

Abstract
An ever increasing number of metalloproteins are being discovered that play essential roles in physiological processes. Inhibitors of these proteins have significant potential for the treatment of human disease, but clinical success of these compounds has been limited. Herein, Zn(II)-dependent metalloprotein inhibitors in clinical use are reviewed, and the potential for using novel metal-binding groups (MBGs) in the design of these inhibitors is discussed. By using human carbonic anhydrase II (hCAII) as a model system, the nuances of MBG-metal interactions in the context of a protein environment can be probed. Understanding how metal coordination influences inhibitor binding may help in the design new therapeutics targeting metalloproteins.
Metalloproteins, those that require a metal ion cofactor for enzymatic activity, are estimated to make up roughly 30% of the human proteome and are involved in a wide variety of physiological processes such as respiration, gene regulation, and protein matrix degradation.1 Metalloproteins have attracted significant attention for the treatment of a host of diseases including cancer, HIV, and hypertension, among others. Table 1 summarizes some, but not all, of the metalloproteins (with an emphasis on Zn(II)-dependent proteins) currently being investigated as therapeutic targets.
Table 1.
Representative metalloproteins that are therapeutic targets.
| Protein | Cofactor | Indication |
|---|---|---|
| Adamalysin | Zn(II) | Cancer |
| Angiotensin Converting Enzyme* | Zn(II) | Hypertension |
| Anthrax Lethal Factor | Zn(II) | Pathogen (Toxin) |
| Botulinum Neurotoxin | Zn(II) | Pathogen (Toxin) |
| Carbonic Anhydrase* | Zn(II) | Glaucoma |
| Farnesyltransferase | Zn(II) | Cancer |
| Histone Deacetlyase* | Zn(II) | Cancer |
| Matrix Metalloprotease* | Zn(II) | Cancer, Inflammation |
| Metallo-β-Lactamase | Zn(II) | Bacterial Infection |
| Neprilysin | Zn(II) | Hypertension |
| TNF-α Converting Enzyme | Zn(II) | Cancer |
| Lipoxygenase* | Fe(II) | Asthma |
| HIV Integrase* | Mg(ll) | Viral Infection |
FDA-approved inhibitors exist for these metalloproteins
The vast majority of metalloprotein inhibitors act by coordinating the active site metal ion. Inhibitors of these enzymes can generally be divided into two functional regions: a metal-binding group (MBG) and a backbone substituent, the latter of which can be optimized to interact with residues around the active site. This forum article will focus on the role of MBGs in metalloprotein inhibitors targeting Zn(II)-dependent human enzymes that have gained approval by the U.S. Food and Drug Administration (FDA). Some inhibitors showing promise in clinical trials will also be discussed. Finally, a brief section of original research will be presented that shows the value of human carbonic anhydrase II (hCAII) as a model protein for investigating and exploring metalloprotein-inhibitor interactions.
Carbonic Anhydrase (CA) Inhibitors
Discovered in 1932 and shown to be dependent on Zn(II) in 1940, carbonic anhydrase (CA) was the first Zn(II)-dependent metalloprotein discovered.2 CA catalyzes the reversible hydration of CO2 to bicarbonate and a proton, an equilibrium crucial to many physiological processes including respiration,3 pH balance,4 and ion transport.5 Shortly after its discovery, aryl sulfonamides were shown to be extremely potent inhibitors of CA.6 The first FDA-approved carbonic anhydrase inhibitor was acetazolamide (trade name Diamox, Figure 1), which was approved for the treatment of epilepsy in 1953. CA inhibitors are also very effective in the treatment of glaucoma, as bicarbonate plays a crucial role in sodium (and therefore water) transport in the eye.7 Second-generation CA inhibitors dichlorphenamide (Daranide) and methazolamide (Neptazane) were approved by the FDA in 1958 and 1959, respectively, for the treatment of glaucoma. Since their initial approval, these inhibitors have also been approved for use as diuretics and in the treatment of altitude sickness. Although effective therapeutics in indications such as glaucoma, oral administration of these drugs leads to side effects due to systemic CA inhibition.8 To combat this, inhibitors compatible with topical administration were developed, including dorzolamide (Trusopt), approved in 1994, and brinzolamide (Azopt) in 1998 (Figure 1).
Figure 1.
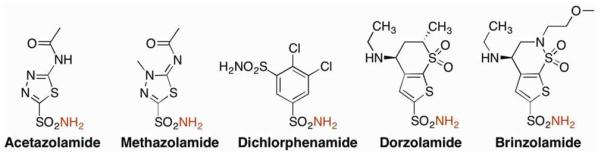
Structures of FDA-approved CA inhibitors. Zn(II)-coordinating nitrogen atoms are highlighted in red.
Although a very effective MBG for CA, the sulfonamide functional group is generally ineffective as a MBG for inhibitors of any other Zn(II)-dependent enzymes.9 The good affinity for the active site Zn(II) ion in CA originates, in part, because the sulfonamide group is ideally positioned to make two hydrogen bonds. Both the hydrogen atom on the Zn(II)-bound nitrogen donor as well as one of the sulfonyl oxygen atoms interact with a nearby threonine residue (Figure 2).10 These interactions are responsible for a large portion of the binding affinity of sulfonamides, illustrated by the fact that even very simple sulfonamides, such as benzene sulfonamide (BSA), are relatively potent CA inhibitor (Ki ≈ 500 nM).6 In fact, these hydrogen bonds are so important that they can also influence metal binding. The crystal structure of a different MBG, acetohydroxamic acid (AHA), reveals that the hydroxamic acid group binds to the CA active site Zn(II) ion through the amine nitrogen, while two oxygen atoms make hydrogen bonds similar to those seen in the case of sulfonamides (Figure 2, right).11 This is a very atypical monodentate binding mode that is not observed in other metalloproteins or small molecule complexes with hydroxamic acid ligands. The `normal' mode of binding by a hydroxamic acid (including AHA) would be bidentate coordination, by the two oxygen atoms, to a metal center (as found in numerous matrix metalloprotease inhibitors).
Figure 2.
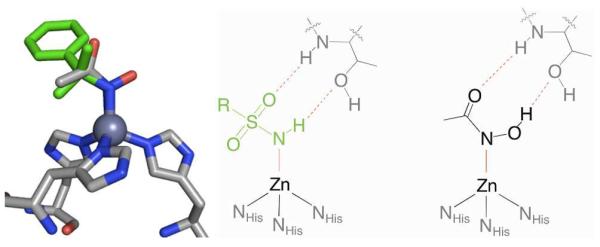
Overlay of BSA (PDB: 2WEJ, green) and AHA (PDB: 1AM6, colored by atom) bound to CA (left). Schematic of the interactions with active site residue Thr199 shown in gray and interactions between the ligands and protein highlighted in red (middle and right).
