

Abstract
Aims
Arrhythmogenic right ventricular Dysplasia/cardiomyopathy (ARVD/C) is an autosomal dominant inherited cardiomyopathy associated with ventricular arrhythmia, heart failure and sudden death. Genetic studies have demonstrated the central role of desmosomal proteins in this disease, where 50% of patients harbor a mutation in a desmosmal gene. However, clinical diagnosis of the disease remains difficult and molecular mechanisms appears heterogeneous and poorly understood. The aim of this study was to characterize the expression profile of desmosomal proteins in explanted ARVD/C heart samples, in order to identify common features of the disease.
Methods and Results
We examined plakophilin-2, desmoglein-2, desmocollin-2, plakoglobin and β-catenin protein expression levels from seven independent ARVD/C heart samples compared to two ischemic, five dilated cardiomyopathy and one healthy heart sample as controls. Ventricular and septum sections were examined by immunoblot analysis of total heart protein extracts and by immunostaining.
Immunoblots indicated significant decreases in desmoglein-2 and desmocollin-2, independent of any known underlying mutations, whereas immune-histochemical analysis showed normal localization of all desmosomal proteins. Quantitative RT-PCR revealed normal DSG2 and DSC2 mRNA transcript levels, suggesting increased protein turn-over rather than transcriptional down regulation.
Conclusion
Reduced cardiac desmoglein-2 and desmocollin-2 levels appear to be specifically associated with ARVD/C, independent of underlying mutations. These findings highlight a key role of desmosomal cadherins in the pathophysiology of ARVD/C. Whether these reductions could be considered as specific markers for ARVD/C requires replication analysis.
Introduction
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is a rare inherited cardiomyopathy characterized by progressive sub-epicardial fibro-fatty replacement of myocardial tissue, mostly in the right ventricle [1,2,3]. The typical clinical presentation associates right ventricular arrhythmias with right ventricular morphological (dilatation and/or wall motion abnormalities) and electrocardiographic abnormalities (epsilon wave, T wave inversion and parietal block in right precordial leads). Left ventricular involvement is not rare ranging from 10 to 28% [4,5]. In a small number of subjects, the worsening of the right or biventricular dysfunction can lead to end-stage heart failure needing heart transplantation [5]. Diagnosis is based on a composite score calculated from electrocardiographic and morphological recordings as well as the familial disease pattern, histology and genetic screening results [6]. However, ARVD/C is clinically heterogeneous, and despite carefully selected criteria, diagnosis remains difficult, especially in moderate or borderline forms of the disease. This clearly highlights the need for an improved understanding of the mechanisms leading to the disease and more selective diagnostic criteria [7].
ARVD/C usually presents with an autosomal dominant mode of inheritance and incomplete penetrance [8,9,10]. A major breakthrough in elucidating the molecular pathogenesis of ARVD/C resulted from the identification of causative heterozygous mutations in genes encoding the cardiac desmosomal proteins; plakophilin-2 (PKP2), desmoglein-2 (DSG2), desmocollin-2 (DSC2), plakoglobin (JUP) and desmoplakin (DSP) [11]. Desmosomal mutations were firstly described in Naxos disease and Carvajal syndrome due to mutations in JUP and DSP, associating ARVD/C with palmoplantar keratosis and woolly hair [12,13]. Overall, mutations have only been identified in 40-60% of ARVD/C patients and, due to incomplete penetrance, do not completely solve the clinical diagnostic challenge. Despite these significant advances in the genetics basis of ARVD/C, how the mutant proteins contribute to the disease physiopathology remains largely unexplained.
Desmosomes are multi-protein complexes expressed in epithelial and cardiac tissues. They ensure strong cell-to-cell adhesion via homophilic and heterophilic interactions between the desmosomal cadherins DSG2 and DSC2 [14,15]. Such interactions are reported to stabilize the cadherins at the membrane [16,17]. Cadherins also interact with partners belonging to the armadillo protein family, such as catenins and plakophilins, described as mediators of intracellular signaling [18]. A few studies have implicated plakoglobin and Wnt/beta-catenin signaling in ARVD/C-related adipogenesis and fibrogenesis, two characteristic histological features of the disease [19,20,21]. In addition, transgenic animal models, which at least partially recapitulate the disease phenotype, have also confirmed a possible role for plakoglobin signaling in ARVD/C [19,22].
More than 40% of the mutations found in ARVD/C are nonsense or frame-shift, indicating that haploinsufficiency is a likely mechanism leading to the disease. Moreover, ARVD/C heart samples reproducibly exhibit smaller desmosomes with increased intermembranous space on electron-micrograph, perhaps resulting from an overall reduction in the length and adhesive properties of the desmosomes [23] [21]. Additional support for this theory comes from recent studies frequently reporting reduced plakoglobin levels at the intercalated discs in ARVD/C cardiac biopsies [24,25]. Based on these observations, we hypothesized that decreased desmosomal protein levels at intercalated disks could be a common feature of ARVD/C. We performed systematic immunolabeling and immunoblotting to assess levels and localization of intercalated disk proteins (JUP, PKP2, DSG2, DSC2 and β-catenin) in a collection of seven heart samples from patients with clinically confirmed ARVD/C. We identified a marked and specific decrease in the expression of cardiac desmosomal cadherins.
