

Background: The LPI-sensitive receptor GPR55 signals through Ca2+.
Results: Activation of sarcolemmal versus intracellular GPR55 mobilizes Ca2+ from distinct pools and associates with cardiomyocyte depolarization and hyperpolarization, respectively.
Conclusion: GPR55 location critically affects LPI-induced modulation of cardiomyocyte function.
Significance: We identify GPR55 as a new receptor regulating cardiac function at two cellular sites.
Keywords: Calcium Imaging, Calcium Intracellular Release, G Protein-coupled Receptors (GPCR), Heart, Lysosomes, Intracellular GPCR, Intracellular Microinjection, Membrane Potential, Ventricular Cardiomyocytes
Abstract
The L-α-lysophosphatidylinositol (LPI)-sensitive receptor GPR55 is coupled to Ca2+ signaling. Low levels of GPR55 expression in the heart have been reported. Similar to other G protein-coupled receptors involved in cardiac function, GPR55 may be expressed both at the sarcolemma and intracellularly. Thus, to explore the role of GPR55 in cardiomyocytes, we used calcium and voltage imaging and extracellular administration or intracellular microinjection of GPR55 ligands. We provide the first evidence that, in cultured neonatal ventricular myocytes, LPI triggers distinct signaling pathways via GPR55, depending on receptor localization. GPR55 activation at the sarcolemma elicits, on one hand, Ca2+ entry via L-type Ca2+ channels and, on the other, inositol 1,4,5-trisphosphate-dependent Ca2+ release. The latter signal is further amplified by Ca2+-induced Ca2+ release via ryanodine receptors. Conversely, activation of GPR55 at the membrane of intracellular organelles promotes Ca2+ release from acidic-like Ca2+ stores via the endolysosomal NAADP-sensitive two-pore channels. This response is similarly enhanced by Ca2+-induced Ca2+ release via ryanodine receptors. Extracellularly applied LPI produces Ca2+-independent membrane depolarization, whereas the Ca2+ signal induced by intracellular microinjection of LPI converges to hyperpolarization of the sarcolemma. Collectively, our findings point to GPR55 as a novel G protein-coupled receptor regulating cardiac function at two cellular sites. This work may serve as a platform for future studies exploring the potential of GPR55 as a therapeutic target in cardiac disorders.
Introduction
A putative role for GPR55 in physiology and disease is beginning to emerge. For instance, accumulating evidence supports its involvement in inflammatory and neuropathic pain (1), bone mass preservation (2), obesity (3), or cancer (4–7). Additional functional implications of GPR55 may be inferred from its widespread distribution throughout the body (8, 9), including the heart (8). Although it serves as target for several cannabinoid ligands, GPR55 appears to be endogenously activated by L-α-lysophosphatidylinositol (LPI)4 (10, 11). Because LPI is generated intracellularly, mainly by the Ca2+-dependent cytoplasmic phospholipase A2 (cPLA2) (12), and because modulation of this enzyme has important pathophysiological implications in the ischemic heart (13, 14), we hypothesized a role for LPI and GPR55 in the myocardium.
Cardiomyocyte physiology is critically regulated by Ca2+ and Ca2+-dependent electrical processes (15). Increases in intracellular Ca2+ concentration as well as alterations in ionic currents and membrane properties have been correlated previously with GPR55 activation (16–18). Considering the emerging paradigm of functional intracellular G protein-coupled receptors (GPCRs) (19, 20), which has also been suggested for the receptors of other lipid mediators (21) and for cannabinoid receptors (22–25), we postulated that a GPR55-dependent signaling pathway may be initiated either at the plasma membrane or within the cell. Thus, we used Ca2+ and voltage imaging and both extracellular and intracellular administration (microinjection) of GPR55 ligands to test the involvement of this receptor in the regulation of cardiomyocyte signaling.
EXPERIMENTAL PROCEDURES
Chemicals
LPI (soy, sodium salt) was purchased from Avanti Polar Lipids (Alabaster, AL), ryanodine and xestospongin C from EMD Millipore (Billerica, MA), ML-193 from Hit2Lead (San Diego, CA), and Ned-19 from Tocris Bioscience (R&D Systems, Minneapolis, MN). Unless stated otherwise, all other chemicals were from Sigma. LPI and ML-193 (100 mm stock solution) were dissolved in dimethyl sulfoxide.
Primary Neonatal Cardiomyocyte Culture
Cardiomyocyte cultures were prepared from 1- to 2-day-old Sprague-Dawley rats (Harlan Laboratories, Indianapolis, IN) by enzymatic digestion. Hearts were excised and placed in a sterile solution containing 116 mm NaCl, 20 mm HEPES, 0.08 mm Na2HPO4, 56 mm glucose, 5.4 mm KCl, and 0.8 mm MgSO4·7H2O (pH 7.35). Blood and connective tissues were removed and ventricles minced and subjected to 5–15 min of enzymatic digestion with collagenase II (Worthington, Lakewood, NJ) and pancreatin. Fibroblasts were removed by preplating for 2 h. Following enrichment, cardiomyocytes were cultured overnight in F-10 medium (Mediatech, Manassas, VA) containing 10% horse serum, 5% FBS, and 1% penicillin/streptomycin/amphotericin B solution (Invitrogen) at 37 °C in a humidified incubator with 5% CO2. Sera were attained from Gemini Bio-Products (West Sacramento, CA). The following day, medium was replaced with F-10 medium containing 5% FBS and 1% PSF.
Real-time PCR
Total RNA was isolated from isolated rat neonatal cardiomyocytes, cortex, and olfactory bulb using an RNeasy Midi kit or RNeasy Fibrous Tissue Midi kit with proteinase K and DNase I digestion (Qiagen). cDNA was synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosystems), and real-time PCR was performed with TaqMan® gene expression master mix (Applied Biosystems) using the StepOne Plus real-time PCR system (Applied Biosystems). The following primer were used: Rn03037213-s1 (GPR55) and Rn01775763-g1 (GAPDH for normalization). They were obtained from Applied Biosystems. All samples were run in triplicate, and data were analyzed using Applied Biosystems Comparative CT Method (ΔΔCT).
Immunocytochemistry and Confocal Imaging Studies
Cultured neonatal ventricular cardiomyocytes transiently transfected with GFP-tagged rat GPR55 receptor and Rab7-red fluorescent protein (RFP) (Addgene, Cambridge MA) 48 h earlier were fixed with paraformaldehyde 4%, washed in PBS, and mounted with DAPI Fluoromont G (Southern Biotech, Birmingham, AL). Cells were imaged using a Carl Zeiss 710 two-photon confocal microscope with a ×63 oil immersion objective using ×1 digital zoom with excitations set for DAPI, GFP, and DsRed at 405 nm, 488 nm, and 561 nm, respectively. Images were analyzed using Zen 2010 (Zeiss) as reported previously (26).
Calcium Imaging
Measurements of [Ca2+]i were performed as described previously (27–29). Cells were incubated with 5 μm Fura-2 AM (Invitrogen) in Hanks' balanced salt solution at room temperature for 45 min, in the dark, washed three times with dye-free Hanks' balanced salt solution, and then incubated for another 45 min to allow for complete de-esterification of the dye. Coverslips (25-mm diameter) were subsequently mounted in an open bath chamber (RP-40LP, Warner Instruments, Hamden, CT) on the stage of an inverted microscope (Nikon Eclipse TiE, Nikon Inc., Melville, NY). The microscope is equipped with a Perfect Focus System and a CoolSnap HQ2 CCD camera (Photometrics, Tucson, AZ). During the experiments, the Perfect Focus System was activated. Fura-2 AM fluorescence (emission = 510 nm), following alternate excitation at 340 and 380 nm, was acquired at a frequency of 0.25 Hz. Images were acquired and analyzed using NIS-Elements AR 3.1 software (Nikon Inc.). The ratio of the fluorescence signals (340/380 nm) was converted to Ca2+ concentrations (30). In Ca2+-free experiments, CaCl2 was omitted. In experiments using KCl, 50 mm NaCl was replaced by 50 mm KCl.
