

Abstract
The effects of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways on proliferation, drug resistance, prevention of apoptosis and sensitivity to signal transduction inhibitors were examined in FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells which are conditionally-transformed to grow in response to Raf and Akt activation. Drug resistant cells were isolated from FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells in the presence of doxorubicin. Activation of Raf-1, in the drug resistant FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells, increased the IC50 for doxorubicin 80-fold, whereas activation of Akt-1, by itself, had no effect on the doxorubicin IC50. However, Akt-1 activation enhanced cell proliferation and clonogenicity in the presence of chemotherapeutic drugs. Thus the Raf/MEK/ERK pathway had profound effects on the sensitivity to chemotherapeutic drugs, and Akt-1 activation was required for the long-term growth of these cells as well as resistance to chemotherapeutic drugs. The effects of doxorubicin on the induction of apoptosis in the drug resistant cells were enhanced by addition of either mTOR and MEK inhibitors. These results indicate that targeting the Raf/MEK/ERK and PI3K/Akt/mTOR pathways may be an effective approach for therapeutic intervention in drug resistant cancers that have mutations activating these cascades.
Keywords: Raf, Akt, signal transduction inhibitors, chemotherapeutic drugs, drug resistance
Introduction
The Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR signaling pathways have been extensively studied over the past few decades.1-6 In this time there have been significant breakthroughs in the discovery of interacting pathway components and insights into how mutations of these components can lead to aberrant signaling and uncontrolled proliferation.1-6 Research has also lead to the development of inhibitors that specifically target critical elements of these pathways.1-6 Many recent studies are directed at increasing cancer patient survival by targeting these and other pathways in cancer stem cells.7-12 Recently these pathways have been shown to play critical roles in the preventing aging and senescence.12-16 Thus these pathways have been targeted to promote longevity by altering the aging process which might be enhanced when these pathways are hyperactivated.17-20
Signaling through the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR cascades is a carefully orchestrated series of events generally starting from the cell surface and leading to controlled gene expression within the nucleus.5 Regulation of these pathways is mediated by a series of kinases, phosphatases and various exchange proteins. Mutations occur in many of these pathway elements leading to uncontrolled regulation and in some cases resistance to chemo- and radiotherapy.4-6,21-24 However, the precise details regarding how these pathways interact to result in therapy resistance is not well elucidated and furthermore, which pathway dominates is not clear. Elucidation of the mechanisms of therapy resistance could enhance cancer therapy.23,24
Proliferation and suppression of apoptosis in many hematopoietic precursor cells is promoted by cytokines such as interleukin-3 (IL-3) and other growth factors.5,25-30 Hematopoietic cell lines have been established which require IL-3 for proliferation and survival.25 The FL5.12 cell line is an IL3-dependent cell line isolated from murine fetal liver and is an in vitro model of early hematopoietic progenitor cells.7,25 Cytokine-deprivation of these cells results in rapid cessation of growth and subsequent death by apoptosis (programmed cell death).26-30 In the presence of IL-3, these cells proliferate continuously, however, they are non-tumorigenic upon injection into immunocompromised mice.26-29 Spontaneous factor-independent cells are rarely recovered from the FL5.12 cell line (< 10−7), making it an attractive model to analyze the effects various genes have on signal transduction, apoptosis and chemotherapeutic drug resistance.22
In the following studies, we sought to determine the effects of Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways on drug resistance and sensitivity to targeted therapy. In order to investigate potential roles, we transformed IL3-dependent FL5.12 cells to proliferate in response to activation of Raf-1 and Akt-1 by introducing conditional ΔRaf-1:AR and ΔAkt-1:ER* constructs into these hematopoietic cells.30 In our conditionally-inducible model, we can investigate the individual and combined contributions these pathways exert on drug resistance and sensitivity to signal transduction inhibitors. This has allowed us to evaluate the potential of combining targeted therapy with classical chemotherapy to treat various cancers.
Results
Isolation of drug resistant FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells
To examine the roles of the Raf/MEK/ERK and PI3K/Akt/mTOR pathways on the induction of chemotherapeutic drug resistance, FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were plated in limiting dilution experiments in 96 well plates in the presence of various concentrations of doxorubicin (Fig. 1). Colonies were isolated that grew in the presence of 10 nM doxorubicin and 4HT + testosterone at a frequency of approximately 1.6 x 10−2 and also in the presence of 25 nM doxorubicin at a frequency of 6.7 x 10−3 that were expanded subsequently into stable cell lines. Clones were also isolated in medium containing 100 nM doxorubicin, 4HT + Test, but the efficiency of recovery of these cells, at the higher doxorubicin concentration, was much lower (A). No colonies were recovered when the medium contained 1,000 nM doxorubicin. Long-term clones were not isolated when FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were plated in doxorubicin and either 4HT or testosterone by themselves indicating that activation of both ΔRaf-1:AR and ΔAkt-1:ER* were required for the long-term growth of the cells in the presence of doxorubicin (data not presented).