Materials and Methods
2.1 Ethics statement
All patients have signed a written informed consent form enabling the use of their tissue for this study. This project was given the approval number 35-06 by the CCPPRB (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale) on behalf of the PHRC (Programme Hospitalier de Recherche Clinique).
2.2 Tissue samples
Cardiac tissue samples were collected between 2006 and 2009 from patients with confirmed ARVD/C (referred to as: ARVD/C 1 to 7) according to validated diagnostic criteria [6,26], and who underwent cardiac transplant. The control samples were derived from the explanted hearts of two patients with ischemic- and five with idiopathic-dilated cardiomyopathy (DCM), and from one donor with no documented heart disease (normal control). All except one of the DCM cases used as controls displayed biventricular dilatation. The remaining case had ischemic DCM and did not present into right ventricular dilatation.
All tissues were immediately frozen in liquid nitrogen or isopentane at -80°C. Mutational screening of the five desmosomal genes PKP2 (NM_004572.3), DSG2 (NM_001943.3), DSC2 (NM_024422.3), DSP (NM_004415.2) and PG (NM_021991.2) was performed in all ARVD/C patients as previously described [8]. Briefly, genomic DNA was extracted from blood cells of each patient and subjected to PCR amplification of each exon and intron- exon junctions from the five genes screened. Sequencing of PCR products was performed using BigDye dideoxy-terminator chemistry (PerkinElmer) on an ABI 3830 DNA sequencer (Applied Biosystems). Analysis of the chromatograms was performed with SeqScape (PE Applied Biosystems). The exons of the five genes were thoroughly analyzed even when a mutation was identified in a given gene. A control group of 300 healthy and unrelated subjects (600 alleles) with Caucasian origin were genotyped as controls to check that mutations were not frequent polymorphisms. Moreover, absence of the identified mutations was also ascertained in the ESV server (URL:http://evs.gs.washington.edu/EVS/).
2.3 Protein extraction and immunoblot analysis
Snap frozen tissues were homogenized in SDS-Urea buffer (1% SDS, 8 mM Urea, 10 mMTris pH7.5, 140 mMNaCl, 5 mM EDTA, 2 mM EGTA) supplemented with a cocktail of protease inhibitors (1:1000) (Sigma-Aldrich®, MO, USA). Proteins were loaded on 9 or 10% polyacrylamide gels and, following electrophoresis, were transferred to nitrocellulose membranes. They were then blocked for two hours in 5% non-fat milk in PBS with 0.1% Tween (PBS-T). Blots were incubated with primary antibodies overnight at 4°C, rinsed and incubated with secondary antibodies conjugated to the infra-red dyes 680LT or 800CW (LI-COR® Biosciences, NE, USA). Blots were imaged using the Odyssey® Imager (LI-COR® Biosciences). Protein signal intensities were quantified with the ImageJ freeware (version 1.41, NIH, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/). For each measurement, the target protein density ratio was calculated using either α-actinin-2 (ACTN2) or cardiac myosin-binding protein C (cMyBP-C) as loading controls. Western blots were performed in duplicate for each heart compartment (ie. septum, left and right ventricles) and for each subject. Two to three independent protein extractions were performed to determine the average relative protein level per compartment and per subject.
2.4: Immunofluorescent labeling
Frozen heart samples were cut into 5 µm sections, and fixed with 4% paraformaldehyde for 10 min at room temperature (RT), followed by incubation in 100% methanol for 10 min at -20°C. Tissues were permeabilized with 0.2% Triton X-100 in phosphate buffered saline (PBS) for 5 minutes at RT, then blocked for 30 min with 5% bovine serum albumin and 0.01% Triton X-100 in PBS, and finally incubated with primary antibodies overnight at 4°C (Table S1). Samples were washed in PBS and labeled with Alexa Fluor® conjugated secondary antibodies (Fisher Scientific, MA, USA) for 1 hour at RT. Tissue sections were imaged with the Olympus® IX50 microscope (Olympus®, Tokyo, Japan), and 3D deconvolution performed using the Metamorph software (Roper Scientific®, NJ, USA), by two independent investigators, who were blinded to the sample origin.
