammation that occur in our model. However, because of the highly reactive nature of O−2, we recognize the possibility that other ROS may be formed quickly and may also play an important role. Fluorogenic spin traps or other more specific methods will be employed in future studies to determine the actual species of ROS responsible for signaling sustained oxidative stress and inflammation within this smoking model. In addition, measuring reduced GSH levels provides a sensitive indication of changes in the redox environment, both intracellularly and extracellularly. Interestingly, GSH levels were increased in response to prolonged (24 h) CSE exposure in primary mouse macrophage cultures (Fig. 1D). This increase is probably a compensatory mechanism to offset the marked increase in ROS that are generated upon CSE exposure.
Discrepancies in the literature exist related to total TSP concentrations used in in vivo smoking experiments [43,44]. TSP concentrations were therefore assessed to determine an appropriate level to evoke an inflammatory response in the 2-week exposure time frame used in our model (Fig. 2A). Total cell counts in the BALF of mice exposed to 0, 25, or 100 μg/m3 TSP were measured and it was determined that a TSP of 100 μg/m3 was required to induce a measurable response (Fig. 2A). All subsequent experiments were performed with this TSP concentration (100 μg/m3). The percentage of neutrophils in the total cell count (BALF cell pellet) was increased in mice exposed to cigarette smoke, indicating that inflammatory cell recruitment signals were turned on. This suggests a potential role for NOX enzymes because they are abundantly expressed in neutrophils (Fig. 2B).
Because ECSOD is substantially expressed in the lung, its role as a protective antioxidant in smoking-induced lung disease is of primary interest in this field of research. Incubation with a SOD mimetic or overexpression of ECSOD sufficiently inhibited cigarette smoke-induced oxidative stress (Fig. 3). These data suggest that ECSOD activity may be an important host defense mechanism associated with prevention of COPD, despite exposure to the concentration of oxidants in cigarette smoke.
In addition to understanding the inherent antioxidant defense mechanisms in place to offset smoke-induced oxidative stress, the source of sustained ROS production is also of interest. Clearly, cigarette smoke exposure induces the recruitment of neutrophils to epithelial lining fluid in vivo (Fig. 2). Because of its high expression in this cell type, NOX activity is a primary candidate for the source of chronic oxidative stress associated with COPD. Inhibition of NOX with inhibitors (DPI and apocynin) showed protection against CSE-induced oxidative stress in primary mouse macrophages (Fig. 5A). Apocynin protected more completely than did DPI and this may be due, in part, to documented scavenging properties of apocynin and/or slight toxicity observed with DPI [45]. We proceeded to assess the effect of CSE in a NOX-deficient system, as superfluous activity of pharmacologic inhibitors (DPI and apocynin) could potentially have an effect on the measurable oxidative stress response [46]. Primary macrophages isolated from NOX knockout mice also exhibited decreased CSE-induced oxidative stress (Figs. 5B and C). These data show that NOX enzyme complexes play an important role in cigarette smoke-induced oxidative stress and suggest a role for the enzymes in COPD.
Interestingly, we did not find ECSOD overexpression to be protective against the formation of free 8-isoprostanes in primary BMMs (Fig. 3D). Based upon this observation, we conclude that cigarette smoke may induce oxidative stress initially at the level of the membrane via a pathway that is not mediated by ECSOD. However, additional data presented here suggest that subsequent oxidative signals are mediated via NOX and are protected against by ECSOD.
Although in vitro studies are useful to elucidate the specific molecules associated with cigarette smoke-induced oxidative stress, there are several changes observed in intact mice exposed to cigarette smoke which cannot necessarily be replicated in cell culture. Therefore, the effects of ECSOD and NOX were measured in vivo by exposing transgenic mice to cigarette smoke and comparing their response to that of wild-type mice. MIP2 is a proinflammatory cytokine that has been shown to increase upon exposure to cigarette smoke [47]. As expected, this cytokine was increased in response to cigarette smoke exposure in wild-type mice, but interestingly, was not altered by smoke exposure in ECSOD overexpressers or NOX KO mice (Fig. 6A). In addition, IFNγ was increased in wild-type mice exposed to CS but not in either of the transgenics (Fig. 6B). These data suggest that the inflammatory signals that regulate cytokine release are triggered by smoke-induced O−2 production.