Intracellular Microinjection
Injections were performed using the Femtotips II, InjectMan NI2, and FemtoJet systems (Eppendorf) as reported previously (24, 28, 29). Pipettes were backfilled with an intracellular solution containing 110 mm KCl, 10 mm NaCl, 20 mm HEPES (pH 7.2), and dimethyl sulfoxide 0.004% (31) or the compounds to be tested. The injection time was 0.4 s at 60 hectopascals with a compensation pressure of 20 hectopascals to maintain the microinjected volume to less than 1% of cell volume, as measured by microinjection of a fluorescent compound (Fura-2 free acid) (31). The intracellular concentration of chemicals was determined on the basis of the concentration in the pipette and the volume of injection.
Measurement of Membrane Potential
The relative changes in membrane potential of single cardiomyocytes were evaluated using bis-(1,3-dibutylbarbituric acid) trimethine oxonol, bis-(1,3-dibutylbarbituric acid) trimethine oxonol, a slow-response, voltage-sensitive dye, as described previously (32–34). Upon membrane hyperpolarization, the dye concentrates in the cell membrane, leading to a decrease in fluorescence intensity, whereas depolarization induces the sequestration of the dye into the cytosol, resulting in an increase of the fluorescence intensity (35). Cultured ventricular cardiomyocytes were incubated for 30 min in Hanks' balanced salt solution containing 0.5 mm bis-(1,3-dibutylbarbituric acid) trimethine oxonol, and fluorescence was monitored at 0.17 Hz (excitation/emission 480 nm/540 nm). Calibration of bis-(1,3-dibutylbarbituric acid) trimethine oxonol fluorescence following background subtraction was performed using the Na+-K+ ionophore gramicidin in Na+-free physiological solution and various concentrations of K+ (to alter membrane potential) and N-methylglucamine (to maintain osmolarity) (35). Under these conditions, the membrane potential is approximately equal to the K+ equilibrium potential determined by the Nernst equation. The intracellular K+ and Na+ concentration were assumed to be 130 mm and 10 mm, respectively.
Electrophysiology
Briefly, pipettes were pulled from tubing with an outer diameter of 1.5 mm (Garner Glass, Claremont, CA). After fire polishing and backfilling, pipette resistances were ∼3–5 MΩ. Membrane potentials from rat neonatal myocytes were recorded using current-clamp configuration with Axopatch-200B patch-clamp amplifier (Axon Instruments, Foster City, CA) and pCLAMP 10 software. The pipette solution consisted of 125 mm KCl, 4 mm MgCl2, 0.06 mm CaCl2, 10 mm HEPES, 5 mm K-EGTA, 3 mm Na2ATP, and 5 mm Na2-creatine phosphate (pH 7.2). The external solution consisted of 132 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 1.8 mm MgCl2, 0.6 mm NaH2PO4, 7.5 mm HEPES, 7.5 mm Na-HEPES, and 5 mm glucose (pH 7.4).
Statistics
Data were expressed as mean ± S.E. One-way analysis of variance, followed by post hoc Bonferroni and Tukey tests, were used to assess significant differences between groups. Mann-Whitney U test was used for comparison of the traces shown in Fig. 4A. p < 0.05 was considered statistically significant.
FIGURE 4.

Ryanodine does not promote Ca2+ leakage from the SR in cultured neonatal cardiomyocytes. A, actual traces depicting the change in Fura-2 AM fluorescence ratio (340/380 nm) in cells treated with thapsigargin (TG, 1 μm in Ca2+-free saline) in the absence (n = 32 cells, top left panel) and presence of ryanodine (Ry, 10 μm, 30-min incubation, n = 29 cells, top right panel). Caffeine (Caff, 10 mm) did not modify basal Ca2+ levels in ryanodine-pretreated cells (n = 26 cells, bottom left panel). A comparison between the averaged traces obtained in the two treatment situations is depicted in the bottom right panel of A (p > 0.05, Mann Whitney U test). B, comparison of the increases in fluorescence ratio (340/380 nm) of Fura-2 AM-incubated neonatal cardiomyocytes in the treatment conditions mentioned in A. C, comparison between the times necessary to reach the maximal effect (τ) and the times in which the fluorescence ratio decreases to the half maximal level (τ/2) in the above-mentioned treatment cases. p > 0.05, one-way analysis of variance followed by Bonferroni and Tukey tests. D, comparison of the averaged areas under the curve of the traces presented in the top panels of A. p > 0.05.
RESULTS
Extracellular Administration of LPI Elicits GPR55-dependent Cytosolic Ca2+ Elevation in Ventricular Cardiomyocytes, Which Is Partially Contingent on Ca2+ Entry
To initially determine whether GPR55 activation can elevate cytosolic Ca2+ in cultured neonatal ventricular myocytes, the cells were stimulated with increasing concentrations of LPI in the absence and presence of a GPR55 antagonist. The cardiomyocytes responded to a bath application of LPI (0.1, 1 and 10 μm) with a concentration-dependent increase in [Ca2+]i of 12 ± 2.6 nm (n = 17 cells), 94 ± 3.8 nm (n = 23), and 727 ± 8.4 nm (n = 19). The effects reached statistical significance (p < 0.05) for concentrations of 1 and 10 μm (Fig. 1A). LPI induced a sustained Ca2+ response that was relatively slow in onset, with 1- to 2-min latency, and reached a maximum within 3–4 min (Fig. 1, B and C). Pretreatment with the GPR55 antagonist ML-193 (30 μm, 10 min) (36) significantly attenuated the effect of LPI (10 μm), supporting GPR55 dependence of the response (Δ[Ca2+]i was 21 ± 6.2 nm; n = 26; Fig. 1, B and C). In the presence of nifedipine (NIF), the effect of extracellular LPI (10 μm) was reduced to 348 ± 7.4 nm, indicating partial involvement of L-type Ca2+ channels (LTCC) (Fig. 1, A and B). Expression of GPR55 in rat neonatal cardiomyocytes was confirmed relative to positive control brain tissue samples via real-time PCR, revealing a substantially lower gene expression level than that found in either cortex or olfactory bulb tissue (Fig. 2), consistent with another report showing high levels of GPR55 expression in various brain regions and low GPR55 mRNA levels in the whole heart (8).
FIGURE 1.
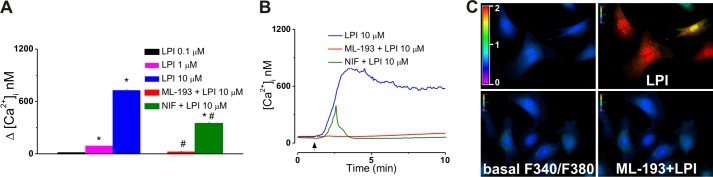
Sarcolemmal GPR55 activation triggers cytosolic Ca2+ increase in cultured neonatal ventricular cardiomyocytes. A, comparison of the increases in [Ca2+]i produced by extracellular administration of LPI (0.1, 1, and 10 μm) and LPI (10 μm) in the presence of GPR55 antagonist ML-193 (30 μm) or of the LTCC blocker nifedipine (NIF). The amplitude of [Ca2+]i was measured at the peak of the response. *, p < 0.05 compared with basal; #, p < 0.05 compared with 10 μm LPI . B, representative recordings of [Ca2+]i in response to LPI (10 μm) in the absence (blue) and presence of ML-193 (30 μm, red) or nifedipine (1 μm, green). C, Fura-2 AM fluorescence ratio (F340 nm/F380 nm) before (left panels) and after (right panels) bath application of LPI (10 μm) in the absence (top panels) and presence (bottom panels) of ML-193 (30 μm).
FIGURE 2.

GPR55 mRNA expression is detectable in rat neonatal cardiomyocytes. Relative expression (log10) of GPR55 mRNA in rat neonatal cortex (black bar) and rat neonatal olfactory bulb (gray bar) tissues with a known high expression of GPR55 in comparison to rat neonatal cardiomyocytes (white bar). Data are expressed as the mean relative quantitation (RQ) value ± RQmin and RQmax from each tissue performed in triplicate.