Angiotensin Converting Enzyme (ACE) Inhibitors
Angiotensin converting enzyme (ACE) is an exopeptidase that acts on several substrates involved in the regulation of vascular resistance. Its active site, similar to CA, contains a Zn(II) ion ligated by three protein residues and a metal-bound hydroxide ion. Instead of three histidine ligands, the coordination sphere of Zn(II) in ACE consists of two histidine ligands and a glutamic acid (Figure 3). The primary substrate of ACE, angiotensin I, is cleaved to yield a decapeptide angiotensin II, a potent vasoconstrictor. Consequently, inhibition of ACE leads to decreased angiotensin II secretion, lowering blood pressure.12 Used in the treatment of hypertension as well as congestive heart failure, ACE inhibitors are the most widely prescribed metalloprotein targeted therapeutics. The first ACE inhibitor approved by the FDA was captopril (Capoten, Figure 3) in 1981, which uses a thiol MBG.13 The free thiol of captopril was implicated in several side effects and relatively poor pharmacokinetics of the drug.14 In 1985, an inhibitor based on a carboxylate MBG, enalapril (Vasotec), was approved. This was followed by several carboxylate-based “me too” drugs over the next decade (Figure 3), with one exception fosinopril (Monopril), which utilizes a phosphate MBG. To achieve better oral bioavailability, all of the approved ACE inhibitors that contain a carboxylate MBG are administered as ester prodrugs, which are hydrolyzed by non-specific esterase activity once absorbed.15 According to the IMS Institute for Healthcare Informatics, lisinopril, a lysine analog of enalapril, was the third-most prescribed drug in 2011, with over 88 million prescriptions.16
Figure 3.
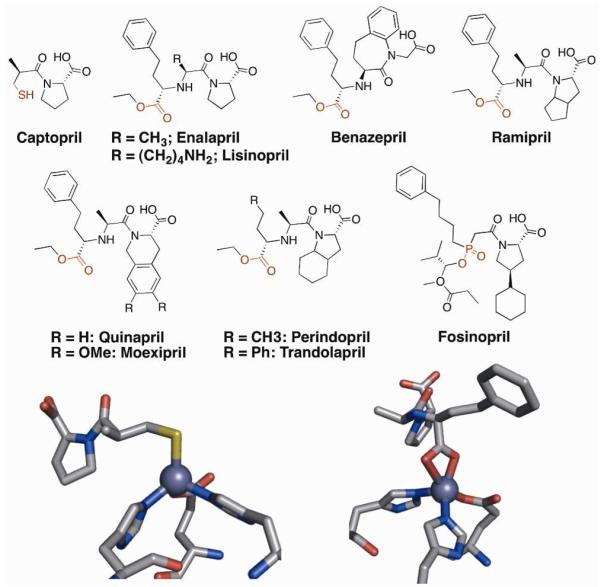
Top: Structures of FDA-approved ACE inhibitors, shown as the ester prodrug where applicable. MBGs are highlighted in red. Bottom: Coordination of the ACE active site Zn(II) ion by captopril (left), and enalaprilal (right).
Using a carboxylate as the MBG in ACE inhibitors has provided several advantages. First, it is not inherently a high-affinity ligand for Zn(II), decreasing the possibility of off-target metalloprotein inhibition.17 Also, carboxylate-based ACE inhibitors generally have higher in vivo stability, with half-lives on the order of 10 hours compared to 2 hours for the thiol- and hydroxamate-based molecules.12 In some cases, analogs using the higher-affinity hydroxamic acid MBG failed to show improvement over the already approved drugs; the potency of one of the more well-studied hydroxamate-based ACE inhibitors (idrapril, IC50 ≈ 10 nM)18 is still less than that of the carboxylate-based drugs (for example lisinopril, IC50 ≈ 1 nM).19 The lack of difference in binding affinity when utilizing the tighter-binding hydroxamate MBG suggests that in certain systems the binding of ACE inhibitors is dominated by the interactions of the peptidomimetic backbone with the protein.
Histone Deacetylase (HDAC) Inhibitors
Gene expression is a tightly controlled process that utilizes a wide variety of transcriptional controls. One of these controls is the covalent modification of histones, the protein complexes around which DNA is wound to form nucleosomes.20 The structure of nucleosomes, and therefore DNA translation, can be altered by chemical modification of lysine residues within the histone proteins. Inhibition of histone deacetylases (HDACs) leads to increased acetylation of these residues, which relaxes the protein-DNA complex and increases gene transcription.21 Although the exact mechanism is unclear, treatment with HDAC inhibitors has been shown to selectively induce growth arrest, differentiation, and apoptosis in cancer cells.22
Vorinostat (Zolinza, Figure 4) was approved by the FDA in 2006 for the treatment of cutaneous T-cell lymphoma (CTCL) that has either recurred or has not responded to at least two other forms of chemotherapy.23 In addition to being the first approved HDAC inhibitor, vorinostat is the only hydroxamate-based metalloprotein inhibitor to gain approval. Romidepsin (Istodax), a natural product initially isolated from Chromobacterium violacium,24 was approved for the treatment of CTCL in patients who have not responded to at least one other form of chemotherapy. Romidepsin is a prodrug that contains an intramolecular disulfide bond that is reduced inside the cell revealing a thiol MBG.
Figure 4.

Structures of HDAC inhibitors. MBGs are highlighted in red.
Vorinostat is currently in Phase III trials for several other malignancies including CTCL, multiple myeloma, and high-grade glioma.25 Romidepsin is being evaluated in Phase III trials for peripheral T-cell lymphoma as well as Phase I/II studies for a variety of other cancers.25 Panobinostat, a hydroxamate-based inhibitor with a greater potency than vorinostat, is also in Phase III trials for a number of cancers. Although they are structurally similar, the two molecules have significantly different stabilities; the half-life of panobinostat (~12 h) is roughly 10× longer than that of vorinostat.26 The vast majority of HDAC inhibitors in development utilize a hydroxamic acid MBG, but there has been progress utilizing other functional groups as well. Currently in Phase II trials, entinostat is significantly more stable in vivo than other HDAC inhibitors.27 With a half-life of 80–100 hours, it can be administered on a weekly basis. The increased half-life of entinostat is attributed, in part, to replacing the hydroxamate functionality with a (2-amino)benzamide MBG (Figure 5). The (2-amino)benzamide MBG has been shown to bind in a bidentate fashion through its aromatic amine and carbonyl oxygen atoms, forming an unusual 7-membered chelate ring (Figure 5).27b
Figure 5.
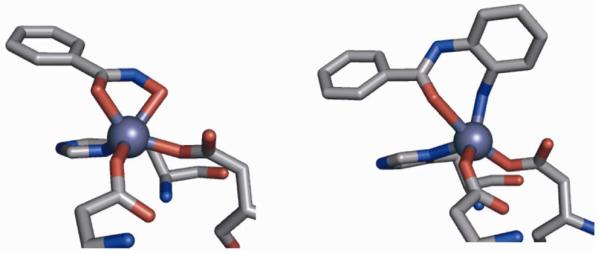
Structure of hydroxamic acid (PDB: 1W22, left) and (2-amino)benzamide MBGs (PDB: 3MAX, right) bound to HDACs. Inhibitor structures are truncated for clarity.