In concordance with data previously reported [48], mice exposed to cigarette smoke showed increased levels of reduced GSH in their BALF. However, ECSOD-overexpressing and NOX KO mice did not exhibit the significant increase observed in wild-type mice (Fig. 6C). These data suggest that there is a O−2-dependent signal that induces increased GSH production in mice exposed to cigarette smoke.
4-HNE, an aldehyde by-product formed by free radical-induced lipid peroxidation, can react with proteins and is therefore utilized as a reliable marker of oxidative stress by immunoblot. Lung tissues from human patients with varying diagnoses of COPD severity were assessed for lipid peroxidation using this method (Fig. 7). 4-HNE levels correlated with severity of COPD. Interestingly, smoking status alone did not predict lipid peroxidation, which suggests that there is something inherently different in COPD patients that makes them susceptible to oxidative lung damage.
These data suggest that the mechanisms underlying the host defense against cigarette smoke-induced oxidative damage and subsequent development of COPD may include endogenous oxidases and antioxidant enzymes. Because not all chronic smokers develop disease, it is essential to understand the mechanisms of endogenous enzymes that regulate oxidative lung damage. Understanding why some individuals continue to produce free radicals and suffer oxidative damage even after smoking cessation, whereas others do not, can provide important insights and potential therapeutic targets for this devastating disease.
Acknowledgments
The authors thank Dr. James Crapo for his input to these studies and for providing the MnTE-2PyP SOD mimetic. In addition the authors thank Tina Watson for assistance with the manuscript, as well as David Goldstrahl and Robin Treuer, and for assisting with experiments. This work was supported by funds provided to Dr. Russ Bowler (Monfort Foundation, National Jewish Health; Flight Attendant Medical Research Institute; supported in part by Colorado CTSA Grant 1 UL1 RR025780 from NCRR/HIH), Dr. Dave Riches (HL55549, HL68628), and Dr. Donna Bratton (AIO58228 and HL34303).
Abbreviations
- 4-HNE
4-hydroxynonenal
- BAL
bronchoalveolar lavage
- BALF
bronchoalveolar lavage fluid
- BMM
bone marrow-derived macrophage
- COPD
chronic obstructive pulmonary disease
- CSE
cigarette smoke extract
- CSF-1
colony-stimulating factor-1
- DCFDA
5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate
- DHE
dihydroethidium
- DMEM
Dulbecco’s modified Eagle medium
- DPI
diphenyleneiodonium
- ECSOD
extracellular superoxide dismutase
- GSH
glutathione
- HRP
horseradish peroxidase
- IFNγ
interferon-γ
- KO
knockout
- MIP2
macrophage inflammatory protein-2
- NF-κB
nuclear factor-κB
- NHLBI
National Heart, Lung, and Blood Institute
- NOX
NADPH oxidase
- O−2
superoxide
- PBS
phosphate-buffered saline
- PVDF
polyvinylidene difluoride
- ROS
reactive oxygen species
- SEM
standard error of the mean
- SOD
superoxide dismutase
- SPC
surfactant protein C
- TNB
2-nitro-5-thiobenzoic acid
- TSP
total suspended particulates
References
- [1].Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic obstructive pulmonary disease surveillance—United States, 1971–2000. MMWR Surveill. Summ. 2002;51:1–16. [PubMed] [Google Scholar]
- [2].American Thoracic Society Cigarette smoking and health. Am. J. Respir. Crit. Care Med. 1996;153:861–865. doi: 10.1164/ajrccm.153.2.8564146. [DOI] [PubMed] [Google Scholar]