2.5 Electron microscopy procedure
Right and left ventricle samples from the ARVD/C patient (ARVD/C 6) were fixed in 2% glutaraldehyde in 0.1M phosphate buffer, pH 7.4at 4° for 4 hours, then incubated in 0.6% glutaraldehyde in phosphate buffer at 4°. Samples were post fixed in 1% osmium tetroxyde in cacodylate buffer during 1 hour, and then colored with uranyl-acetate for 1 hour. Tissues were then dehydrated in a series of alcohol baths from 30% to 100% then in 100% propylene oxyde and embedded in
2.5. RNA extraction Epon 812 resin before electron microscopy imaging (HITACHI 120kV HT 7700). and quantitative RT-PCR
Total mRNA was isolated from snap frozen hearts with TRIzol (Life Technologies®, CA, USA) and concentrated with the RNeasy® minikit (Qiagen®, Hilden, Germany). cDNA was synthesized from 500 ng of total mRNA with oligo[dT]12-18 priming and the SuperScript™ III reverse transcriptase (Life TechnologiesTM). Quantitative real-time PCR (RT-PCR) was performed using the LightCycler® 480 System (Roche Diagnostics, IN, USA) and the Brilliant II SYBR® Green qPCR Master Mix (Agilent technologies, CA, USA), according to the manufacturer’s instructions. Specific primers were designed to amplify DSG2, DSC2 and ACTN2 cDNAs (DSG2-F: 5’ atgacggctaggaacaccac3’; DSG2-R: 5’ gggtcagtttgtggctgact3’; DSC2-F: 5’ ttggagcatcaaacaaaggtc3’; DSC2-R: 5’ atagttttgggccgtgtcag3’; ACTN2-F: 5’ cagaggaagaccttcactgc3’; ACTN2-R: 5’ caattttgtggaaccgcatt3’). All data were normalized to ACTN2 mRNA level using the 2-ΔΔCT method [27].
2.6 Statistics
Differences between the two groups (ARVD/C and controls) were analyzed using the non-parametric Mann-Whitney Rank Sum test. A p value < 0.05 was considered as statistically significant. Data are expressed as the mean value ± standard error (SEM).
Results
3.1 Patient characteristics
Patient clinical and genetic details are presented in Table 1. All seven ARVD/C patients displayed severe disease with multifocal ventricular tachycardia and severe right ventricular or biventricular heart failure. They underwent cardiac transplantation at ages ranging from 14 to 74 years old. The presence of extensive fibro-fatty myocyte replacement, predominantly seen in the right ventricle, but also in the left ventricle, confirmed the ARVD/C diagnosis in all patients (Figures S1 and S2). Three of the patients (ARVD/C1, 2 and 6) carried heterozygous DSG2 mutations as previously described (Table 1) [8]. No causative mutations were identified in the remaining four ARVD/C patients.
Table 1. Clinical and histopathological details of patients ARVD 1 to 7 included in this study.
| ARVD/CPatient | Desmosomal mutation |
Demographics
|
Familial history | ECG |
Arrhythmias
|
Morphology
|
Histology
|
Major/Minor criteria | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sex | age at diagnosis | Age at Ht Tx | Ventricular arrhythmias | Atrial arrhythmias | ICD | RV | RVEF | LV involment | LVEF | RV | LV | lymphocytes | |||||
| 1 | DSG2 (c.690+1 G>A) | M | 50 | 73 | none | AV block, paced | Multifocal VT from RV** and LV | none | yes | major RV dilatation and hypokinesia** | <25% | yes | 30% | Extensive fibro-fatty replacement ** | Mild fibrosis | - | 3/0 |
| 2 | DSG2 (p.Arg49His) | M | 13 | 14 | Grandfather: severe ARVD leading to Ht Tx* | iRBBB, epsilon wave** | Multifocal VT from RV** | none | no | major RV dilatation and hypokinesia** | 13% | yes | 35% | Extensive sub-epicardial fibro-fatty replacement ** | Large regions with sub-epicardial fibro-fatty replacement Extensive fibrosis | + | 4/1 |
| 3 | none | M | 46 | 53 | none | RBBB, TWI V1-V4*, epsilon wave** | Multifocal VT from RV**, FV | AF | yes | major RV dilatation, anterior akinesia severe hypokinesia** | <25% | none | 55% | Extensive fibro-fatty replacement ** | Mild sub-epicardial fibro-fatty replacement | + | 4/1 |
| 4 | none | F | 21 | 30 | none | RBBB, epsilon wave**, TWI V1-V4* | Multifocal VT from RV** | none | yes | major RV dilatation and hypokinesia** | 27% | none | 65% | Extensive fibro-fatty replacement ** | Mild sub-epicardial fibro-fatty replacement | - | 4/1 |
| 5 | none | F | 38 | 39 | none | epsilon wave**, iRBBB, | 11008 bimorphic VEc/24H*, polymorphic NSVT | AF | yes | major RV dilatation anterior akinesia and global hypokinesia** | 28% | yes | 45% | Extensive fibro-fatty replacement of RV anterior wall ** | Large regions with fibrosis Mild fatty infiltration | + | 3/1 |
| 6 | DSG2 (p.Arg46Trp) | M | 39 | 48 | Brother : SD at 22* | iRBBB, epsilon wave**, TWI V1-V6** | Multifocal VT from RV** | None | yes | major RV dilatation and hypokinesia** | 13% | yes | 45% | Extensive fibro-fatty replacement ** | Mild fibrosis | + | 5/1 |
| 7 | none | M | 46 | 49 | none | iRBBB, TWI V1 to V6**, epsilon wave** | Multifocal VT from RV** | None | yes | major RV dilatation and hypokinesia** | <25% | yes | 26% | Extensive fibro-fatty replacement ** | Large regions with sub-epicardial fibro-fatty replacement | + | 5/0 |
HtTx: Heart Transplantation; AV: Atrioventricular block; RBBB: right bundle branch block; iRRRB: incomplete RBBB; TWI: T-wave inversion; VT: ventricular tachycardia; NSVT: nonsustained VT; Vec: ventricular ectopies; FV: ventricular fibrillation; RV: right ventricular; LV: left ventricular; EF: ejection fraction; SD: sudden death; AF: atrial fibrillation; ICD: implantable cardiac defibrillator; F: female; M: male; *: minor criteria; **: major criteria.