Other Inhibitors in Advanced Clinical Trials
Along with the previously discussed ACE inhibitors, neprilysin inhibitors have shown potential in the treatment of hypertension and chronic heart failure. Neprilysin is a Zn(II)-dependent endopeptidase that degrades many signal peptides including endothelins, a family of potent vasoconstrictors.28 Inhibition of neprilysin is therefore complimentary to inhibiting ACE; instead of lowering secretion of a vasoconstrictor, it prevents the degradation of vasodilators. LCZ-696 is currently in Phase III trials, to compare its effect to enalapril (ACE inhibitor), on patient outcomes in cases of chronic heart failure. LCZ-696 is a mixture of two therapeutics: valsartan, an FDA-approved angiotensin II receptor blocker, and AHU-377, a neprilysin inhibitor. AHU-377 utilizes a carboxylate MBG, and like the carboxylate-based ACE inhibitors, is administered as an ester prodrug (Figure 6).
Figure 6.
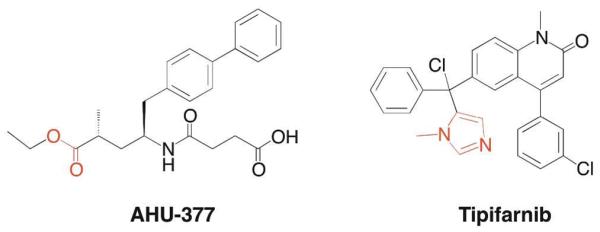
Structures of neprilysin and FTAse inhibitors. MBGs are highlighted in red.
Another metalloprotein target of clinical interest is Ras farnesyltranferase (FTAse), which catalyzes the addition of a prenyl group onto the Ras family of GTPases. Because this modification is necessary for function and activating mutations in the Ras genes are found in many forms of cancer, FTAse inhibitors have potential as effective oncology therapeutics.29 Tipifarnib (Zarnestra), which utilizes a methylimidazole MBG, was first tested on patients over 65 with acute myeloid leukemia (AML), but showed no benefit to patient outcome.30 It is currently in Phase III trials to determine if it is effective in preventing the recurrence of AML.
The Need For New MBGs in Drug Discovery
The discussion above highlights the observation that a relatively narrow scope of MBGs (e.g. carboxylates, hydroxamates, thiols, phosphonates) has been deeply explored for use in metalloprotein inhibitors. The need for new MBGs in drug discovery is best illustrated by the case of the matrix metalloproteases (MMPs). Having been implicated in a wide variety of diseases including tumor proliferation,31 arthritis,32 and ischemic damage following stroke,33 MMPs were among the most popular metalloprotein targets of the past several decades. Despite performing well both in vitro and in animal models, essentially no MMP inhibitor has proven effective in Phase III clinical trials (Figure 7).34 One exception is doxycycline, a molecule that inhibits the expression of MMPs but is a relatively weak direct inhibitor of MMPs. Doxycycline (Periostat) is approved for the treatment of periodontal disease, but the exact mechanism of MMP inhibition by doxycycline is still unclear.35 Reasons for the lack of clinical success of MMP inhibitors have been reviewed elsewhere,34b but the emergence of dose-limiting side effects seems to indicate that lack of target specificity may play a role. The large number of isoforms (>25 discovered to date) and their involvement in many important physiological processes suggest a need for the development of isoform-specific inhibitors.36 It has been demonstrated that, when combined with backbone structure optimization, varying the MBG can modulate inhibitor selectivity.37 Although much of the current research into MMP inhibitors still focuses on backbone optimization appended to traditional MBGs, promising inhibitors have been developed using pyrimidine- and hydroxypyrone-based MBGs.34a
Figure 7.
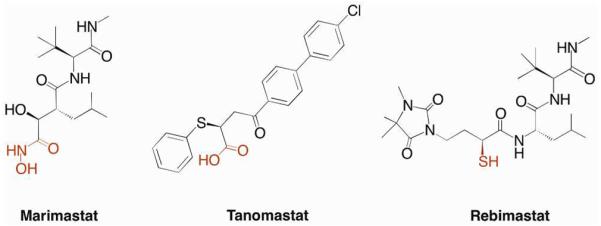
Representative examples of MMP inhibitors that have entered, but failed in, Phase III clinical trials. MBGs are highlighted in red.
Despite being the most popular MBG in the design of metalloprotein-targeted therapeutics, hydroxamic acids are found in few FDA-approved drugs.38 Partly to blame is poor pharmacokinetics and oral bioavailability.39 Also, due to their relatively small size and good affinity for many metal ions, hydroxamates may be capable of binding to a wide variety of metalloprotein active sites. It would seem that in many cases, backbone optimization coupled to a “standard” hydroxamic acid MBG is not an efficient strategy for metalloprotein inhibitor design. In the most therapeutically successful class of metalloprotein inhibitors, that of ACE inhibitors, there are no isoforms or structurally related proteins for which off-target inhibition is a concern.40 This limits side effects and decreases the importance of the MBG. In the case of CA, the sulfonamide functional group appears ideally suited as an MBG for the CA active site, which makes off-target metalloprotein inhibition essentially negligible. Both ACE and CA are unique because the normal, physiological function of the enzyme is the target of therapeutic intervention. Most metalloproteins, including MMPs and HDACs, require inhibition of enzyme activity at sites of disease, while sparing their role in normal processes. In order to permit basal function of some isoforms while inhibiting those related to disease, highly specific inhibitors are needed. Because many of these proteins are structurally similar, inhibitors should optimize interactions of the MBG and backbone components. In order to exploit both metal-ligand binding as well as interactions with the surrounding protein environment, a more diverse collection of MBGs should be considered.41
To aid in this process, a library of small molecules capable of binding metal ions (chelator fragment library, CFL-1.1) was assembled.9 Using the principles of fragment-based lead discovery (FBLD), this library has been used to successfully design inhibitors of several metalloproteins based on novel MBGs.42 The ability of MBGs to modulate inhibitor selectivity is illustrated by two inhibitors that have been developed based on fragments from CFL-1.1 (Figure 8). AM-2S is an inhibitor that uses a 2-hydroxythiopyrone MBG and is effective against the Zn(II)-dependent anthrax lethal factor (LF). In addition to its activity against LF (IC50 = 14 μM),43 AM-2S is a modest inhibitor of several MMPs (IC50 ≈ 5 μM).44 However, when the same inhibitor backbone is combined with a nearly identical MBG, where only a sulfur donor is changed to an oxygen atom (AM-2, Figure 8), the inhibitor becomes selective (>400-fold) for MMPs over LF. The fact that such a drastic difference is caused by changing one atom of the MBG demonstrates how powerful the strategy of MBG optimization in drug design can be.
Figure 8.