- [3].Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985;64:111–126. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McCord JM, Fridovich I. The utility of superoxide dismutase in studying free radical reactions. I. Radicals generated by the interaction of sulfite, dimethyl sulfoxide, and oxygen. J. Biol. Chem. 1969;244:6056–6063. [PubMed] [Google Scholar]
- [5].Oury TD, Chang LY, Marklund SL, Day BJ, Crapo JD. Immunocytochemical localization of extracellular superoxide dismutase in human lung. Lab. Invest. 1994;70:889–898. [PubMed] [Google Scholar]
- [6].Oury TD, Day BJ, Crapo JD. Extracellular superoxide dismutase in vessels and airways of humans and baboons. Free Radic. Biol. Med. 1996;20:957–965. doi: 10.1016/0891-5849(95)02222-8. [DOI] [PubMed] [Google Scholar]
- [7].Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic. Biol. Med. 2003;35:236–256. doi: 10.1016/s0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- [8].Svensk AM, Soini Y, Paakko P, Hiravikoski P, Kinnula VL. Differential expression of superoxide dismutases in lung cancer. Am. J. Clin. Pathol. 2004;122:395–404. doi: 10.1309/A45Q-HB0Q-RRX6-CT9A. [DOI] [PubMed] [Google Scholar]
- [9].Yoo DG, Song YJ, Cho EJ, Lee SK, Park JB, Yu JH, Lim SP, Kim JM, Jeon BH. Alteration of APE1/ref-1 expression in non-small cell lung cancer: the implications of impaired extracellular superoxide dismutase and catalase antioxidant systems. Lung Cancer. 2008;60:277–284. doi: 10.1016/j.lungcan.2007.10.015. [DOI] [PubMed] [Google Scholar]
- [10].Bowler RP, Nicks M, Warnick K, Crapo JD. Role of extracellular superoxide dismutase in bleomycin-induced pulmonary fibrosis. Am. J. Physiol. 2002;282:L719–L726. doi: 10.1152/ajplung.00058.2001. [DOI] [PubMed] [Google Scholar]
- [11].Kinnula VL, Hodgson UA, Lakari EK, Tan RJ, Sormunen RT, Soini YM, Kakko SJ, Laitinen TH, Oury TD, Paakko PK. Extracellular superoxide dismutase has a highly specific localization in idiopathic pulmonary fibrosis/usual interstitial pneumonia. Histopathology. 2006;49:66–74. doi: 10.1111/j.1365-2559.2006.02470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McCord JM, Gao B, Leff J, Flores SC. Neutrophil-generated free radicals: possible mechanisms of injury in adult respiratory distress syndrome. Environ. Health Perspect. 1994;102(Suppl. 10):57–60. doi: 10.1289/ehp.94102s1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gongora MC, Lob HE, Landmesser U, Guzik TJ, Martin WD, Ozumi K, Wall SM, Wilson DS, Murthy N, Gravanis M, Fukai T, Harrison DG. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. Am. J. Pathol. 2008;173:915–926. doi: 10.2353/ajpath.2008.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Juul K, Tybjaerg-Hansen A, Marklund S, Lange P, Nordestgaard BG. Genetically increased antioxidative protection and decreased chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006;173:858–864. doi: 10.1164/rccm.200509-1387OC. [DOI] [PubMed] [Google Scholar]
- [15].Dahl M, Bowler RP, Juul K, Crapo JD, Levy S, Nordestgaard BG. Superoxide dismutase 3 polymorphism associated with reduced lung function in two large populations. Am. J. Respir. Crit. Care Med. 2008;178:906–912. doi: 10.1164/rccm.200804-549OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ueda J, Starr ME, Takahashi H, Du J, Chang LY, Crapo JD, Evers BM, Saito H. Decreased pulmonary extracellular superoxide dismutase during systemic inflammation. Free Radic. Biol. Med. 2008;45:897–904. doi: 10.1016/j.freeradbiomed.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gao F, Koenitzer JR, Tobolewski JM, Jiang D, Liang J, Noble PW, Oury TD. Extracellular superoxide dismutase inhibits inflammation by preventing oxidative fragmentation of hyaluronan. J. Biol. Chem. 2008;283:6058–6066. doi: 10.1074/jbc.M709273200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. 2008;8:183–192. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- [19].Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J. Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- [20].Hunninghake GW, Crystal RG. Cigarette smoking and lung destruction: accumulation of neutrophils in the lungs of cigarette smokers. Am. Rev. Respir. Dis. 1983;128:833–838. doi: 10.1164/arrd.1983.128.5.833. [DOI] [PubMed] [Google Scholar]