3.2 Immunoblot quantitation
An immunoblotting study was performed on left and right ventricle, and in septum heart samples to identify potential ARVD/C-specific cardiac protein expression patterns.
As shown in Figure 1, total β-catenin, JUP and PKP2 protein levels were similar in ARVD/C and controls heart samples (all compartment together). There was neither any statistical difference in any compartments when assessed independently (data not shown). Conversely, the total DSG2 level was significantly decreased in all ARVD/C samples, compared to controls and normal heart, regardless of the disease-causing mutation (Figure 2a). Replicated densitometric measurement revealed an overall 74% decrease of desmoglein-2 signal in ARVD/C compared to controls samples, the ratio of DSG2 to α-actinin-2 (ACTN2) was 17.2 ± 6.6 in ARVD/C, 67.1 ± 9.8 in DCM samples (p<0.001, Mann Whitney Test) and 84.9 in the normal heart sample. Decreased DSG2 was observed for all patients (Figure S3) and in all cardiac compartments examined, but was more drastic in the left and right ventricles compared to the septum (Figure 2a).
Figure 1. Quantitative immunoblotting of selected intercalated disk proteins; plakophilin-2, plakoglobin and β-catenin.

Representative immunoblots obtained for (a) plakoglobin (left Ventricle), (b) β-catenin (septum) and (c) plakophilin-2 (septum). Bar graphs indicate the mean ± SEM following quantification of protein signals from all blots from the seven patient tissue samples and all compartments after normalization to the cardiac protein α-actinin-2 (ACTN2) or cardiac myosin-binding protein C (cMyBP-C) and to the control with the strongest signal level. NS for p>0.05, Mann Whitney test.
Figure 2. Quantitative immunoblotting of desmosomal cadherins desmoglein-2 and desmocollin-2.

Representative images of western blots obtained for desmoglein-2 (a) and desmocollin-2 (b) in the right ventricle. Bar graphs indicate the mean expression ± SEM for each heart compartment from the seven patients, following normalization to the cardiac protein α-actinin-2 (ACTN2) and the control with the strongest signal level. *p<0.001, Mann Whitney Test.
Additionally, using electron microscopy on ARVD/C 6 patient, we have observed a clear reduction in desmosomal dense material at intercellular junctions; this was observed in both ventricles. Moreover we also observed opening inter-membrane vacuoles suggesting cellular disjunction (Figure 3).
Figure 3. Electron microscopy of heart samples from ARVD/C 6.
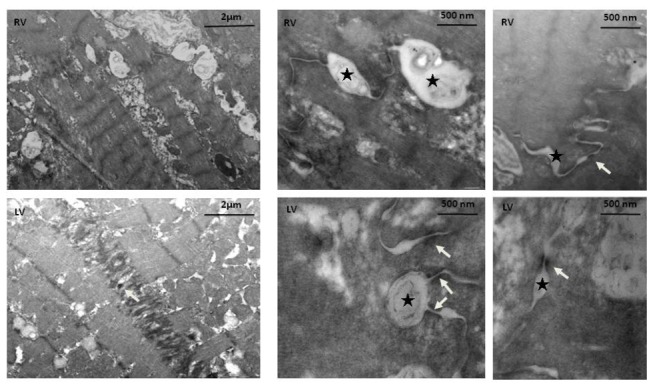
The electronic microscopy study of heart tissue from the p.Arg46Trp mutation-carrier (ARVD/C6) showed the presence of small desmosomes (white arrows) associated with abnormal inter-membrane vacuoles (black stars) in both ventricles (LV; left ventricle, RV; right ventricles).
Total DSC2 protein was also measured by immunoblot in the septum and right ventricle of only four ARVD/C patient samples due to the limited quantity of available tissue. The total DSC2 protein level was decreased (-43%) compared to controls. The ratio of DSC2 to ACTN2 was 38.7 ± 13.8 in ARVD/C, 68.1 ± 12.9 in controls samples (p<0.001, Mann Whitney Test) and 122.8 in the normal heart sample (Figure 2b). This decrease was statistically significant only in the right ventricle.