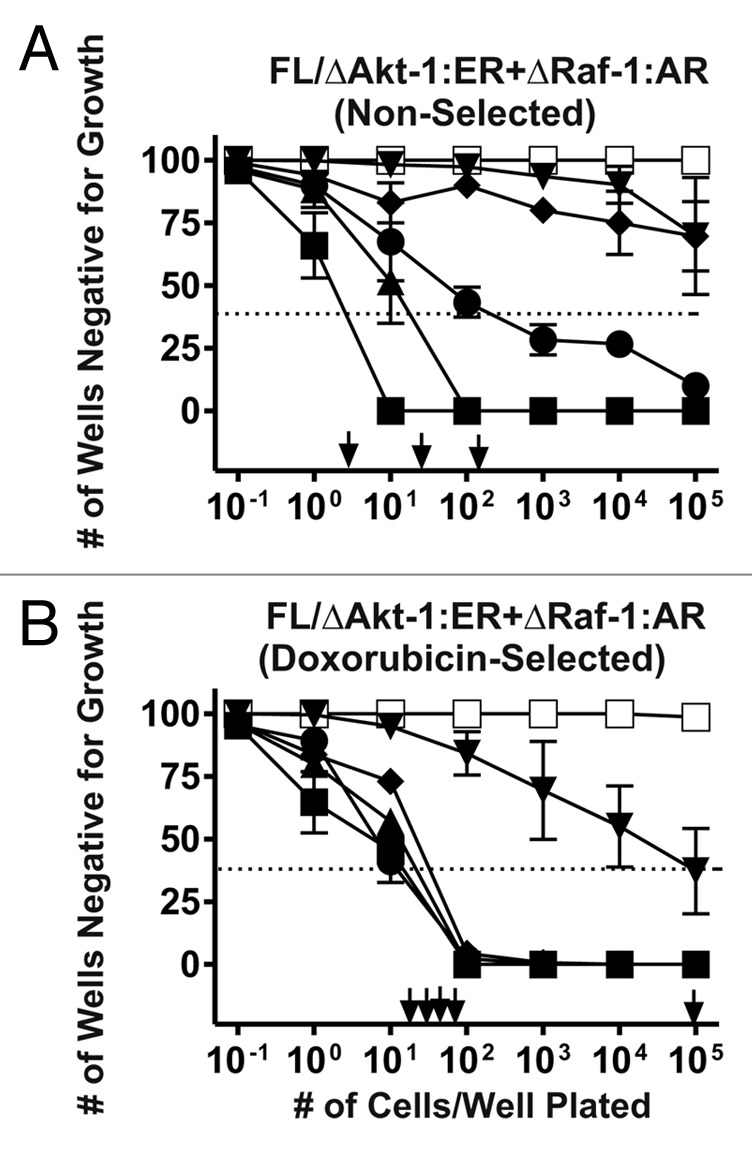
Figure 1. Isolation of doxorubicin resistant FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR Cells. FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were plated in medium containing 4HT + Test at different cell concentrations in the indicated concentrations of doxorubicin (A). Medium with fresh 4HT + Test and the indicated concentrations of doxorubicin were added every three days. At the end of approximately 3 weeks, the number of wells positive for growth were determined and the cloning efficiency in doxorubicin estimated by Poisson statistical analysis. Clones were isolated from the wells that were positive at the lowest cell dilution and expanded into larger cultures. The cloning efficiency of these doxorubicin-selected cells in doxorubicin was examined subsequently (B). These experiments were repeated 6 times and averaged together. Symbols: solid squares (■) = 0 nM doxorubicin, solid triangles (▲) = 10 nM doxorubicin, solid circles (●) = 25 nM doxorubicin, solid diamonds (◆) = 50 nM doxorubicin, solid downward triangles (▼) = 100 nM doxorubicin, open squares (□) = 1000 nM doxorubicin. A dotted line is drawn at 37% of wells negative for growth that can be used by Poisson statistical analysis to indicate the cloning efficiency. This is the concentration of cells required to yield 1 colony/well.