Activation of GPR55 at the Plasma Membrane Mobilizes Ca2+ from the sarcoplasmic reticulum
When cultured neonatal cardiomyocytes were incubated with Ca2+-free saline, extracellular application of LPI (10 μm) produced a transient increase in [Ca2+]i, by 314 ± 6.1 nm (n = 19 Fig. 3A, B), which was sensitive to IP3R blockade with heparin and xestospongin C (Δ[Ca2+]i = 8 ± 1 nm, n = 31). The response to LPI was largely unaffected by pretreatment with Ned-19 (1 μm, 15 min), which blocks two-pore channels (TPCs) and subsequent NAADP-dependent Ca2+ release from the endolysosomes (37) (Δ[Ca2+]i = 308 ± 6.7 nm, n = 16, Fig. 3). In the presence of ryanodine (10 μm, 30-min incubation), the response to LPI was reduced but not abolished (Δ[Ca2+]i = 126 ± 3.7 nm, n = 24), indicating an amplification of the IP3-dependent signal via Ca2+-induced Ca2+ release (CICR) and ryanodine receptors (RyR) (Fig. 3).
FIGURE 3.

Extracellular application of LPI mobilizes Ca2+ from internal stores via IP3R and RyR in cultured neonatal ventricular cardiomyocytes. A, representative traces illustrating the Ca2+ responses to bath application of LPI (10 μm) to ventricular myocytes incubated with Ca2+-free saline in the absence (top left panel) and presence of Ned-19 (inhibitor of lysosomal Ca2+ release, top right panel), xestospongin C (XeC), and heparin (Hep) (IP3R blockers, bottom left panel) or ryanodine (Ry, RyR antagonist, bottom right panel). B, comparison of the [Ca2+]i increases in response to extracellular administration of LPI (10 μm) in Ca2+ free saline in the absence and presence of the indicated antagonists. *, p < 0.05 compared with LPI.
Ryanodine inhibits Ca2+ mobilization either by blocking Ca2+ release channels or by promoting the leakage of Ca2+ from the SR (38). To distinguish between these possibilities, we treated neonatal cardiomyocytes with thapsigargin (1 μm in Ca2+-free saline), an inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase, conditions which deplete both ryanodine- and IP3-sensitive Ca2+ pools (39). As shown in Fig. 4, A–D, application of thapsigargin (1 μm) increased the fluorescence ratio (340/380 nm) of Fura-2 AM-loaded neonatal cardiomyocytes with similar amplitudes and kinetics in the absence and presence of ryanodine (10 μm, 30 min). Should ryanodine promote the basal Ca2+ leak from the SR, this would result in a reduced SR Ca2+ store level, further manifested as a significantly lower Ca2+ response to thapsigargin in ryanodine-treated versus untreated cells. Moreover, in Ca2+-free saline, neonatal cardiomyocytes pretreated with ryanodine (10 μm, 30 min) did not respond to caffeine (10 mm) with an increase in [Ca2+]i (Fig. 4, A and B). Caffeine is known to release intracellular Ca2+ by targeting RyRs in various cell types, including neonatal cardiomyocytes (40). In view of these findings, we conclude that the effects of ryanodine (10 μm, 30 min) observed in our study are due to inhibition of ryanodine receptors.
Several control experiments (n = 6 cells/experiment) were performed to ensure that inhibition of LPI-induced Ca2+ responses was due to ML-193 antagonism at GPR55 rather than off-target effects at Ca2+ entry or Ca2+ release channels. High concentrations of KCl produce depolarization-induced activation of LTCCs in cardiomyocytes (41). As shown in Fig. 5,A and B, the Ca2+ response of cultured neonatal ventricular cardiomyocytes to 50 mm KCl was largely identical in shape and amplitude in the absence (the Δ[Ca2+]i was 509 ± 6.7 nm) and presence of 30 μm ML-193 (the Δ[Ca2+]i was 523 ± 5.4 nm). Angiotensin II (100 nm), which activates IP3 production in the heart (42), also induced similar Ca2+ responses in the two paradigms: the Δ[Ca2+]i was 192 ± 2.7 nm in the absence of ML-193 and 188 ± 3.1 nm in the presence of ML-193 (30 μm) (Fig. 5, A and B). Cardiac activation of β-adrenoreceptors with isoproterenol has recently been shown to produce cyclic ADP-ribose and NAADP (43), second messengers that activate RyRs and TPCs, respectively (44, 45). Isoproterenol (100 nm) elevated the [Ca2+]i of neonatal ventricular myocytes with similar amplitudes in the absence (the Δ[Ca2+]i was 283 ± 4.8 nm) and presence of 30 μm ML-193 (the Δ[Ca2+]i was 267 ± 4.1 nm, Fig. 5, A and B). Thus, the absence of the LPI-induced Ca2+ response in cultured cardiomyocytes treated with ML-193 is not due to off-target effects at LTCCs, IP3Rs, RyRs, or TPCs.
FIGURE 5.

ML-193 has no off-target effects at LTCCs, IP3Rs, RyRs, or TPCs. A, representative Ca2+ responses of ventricular myocytes to KCl (50 mm, a depolarizing agent), angiotensin II (Ang II, 100 nm, activates IP3 production), and isoproterenol (Iso, 100 nm, activates formation of second messengers cyclic ADP-ribose and NAADP) in the absence and presence of ML-193 (30 μm). B, comparison of the [Ca2+]i increases in response to the mentioned compounds in the absence and presence of ML-193.
Microinjection of LPI Elevates the [Ca2+]i of Cultured Neonatal Ventricular Cardiomyocytes upon Activation of Intracellular GPR55
To examine the presence and functionality of intracellular GPR55 in cultured ventricular cardiomyocytes, cells underwent intracellular microinjection with LPI in the absence or presence of ML-193. The [Ca2+]i of ventricular myocytes was not modified significantly by control buffer microinjection (the Δ[Ca2+]i was 34 ± 6 nm; n = 6; Fig. 6, A–C), whereas intracellular administration of LPI (0.01, 0.1 and 1 μm) triggered [Ca2+]i elevations of 36 ± 4 nm, 83 ± 6.9 nm (p < 0.05), and 492 ± 7 nm (p < 0.05), respectively (n = 6 for each concentration tested, Fig. 6A). LPI produced a fast, robust, and transitory increase in [Ca2+]i (Fig. 6, B and D), sensitive to intracellular blockade of GPR55 with ML-193 (3 μm; the Δ[Ca2+]i was 57 ± 11 nm; n = 6; Fig. 6, A, B, and E). To exclude any possible involvement of plasmalemmal GPR55 in LPI-induced responses, ML-193 (30 μm, 1-min incubation) was also present in the extracellular solution. Blockade of intracellular GPR55 was a mandatory step to prevent the effect of microinjected LPI because the extent to which ML-193 crosses the sarcolemma even after 10 min of incubation is not sufficient to completely abolish the Ca2+ response of cultured neonatal ventricular myocytes to intracellular LPI. 10-min incubation with ML-193 reduces the LPI-induced effect on [Ca2+]i from 492 ± 7 nm to 213 ± 44 nm, n = 6 cells (Fig. 6, F and G). Conversely, a 10-min incubation with ML-193 was sufficient to block the effect of extracellular LPI (Fig. 1).
FIGURE 6.

Intracellular GPR55 stimulation elevates [Ca2+]i in cultured neonatal ventricular cardiomyocytes. A, comparison of the increases in [Ca2+]i produced by the microinjection of control buffer LPI (0.01, 0.1, and 1 μm) and of LPI (1 μm) in the presence of ML-193 (3 μm). *, p < 0.05 compared with control; #, p < 0.05 compared with 1 μm LPI . B, averaged Ca2+ responses (n = 6) of ventricular myocytes to intracellular administration of control buffer (black) or of LPI (1 μm) in the absence (blue) or presence of ML-193 (3 μm, red). C–E, typical fluorescence images of Fura-2 AM-loaded cardiomyocytes before (left panels), during (center panels), and 6 min after (right panels) intracellular administration of control buffer (C), 1 μm LPI alone (D), or in presence of ML-193 (E). The arrows indicate the injected cells. The fluorescence scale (0–3) is illustrated in each panel and magnified in the first panel of C. F, comparison of the increases in [Ca2+]i produced by microinjected LPI (1 μm) alone or after 10-min incubation with ML-193 (3 μm). G, averaged Ca2+ responses (n = 6 cells) of ventricular myocytes to intracellular administration of LPI (1 μm) in the absence (black) or presence of extracellular ML-193 (30 μm, 10-min incubation, red).