3.3 Intercalated disc protein Localization
To delve further into the characterization of the cadherin protein reduction, we performed immunohistochemical studies on the left and right ventricles (Figure 4). In addition to four desmosomal proteins found mutated in ARVD/C, (PKP2, DSG2, DSC2 and JUP), we also labeled β-catenin because of its possible functional implication in ARVD/C [28,29]. To control for tissue quality and localize cardiomyocytes, immunolabeling for ACTN2 or cardiac myosin-binding protein C (cMyBP-C) was also performed. All desmosomal proteins (PKP2, JUP, DSG2 and DSC2) and β-catenin showed normal localization at intercalated disks in both ARVD/C and control samples (Figure 4). Using the limit dilution of 1/300 for the antibody directed against DSG2, we observed a complete loss of immunolabeling for the 3 samples displaying the weakest amount of DSG2 on immunoblot (ARVD/C 1, 5 and 6, Figure S3), suggesting a correlation between the two measurement techniques (Figure S4).
Figure 4. Representative immunolocalization of intercalated disc proteins in ARVD/C patient right ventricles.

Representative pictures of plakophilin-2, β-catenin, plakoglobin, desmoglein-2 and desmocollin-2 labeling are shown (in green). Cardiac myosin-binding protein C (cMyBP-C) and α-actinin-2 (both red labeling) were used as cardiomyocyte specific markers. The figure shows heart tissue from two patients with ARVD/C compared to a representative (non ARVD/C) control.
3.4 DSG2 and DSC2 mRNA transcripts in ARVD/C.
The decrease in desmosomal cadherins observed in ARVD/C samples could either be the consequence of transcriptional down-regulation or increased protein turnover. To answer this question, we studied DSG2 and DSC2 mRNA transcript level in four ARVD/C patient right ventricle samples (ARVD/C1 to 4) and in six septum samples (all patients except ARVD/C 3) by quantitative RT-PCR. ACTN2 expression was used to standardize for cardiomyocyte specific mRNA input. As shown in Figure 5, there was no significant difference in DSG2 and DSC2 mRNA transcript level in ARVD/C compared to DCM control samples. This indicated that reduced levels of DSG2 and DSC2 in ARVD/C are not a consequence of mRNA down-regulation, but rather suggest an increased degradation or translation defect.
Figure 5. Quantitative RT-PCR analysis of DSG2 and DSC2 relative to ACTN2 mRNA transcripts.

Relative quantification was performed using RNA isolated from the septum or right ventricle of heart samples. Bar graphs show the mean ± SEM of six samples (2-ΔΔCT method). p>0.05, Mann Whitney Test.
Discussion
We characterized desmosomal protein expression in heart tissue samples from end-stage ARVD/C patients with or without mutations in desmosomal protein genes. We observed a significant decrease in DSG2 and DSC2 protein levels in all ARVD/C samples, regardless of the genetic status, but no mislocalization of desmosomal proteins, in comparison to the seven controls.
Correlation between gene and protein expression
Mutations were not identified in the coding sequences of desmosomal genes (PKP2, DSG2, DSC2, DSP and JUP) in four ARVD/C cases, suggesting that the observed desmosomal cadherin decrease is an indicator of ARVD/C which is independent of the causative mutation. Nevertheless, it still remains unexplained why DSG2 from transplanted ARVD/C subjects seems more frequently mutated in our patients (three of seven) compared to that which has been reported in previous ARVD/C screens (around 10% [8,30]), and to PKP2, the major gene in ARVD/C (around 30% [8]). It may be that DSG2 mutations are associated with a more severe phenotype, thus requiring transplantation, as has already been reported by our team in a clinical analysis of 135 genotyped ARVD/C patients [8]. Confirming this observation with a larger study would be of great interest in defining phenotype-genotype correlations in ARVD/C.
Specificity of desmosomal cadherin decrease in ARVD/C
DSG2 and DSC2 were the only desmosomal proteins shown to be significantly reduced by immunoblot in our study. This result suggests that desmosomal cadherin decrease is a specific feature of late-stage ARVD/C, rather than a general marker of cardiomyopathy-induced cardiac remodeling. None of the desmosomal proteins appeared altered by immunohistochemistry, suggesting no abnormal relocalization of them.
Although it was not our intention to quantify the signal on tissue immunostaining, we did not observe systematic decreased intensity of any specific desmosomal protein in all ARVD/C patients compared to controls. In order to increase our sensitivity to detect a decreased signal specific for DSG2 we applied a dilution protocol for the antibody directed against DSG2 as proposed by Asimaki et al. [24]. At maximum dilution only 3 out of 6 ARVD/C samples showed clear loss of fluorescent signal (Figure S4). Interestingly, these samples correlated to ARVD/C samples 1, 5 and 6 which display the most drastic decrease in DSG2 as measured by immunoblotting (Figure S3). Therefore this suggests a correlation between immunoblotting and immunolabeling that was only partial due to a threshold effect and insufficient sensitivity of our antibody. We also anticipated a reduced plakoglobin labeling, as described by Asimaki et al. [24]. However our immunolabeling protocol was slightly different and perhaps the primary antibody dilution we used may not have been adequately sensitive [24]. Therefore, the described decrease of plakoglobin at intercalated discs of ARVD/C cardiomyocytes, together with our immunoblot results, showing stable overall levels of plakoglobin protein, suggest a relocalization of the protein rather than a degradation, as was proposed by Asimaki et al. [24,25].