The subcloning efficiency of the doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells isolated in medium containing 10 nM doxorubicin and 4HT + Test was further examined in different concentrations of doxorubicin (B). These cells also had an approximately 2–10-fold higher subcloning efficiency than the initial cell line when plated in 10, 25, 50 or 100 nM doxorubicin. However, no clones were recovered when the cells were plated in medium containing 1,000 nM doxorubicin.
Elevated chemotherapeutic drug IC50s in doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells
The sensitivities of the non-selected and doxorubicin-selected FL/ΔAkt-1:ER* + ΔRaf-1:AR cell lines to five different chemotherapeutic drugs were compared (Fig. Two and Table 1). The doxorubicin-selected FL/ΔAkt-1:ER* + ΔRaf-1:AR cells had approximately 2-fold higher IC50s for doxorubicin (A) and daunorubicin (B) and approximately 4-fold higher IC50 for paclitaxel (C) than the non-selected FL/ΔAkt-1:ER* + ΔRaf-1:AR cells. In contrast, non-selected and doxorubicin-selected FL/ΔAkt-1:ER* + ΔRaf-1:AR cells did not have significant differences in their IC50s when plated in medium containing either cisplatin or 5-flurouracil (Table 1).
Table 1. Differences in chemotherapeutic drug IC50s in non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells1.
| Cell line→ | Non-selected | Doxorubicin-selected | Fold difference |
|---|---|---|---|
|
Drug↓ |
|
|
|
| Doxorubicin |
25 ± 6 nM |
65 ± 11 nM |
2.6X |
| Daunorubicin |
9 ± 3 nM |
18 ± 5 nM |
2.0X |
| Paclitaxel |
3 ± 0.8 nM |
13 ± 2 nM |
4.3X |
| Cisplatin |
80,000 ± 9,000 |
90,000 ± 11,000 nM |
1.1X |
| 5-Flurouracil | 1,000 ± 200 nM | 1,200 ± 100 nM | 1.2X |
1 Determined by plating 2,500 cells/well in 96 well plates in 500 nM 4HT + 100 nM as described in materials and methods.
Lack of elevated drug transport activity in doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells
Many drug resistant cancers display elevated transport (efflux) of chemotherapeutic drugs.31-33 Drug resistant phenotypes can be due to many different biochemical phenomenon, a common mechanism is elevated MDR-1/MRP-1 expression. To determine if the non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells differed in rates of drug efflux, their abilities to efflux daunorubicin or rhodamine 123 were compared. Daunorubicin and rhodamine 123 are both substrates for MDR-1/MRP-1.31-33 No significant differences in their abilities to efflux these two drugs were observed. Furthermore, addition of verapmil, which inhibits drug efflux mediated by MDR-1/MRP-1 did not alter the efflux of these compounds (data not presented). Thus the drug resistance of these cells was not likely mediated by increased activity of these two drug transporters.
Increased endogenous Akt and p53 activation in doxorubicin-selected cells
The expression of the Raf/MEK/ERK, PI3K/Akt and p53 pathways in response to doxorubicin was examined in the non-selected and doxorubicin-selected FL/ΔAkt-1:ER* + ΔRaf-1:AR cells after treatment of the cells with different concentrations of doxorubicin for 4 h (Fig. 3). As a control, the levels of total ERK1,2 were examined and relatively equal levels were detected in the two different cell types over the doxorubicin dose curve performed. Treatment with 1 to 10 nM doxorubicin resulted in an increase in the detection of activated ERK1,2 and p38MAPK in both non-selected and doxorubicin-selected cells. Activated p38MAPK was detected after 10 nM doxorubicin treatment of the non-selected cells and after 1–10 nM treatment of the doxorubicin selected cells. A slight increase in activated JNK was detected in the doxorubicin-selected cells.

Figure 3. Doxorubicin-induced gene expression in non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells. The effects of different concentrations of doxorubicin on MAPK, Akt, p53 and Bcl-XL expression were examined in non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells after they were cultured in the presence of 4HT + Test for 24 h in the absence of doxorubicin. Cells were then stimulated with the indicated concentrations of doxorubicin for 4 h and then western blot analysis was performed.