The Ca2+ Release Triggered by Intracellular Microinjection of LPI is NAADP-dependent
In the absence of extracellular Ca2+, intracellular injection of LPI (1 μm) into cultured ventricular myocytes elicited a fast and transient elevation of [Ca2+]i, by 477 ± 11 nm, insensitive to IP3R blockade with heparin and xestospongin C ([Ca2+]i = 468 ± 16 nm), diminished by ryanodine pretreatment (10 μm, 30 min, Δ[Ca2+]i = 184 ± 6 nm), and largely abolished in the presence of Ned-19 (1 μm, 15 min, Δ[Ca2+]i = 36 ± 7 nm) (Fig. 7, n = 6 cells/experiment). These findings suggest that NAADP-dependent lysosomal Ca2+ stores are critically involved in LPI signaling upon activation of intracellularly located GPR55. This signal is further amplified by CICR via RyR.
FIGURE 7.

LPI injection releases Ca2+ from acidic stores and sarcoplasmic reticulum in ventricular cardiomyocytes. A, averaged Ca2+ responses (n = 6) to LPI (1 μm) microinjection to ventricular myocytes incubated with Ca2+-free saline in the absence (top left panel) and presence of Ned-19 (inhibitor of lysosomal Ca2+ release, top right panel), xestospongin C (XeC), and heparin (Hep) (IP3R blockers, bottom left panel) or ryanodine (Ry, RyR antagonist, bottom right panel). B, comparison of the [Ca2+]i increases in response to intracellular administration of LPI (1 μm) in Ca2+ free saline in the absence and presence of the indicated antagonists. *, p < 0.05 compared with LPI.
Intracellular GPR55 Is Located at Endolysosomes in Neonatal Rat Cardiomyocytes
Extensive colocalization was observed when GFP-tagged GPR55 was coexpressed with RFP-tagged Rab7, a small GTPase associated with both endosomes and lysosomes (46). When cells where incubated with bafilomycin A1, a V-type ATPase that prevents lysosomal acidification (47), the GPR55 and Rab7 signal was abolished, confirming endolysosomal location of intracellular GPR55 (Fig. 8).
FIGURE 8.
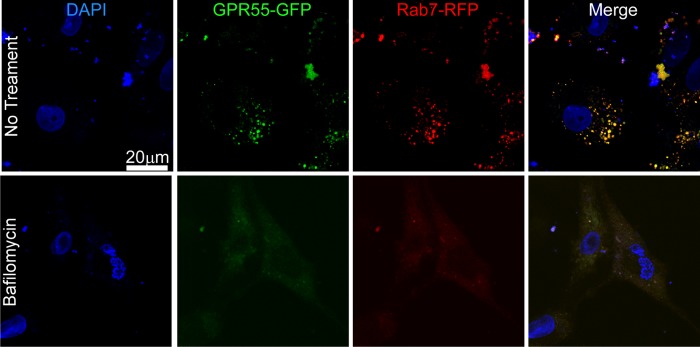
Intracellular GPR55 is endolysosomally located in rat neonatal cardiomyocytes. Top panels, extensive colocalization of GFP-tagged GPR55 and RFP-tagged Rab7, an endolysosomal marker, in cultured neonatal ventricular myocytes. The nuclei are labeled with DAPI. Bottom panels, lysosomal disruption with bafilomycin A1 markedly reduces immunostaining of the GPR55 and Rab7.
Antipodal Changes in Resting Membrane Potential in Response to Intra- versus Extracellular LPI Administration to Cultured Neonatal Ventricular Myocytes and Relation to LPI-induced Changes in [Ca2+]i
Cultured cardiomyocytes had a mean resting membrane potential of −72.7 ± 0.05 mV. An extracellular application of LPI (10 μm) produced a relatively long-lasting membrane depolarization with a mean amplitude of 12.6 ± 0.7 mV (n = 17) that was abolished by pretreatment with ML-193 (30 μm, ΔVm = 0.9 ± 0.3 mV, Fig. 9, A and B) (n = 11). Conversely, intracellular microinjection of LPI (1 μm) to ventricular myocytes resulted in a rapid, short-lived hyperpolarization with a mean amplitude of −7.3 ± 0.5 mV (Fig. 9, A and B) (n = 6). Similar to the plasma membrane-initiated response, the effect of microinjected LPI was sensitive to GPR55 blockade with ML-193 (3 μm). In the presence of the GPR55 antagonist, microinjection of LPI (1 μm) elicited an insignificant response (ΔVm = −1.2 ± 0.3 mV, n = 6) largely similar to that of control buffer microinjection (ΔVm = −0.9 ± 0.2 mV, n = 6, Fig. 9, A and B). To evaluate how the GPR55-dependent Ca2+ signaling correlates with the voltage changes observed in this study, cells were loaded with BAPTA-AM (20 μm, 20 min), a potent Ca2+ chelator. As expected, BAPTA-AM-treated cells did not elevate their [Ca2+]i in response to extracellular or intracellular administration of LPI. As shown in Fig. 9, C and D, incubation with BAPTA-AM markedly reduced the cardiomyocyte Ca2+ response to extracellularly applied LPI from 727 ± 8.4 nm to 14 ± 2.1 nm (n = 19) and that of microinjected LPI from 492 ± 7 nm (n = 6) to 11 ± 2.7 (n = 6 cells). However, even in the presence of BAPTA-AM, a bath application of LPI (10 μm) resulted in a depolarization of 12.79 ± 0.8 mV (n = 12) similar to that observed in the absence of BAPTA-AM pretreatment (Fig. 9, A and B). Conversely, LPI (1 μm) microinjection to BAPTA-AM-incubated neonatal cardiomyocytes did not significantly modify sarcolemmal potential (the ΔVm was −1.1 ± 0.3 mV; n = 9 cells; Fig. 9, A and B). In patch-clamp experiments, a bath application of LPI (10 μm) did not trigger action potential but produced a 10.2 ± 3.2 mV (n = 6 cells) depolarization of cardiomyocyte membrane similar to that observed using voltage-sensitive dyes (Fig. 9, E and F).
FIGURE 9.

Opposite membrane potential modifications in response to extracellular versus intracellular administration of LPI to neonatal ventricular cardiomyocytes. Relation to LPI-induced Ca2+ effects. A, representative recordings depicting membrane potential modification of ventricular myocytes upon bath application of LPI (10 μm) in the absence (blue) and presence of ML-193 (30 μm, red) of the Ca2+ chelator BAPTA-AM (20 μm, green) (left panel) or upon intracellular injection of control buffer (black) or LPI (1 μm) in the absence (blue) and presence of ML-193 (3 μm, red) or BAPTA-AM (20 μm, green) (right panel). Ca2+ chelation does not affect the depolarization induced by extracellular LPI but prevents the hyperpolarization triggered by microinjected LPI. This indicates Ca2+ dependence in the latter case but not in the former. B, extracellular application of LPI (10 μm) depolarized ventricular myocytes with a mean value of 12.6 ± 0.7 mV (n = 17), a response sensitive to GPR55 inhibition with ML-193 (30 μm) but not modified by BAPTA-AM (20 μm, left panel). Intracellular administration of LPI (1 μm) hyperpolarized ventricular myocytes with a mean value of −7.3 ± 0.5 mV (n = 6) in the absence but not in the presence of ML-193 (30 μm) or BAPTA-AM (20 μm, right panel). *, p < 0.05 compared with basal levels/control microinjection; #, p < 0.05 compared with LPI administration alone. C, representative recordings of [Ca2+]i in response to LPI (10 μm) in the absence (blue) and presence of BAPTA-AM (20 μm, green). D, comparison of the increases in [Ca2+]i produced by extracellular administration of LPI (10 μm) in the absence and presence of BAPTA-AM (20 μm). *, p < 0.05 compared with LPI alone. E, representative continuous current-clamp recording of a neonatal cardiomyocyte depolarized by bath-applied LPI (10 μm). F, in electrophysiological recordings, superfusion of LPI (10 μm) depolarized cultured neonatal ventricular myocytes with a mean amplitude of 10.2 ± 3.2 mV. *, p < 0.05 compared with basal levels.