We also observed that among the desmosomal proteins tested, only cadherins were specifically decreased. This result is partly supported by another study performed on an isolated case with compound heterozygous mutations in DSC2 and DSG2, showing decreased DSG2 expression with unchanged plakoglobin and PKP2 [31]. Compare to DSG2, the decrease in DSC2 was less significant and restricted to right ventricle. This is possibly correlated with increased severity of the phenotype in this compartment which is primarily altered in ARVD/C. Similar observation was made by Gehmlich et al. reporting a complete DSG2 extinction associated with a strong but incomplete decrease in DSC2 in a single explanted heart from a human ARVD/C case carrying a heterozygous DSG2 null mutation [32].
We studied samples with advanced forms of ARVD/C from patients carrying DSG2 or other undetected mutations. It remains to be determined if the desmosomal cadherins decrease, particularly of DSG2, is also observed in patients with mutations in genes encoding desmosomal proteins other than cadherin, and if this reduction is discernible in earlier forms of ARVD/C.
Physiopathological mechanisms of desmosomal cadherin decrease in ARVD/C
Desmosomal cadherin decrease is likely due to increased protein degradation or to translational defects, as the levels of DSG2 and DSC2 transcripts were similar to those measured in controls. Normal levels of cadherin mRNA in the right ventricle of ARVD/C patients has also been previously reported [33]. This is correlated with electronic microscopy studies on ARVD/C biopsies [23,30,34] which clearly indicate a progressive decrease in desmosomal length associated with disrupted intercellular junctions. In our study, desmosomal cadherins appear central to the pathophysiological mechanism of ARVD/C. However, the exact mechanism of the reduction remains unclear, and needs to be confirmed in larger studies. An attractive hypothesis could be that non-functional desmosomes (either due to mutant protein incorporation acting through a dominant negative effect, or to decreased expression of key structural components such as PKP2), are internalized with subsequent selective degradation of the cadherins, while other desmosomal components are recycled. Accordingly, via electron microscopy we observed such reduction in desmosomal materials in one of our studied explanted tissue (ARVD/C6). Such a mechanism is observed for desmoglein-3 in the life-threatening dermatosis pemphigus vulgaris, where the inhibition of cadherin adhesion (resulting from interaction of anti-cadherin auto-antibodies) leads to cadherin internalization and degradation, while plakoglobin steady state levels remain unaltered [16,35].
Altogether, our results suggest that a key aspect of ARVD/C pathophysiology could be increased degradation of cardiac cadherins, leading to desmosomal disorganization and disrupted cardiomyocyte junctions, and which may be a marker of ARVD/C. In order to gain insight into ARVD/C physiopathology, further studies are required to confirm this finding and identify the downstream pathways affected by desmosomal cadherin loss.
Study limitations
Decreased cadherin expression is observed in all end-stage ARVD/C patient heart samples and not in the normal heart or dilated cardiomyopathy samples. We had access to seven heart tissue samples from ARVD/C patients and only one normal heart, low numbers which weaken our conclusions. Additionally, mutations in desmosomal genes have been recently reported in 5% of DCM cases [36], and we cannot exclude that the explanted DCM heart samples originated from donors carrying desmosomal gene mutations. However, given the low prevalence, the probability that one DCM donor carried a desmosomal gene mutation is small.
Finally, we cannot exclude that patients with no identified mutations, may carry undetected genetic abnormalities such as large deletions in DSG2 and DSC2, or mutations in unknown genes which could affect DSC2 and DSG2 expression.
Supporting Information
Representative histology images of right ventricles from ARVD/C1-4. Hematoxylin and eosin-stained section of the right ventricular myocardium showing mostly fatty replacement in all ARVD/C patients. Two magnifications are represented (2X and 10X).
(TIF)
Representative histology images of right ventricles from ARVD/C5-7. Hematoxylin and eosin-stained section of the right ventricular myocardium showing mostly fatty replacement in all ARVD/C patients. Two magnifications are represented (2X and 10X).
(TIF)
Quantitative immunoblotting of desmoglein-2 in all ARVD/C and control patients. Representative images of western blots obtained for desmoglein-2 in the right, left ventricle and septum for each patient. Bar graphs indicate the mean ± SEM following normalization to the cardiac protein α-actinin-2 (ACTN2).
(TIF)
Representative images of immunofluorescent staining in ARVD/C and control patients after maximum dilution of anti-desmoglein-2 antibody. Immunofluorescent staining of desmoglein-2 (DSG2) was performed in all ARVD/C and control samples as described in the material and methods section of the manuscript. We used the maximum dilution (before total extinction of the signal in controls) of anti-DSG2 antibody (1/300). The labeling of three samples, ARVD/C1, 5 and 6, appeared reduced compare to the others (ARVD/C2, 3, 4 and controls 1 to 6). These three samples match the samples for which the immunoblot based quantification of DSG2 appeared lower (Figure S3).
(TIF)
Primary antibodies used in study.