When these blots were probed with an antibody that recognized Akt, two bands were detected, the endogenous (E)-derived Akt protein and the vector (V)-derived (ΔAkt-1:ER*) protein. Higher levels of activated E-Akt were detected constitutively in the doxorubicin-selected cells than in the non-selected cells when the blots were probed with the antibody which recognizes activated S-473 phosphorylated Akt. In contrast, relatively equal levels of V- and E-Akt were detected in the non-selected and doxorubicin-selected cells when the blots were probed with an antibody which recognizes total (T) Akt. Thus in the doxorubicin-selected FL/ΔAkt-1:ER* + ΔRaf-1:AR cells, there was constitutive expression of activated Akt. This band is highlighted with an arrow for clarity sake.
One of the key downstream effects of doxorubicin in cells containing WT p53 is the activation of p53. Higher levels of activated p53 were detected in the doxorubicin-selected cells, at lower doxorubicin concentrations than in the non-selected cells, although both cell types displayed similar levels of p53 when treated with 10 and 100 nM doxorubicin.
The expression of the cytokine-regulated Bcl-XL gene was examined in the non-selected and doxorubicin-selected cells. Interestingly in the non-selected cells, the expression of Bcl-XL was detected at similar levels after 0 to 100 nM treatment. However, the doxorubicin-selected cells displayed a different pattern of Bcl-XL expression in response to doxorubicin. Bcl-XL expression was not observed in the doxorubicin-resistant cells in absence of doxorubicin but was seen when they were treated with doxorubicin concentrations 1 nM and higher suggesting that the cells had developed a dependency on doxorubicin for the expression of certain survival factors.
Importance of Raf-1 and Akt-1 in promoting proliferation and drug resistance
The importance of Raf-1 and Akt-1 in promoting proliferation in the presence of chemotherapeutic drugs was examined in the non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells (Fig. 4). In these 4 d proliferation assays, activation of ΔRaf-1:AR was necessary for the proliferation of these cells in the absence of chemotherapeutic drugs (Fig. 4A and B). In contrast, only minimal proliferation was observed with either cell line was treated with no supplement or 4HT (A and B). In the absence of either doxorubicin or paclitaxel, the doxorubicin-sensitive and doxorubicin-resistant cells had similar growth rates in the presences of 4HT + Test (C).

Figure 4. Dominance of Raf-1 in promoting proliferation and drug resistance. Growth was measured over a period of 4 d in the presence of no drugs (A–C), 25 nM doxorubicin (D–F) or 10 nM paclitaxel (G–I). Symbols: solid squares (■) = no supplement, solid downward triangles (▼) = 100 nM Test, solid upward triangles (▲) = 500 nM 4HT, solid diamonds (◆) = 500 nM 4HT + 100 nM Test. Each data point is the average of 8 replicates.
The proliferation of the cells was also examined in the presence of 25 nM doxorubicin (D–F) or 10 nM paclitaxel (G–I)
While activation of ΔAkt-1:ER* by itself did not promote proliferation, it enhanced the level of proliferation when the non-selected and doxorubicin-selected cells were cultured in either doxorubicin or paclitaxel (D and E and G and H, respectively). Similar results were observed with daunorubicin (data not presented). While the -non-selected and doxorubicin-selected cells displayed similar growth rates in the absence of drugs (C), the doxorubicin-selected cells showed enhanced growth rates, compared with the non-selected cells, in the presence of 4HT + Test and either doxorubicin (F) or paclitaxel (I). These results indicated that the doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells remained dependent on ΔRaf-1:AR for proliferation and activation of ΔAkt-1:ER* enhanced their proliferation in the presence of chemotherapeutic drugs.
A dose response curve was also performed with the non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells (Fig. 5). Activation of ΔRaf-1:AR was essential for the elevated doxorubicin IC50 of both non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells. As presented previously (Fig. 2), doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells had an approximately 2-fold higher IC50 for doxorubicin than non-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells.

Figure 5. Dominance of Raf-1 in increasing doxorubicin IC50. The effects of Raf-1 and Akt-1 activation on the drug resistance were examined by culturing the non-selected and doxorubicin-selected cells in the presence of the different concentrations of doxorubicin for 4 d and then determining their relative growth by MTT analysis. A dotted line at 50% relative growth on the Y-axis is presented from which the IC50s on the X-axis can be estimated which are indicated by arrows. (A) non-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells, and (B) doxorubicin-selected resistant FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells. Symbols: solid squares (■) = no supplement, solid downward triangles (▼) = 100 nM Test, solid upward triangles (▲) = 500 nM 4HT and solid diamonds (◆) = 500 nM 4HT + 100 nM Test.