- [21].Sato E, Koyama S, Takamizawa A, Masubuchi T, Kubo K, Robbins RA, Nagai S, Izumi T. Smoke extract stimulates lung fibroblasts to release neutrophil and monocyte chemotactic activities. Am. J. Physiol. 1999;277:L1149–L1157. doi: 10.1152/ajplung.1999.277.6.L1149. [DOI] [PubMed] [Google Scholar]
- [22].Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:S29–S32. doi: 10.1164/ajrccm.160.supplement_1.9. [DOI] [PubMed] [Google Scholar]
- [23].Gamble E, Grootendorst DC, Hattotuwa K, O’Shaughnessy T, Ram FS, Qiu Y, Zhu J, Vignola AM, Kroegel C, Morell F, Pavord ID, Rabe KF, Jeffery PK, Barnes NC. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur. Respir. J. 2007;30:467–471. doi: 10.1183/09031936.00013006. [DOI] [PubMed] [Google Scholar]
- [24].Montuschi P, Barnes PJ. Analysis of exhaled breath condensate for monitoring airway inflammation. Trends Pharmacol. Sci. 2002;23:232–237. doi: 10.1016/s0165-6147(02)02020-5. [DOI] [PubMed] [Google Scholar]
- [25].Louhelainen N, Rytila P, Haahtela T, Kinnula VL, Djukanovic R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulm. Med. 2009;9:25. doi: 10.1186/1471-2466-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bowler RP, Crapo JD. Oxidative stress in airways: is there a role for extracellular superoxide dismutase? Am. J. Respir. Crit. Care Med. 2002;166:S38–S43. doi: 10.1164/rccm.2206014. [DOI] [PubMed] [Google Scholar]
- [27].DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J. Leukoc. Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- [28].Oury TD, Ho YS, Piantadosi CA, Crapo JD. Extracellular superoxide dismutase, nitric oxide, and central nervous system O2 toxicity. Proc. Natl Acad. Sci. USA. 1992;89:9715–9719. doi: 10.1073/pnas.89.20.9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Riches DW, Underwood GA. Expression of interferon-beta during the triggering phase of macrophage cytocidal activation: evidence for an autocrine/paracrine role in the regulation of this state. J. Biol. Chem. 1991;266:24785–24792. [PubMed] [Google Scholar]
- [30].Thaikoottathil JV, Martin RJ, Zdunek J, Weinberger A, Rino JG, Chu HW. Cigarette smoke extract reduces VEGF in primary human airway epithelial cells. Eur. Respir. J. 2009;33:835–843. doi: 10.1183/09031936.00080708. [DOI] [PubMed] [Google Scholar]
- [31].Anderson ME. Determination of glutathione and glutathione disulfide in biological samples. Meth. Enzymol. 1985;113:548–555. doi: 10.1016/s0076-6879(85)13073-9. [DOI] [PubMed] [Google Scholar]
- [32].Tan RJ, Lee JS, Manni ML, Fattman CL, Tobolewski JM, Zheng M, Kolls JK, Martin TR, Oury TD. Inflammatory cells as a source of airspace extracellular superoxide dismutase after pulmonary injury. Am. J. Respir. Cell Mol. Biol. 2006;34:226–232. doi: 10.1165/rcmb.2005-0212OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Arcaroli JJ, Hokanson JE, Abraham E, Geraci M, Murphy JR, Bowler RP, Dinarello CA, Silveira L, Sankoff J, Heyland D, Wischmeyer P, Crapo JD. Extracellular superoxide dismutase haplotypes are associated with acute lung injury and mortality. Am. J. Respir. Crit. Care Med. 2009;179:105–112. doi: 10.1164/rccm.200710-1566OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ganguly K, Depner M, Fattman C, Bein K, Oury TD, Wesselkamper SC, Borchers MT, Schreiber M, Gao F, von Mutius E, Kabesch M, Leikauf GD, Schulz H. Superoxide dismutase 3, extracellular (SOD3) variants and lung function. Physiol. Genomics. 