(DOC)
Acknowledgments
We thank Myobank – AFM (Institut de Myologie Hôpital Pitié-Salpêtrière, Paris, France) for the collection of heart samples from ARVD/C patients. We thank Dr Dominique Langui for technical help in electron microscopy. We also thank Dr Rachel Peat for critical reading of the manuscript.
Funding Statement
This work was supported by grants from Assistance Publique – Hôpitaux de Paris (PHRC programme hospitalier de recherche clinique AOM05073), "Fédération Française de Cardiologie,” “Société française de cardiologie,” and “Ligue contre la Cardiomyopathie”. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fontaine G, Fontaliran F, Hébert JL, Chemla D, Zenati O et al. (1999) Arrhythmogenic right ventricular dysplasia. Annu Rev Med 50: 17-35. doi:10.1146/annurev.med.50.1.17. PubMed: 10073261. [DOI] [PubMed] [Google Scholar]
- 2. Basso C, Corrado D, Marcus FI, Nava A, Thiene G (2009) Arrhythmogenic right ventricular cardiomyopathy. Lancet 373: 1289-1300. doi:10.1016/S0140-6736(09)60256-7. PubMed: 19362677. [DOI] [PubMed] [Google Scholar]
- 3. Corrado D, Thiene G (2006) Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation 113: 1634-1637. doi:10.1161/CIRCULATIONAHA.105.616490. PubMed: 16585401. [DOI] [PubMed] [Google Scholar]
- 4. Quarta G, Muir A, Pantazis A, Syrris P, Gehmlich K et al. (2011) Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation 123: 2701-2709. doi:10.1161/CIRCULATIONAHA.110.976936. PubMed: 21606390. [DOI] [PubMed] [Google Scholar]
- 5. Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G (2004) Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 110: 1879-1884. doi:10.1161/01.CIR.0000143375.93288.82. PubMed: 15451782. [DOI] [PubMed] [Google Scholar]
- 6. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B et al. (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 31: 806-814. doi:10.1093/eurheartj/ehq025. PubMed: 20172912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fontaine GH (2011) The multiple facets of right ventricular cardiomyopathies. Eur Heart J 32: 1049-1051. doi:10.1093/eurheartj/ehr088. PubMed: 21460364. [DOI] [PubMed] [Google Scholar]
- 8. Fressart V, Duthoit G, Donal E, Probst V, Deharo JC et al. (2010) Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace 12: 861-868. doi:10.1093/europace/euq104. PubMed: 20400443. [DOI] [PubMed] [Google Scholar]
- 9. Awad MM, Calkins H, Judge DP (2008) Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med 5: 258-267. doi:10.1038/ncpcardio1182. PubMed: 18382419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Christensen AH, Benn M, Bundgaard H, Tybjaerg-Hansen A, Haunso S et al. (2010) Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. J Med Genet 47: 736-744. doi:10.1136/jmg.2010.077891. PubMed: 20864495. [DOI] [PubMed] [Google Scholar]
- 11. van Tintelen JP, Hofstra RM, Wiesfeld AC, van den Berg MP, Hauer RN et al. (2007) Molecular genetics of arrhythmogenic right ventricular cardiomyopathy: emerging horizon? Curr Opin Cardiol 22: 185-192. doi:10.1097/HCO.0b013e3280d942c4. PubMed: 17413274. [DOI] [PubMed] [Google Scholar]
- 12. McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A et al. (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355: 2119-2124. doi:10.1016/S0140-6736(00)02379-5. PubMed: 10902626. [DOI] [PubMed] [Google Scholar]
- 13. Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J et al. (2000) Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 9: 2761-2766. doi:10.1093/hmg/9.18.2761. PubMed: 11063735. [DOI] [PubMed] [Google Scholar]
- 14. Getsios S, Huen AC, Green KJ (2004) Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol 5: 271-281. doi:10.1038/nrm1356. PubMed: 15071552. [DOI] [PubMed] [Google Scholar]
- 15. Le TL, Yap AS, Stow JL (1999) Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J Cell Biol 146: 219-232. doi:10.1083/jcb.146.1.219. PubMed: 10402472. [PMC free article] [PubMed] [Google Scholar]
- 16. Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K et al. (2006) Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem 281: 7623-7634. doi:10.1074/jbc.M512447200. PubMed: 16377623. [DOI] [PubMed] [Google Scholar]
- 17. Delmar M, McKenna WJ (2010) The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 107: 700-714. doi:10.1161/CIRCRESAHA.110.223412. PubMed: 20847325. [DOI] [PubMed] [Google Scholar]
- 18. Thomason HA, Scothern A, McHarg S, Garrod DR (2010) Desmosomes: adhesive strength and signalling in health and disease. Biochem J 429: 419-433. doi:10.1042/BJ20100567. PubMed: 20626351. [DOI] [PubMed] [Google Scholar]
- 19. Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD et al. (2006) Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 116: 2012-2021. doi:10.1172/JCI27751. PubMed: 16823493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lombardi R, da Graca Cabreira-Hansen M, Bell A, Fromm RR, Willerson JT et al. (2011) Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res 109: 1342-1353. doi:10.1161/CIRCRESAHA.111.255075. PubMed: 22021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim C, Wong J, Wen J, Wang S, Wang C et al. (2013) Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494: 105-110. doi:10.1038/nature11799. PubMed: 23354045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pilichou K, Bezzina CR, Thiene G, Basso C (2011) Arrhythmogenic cardiomyopathy: transgenic animal models provide novel insights into disease pathobiology. Circ Cardiovasc Genet 4: 318-326. doi:10.1161/CIRCGENETICS.110.959031. PubMed: 21673311. [DOI] [PubMed] [Google Scholar]