DISCUSSION
GPCR signaling in the heart has important physiological and pathological implications (48). Cardiac cell processes such as contractility, response to ischemia, proliferation, apoptosis, and gene expression are GPCR-regulated, making GPCRs an important therapeutic target in a variety of heart diseases (49, 50). Importantly, in addition to the “classical” GPCRs involved in heart function, such as those for angiotensin II, endothelin 1, or catecholamines (48, 49), a wide variety of recently deorphanized GPCRs are expressed in cardiac cells (50). Defining the signaling pathways coupled with these newly recognized cardiac GPCRs may yield critical insights into cardiac cell function and is imperative if new strategies are to be employed in the treatment of heart disorders (48, 50).
The expression of the LPI/cannabinoid-sensitive receptor GPR55 has been detected in the whole heart by others (8) and in rat neonatal cardiomyocytes in this study, suggesting that GPR55 may play a role in cardiac cell function. Given that GPR55 is a Gq-coupled receptor that signals through Ca2+ (17, 51) and that Ca2+-dependent signaling is critical in cardiomyocytes (15, 52), in a first series of experiments we tested whether GPR55 activation at the sarcolemma elicits [Ca2+]i elevation. Indeed, extracellular application of LPI increased [Ca2+]i in cultured neonatal ventricular myocytes, a response completely sensitive to GPR55 antagonism.
In cardiomyocytes, Ca2+ signaling regulates the coupling of excitation with contraction and transcription (15, 52). In excitation-contraction coupling, membrane depolarization triggers Ca2+ entry through plasma membrane LTCC, which further induces CICR via RyRs and a global rise in [Ca2+]i that activates myofilaments to produce cardiac contraction. An increase in [Ca2+]i may also induce gene expression and transcription, eliciting hypertrophic responses, a process occurring on a longer time scale and known as excitation-transcription coupling (15). In both processes, a major role is played by Ca2+ release from internal stores (52), although it was shown recently that the latter is also triggered by Ca2+ influx (53). Thus, identification of the Ca2+ pools mobilized by LPI is critical for the characterization of the GPR55-initiated signaling pathways in ventricular cardiomyocytes.
We noticed that LTCC blockade reduced to half the effect of bath-applied LPI on [Ca2+]i. Moreover, the response was rather brief and no longer showed a plateau phase. Likewise, in the absence of extracellular Ca2+, LPI application induced a transient [Ca2+]i rise with a decreased amplitude. Thus, both Ca2+ entry and Ca2+ release occur downstream of sarcolemmal GPR55 activation in cultured neonatal ventricular cardiomyocytes. In addition to the endo/sarcoplasmic reticulum, which expresses Ca2+ release channels such as IP3R and RyR (54), the endolysosomal system also functions as a Ca2+ store involved in Ca2+ mobilization (45, 55).
Using a pharmacological approach, we demonstrate that Ca2+ release occurs primarily via IP3Rs and is further amplified by a RyR-dependent CICR mechanism, whereas the endolysosomal NAADP-dependent Ca2+ stores are not involved. The IP3 dependence of the response is not surprising because GPR55 has been shown to couple with Gq (51). Compared with RyR, IP3R expression is scarce in ventricular myocytes, indicating a rather minor physiological role. While IP3R activation may be beneficial in that it serves as an inotropic support for ventricles, its predominant effect is to promote arrhythmias (56, 57). To explain the increased arrhythmogenicity of IP3R stimulation, it has been suggested that Ca2+ release via IP3Rs may activate additional currents such as the store-operated Ca2+ currents or the Na+/Ca2+ exchange current (56). These currents may be partially responsible for the plateau phase of the Ca2+ response elicited by extracellular application of LPI to cultured neonatal ventricular myocytes incubated with Ca2+-containing saline. On a different note, studies on adult and neonatal cardiomyocytes correlated both Ca2+ influx via LTCC (53) and the IP3-dependent Ca2+ release with cardiac hypertrophy and heart failure (52, 57–59), suggesting that sarcolemmal GPR55 activation may produce hypertrophic signals.
Because cardiovascular cell function is also regulated by intracellularly expressed GPCRs (60, 61) and preliminary data from our laboratory suggested functionality of intracellular GPR55 (23), we evaluated the effect of LPI microinjection in cultured neonatal ventricular myocytes. Similar to the response elicited at the plasma membrane, the [Ca2+]i elevation triggered by intracellular injection of LPI was GPR55-dependent. However, the Ca2+ response pattern was strikingly different in the two paradigms. Extracellular administration of LPI resulted in a relatively slow response that reached a peak within 1–2 min and continued with a plateau phase, whereas LPI microinjection induced a fast and transitory effect similar to that reported upon activation of other intracellular GPCRs, such as those for angiotensin II (29, 33, 62) or endothelin 1 (28). We have noticed likewise discrepancies in the plasmalemmal-initiated versus intracellularly initiated Ca2+ responses by the G protein-coupled estrogen receptor GPER/GPR30 (34, 63), which is reportedly expressed at both sites (64, 65). LPI is a lipid messenger, expected to get partitioned in the lipid bilayer and reach the other side of the sarcolemma. However, LPI cannot passively diffuse across the plasma membrane. Rather, after its intracellular synthesis by the phospholipase A2, LPI is pumped out of the cell via the multidrug resistance-associated protein 1 (Mrp1/ABCC1) (7). Mrp1 is also expressed in the sarcolemma of rodent heart (66, 67).
Furthermore, we found that the [Ca2+]i rise produced by intracellular administration of LPI is completely contingent on Ca2+ mobilization from internal stores. Importantly, the Ca2+ is released from the endolysosomes via the NAADP-sensitive TPCs (45, 55), a signal further amplified by RyR-dependent CICR from the sarcoplasmic reticulum. NAADP-dependent Ca2+ release from acidic stores has been shown to elevate the sarcoplasmic reticulum Ca2+ load, thus enhancing cardiomyocyte contraction (68). Moreover, β-adrenoreceptors in ventricular myocytes can signal through NAADP (43, 68). In line with these observations, intracellular GPR55 may play a role in the regulation of cardiomyocyte contraction.
The accumulating evidence pointing to endolysosomes as intracellular locations where GPCRs initiate signaling (19, 24, 28, 69) prompted us to probe for colocalization of GFP-tagged GPR55 and RFP-tagged Rab7, an endolysosomal-associated small GTPase (46). We noticed extensive colocalization of GPR55 and Rab7. In addition, lysosomal disruption with bafilomycin A1 markedly reduced the fluorescence intensity of both GPR55 and Rab7, suggesting that GPR55 localizes intracellularly at the endolysosomes. GPR55 localization at this site correlates well with its ability to trigger NAADP-dependent Ca2+ responses because NAADP-generating enzymes (70, 71) have been also reported to localize at the membrane of acidic organelles (72).
An important discrepancy in the responses initiated by sarcolemmal versus intracellular GPR55 stimulation is that the former depolarized, whereas the latter hyperpolarized the membrane of neonatal ventricular cardiomyocytes. The depolarizing effect of bath-applied LPI was independent of Ca2+ signaling, whereas LPI induced GPR55-dependent Ca2+ influx via LTCC. A direct effect of LPI on LTCC was considered less likely because it has been reported that, similar to other phospholipids, LPI slightly suppresses LTCC activity (73). The LPI-induced depolarization was not large enough to generate action potentials and, thus, activate LTCC. Rather, the explanation of the LPI-induced effects sits in the fact that cardiac LTCC can be directly activated by Gq (74) because GPR55 is Gq-coupled (17). Conversely, hyperpolarization in response to intracellular GPR55 activation was short-lived and Ca2+-dependent. Ca2+ extrusion is a fast process, explaining why the hyperpolarization is transient. Similarly, intracellular microinjection of an acknowledged intracrine, angiotensin II, hyperpolarizes ventricular myocytes via intracellular AT1 receptors (75). Although hyperpolarization may be beneficial in that it may prevent arrhythmias, it could also trigger If/Ih. If/Ih is typically a Na+/K+ inward current associated with hyperpolarization-activated cyclic nucleotide-gated channels, playing a pivotal role in cardiac pacemaking but associated with cardiac hypertrophy and failure when expressed in the ventricles (76). LPI has been shown to have GPR55-independent hyperpolarizing effects on the endothelial cell membrane (77). However, in the presence of the GPR55 antagonist ML-193 (36), LPI administration to neonatal ventricular cardiomyocytes produced non-significant responses, supporting GPR55-dependence of the LPI effect in our paradigm.