2009;37:260–267. doi: 10.1152/physiolgenomics.90363.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Young RP, Hopkins R, Black PN, Eddy C, Wu L, Gamble GD, Mills GD, Garrett JE, Eaton TE, Rees MI. Functional variants of antioxidant genes in smokers with COPD and in those with normal lung function. Thorax. 2006;61:394–399. doi: 10.1136/thx.2005.048512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yao H, Arunachalam G, Hwang JW, Chung S, Sundar IK, Kinnula VL, Crapo JD, Rahman I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA. 2010;107:15571–15576. doi: 10.1073/pnas.1007625107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Petersen SV, Oury TD, Ostergaard L, Valnickova Z, Wegrzyn J, Thogersen IB, Jacobsen C, Bowler RP, Fattman CL, Crapo JD, Enghild JJ, et al. Extracellular superoxide dismutase (EC-SOD) binds to type I collagen and protects against oxidative fragmentation. J. Biol. Chem. 2004;279:13705–13710. doi: 10.1074/jbc.M310217200. [DOI] [PubMed] [Google Scholar]
- [38].Bowler RP, Nicks M, Tran K, Tanner G, Chang LY, Young SK, Worthen GS. Extracellular superoxide dismutase attenuates lipopolysaccharide-induced neutrophilic inflammation. Am. J. Respir. Cell Mol. Biol. 2004;31:432–439. doi: 10.1165/rcmb.2004-0057OC. [DOI] [PubMed] [Google Scholar]
- [39].Cheng SE, Lee IT, Lin CC, Kou YR, Yang CM. Cigarette smoke particle-phase extract induces HO-1 expression in human tracheal smooth muscle cells: role of the c-Src/NADPH oxidase/MAPK/Nrf2 signaling pathway. Free Radic. Biol. Med. 2010;48:1410–1422. doi: 10.1016/j.freeradbiomed.2010.02.026. [DOI] [PubMed] [Google Scholar]
- [40].Wan CP, Myung E, Lau BH. An automated micro-fluorometric assay for monitoring oxidative burst activity of phagocytes. J. Immunol. Meth. 1993;159:131–138. doi: 10.1016/0022-1759(93)90150-6. [DOI] [PubMed] [Google Scholar]
- [41].Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat. Protoc. 2008;3:8–21. doi: 10.1038/nprot.2007.473. [DOI] [PubMed] [Google Scholar]
- [42].Myhre O, Andersen JM, Aarnes H, Fonnum F. Evaluation of the probes 2′, 7′-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem. Pharmacol. 2003;65:1575–1582. doi: 10.1016/s0006-2952(03)00083-2. [DOI] [PubMed] [Google Scholar]
- [43].Essenberg JM, Leavitt AM, Gaffney E. The effect of arsenic in tobacco on primary neoplasms of the lungs of albino mice. West. J. Surg. Obstet. Gynecol. 1956;64:35–36. [PubMed] [Google Scholar]
- [44].Witschi H. Tobacco smoke as a mouse lung carcinogen. Exp. Lung Res. 1998;24:385–394. doi: 10.3109/01902149809087375. [DOI] [PubMed] [Google Scholar]
- [45].Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- [46].Castor LR, Locatelli KA, Ximenes VF. Pro-oxidant activity of apocynin radical. Free Radic. Biol. Med. 2010;48:1636–1643. doi: 10.1016/j.freeradbiomed.2010.03.010. [DOI] [PubMed] [Google Scholar]
- [47].Smith KR, Uyeminami DL, Kodavanti UP, Crapo JD, Chang LY, Pinkerton KE. Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic. Biol. Med. 2002;33:1106–1114. doi: 10.1016/s0891-5849(02)01003-1. [DOI] [PubMed] [Google Scholar]
- [48].Qamar W, Sultana S. Farnesol ameliorates massive inflammation, oxidative stress and lung injury induced by intratracheal instillation of cigarette smoke extract in rats: an initial step in lung chemoprevention. Chem. Biol. Interact. 2008;176:79–87. doi: 10.1016/j.cbi.2008.08.011. [DOI] [PubMed] [Google Scholar]