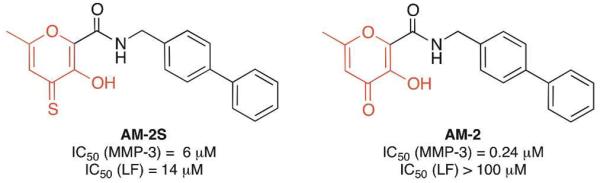
Example of MBG-dependent target selectivity of metalloprotein inhibitors. MBGs are highlighted in red.
Carbonic Anhydrase as a Model System
As described above, MBGs themselves can impart selectivity and/or potency for certain targets independent of the inhibitor backbone. In order to optimize metalloprotein inhibitor design, MBG-protein interactions must be more thoroughly understood. The affinity of an MBG for a metalloprotein active site can be influenced by at least three factors: 1) the identity of the active site metal ion(s), 2) the coordination environment of the active site metal ion(s), and 3) the surrounding protein residues which interact with the MBG upon binding. Synthetic model complexes have been used to determine metal-ligand bond parameters (lengths, angles, etc.) and relative affinities for MBG-metalloprotein interactions,45 but these lack the surrounding protein environment that can influence metal binding.46 A next step in understanding these interactions is to systematically study the forces that drive MBG binding in a protein environment.
The most abundant form of CA in the body, human carbonic anhydrase II (hCAII), has many characteristics that make it an ideal model system for such a study, including its stability, solubility, and ease of crystallization. For these reasons and more, hCAII has been used extensively in biophysical studies of protein-ligand interactions.47 The active site of hCAII consists of a Zn(II) ion coordinated in a tetrahedral geometry by three histidine residues (His94, His96, His119), and a water molecule. This water is deprotonated at physiological pH to yield the active hydroxide nucleophile. The active site is relatively solvent-exposed, sitting at the bottom of a cone-shaped depression roughly 15 Å wide and 15 Å deep. One side of the active site is predominantly hydrophobic while the other is hydrophilic. A threonine residue on the hydrophilic side of the active site (Thr199) accepts a hydrogen bond from the bound hydroxide ion, which serves to orient the nucleophile towards the hydrophobic substrate binding site (Figure 9). In addition to CO2 hydration, hCAII is capable of catalyzing the hydrolysis of a variety of esters through a common mechanism.
Figure 9.
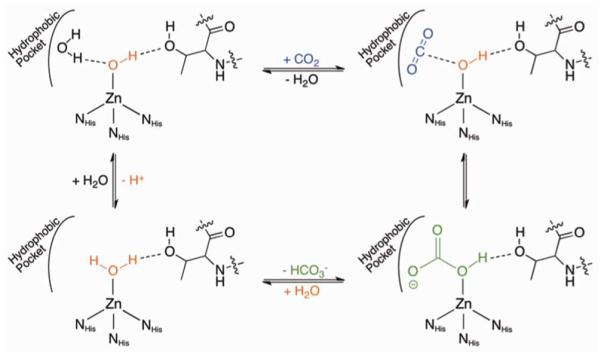
Mechanism of hCAII hydration of CO2.
Fierke, Christanson, and Whitesides, among others, published many pioneering studies using CA as a model system for Zn(II) metalloproteins. Mutagenesis studies examined which aspects of the enzyme's active site are responsible for its high metal affinity and rapid turnover rate. Not surprisingly, the three Zn(II)-binding histidine residues play a major role in both metal binding and catalytic activity. The identity of the metal-coordinating residues themselves is crucial, but several second-sphere interactions are also critical in both metal binding and catalysis. Notably, the residues that accept hydrogen bonds from the Zn(II)-bound imidazole groups play a role not only in pre-organizing the ligands for tetrahedral coordination but also in electrostatic stabilization of the metal-ligand bond.48 In addition, studies of transition metal binding to both WT hCAII and active site variants has shed light on what factors influence metal binding in the protein active site environment.49 Using the previously described sulfonamide ZBG as an anchor, extensive structure-activity studies, both with variations in ligand structure and using active site mutagenesis, have been performed to understand the origins of inhibitor potency.47
Effect of Coordination Environment on Inhibitor Affinity
The previously described work showed that some mutations in the amino acids coordinated to the Zn(II) ion of hCAII result in proteins that retain enzymatic activity, although in a significantly decreased capacity.50 While the catalytic activity of these mutants was thoroughly explored, their inhibition was not. Because the surrounding protein environment remains the same, comparing MBG binding in several hCAII variants allows the variable of the Zn(II) coordination sphere to be isolated and its effect on inhibitor binding to be studied. Using a previously reported assay that capitalizes on the esterase activity of hCAII, the inhibition of WT, H94D, and H94C mutants by the fragments in CFL-1.1 (Figure S2) at a concentration of 500 μM was evaluated (Figure 10).51 Percent inhibition values for all compounds in CFL-1.1 can be found in the Supporting Information (Figure S3).
Figure 10.

Thermal plot representing the results of screening CFL-1.1 fragments against hCAII variants. Cells are color-coded by percent inhibition at 500 μM: black (<20%), red (21–40%), orange (41–60%), yellow (>60%). Gray cells indicate that compounds were not tested due to interference with the assay (1g) or lack of solubility under the assay conditions (all others).
Given its relatively open active site, it is surprising how few compounds from CFL-1.1 significantly inhibit WT hCAII; only two compounds show >40% inhibition at 500 μM. This number greatly increases for the H94C mutant (seven) and even more so in the case of H94D (twelve). As expected, toluenesulfonamide (7h) is the most potent WT inhibitor from the library, showing complete inhibition of WT hCAII at 500 μM. The activity of the sulfonamide 7h is drastically decreased against hCAII H94D (~50% inhibition at 500 μM) and shows almost no activity against the H94C variant (vide infra). Other molecules that inhibit WT hCAII are 2d (3-hydroxypyran-4-thione, 73% inhibition) and to a lesser extent its 3-methyl derivative (5d), as well as 2-mercaptopyridine N-oxide (2e), which contains a similar O,S donor set as 2d and 5d. For all three O,S donor molecules, slightly greater inhibition is seen in the mutant enzymes compared to WT. Interestingly, substitution of the heterocyclic oxygen of the 2-hydroxythiopyrones with an alkyl amine (e.g. 7e vs. 5d) abolishes inhibition against the WT enzyme while only slightly changing the activity against the two mutants. Although this modification does not change the O,S donor set, the pKa of the hydroxyl group is increased, which impacts metal binding. When the sulfur atom of these molecules is switched to an oxygen donor (e.g. 4e vs. 12e), the compounds lose their activity against the H94C variant but maintain weak inhibition against the H94D variant. The only exception to this is 2-hydroxypyridine N-oxide (1e), which shows slightly greater activity against the H94C mutant. The modest susceptibility of the H94D variant for other O,O donors is also seen with tropolone (11g), which does not show significant activity against WT or H94C hCAII. Overall, these results show that the coordination environment of a metalloprotein active site can influence the affinity of inhibitors. Further studies are needed to fully understand how the coordinating residues are producing this effect.