In animal models of ischemia-reperfusion injury, deletion of the main LPI-synthesizing enzyme cPLA2α has been shown to be either cardioprotective (13) or deleterious to the heart (14). In light of our current findings, the discrepancy between these results may be correlated with the ability of LPI to activate GPR55 either at the sarcoplasmic membrane or within the cardiomyocyte and trigger distinct mechanisms with opposite outcomes.
Collectively, our study shows for the first time that the LPI-responsive receptor GPR55 is involved in the regulation of cardiomyocyte Ca2+ signaling. Furthermore, functional GPR55 can elicit signaling both at the sarcolemma and at the endolysosomal compartment, and, as shown in Fig. 10, the downstream cascades are distinct. Activation of plasmalemmal GPR55 triggers Ca2+ influx through LTCC and IP3-dependent Ca2+ release that is amplified by CICR via RyR. Independently of this Ca2+ signaling cascade, plasmalemmal GPR55 couples to depolarization of the cardiomyocyte membrane. Intracellular GPR55 activation associates with NAADP-dependent Ca2+ mobilization from acidic-like Ca2+ stores, which is amplified by CICR via RyR, converging to membrane hyperpolarization. This work may serve as a platform for future studies exploring the potential of GPR55 as a therapeutic target in cardiac disorders.
FIGURE 10.
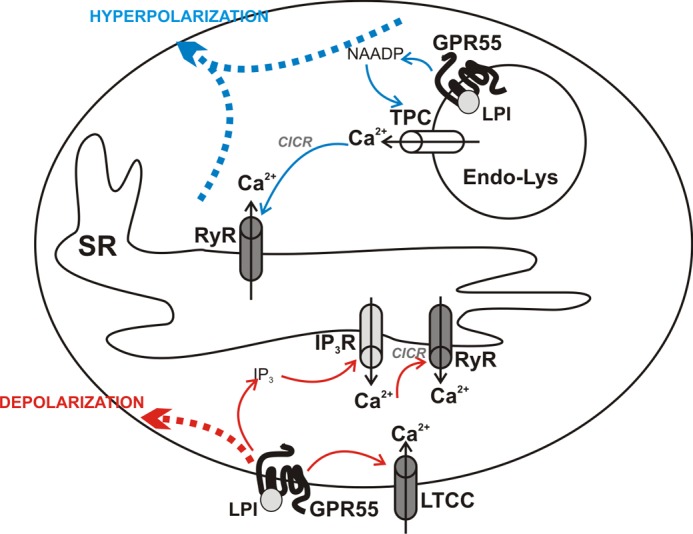
Proposed mechanisms initiated by LPI at the sarcolemma or within the neonatal ventricular cardiomyocyte via GPR55. LPI activates GPR55 located on the membrane of endolysosomes (Endo-Lys) to promote NAADP-dependent Ca2+ release from these organelles, which further induces CICR via RyRs located at the SR and converging to membrane hyperpolarization (top, blue). GPR55 stimulation at the sarcoplasmic membrane results, on one hand, in Ca2+ entry through LTCCs and, on the other, in IP3-dependent Ca2+ release from the intracellular stores, a signal further amplified by CICR via RyRs. Independently of Ca2+ signaling, GPR55 activation results in membrane depolarization (bottom, red).
This work was supported, in whole or in part, by National Institutes of Health Grants HL090804 (to E. B.), HL105414 (to D. G. T.), HL086699 (to M. M.), and DA023204 (to M. E. A.).
- LPI
- L-α-lysophosphatidylinositol
- GPCR
- G protein-coupled receptor
- LTCC
- L-type Ca2+ channel
- IP3R
- inositol 1,4,5-trisphosphate receptor
- TPC
- two-pore channel
- CICR
- Ca2+-induced Ca2+ release
- RyR
- ryanodine receptor
- SR
- sarcoplasmic reticulum
- IP3
- inositol 1,4,5-trisphosphate
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- [Ca2+]i
- intracellular concentration of Ca2+ ions
- NAADP
- nicotinic acid adenine dinucleotide phosphate
- RFP
- red fluorescent protein.
REFERENCES
- 1. Staton P. C., Hatcher J. P., Walker D. J., Morrison A. D., Shapland E. M., Hughes J. P., Chong E., Mander P. K., Green P. J., Billinton A., Fulleylove M., Lancaster H. C., Smith J. C., Bailey L. T., Wise A., Brown A. J., Richardson J. C., Chessell I. P. (2008) The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain 139, 225–236 [DOI] [PubMed] [Google Scholar]
- 2. Whyte L. S., Ryberg E., Sims N. A., Ridge S. A., Mackie K., Greasley P. J., Ross R. A., Rogers M. J. (2009) The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 16511–16516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moreno-Navarrete J. M., Catalán V., Whyte L., Díaz-Arteaga A., Vázquez-Martínez R., Rotellar F., Guzmán R., Gómez-Ambrosi J., Pulido M. R., Russell W. R., Imbernón M., Ross R. A., Malagón M. M., Dieguez C., Fernández-Real J. M., Frühbeck G., Nogueiras R. (2012) The L-α-lysophosphatidylinositol/GPR55 system and its potential role in human obesity. Diabetes 61, 281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andradas C., Caffarel M. M., Pérez-Gómez E., Salazar M., Lorente M., Velasco G., Guzmán M., Sánchez C. (2011) The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 30, 245–252 [DOI] [PubMed] [Google Scholar]
- 5. Ford L. A., Roelofs A. J., Anavi-Goffer S., Mowat L., Simpson D. G., Irving A. J., Rogers M. J., Rajnicek A. M., Ross R. A. (2010) A role for L-α-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br. J. Pharmacol. 160, 762–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hu G., Ren G., Shi Y. (2011) The putative cannabinoid receptor GPR55 promotes cancer cell proliferation. Oncogene 30, 139–141 [DOI] [PubMed] [Google Scholar]
- 7. Piñeiro R., Maffucci T., Falasca M. (2011) The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 30, 142–152 [DOI] [PubMed] [Google Scholar]
- 8. Henstridge C. M., Balenga N. A., Kargl J., Andradas C., Brown A. J., Irving A., Sanchez C., Waldhoer M. (2011) Minireview. Recent developments in the physiology and pathology of the lysophosphatidylinositol-sensitive receptor GPR55. Mol. Endocrinol. 25, 1835–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryberg E., Larsson N., Sjögren S., Hjorth S., Hermansson N. O., Leonova J., Elebring T., Nilsson K., Drmota T., Greasley P. J. (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 152, 1092–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henstridge C. M. (2012) Off-target cannabinoid effects mediated by GPR55. Pharmacology 89, 179–187 [DOI] [PubMed] [Google Scholar]
- 11. Zhao P., Abood M. E. (2012) GPR55 and GPR35 and their relationship to cannabinoid and lysophospholipid receptors. Life Sci. 92, 453–457 [DOI] [PubMed] [Google Scholar]
- 12. Piñeiro R., Falasca M. (2012) Lysophosphatidylinositol signalling. New wine from an old bottle. Biochim. Biophys. Acta 1821, 694–705 [DOI] [PubMed] [Google Scholar]
- 13. Kerkelä R., Boucher M., Zaka R., Gao E., Harris D., Piuhola J., Song J., Serpi R., Woulfe K. C., Cheung J. Y., O'Leary E., Bonventre J. V., Force T. (2011) Cytosolic phospholipase A(2)α protects against ischemia/reperfusion injury in the heart. Clin. Transl. Sci. 4, 236–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saito Y., Watanabe K., Fujioka D., Nakamura T., Obata J. E., Kawabata K., Watanabe Y., Mishina H., Tamaru S., Kita Y., Shimizu T., Kugiyama K. (2012) Disruption of group IVA cytosolic phospholipase A(2) attenuates myocardial ischemia-reperfusion injury partly through inhibition of TNF-α-mediated pathway. Am. J. Physiol. Heart Circ. Physiol. 302, H2018–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bers D. M., Guo T. (2005) Calcium signaling in cardiac ventricular myocytes. Ann. N.Y. Acad. Sci. 1047, 86–98 [DOI] [PubMed] [Google Scholar]
- 16. Bondarenko A., Waldeck-Weiermair M., Naghdi S., Poteser M., Malli R., Graier W. F. (2010) GPR55-dependent and -independent ion signalling in response to lysophosphatidylinositol in endothelial cells. Br. J. Pharmacol. 161, 308–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lauckner J. E., Jensen J. B., Chen H. Y., Lu H. C., Hille B., Mackie K. (2008) GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. U.S.A. 105, 2699–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Waldeck-Weiermair M., Zoratti C., Osibow K., Balenga N., Goessnitzer E., Waldhoer M., Malli R., Graier W. F. (2008) Integrin clustering enables anandamide-induced Ca2+ signaling in endothelial cells via GPR55 by protection against CB1-receptor-triggered repression. J. Cell Sci. 121, 1704–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Irannejad R., Tomshine J. C., Tomshine J. R., Chevalier M., Mahoney J. P., Steyaert J., Rasmussen S. G., Sunahara R. K., El-Samad H., Huang B., von Zastrow M. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jalink K., Moolenaar W. H. (2010) G protein-coupled receptors. The inside story. BioEssays 32, 13–16 [DOI] [PubMed] [Google Scholar]