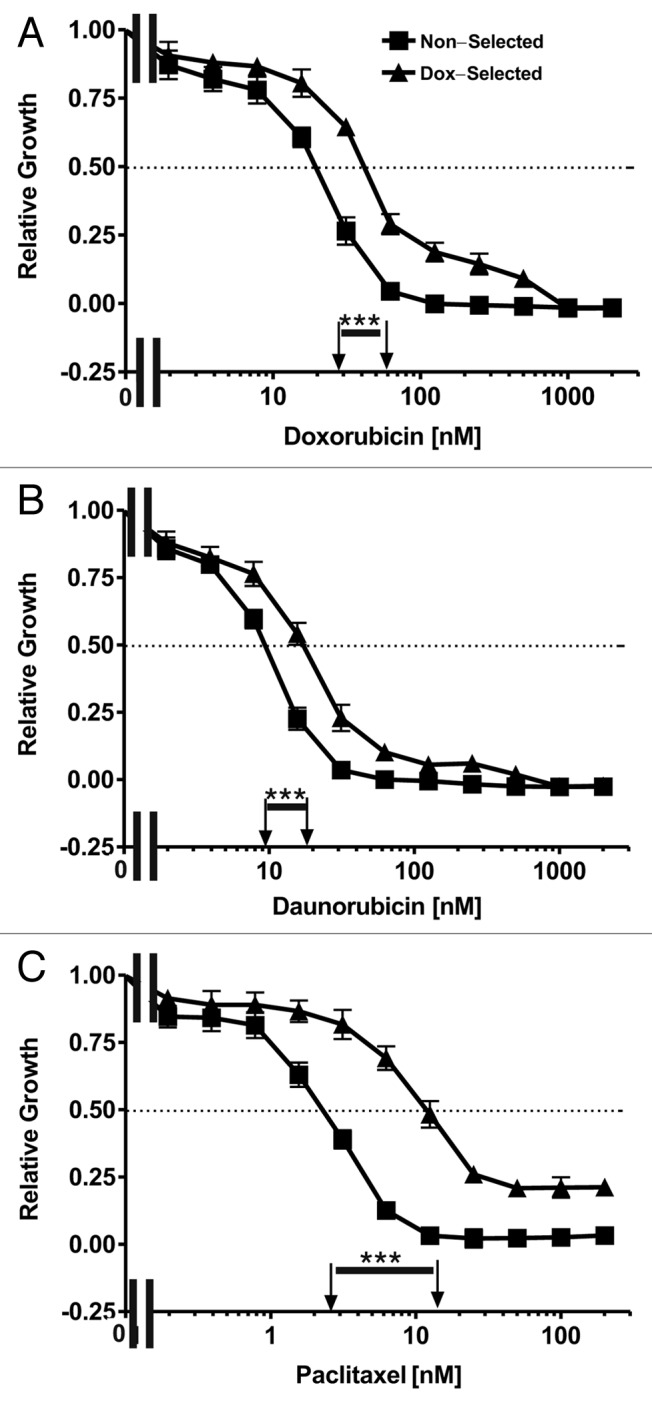
Figure 2. Sensitivities of non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells to chemotherapeutic drugs. The sensitivities of non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells to three chemotherapeutic drugs was examined by MTT analysis. Cells were grown in medium containing 500 nM 4HT + 100 nM Test. Symbols: solid squares (■) = non-selected cells, solid upward triangles (▲) = doxorubicin-selected cells. A dotted line at 50% relative growth on the Y-axis is presented from which the IC50s on the X-axis can be estimated which are indicated by arrows. These experiments were repeated 5-times and similar results were observed. Differences between the IC50s in the drug sensitive and resistant cells were statistically examined by the Student’s t test and statistical significance is indicated above the line with ***(p < 0.001).
Effects of Raf-1 and Akt-1 on the prevention of apoptosis in non-selected and doxorubicin-selected cells
The effects of IL-3, ΔRaf-1:AR or ΔAkt-1:ER* activation on the sensitivity to doxorubicin-induced apoptosis were examined in non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells by annexin V/PI experiments (Fig. 6A and B). Activation of ΔRaf-1:AR increased the IC50 for doxorubicin-induced apoptosis, compared with untreated cells, approximately 10-fold in the non-selected cells (A) and approximately 80-fold in doxorubicin-selected cells (B). In contrast, activation of ΔAkt-1:ER*, did not alter the IC50 for doxorubicin as the IC50 for doxorubicin induced apoptosis was similar in cells treated with no supplement or treated with 500 nM 4HT. Furthermore in these annexin V apoptosis assays, activation of Akt-1 in the presence of Raf-1 activation did not further increase the resistance of either the non-selected and doxorubicin selected cells to doxorubicin-induced apoptosis (compare when the cells were treated with Test vs. 4HT + Test).