Effect of Coordination Environment on Inhibitor Binding Geometry
To understand the origin of the changes in affinity observed in the library screen, four inhibitor fragments were chosen for a more detailed analysis (Figure 12). These fragments were chosen because they have been crystallographically characterized with WT hCAII and represent four distinct binding modes to the protein active site. The inhibition constant (Ki) was determined for each molecule against the three hCAII variants, and crystal structures of their adducts with the enzymes were compared. Benzenesulfonamide (BSA, 1), a representative of the arylsulfonamide class of CA inhibitors (e.g. 7h), is a monodentate ligand that binds through the nitrogen atom of a deprotonated sulfonamide.
Figure 12.
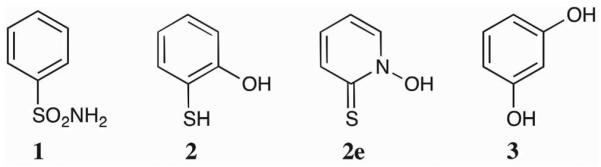
Structures of compounds 1–3.
2-Mercaptophenol (2), which is the hydroxyl derivative of 10g from CFL-1.1, has been shown to act as a monodentate ligand to the Zn(II) ion of hCAII through its thiol functionality. Aromatic thiols are relatively potent hCAII inhibitors, with inhibition constants in the low micromolar range.52 While CFL-1.1 contains many aromatic thiols (4c, 5c, 7c, and 9c), these compounds contain a pyrimidine ring. It has been seen that having a pyridyl nitrogen alpha- to an aromatic thiol can alter the binding mode of the thiol (data not shown). Compound 10g was too insoluble to screen at 500 μM; screening at 50 μM showed <20% inhibition against all three mutants. Examination of the crystal structure of 2 bound to WT hCAII (PDB: 2OSM) shows that the hydroxyl group is pointed towards the hydrophobic wall of the active site, and that the addition of a methyl group (as in 10g) may cause a steric clash with those residues.
Although 2e from CFL-1.1 contains a binding motif similar to that of 2, the 2-mercaptopyridine N-oxide moiety acts as a bidentate ligand in its adduct with WT hCAII.53 Finally, resorcinol (3) has been shown to inhibit hCAII by hydrogen bonding to the Zn(II)-bound hydroxide, and does not engage in metal ion binding.54 Inhibition constants and previously reported binding modes for these molecules are summarized in Table 2.
Table 2.
Ki values (μM) of compounds 1–3 against hCAII variants.
| Coordination Mode | Ki (WT) | Ki (H94D) | Ki (H94C) | |
|---|---|---|---|---|
| 1 | Monodentate (N−) | 0.490 ± 0.10 | 11 ± 2 | 5000 ± 1100 |
| 2 | Monodentate (S−) | 3.1 ± 0.7 | 30 ± 7 | 14 ± 5 |
| 2e | Bidentate | 850 ± 90 | 500 ± 60 | 270 ± 40 |
| 3 | Hydrogen Bonding | 4700 ± 300 | 2300 ± 900 | 1100 ± 400 |
The affinity of 1 for hCAII decreases dramatically in the mutants, particularly the H94C variant. The metal-ligand interaction for this class of molecules includes a strong electrostatic component between the negatively charged sulfonamide and the positive charge on the metal ion. Because this positive charge is decreased upon introduction of anionic cysteine or aspartate donors to the metal center, a loss of affinity is expected. However, charge neutralization alone does not explain the large difference in binding affinity between the H94D and H94C mutants. Compared to the drastic changes seen for 1, both 2 and 2e show smaller differences in affinity for the mutants relative to the WT enzyme. As opposed the N–Zn bond formed by 1, which is electrostatic in nature, the S–Zn bonds formed by 2 and 2e are more covalent, so a change in charge at the Zn(II) ion should have a less pronounced effect. The slight decrease in affinity of 2 for both mutants is likely also caused by a weaker electrostatic interaction. Because the S–Zn bond is more covalent, the reduction in binding for 2 is less pronounced than for 1. The affinity of 2e for all three mutants shows even less variability, which suggests that bidentate coordination (vide infra) makes the metal-ligand interaction more stable with respect to changes in the metal coordination environment. Although 3 does not interact directly with the metal ion, its affinity is slightly higher for the two mutants than for the WT enzyme. This difference is likely due to change in pKa of the Zn(II)-bound water molecule; both the H94C and H94D mutants have a pKa of over 9.5 compared to 6.8 for the WT enzyme.50 Since the affinity increases when the water molecule becomes more basic, it is likely that it is acting as a hydrogen bond donor to the hydroxyl group of 3.
In addition to the Ki data obtained, the first crystal structures of inhibitor fragments bound across a series of hCAII mutants were obtained in order to determine if any differences in binding mode are brought on by the change in metal coordination environment. First, the crystal structures of inhibitor-free hCAII H94D and H94C were analyzed. Although both variants have been structurally characterized previously, the reported structures are of relatively low resolution (~2.2 Å).55 In order to have better insight into the bond distances in the inhibitor-protein complexes, higher resolution data was obtained.
The crystal structure of inhibitor-free hCAII H94D at a resolution of 1.55 Å shows a tetrahedral Zn(II) ion bound by Asp94, His96, His119, and a water molecule (Figure 13). Asp94 acts as a monodentate ligand with a Zn–O bond distance of 2.00 Å, while its unbound carboxylate oxygen makes contacts with both an active site water molecule (2.67 Å) and Gln92 (2.89 Å). The NHis96 and NHis119 bond lengths (2.05 and 2.09 Å, respectively) are ~0.03 Å longer than in the WT enzyme. The bond between the Zn(II) ion and the catalytic water (2.02 Å) is over 0.1 Å longer than that in the WT structure, consistent with binding as neutral water as opposed to a hydroxide ion. The hydrogen bond with Thr199 is maintained at a distance of 2.7 Å.
Figure 13.
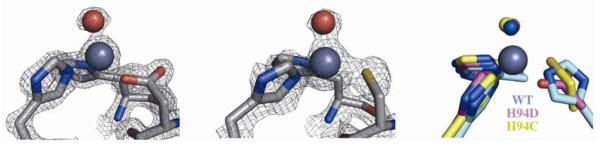
Inhibitor-free structures of the hCAII H94D (left) and H94C (middle) active sites. Electron density maps are contoured at 2σ. An overlay with the WT enzyme active site is shown on the right.
The structure of hCAII H94C at a resolution of 1.90 Å shows that the Zn(II) ion is coordinated in a tetrahedral geometry by Cys94, His96, His119, and a water molecule (Figure 13). The His-Zn distances are similar to those in the WT enzyme, and the SCys–Zn bond length is 2.16 Å. As in the H94D variant, the bond between Zn(II) and the Zn(II)-bound water is long (2.04 Å), but the hydrogen bond with Thr199 is also lengthened (2.84 Å).