- 21. Zhu T., Gobeil F., Vazquez-Tello A., Leduc M., Rihakova L., Bossolasco M., Bkaily G., Peri K., Varma D. R., Orvoine R., Chemtob S. (2006) Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors. A paradigm based on PGE2, PAF, and LPA1 receptors. Can. J. Physiol. Pharmacol. 84, 377–391 [DOI] [PubMed] [Google Scholar]
- 22. Bénard G., Massa F., Puente N., Lourenço J., Bellocchio L., Soria-Gómez E., Matias I., Delamarre A., Metna-Laurent M., Cannich A., Hebert-Chatelain E., Mulle C., Ortega-Gutiérrez S., Martín-Fontecha M., Klugmann M., Guggenhuber S., Lutz B., Gertsch J., Chaouloff F., López-Rodríguez M. L., Grandes P., Rossignol R., Marsicano G. (2012) Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat. Neurosci. 15, 558–564 [DOI] [PubMed] [Google Scholar]
- 23. Brailoiu E., Brailoiu G. C., Deliu E., Sharir H., Zhao P., Abood M. E. (2012) 22nd Annual Symposium on the Cannabinoids, p. 25, International Cannabinoid Research Society, Research Triangle Park, NC [Google Scholar]
- 24. Brailoiu G. C., Oprea T. I., Zhao P., Abood M. E., Brailoiu E. (2011) Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J. Biol. Chem. 286, 29166–29174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. den Boon F. S., Chameau P., Schaafsma-Zhao Q., van Aken W., Bari M., Oddi S., Kruse C. G., Maccarrone M., Wadman W. J., Werkman T. R. (2012) Excitability of prefrontal cortical pyramidal neurons is modulated by activation of intracellular type-2 cannabinoid receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 3534–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kato K., Lillehoj E. P., Park Y. S., Umehara T., Hoffman N. E., Madesh M., Kim K. C. (2012) Membrane-tethered MUC1 mucin is phosphorylated by epidermal growth factor receptor in airway epithelial cells and associates with TLR5 to inhibit recruitment of MyD88. J. Immunol. 188, 2014–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brailoiu G. C., Arterburn J. B., Oprea T. I., Chitravanshi V. C., Brailoiu E. (2013) Bradycardic effects mediated by activation of G protein-coupled estrogen receptor in rat nucleus ambiguus. Exp. Physiol. 98, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deliu E., Brailoiu G. C., Mallilankaraman K., Wang H., Madesh M., Undieh A. S., Koch W. J., Brailoiu E. (2012) Intracellular endothelin type B receptor-driven Ca2+ signal elicits nitric oxide production in endothelial cells. J. Biol. Chem. 287, 41023–41031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deliu E., Tica A. A., Motoc D., Brailoiu G. C., Brailoiu E. (2011) Intracellular angiotensin II activates rat myometrium. Am. J. Physiol. Cell Physiol. 301, C559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 31. Guse A. H., Berg I., da Silva C. P., Potter B. V., Mayr G. W. (1997) Ca2+ entry induced by cyclic ADP-ribose in intact T-lymphocytes. J. Biol. Chem. 272, 8546–8550 [DOI] [PubMed] [Google Scholar]
- 32. Brailoiu G. C., Brailoiu E., Chang J. K., Dun N. J. (2008) Excitatory effects of human immunodeficiency virus 1 Tat on cultured rat cerebral cortical neurons. Neuroscience 151, 701–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brailoiu G. C., Gurzu B., Gao X., Parkesh R., Aley P. K., Trifa D. I., Galione A., Dun N. J., Madesh M., Patel S., Churchill G. C., Brailoiu E. (2010) Acidic NAADP-sensitive calcium stores in the endothelium. Agonist-specific recruitment and role in regulating blood pressure. J. Biol. Chem. 285, 37133–37137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tica A. A., Dun E. C., Tica O. S., Gao X., Arterburn J. B., Brailoiu G. C., Oprea T. I., Brailoiu E. (2011) G protein-coupled estrogen receptor 1-mediated effects in the rat myometrium. Am. J. Physiol. Cell Physiol. 301, C1262–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bräuner T., Hülser D. F., Strasser R. J. (1984) Comparative measurements of membrane potentials with microelectrodes and voltage-sensitive dyes. Biochim. Biophys. Acta 771, 208–216 [DOI] [PubMed] [Google Scholar]
- 36. Heynen-Genel S., Dahl R., Shi S., Milan L., Hariharan S., Sergienko E., Hedrick M., Dad S., Stonich D., Su Y., Vicchiarelli M., Mangravita-Novo A., Smith L. H., Chung T. D. Y., Sharir H., Caron M. G., Barak L. S., Abood M. E. (2010) Screening for selective ligands for GPR55 antagonists in Probe Reports from the NIH Molecular Libraries Program (Internet), National Center for Biotechnology Information, Bethesda, MD: [PubMed] [Google Scholar]
- 37. Naylor E., Arredouani A., Vasudevan S. R., Lewis A. M., Parkesh R., Mizote A., Rosen D., Thomas J. M., Izumi M., Ganesan A., Galione A., Churchill G. C. (2009) Identification of a chemical probe for NAADP by virtual screening. Nat. Chem. Biol. 5, 220–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vites A. M., Pappano A. J. (1994) Distinct modes of inhibition by ruthenium red and ryanodine of calcium-induced calcium release in avian atrium. J. Pharmacol. Exp. Ther. 268, 1476–1484 [PubMed] [Google Scholar]
- 39. Stutzmann G. E., Mattson M. P. (2011) Endoplasmic reticulum Ca2+ handling in excitable cells in health and disease. Pharmacol. Rev. 63, 700–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Janowski E., Berríos M., Cleemann L., Morad M. (2010) Developmental aspects of cardiac Ca2+ signaling. Interplay between RyR- and IP(3)R-gated Ca2+ stores. Am. J. Physiol. Heart Circ. Physiol. 298, H1939–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saini H. K., Dhalla N. S. (2006) Modification of intracellular calcium concentration in cardiomyocytes by inhibition of sarcolemmal Na+/H+ exchanger. Am. J. Physiol. Heart Circ Physiol. 291, H2790–2800 [DOI] [PubMed] [Google Scholar]
- 42. Goutsouliak V., Rabkin S. W. (1998) Comparison of angiotensin II type-1 and type-2 receptor antagonists on angiotensin II-induced IP3 generation in cardiomyocytes. Gen. Pharmacol. 30, 367–372 [DOI] [PubMed] [Google Scholar]
- 43. Lewis A. M., Aley P. K., Roomi A., Thomas J. M., Masgrau R., Garnham C., Shipman K., Paramore C., Bloor-Young D., Sanders L. E., Terrar D. A., Galione A., Churchill G. C. (2012) β-Adrenergic receptor signaling increases NAADP and cADPR levels in the heart. Biochem. Biophys. Res. Commun. 427, 326–329 [DOI] [PubMed] [Google Scholar]
- 44. Lee H. C. (2001) Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu. Rev. Pharmacol. Toxicol. 41, 317–345 [DOI] [PubMed] [Google Scholar]
- 45. Patel S., Marchant J. S., Brailoiu E. (2010) Two-pore channels. Regulation by NAADP and customized roles in triggering calcium signals. Cell Calcium 47, 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang T., Ming Z., Xiaochun W., Hong W. (2011) Rab7. Role of its protein interaction cascades in endo-lysosomal traffic. Cell. Signal. 23, 516–521 [DOI] [PubMed] [Google Scholar]