Figure 6. Dominance of Raf-1 in the prevention of apoptosis induced by doxorubicin. The effects of IL-3, Raf-1 and Akt-1 activation on the prevention of apoptosis were examined by culturing the non-selected (A) and doxorubicin-selected (B) FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells in the presence of the different concentrations of doxorubicin for 3 d and then determining the extent of apoptosis induction by annexin. V/PI staining. (A) non-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells, and (B) doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells. Symbols: solid squares (■) = no supplement, solid downward triangles (▼) = 100 nM Test, solid upward triangles (▲) = 500 nM 4HT, solid diamonds (◆) = 500 nM 4HT and 100 nM Test, solid circles (●) IL-3 (10% WCM).
To further examine the effects of these pathways on the induction of apoptosis, Caspase 3 activation assays were performed (Fig. 7). In these experiments, we also included cytokine-dependent FL5.12 cells as a control as they are very sensitive to cytokine withdrawal. FL5.12 cells are the parental cell line from which the non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were derived. When FL5.12 cells were treated with either doxorubicin or paclitaxel, a dose dependent induction of apoptosis was observed when the cells were cultured with IL-3 (A and E). When FL5.12 cells were cultured in the absence of IL-3 and either doxorubicin (B–D) or paclitaxel (F–H), over 90% of the cells registered positive for activation of Caspase 3, similar results were observed with Caspase 9 activation (data not presented).

Figure 7. Dominance of Raf-1 in the prevention of Caspase 3 activation induced by chemotherapeutic drugs. The effects of IL-3, Raf-1 and Akt-1 activation on the prevention of caspase 3 activation mediated by doxorubicin (A–D) or paclitaxel (E–H) were examined. Cells were incubated in the presence of the different concentrations of doxorubicin or paclitaxel for 3 d and then caspase 3 activation was determined by flow cytometic analysis. Cells were incubated with IL-3 (10% WCM) (A and E), 500 nM 4HT (B and F), 100 nM Test (C and G) or 500 nM 4HT + 100 nM Test (D and H). Symbols: solid squares (■) = cytokine dependent FL5.12 cells, solid upward triangles (▲) = non-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells, and solid downward triangles (▼) = doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells.
Overall, less caspase 3 activation was detected in the doxorubicin-selected than in the non-selected cells in response to either doxorubicin or paclitaxel treatment. More caspase 3 activation was detected when either the non-selected or doxorubicin-selected cells were treated with 4HT which activated ΔAkt-1:ER (B and F), than when the cells were cultured with Test which activated ΔRaf-1:ER (C and G).
Effects of signal transduction inhibitors on the sensitivity to doxorubicin
The effects of signal transduction inhibitors on non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were examined (Fig. 8). Both non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were sensitive to the MEK1 inhibitor U0126 at concentrations greater than 1 μM. The doxorubicin-selected FL/ΔAkt:ER*(Myr+) + ΔRaf-1:AR cells were more resistant to the MEK1 inhibitor UO126 than the non-selected cells. Likewise both non-selected and doxorubicin-selected cells were sensitive to the mTOR inhibitor rapamycin. The response to rapamycin occurred at very low concentrations (< 1 nM) and increasing the dose of rapamycin did not increase the percentage of apoptotic cells which hovered around 50%. Inhibiting both MEK1 and mTOR resulted in an increase in cell death in both cell lines as indicated in Figure 8.