After examination of inhibitor-free WT and mutant hCAII variants, co-crystals of these enzymes with inhibitor fragments were obtained. The coordination of 1 is nearly identical in all three variants despite the large differences in binding affinity, resulting in a distorted tetrahedral coordination sphere around the Zn(II) ion (Figure 14). The H94D-1 complex has nearly the same bond length as that of the WT complex (1.96 vs. 1.95 Å) and, as in the inhibitor-free structure, Asp94 acts as a monodentate ligand, with the OAsp–Zn bond contracted by 0.06 Å, accompanied by a lengthening of the hydrogen bond between Asp94 and Gln92 by ~0.1 Å. Corresponding to the substantially weaker inhibition of hCAII H94C by 1, the Zn–N bond length is longer than the other two variants (2.03 Å) and the SCys–Zn bond is lengthened by ~0.1 Å compared to the inhibitor-free structure. The slightly longer Zn–N bond length in the H94C-1 complex is accompanied by a closer interaction between the sulfonyl oxygen of 1 and the backbone amide of Thr199 (O–N distance of 2.8 Å compared to ~2.9 Å for WT and H94D structures, respectively). However, in both mutants, the hydrogen bonding between 1 and Thr199 is maintained (Figure 2), suggesting the loss of inhibitory activity is largely due to the reduced electrostatic interaction between the ligand fragment and the metal ion.
Figure 14.
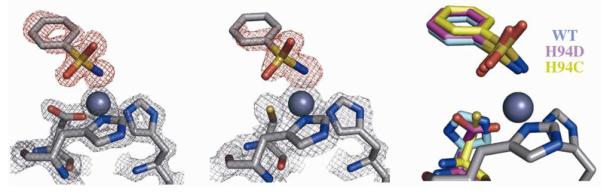
Crystal structures of 1 bound to H94D (left) and H94C (middle) variants. Fobs maps (2σ) for the protein are shown in gray while Fo-Fc omit maps (3σ for H94D, 2.5σ for H94C) are shown in red. An overlay with the WT structure shows little change in binding geometry (right).
Unfortunately, the structures of 2 bound to hCAII H94C and H94D could not be fully refined. In both cases, an atom was observed roughly 2.3 Å from the Zn(II) ion, much further than the Zn-water distance seen in the inhibitor-free structure. This distance is similar to that expected for a Zn–S bond, and the atom results in tetrahedral geometry around the metal ion. These observations are consistent with 2 binding the mutants in a monodentate fashion similar to that observed in the WT enzyme, but additional crystallographic data is required to definitively assign the mode of binding.
The previously reported crystal structure of 2e bound to WT hCAII (PDB ID 3M1K) reveals bidentate coordination, which results in trigonal bipyramidal geometry around the Zn(II) ion with the sulfur atom of 2e and His96 acting as axial ligands.53 This geometry is maintained in the complex of 2e with hCAII H94D (Figure 15). Corresponding with the increase in affinity observed in the inhibitory studies, the Zn–S and Zn–O bond lengths in the H94D-2e complex are both ~0.17 Å shorter than in the WT adduct. While the OAsp–Zn bond length is relatively unchanged upon binding 2e, the NHis96–Zn and NHis119–Zn bond lengths are significantly longer than in the inhibitor free structure (2.26 vs. 2.05 and 2.43 vs. 2.09 Å, respectively). The B-factor of the Zn(II) ion in the H94D variant bound to 2e is significantly higher than in the inhibitor)free structure (22.6 vs. 8.0) while those of the coordinating histidine and aspartate ligands do not change significantly, suggesting that the site is likely only partially occupied. In fact, when similar ligand soaking conditions were applied to crystals of hCAII H94C, which has a slightly higher zinc Kd, the active site was observed without a Zn(II) ion bound (data not shown); however, removal of metal ions from the active site is not likely to be the mechanism by which these molecules inhibit the enzymes.
Figure 15.
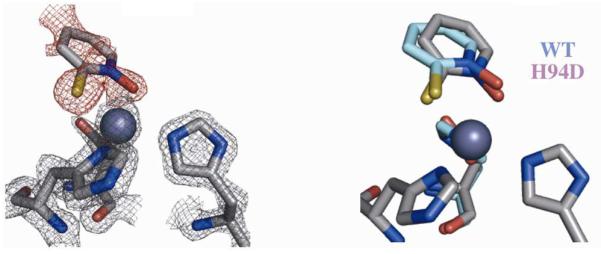
Crystal structure of 2e bound to hCAII H94D (left). The Fobs map (2σ) for the protein is shown in gray while the Fo-Fc omit map (2σ) for 2e is shown in red. An overlay with the WT structure shows little change in binding geometry (right).
Conclusions
Metalloproteins are attractive targets for therapeutics treating a wide variety of diseases. While there are some important success stories, the development of metalloprotein inhibitors as drugs remains challenging. Exploration of the chemical space encompassing MBGs may be a way to overcome some of these challenges, but before a wider variety of MBGs can efficiently aid drug design, a better understanding of the forces that drive metal binding in protein environments is needed. The original research presented here represents the first systematic study of how the coordination environment of a catalytic metal ion affects the binding of inhibitors. Without changing the residues that interact directly with inhibitors, altering the coordinating residues in hCAII results in large differences in their binding affinity, but without corresponding changes in the metal-binding geometry. The implications for these findings in the design of metalloprotein inhibitors will require substantially more studies, but the preliminary findings here lay the foundation for a deeper understanding of these important metal)ligand interactions within a protein environment. While CA has been used in the past to show that subtle changes in inhibitor binding can lead to drastic effects on potency, it can also be used to understand how subtle changes in metal environment can have the same effect.
Experimental
hCAII Activity Assay
WT hCAII, along with H94D and H94C variants, was expressed and purified as previously reported; details can be found in the Supporting Information. Assays were run in 50 mM HEPES pH 8.0 containing 5% DMSO with Na2SO4 added to an ionic strength of 100 mM. For the screening of CFL-1.1, a BioTek Precision XS microplate sample processor was utilized. Compounds from CFL)1.1 (500 μM final concentration) were incubated with protein (final concentrations of 100 nM for WT hCAII, 1 μM for H94D and H94C variants) for 10 minutes at 25 °C. Substrate (p-nitrophenylacetate, final concentration of 500 μM for WT and H94D, 1 mM for H94C) was added and hCAII-catalyzed cleavage was monitored by the increase in absorbance at 405 nm corresponding to the formation of the p-nitrophenolate anion. The initial linear reaction rate was compared to that of wells containing no inhibitor (0% inhibition) and no protein (100% inhibition). The rate of non-hCAII catalyzed PNPA hydrolysis in the presence of inhibitor was subtracted from each trial before determining percent inhibition. Details for determination of Kis can be found in the Supporting Information.
hCAII Crystallization
All three variants were crystallized using vapor diffusion; full details can be found in the Supporting Information. Briefly, 3 μL of a protein solution containing 20 mg/mL hCAII and 1 mM 4-chloromercuribenzoic acid was combined with 2.5)4.0 μL of a precipitant solution containing 2.7–3.0 M (NH4)2SO4 and allowed to equilibrate against 750 μL of precipitant solution. Crystals generally formed within 3–10 d. Crystallization of the H94C variant required the presence of dithiothreitol (DTT) and plates had to be prepared under inert atmosphere. Once formed, crystals were transferred to precipitant solutions containing inhibitor and 5% glycerol as a cryoprotectant and allowed to soak with saturated solutions of inhibitor fragments for between 8 h and 5 d. Crystals were taken directly from soak solutions for data collection. Details of data collection and structure refinement can be found in the Supporting Information.