- 47. Bowman E. J., Siebers A., Altendorf K. (1988) Bafilomycins. A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. U.S.A. 85, 7972–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salazar N. C., Chen J., Rockman H. A. (2007) Cardiac GPCRs. GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta 1768, 1006–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang M., Chung K. Y., Walker J. W. (2007) G-protein coupled receptor signaling in myocardium. Not for the faint of heart. Physiology 22, 174–184 [DOI] [PubMed] [Google Scholar]
- 50. Tang C. M., Insel P. A. (2004) GPCR expression in the heart. “New” receptors in myocytes and fibroblasts. Trends Cardiovasc. Med. 14, 94–99 [DOI] [PubMed] [Google Scholar]
- 51. Sharir H., Abood M. E. (2010) Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol. Ther. 126, 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bers D. M. (2008) Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49 [DOI] [PubMed] [Google Scholar]
- 53. Gao H., Wang F., Wang W., Makarewich C. A., Zhang H., Kubo H., Berretta R. M., Barr L. A., Molkentin J. D., Houser S. R. (2012) Ca2+ influx through L-type Ca2+ channels and transient receptor potential channels activates pathological hypertrophy signaling. J. Mol. Cell Cardiol. 53, 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berridge M. J. (2002) The endoplasmic reticulum. A multifunctional signaling organelle. Cell Calcium 32, 235–249 [DOI] [PubMed] [Google Scholar]
- 55. Patel S., Ramakrishnan L., Rahman T., Hamdoun A., Marchant J. S., Taylor C. W., Brailoiu E. (2011) The endo-lysosomal system as an NAADP-sensitive acidic Ca2+ store. Role for the two-pore channels. Cell Calcium 50, 157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ju Y. K., Woodcock E. A., Allen D. G., Cannell M. B. (2012) Inositol 1,4,5-trisphosphate receptors and pacemaker rhythms. J. Mol. Cell Cardiol. 53, 375–381 [DOI] [PubMed] [Google Scholar]
- 57. Kockskämper J., Zima A. V., Roderick H. L., Pieske B., Blatter L. A., Bootman M. D. (2008) Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J. Mol. Cell Cardiol. 45, 128–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Luo D. L., Gao J., Lan X. M., Wang G., Wei S., Xiao R. P., Han Q. D. (2006) Role of inositol 1,4,5-trisphosphate receptors in α1-adrenergic receptor-induced cardiomyocyte hypertrophy. Acta Pharmacol. Sin. 27, 895–900 [DOI] [PubMed] [Google Scholar]
- 59. Nakayama H., Bodi I., Maillet M., DeSantiago J., Domeier T. L., Mikoshiba K., Lorenz J. N., Blatter L. A., Bers D. M., Molkentin J. D. (2010) The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ. Res. 107, 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cook J. L., Re R. N. (2012) Lessons from in vitro studies and a related intracellular angiotensin II transgenic mouse model. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R482–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tadevosyan A., Vaniotis G., Allen B. G., Hébert T. E., Nattel S. (2012) G protein-coupled receptor signalling in the cardiac nuclear membrane. Evidence and possible roles in physiological and pathophysiological function. J. Physiol. 590, 1313–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Haller H., Lindschau C., Erdmann B., Quass P., Luft F. C. (1996) Effects of intracellular angiotensin II in vascular smooth muscle cells. Circ. Res. 79, 765–772 [DOI] [PubMed] [Google Scholar]
- 63. Deliu E., Brailoiu G. C., Arterburn J. B., Oprea T. I., Benamar K., Dun N. J., Brailoiu E. (2012) Mechanisms of G protein-coupled estrogen receptor-mediated spinal nociception. J. Pain 13, 742–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Revankar C. M., Cimino D. F., Sklar L. A., Arterburn J. B., Prossnitz E. R. (2005) A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307, 1625–1630 [DOI] [PubMed] [Google Scholar]
- 65. Thomas P., Pang Y., Filardo E. J., Dong J. (2005) Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146, 624–632 [DOI] [PubMed] [Google Scholar]
- 66. Costa V. M., Ferreira L. M., Branco P. S., Carvalho F., Bastos M. L., Carvalho R. A., Carvalho M., Remio F. (2009) Cross-functioning between the extraneuronal monoamine transporter and multidrug resistance protein 1 in the uptake of adrenaline and export of 5-(glutathion-S-yl)adrenaline in rat cardiomyocytes. Chem. Res. Toxicol. 22, 129–135 [DOI] [PubMed] [Google Scholar]
- 67. Jungsuwadee P., Nithipongvanitch R., Chen Y., Oberley T. D., Butterfield D. A., St Clair D. K., Vore M. (2009) Mrp1 localization and function in cardiac mitochondria after doxorubicin. Mol. Pharmacol. 75, 1117–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Macgregor A., Yamasaki M., Rakovic S., Sanders L., Parkesh R., Churchill G. C., Galione A., Terrar D. A. (2007) NAADP controls cross-talk between distinct Ca2+ stores in the heart. J. Biol. Chem. 282, 15302–15311 [DOI] [PubMed] [Google Scholar]
- 69. Calebiro D., Nikolaev V. O., Lohse M. J. (2010) Imaging of persistent cAMP signaling by internalized G protein-coupled receptors. J. Mol. Endocrinol. 45, 1–8 [DOI] [PubMed] [Google Scholar]
- 70. Cosker F., Cheviron N., Yamasaki M., Menteyne A., Lund F. E., Moutin M. J., Galione A., Cancela J. M. (2010) The ecto-enzyme CD38 is a nicotinic acid adenine dinucleotide phosphate (NAADP) synthase that couples receptor activation to Ca2+ mobilization from lysosomes in pancreatic acinar cells. J. Biol. Chem. 285, 38251–38259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim S. Y., Cho B. H., Kim U. H. (2010) CD38-mediated Ca2+ signaling contributes to angiotensin II-induced activation of hepatic stellate cells. Attenuation of hepatic fibrosis by CD38 ablation. J. Biol. Chem. 285, 576–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Davis L. C., Morgan A. J., Ruas M., Wong J. L., Graeff R. M., Poustka A. J., Lee H. C., Wessel G. M., Parrington J., Galione A. (2008) Ca2+ signaling occurs via second messenger release from intraorganelle synthesis sites. Curr. Biol. 18, 1612–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ben-Zeev G., Telias M., Nussinovitch I. (2010) Lysophospholipids modulate voltage-gated calcium channel currents in pituitary cells. Effects of lipid stress. Cell Calcium 47, 514–524 [DOI] [PubMed] [Google Scholar]
- 74. Weiss S., Doan T., Bernstein K. E., Dascal N. (2004) Modulation of cardiac Ca2+ channel by Gq-activating neurotransmitters reconstituted in Xenopus oocytes. J. Biol. Chem. 279, 12503–12510 [DOI] [PubMed] [Google Scholar]
- 75. De Mello W. C. (2011) Intracrine action of angiotensin II in the intact ventricle of the failing heart. Angiotensin II changes cardiac excitability from within. Mol. Cell Biochem. 358, 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stillitano F., Sartiani L., DePaoli P., Mugelli A., Cerbai E. (2008) Expression of the hyperpolarization-activated current, I(f), in cultured adult rat ventricular cardiomyocytes and its modulation by hypertrophic factors. Pharmacol. Res. 57, 100–109 [DOI] [PubMed] [Google Scholar]
- 77. Bondarenko A. I., Malli R., Graier W. F. (2011) The GPR55 agonist lysophosphatidylinositol acts as an intracellular messenger and bidirectionally modulates Ca2+-activated large-conductance K+ channels in endothelial cells. Pflugers Arch. 461, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]