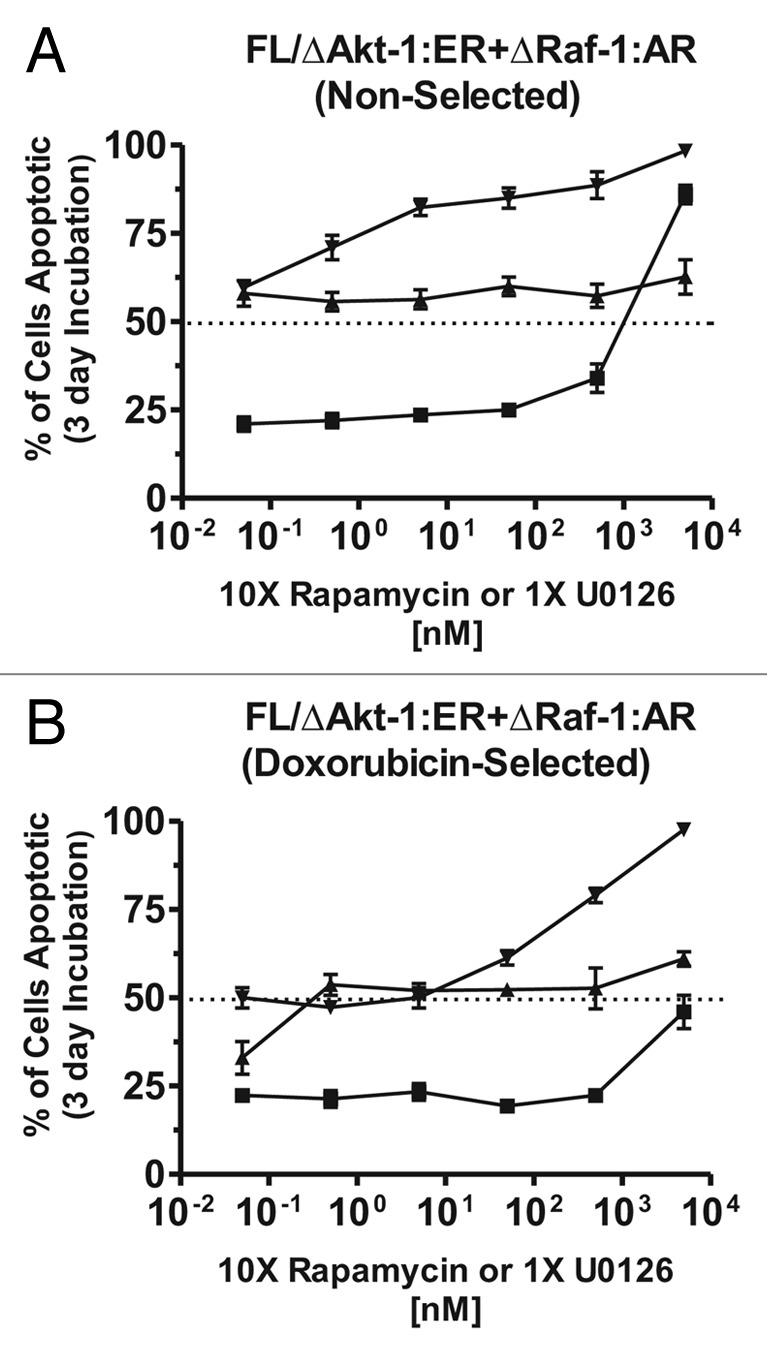
Figure 8. Effects of MEK and mTOR inhibitors on the induction of apoptosis. The effects of MEK1 and mTOR inhibitors on the induction of apoptosis were examined by culturing the indicated cells in the presence of different concentrations of the inhibitors for 3 d and then determining the extent of apoptosis induction by annexin V/PI staining. Cells were cultured in medium containing 500 nM 4HT + 100 nM Test and the indicated concentrations of the inhibitors. (A) non-selected FL/ΔAkt:ER*(Myr+) + ΔRaf-1:AR cells, and (B) doxorubicin-selected FL/ΔAkt:ER*(Myr+) + ΔRaf-1:AR cells. Symbols: solid squares (■) = U0126 MEK inhibitor (5,000, 500, 50, 5, 0.5 and 0.05 nM), solid triangles. (▲) = Rapamycin mTOR inhibitor (500, 50, 5, 0.5, 0.05 and 0.005 nM), solid downward triangle (▼) = combination of rapamycin (500, 50, 5, 0.5, 0.05 and 0.005 nM) and U0126 (5,000, 500, 50, 5, 0.5 and 0.05 nM).
Interactions between doxorubicin and targeted therapy in the induction of apoptosis
The ability of targeted therapy to enhance chemotherapy was examined (Fig. 9). Increased induction of apoptosis was more readily detected between doxorubicin and either the MEK or the mTOR inhibitors in the non-selected cells (A). Increased apoptosis also occurred when the mTOR inhibitor and doxorubicin were added together in the doxorubicin-selected cells (B). Increased apoptosis was observed when the mTOR and MEK inhibitors were combined. Treatment of both cell lines with MEK, mTOR and doxorubicin resulted in enhanced apoptosis. These results indicate that targeting these signaling pathways may increase the effectiveness of chemotherapeutic drugs to induce cell death.