Supplementary Material
Synopsis.
Human carbonic anhydrase II (hCAII), a Zn(II)-dependent hydrolase, has been widely studied to understand ligand-protein binding such as hydrogen bonding and hydrophobic interactions. This report expands the use of hCAII as a model system by examining how ligand binding is effected by mutations of residues within the Zn(II) ion coordination sphere. In this way, we can begin to understand how metal-binding group selectivity and affinity is perturbed by the composition of the ligating active site residues.
Figure 11.
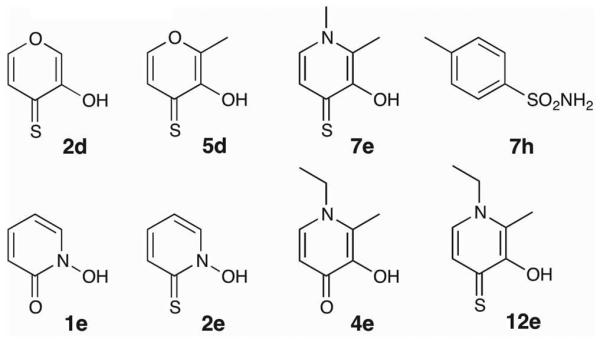
Structure of select molecules from CFL-1.1.
Acknowledgement
This work was funded by a grant from the National Institutes of Health through the National Institute of General Medical Sciences NIH (R01 GM098435). We thank Prof. Arnold Rheingold and Dr. Curtis Moore (UCSD) for assistance with crystallographic data collection and structure determination.
References
- 1.Holm RH, Kennepohl P, Solomon EI. Chem. Rev. 1996;96(7):2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 2.(a) Meldrum NU, Roughton FJW. J. Physiol. 1933;80(2):113–142. doi: 10.1113/jphysiol.1933.sp003077. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Keilin D, Mann T. Biochem. J. 1940;34(8–9):1163–1176. doi: 10.1042/bj0341163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henry RP, Swenson ER. Resp. Physiol. 2000;121:1–12. doi: 10.1016/s0034-5687(00)00110-9. [DOI] [PubMed] [Google Scholar]
- 4.Shah GN, Ulmasov B, Waheed A, Becker T, Makani S, Svichar N, Chesler M, Sly WS. Proc. Nat. Acad. Sci. 2005;102(46):16771–16776. doi: 10.1073/pnas.0508449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henry RP. Annu. Rev. Physiol. 1996;58:523–538. doi: 10.1146/annurev.ph.58.030196.002515. [DOI] [PubMed] [Google Scholar]
- 6.Krebs HA. Biochem. J. 1948;43(4):525–528. doi: 10.1042/bj0430525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wood AJJ. New Engl. J. Med. 1998;339(18):1298–1307. doi: 10.1056/NEJM199810293391808. [DOI] [PubMed] [Google Scholar]
- 8.Epstein DL, Grant WM. Arch. Ophthamol. 1977;95:1378–1382. doi: 10.1001/archopht.1977.04450080088009. [DOI] [PubMed] [Google Scholar]
- 9.Jacobsen JA, Fullagar JL, Miller MT, Cohen SM. J. Med. Chem. 2011;54(2):591–602. doi: 10.1021/jm101266s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boriack-Sjodin PA, Zeitlin S, Christianson DW, Chen H-H, Crenshaw L, Gross S, Dantanarayana A, Delgado P, May JA, Dean T. Prot. Sci. 1998;7(12):2483–2489. doi: 10.1002/pro.5560071201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scolnick LR, Clements AM, Liao J, Crenshaw L, Hellberg M, May J, Dean TR, Christianson DW. J. Am. Chem. Soc. 1997;119(4):850–851. [Google Scholar]
- 12.Brown NJ, Vaughan DE. Circulation. 1998;97:1411–1420. doi: 10.1161/01.cir.97.14.1411. [DOI] [PubMed] [Google Scholar]
- 13.Smith CG, Vane JR. FASEB J. 2003;17(8):788–789. doi: 10.1096/fj.03-0093life. [DOI] [PubMed] [Google Scholar]
- 14.Gavras H, Gavras I. Hypertension. 1988;11(3):II37–II41. doi: 10.1161/01.hyp.11.3_pt_2.ii37. [DOI] [PubMed] [Google Scholar]
- 15.Grima M, Welsch C, Michel B, Barthelmebs M, Imbs JL. Hypertension. 1991;17(4):492–496. doi: 10.1161/01.hyp.17.4.492. [DOI] [PubMed] [Google Scholar]
- 16. [(accessed Dec. 18, 2012)];Top Products by U.S. Dispensed Prescriptions. http://www.imshealth.com.
- 17.(a) Ryde U. Biophys. J. 1999;77(5):2777–2787. doi: 10.1016/S0006-3495(99)77110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ducháčková L, Roithová J. Chem. Eur. J. 2009;15:13399–13405. doi: 10.1002/chem.200901645. [DOI] [PubMed] [Google Scholar]
- 18.Subissi A, Criscuoli M, Sardelli G, Guelfi M, Giachetti A. J. Cardiovasc. Pharm. 1992;20:139–146. [PubMed] [Google Scholar]
- 19.Patchett AA. J. Med. Chem. 1993;36(15):2051–2058. doi: 10.1021/jm00067a001. [DOI] [PubMed] [Google Scholar]
- 20.Strahl BD, Allis CD. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 21.Grunstein M. Nature. 1997;389(6649):349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 22.Bolden JE, Peart MJ, Johnstone RW. Nat. Rev. Drug Disc. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 23.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. Oncologist. 2007;12(10):1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 24.Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M. J. Antibiot. 1994;47(3):301–310. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]
- 25. [(accessed December 12, 2012)]; http://www.clinicaltrials.gov.