- 23. Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G et al. (2006) Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J 27: 1847-1854. doi:10.1093/eurheartj/ehl095. PubMed: 16774985. [DOI] [PubMed] [Google Scholar]
- 24. Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S et al. (2009) A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 360: 1075-1084. doi:10.1056/NEJMoa0808138. PubMed: 19279339. [DOI] [PubMed] [Google Scholar]
- 25. Munkholm J, Christensen AH, Svendsen JH, Andersen CB (2012) Usefulness of immunostaining for plakoglobin as a diagnostic marker of arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 109: 272-275. doi:10.1016/j.amjcard.2011.08.044. PubMed: 22036107. [DOI] [PubMed] [Google Scholar]
- 26. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C et al. (1994) Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J 71: 215-218. doi:10.1136/hrt.71.3.215. PubMed: 8142187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408. doi:10.1006/meth.2001.1262. PubMed: 11846609. [DOI] [PubMed] [Google Scholar]
- 28. Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G et al. (2004) Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol 13: 26-32. doi:10.1016/S1054-8807(03)00107-8. PubMed: 14761782. [DOI] [PubMed] [Google Scholar]
- 29. Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C et al. (2004) Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 1: 3-11. doi:10.1016/j.hrthm.2004.03.002. PubMed: 15851108. [DOI] [PubMed] [Google Scholar]
- 30. Pilichou K, Nava A, Basso C, Beffagna G, Bauce B et al. (2006) Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113: 1171-1179. doi:10.1161/CIRCULATIONAHA.105.583674. PubMed: 16505173. [DOI] [PubMed] [Google Scholar]
- 31. Gehmlich K, Lambiase PD, Asimaki A, Ciaccio EJ, Ehler E et al. (2011) A novel desmocollin-2 mutation reveals insights into the molecular link between desmosomes and gap junctions. Heart Rhythm, 8: 711–8. PubMed: 21220045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gehmlich K, Syrris P, Reimann M, Asimaki A, Ehler E et al. (2012) Molecular changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. Cardiovasc Pathol 21: 275-282. doi:10.1016/j.carpath.2011.09.005. PubMed: 22036071. [DOI] [PubMed] [Google Scholar]
- 33. Gaertner A, Schwientek P, Ellinghaus P, Summer H, Golz S et al. (2012) Myocardial transcriptome analysis of human arrhythmogenic right ventricular cardiomyopathy. Physiol Genomics 44: 99-109. doi:10.1152/physiolgenomics.00094.2011. PubMed: 22085907. [DOI] [PubMed] [Google Scholar]
- 34. Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE et al. (2007) A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 81: 964-973. doi:10.1086/521633. PubMed: 17924338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M et al. (2010) p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem 285: 8936-8941. doi:10.1074/jbc.M109.087999. PubMed: 20093368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elliott P, O’Mahony C, Syrris P, Evans A, Rivera Sorensen C et al. (2010) Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 3: 314-322. doi:10.1161/CIRCGENETICS.110.937805. PubMed: 20716751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative histology images of right ventricles from ARVD/C1-4. Hematoxylin and eosin-stained section of the right ventricular myocardium showing mostly fatty replacement in all ARVD/C patients. Two magnifications are represented (2X and 10X).
(TIF)
Representative histology images of right ventricles from ARVD/C5-7. Hematoxylin and eosin-stained section of the right ventricular myocardium showing mostly fatty replacement in all ARVD/C patients. Two magnifications are represented (2X and 10X).
(TIF)
Quantitative immunoblotting of desmoglein-2 in all ARVD/C and control patients. Representative images of western blots obtained for desmoglein-2 in the right, left ventricle and septum for each patient. Bar graphs indicate the mean ± SEM following normalization to the cardiac protein α-actinin-2 (ACTN2).
(TIF)
Representative images of immunofluorescent staining in ARVD/C and control patients after maximum dilution of anti-desmoglein-2 antibody. Immunofluorescent staining of desmoglein-2 (DSG2) was performed in all ARVD/C and control samples as described in the material and methods section of the manuscript. We used the maximum dilution (before total extinction of the signal in controls) of anti-DSG2 antibody (1/300). The labeling of three samples, ARVD/C1, 5 and 6, appeared reduced compare to the others (ARVD/C2, 3, 4 and controls 1 to 6). These three samples match the samples for which the immunoblot based quantification of DSG2 appeared lower (Figure S3).
(TIF)
Primary antibodies used in study.
(DOC)