- 26.Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R, Mita M, Beck J, Burris H, Ryan C, Atadja P, Butterfoss D, Dugan M, Culver K, Johnstone R, Prince HM. Clin. Cancer Res. 2008;14(14):4500–4510. doi: 10.1158/1078-0432.CCR-07-4262. [DOI] [PubMed] [Google Scholar]
- 27.(a) Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. Proc. Nat. Acad. Sci. 1999;96:4592–4597. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, Jong RD, O'Connell S, Grimshaw CE, Navre M, Gangloff AR. Bioorg. Med. Chem. Lett. 2010;20(10):3142–3145. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- 28.Roques BP, Noble F, Daugé V, Fournié-Zaluski M-C, Beaumont A. Pharmacol. Rev. 1993;45(1):87–146. [PubMed] [Google Scholar]
- 29.Berndt N, Hamilton AD, Sebti SM. Nat. Rev. Cancer. 2011;11(11):775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harousseau JL, Martinelli G, Jedrejczak WW, Brandwein JM, Bordessoule D, Masszi T, Ossenkoppele GJ, Alexeeva JA, Beutel G, Maertens J, Vidriales MB, Dombret H, Thomas X, Burnett AK, Robak T, Khuageva NK, Golenkov AK, Tothova E, Mollgard L, Park YC, Bessems A, DePorre P, Howes AJ. Blood. 2009;114(6):1166–1173. doi: 10.1182/blood-2009-01-198093. [DOI] [PubMed] [Google Scholar]
- 31.(a) Egeblad M, Werb Z. Nat. Rev. Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]; (b) Deryugina EI, Quigley JP. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 32.Murphy G, Nagase H. Nat. Clin. Pract. Rheumatol. 2008;4(3):128–135. doi: 10.1038/ncprheum0727. [DOI] [PubMed] [Google Scholar]
- 33.(a) Zhao B-Q, Wang S, Kim H-Y, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Nat. Med. 2006;12(4):441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]; (b) Lee S-R, Tsuji K, Lee S-R, Lo EH. J. Neurosci. 2004;24(3):671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Fisher JF, Mobashery S. Cancer Metastasis Rev. 2006;25:115–136. doi: 10.1007/s10555-006-7894-9. [DOI] [PubMed] [Google Scholar]; (b) Coussens LM, Fingleton B, Matrisian LM. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 35.Griffin MO, Ceballos G, Villarreal FJ. Pharmacol. Res. 2011;63(2):102–107. doi: 10.1016/j.phrs.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Overall CM, Lopez-Otin C. Nat. Rev. Cancer. 2002;2(9):657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 37.(a) Agrawal A, Romero-Perez D, Jacobsen JA, Villarreal FJ, Cohen SM. ChemMedChem. 2008;3(5):812–820. doi: 10.1002/cmdc.200700290. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rouffet M, Oliveira C. A. F. d., Udi Y, Agrawal A, Sagi I, McCammon JA, Cohen SM. J. Am. Chem. Soc. 2010;132(24):8232–8233. doi: 10.1021/ja101088j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rubino MT, Agamennone M, Campestre C, Fracciolla G, Laghezza A, Loiodice F, Nuti E, Rossello A, Tortorella P. ChemMedChem. 2009;4(3):352–362. doi: 10.1002/cmdc.200800324. [DOI] [PubMed] [Google Scholar]
- 38.Muri EMF, Nieto MJ, Sindelar RD, Williamson JS. Curr. Med. Chem. 2002;9(17):1631–1653. doi: 10.2174/0929867023369402. [DOI] [PubMed] [Google Scholar]
- 39.Weisburger JH, Weisburger EK. Pharmacol. Rev. 1973;25(1):1–66. [PubMed] [Google Scholar]
- 40.Turner AJ, Hooper NM. Trends Pharm. Sci. 2002;23(4):177–183. doi: 10.1016/s0165-6147(00)01994-5. [DOI] [PubMed] [Google Scholar]
- 41.Rouffet M, Cohen SM. Dalton Trans. 2011;40:3445–3454. doi: 10.1039/c0dt01743d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Puerta DT, Griffin MO, Lewis JA, Romero-Perez D, Garcia R, Villarreal FJ, Cohem SM. J. Biol. Inorg. Chem. 2006;11(2):131–138. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]; (b) Agrawal A, Oliveira C. A. d., Cheng Y, Jacobsen JA, McCammon JA, Cohen SM. J. Med. Chem. 2009;52(4):1063–1074. doi: 10.1021/jm8013212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Garner AL, Struss AK, Fullagar JL, Agrawal A, Moreno AY, Cohen SM, Janda KD. ACS Med. Chem. Lett. 2012;3(8):668–672. doi: 10.1021/ml300128f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis JA, Mongan J, McCammon JA, Cohen SM. ChemMedChem. 2006;1:694–697. doi: 10.1002/cmdc.200600102. [DOI] [PubMed] [Google Scholar]
- 44.Yan Y-L, Miller MT, Cao Y, Cohen SM. Bioorg. Med. Chem. Lett. 2009;19:1970–1976. doi: 10.1016/j.bmcl.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.(a) Puerta DT, Cohen SM. Inorg. Chem. 2003;42:3423–3430. doi: 10.1021/ic026029g. [DOI] [PubMed] [Google Scholar]; (b) Puerta DT, Lewis JA, Cohen SM. J. Am. Chem. Soc. 2004;126:8388–8389. doi: 10.1021/ja0485513. [DOI] [PubMed] [Google Scholar]
- 46.Parkin G. Chem. Rev. 2004;104(2):699–768. doi: 10.1021/cr0206263. [DOI] [PubMed] [Google Scholar]
- 47.Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB, Whitesides GM. Chem. Rev. 2008;108(3):946–1051. doi: 10.1021/cr050262p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.(a) Kiefer LL, Paterno SA, Fierke CA. J. Am. Chem. Soc. 1995;117(26):6831–6837. [Google Scholar]; (b) Lesburg CA, Christianson DW. J. Am. Chem. Soc. 1995;117(26):6838–6844. [Google Scholar]
- 49.McCall KA, Fierke CA. Biochemistry. 2004;43(13):3979–3986. doi: 10.1021/bi0498914. [DOI] [PubMed] [Google Scholar]
- 50.Kiefer LL, Fierke CA. Biochemistry. 1994;33(51):15233–15240. doi: 10.1021/bi00255a003. [DOI] [PubMed] [Google Scholar]
- 51.Verpoorte JA, Mehta S, Edsall J. J. Biol. Chem. 1967;242(18):4421–4229. [PubMed] [Google Scholar]
- 52.A. A. B., Genis C, Fisher SZ, Orwenyo JN, Kumara MT, Dutta SK, Phillips E, Kiddle JJ, Tu C, Silverman DN, Covindasamy L, Agbandje-McKenna M, McKenna R, Tripp BC. Biochemistry. 2008;47(10):3174–3184. doi: 10.1021/bi702385k. [DOI] [PubMed] [Google Scholar]
- 53.Wischeler JS, Innocenti A, Vullo D, Agrawal A, Cohen SM, Heine A, Supuran CT, Klebe G. ChemMedChem. 2010;5:1609–1615. doi: 10.1002/cmdc.201000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin DP, Cohen SM. Chem. Commun. 2012;48:5259–5261. doi: 10.1039/c2cc32013d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.(a) Ippolito JA, Christianson DW. Biochemistry. 1994;33(51):15241–15249. [PubMed] [Google Scholar]; (b) Kiefer LL, Ippolito JA, Fierke CA, Christianson DW. J. Am. Chem. Soc. 1993;115(26):12581–12582. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.