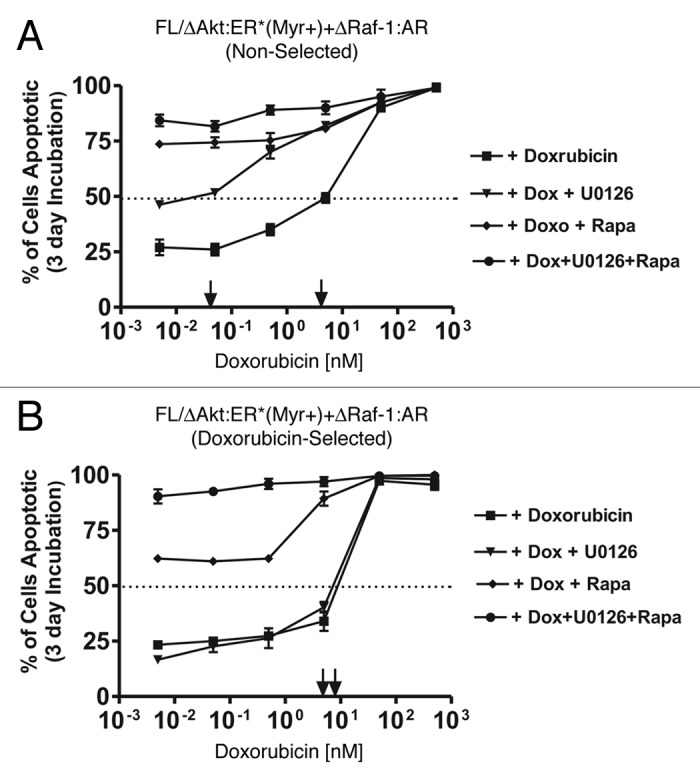
Figure 9. Effects of MEK and mTOR inhibitors on the induction of apoptosis induced by doxorubicin. The effects of MEK1 and mTOR inhibitors on doxorubicin IC50 was examined by culturing the indicated cells in the presence of the different concentrations of doxorubicin for three days and then determining the extent of apoptosis induction by annexin V/PI staining. Cells were cultured in medium containing 500 nM 4HT + 100 nM Test and the indicated concentrations of the inhibitors. (A) non-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells, and (B) doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells. Symbols: solid squares (■) = doxorubicin, solid downward triangles (▼) = indicated concentrations of doxorubicin and 5,000, 500, 50, 5, 0.5 and 0.05 nM MEK inhibitor, solid diamonds (◆) = indicated concentrations of doxorubicin and 500, 50, 5, 0.5, 0.05 and 0.005 nM mTOR inhibitors, and solid circles (●) = indicated concentrations of doxorubicin and 5,000, 500, 50, 5, 0.5 and 0.05 nM MEK and mTOR inhibitors.
Discussion
In this study, the roles of Raf and Akt in resistance to chemotherapeutic drugs and sensitivity to targeted therapy were examined. We have previously demonstrated the individual roles of Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades in drug resistance and cell cycle progression.22,30,36 In this current study, we compared the roles of these pathways in drug resistance in hematopoietic cells where we can conditionally control the expression of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways. Raf-1 was dominant in terms of promoting both growth and drug resistance when compared with Akt-1. These results indicated that targeting Raf or downstream MEK may be an effective means to combat the growth of certain drug resistant leukemias.
Interestingly in the doxorubicin-selected cells, they did not display grossly elevated activated ERK, p38MAPK or JNK, however, activation of ERK occurred at lower doxorubicin concentrations in the doxorubicin-selected cells than in the non-selected cells. Also the doxorubicin-selected cells displayed elevated activated endogenous Akt and p53 compared with the non-selected cells. Furthermore, Bcl-XL expression was dependent on the presence of doxorubicin concentrations greater than 1 nM indicating that the expression of this survival protein had become doxorubicin-dependent in the doxorubicin-selected cells. The mechanisms resulting in the doxorubicin-regulated expression of the Bcl-XL gene in the drug selected cells is under further investigation. The drug resistance of the doxorubicin-selected cells did not appear to be due to a lack of activated JNK expression, which has been observed in some drug resistant leukemias.37
Our studies indicate the critical roles of the Raf/MEK/ERK pathway in the promotion of cell growth, chemotherapeutic drug resistance and sensitivity to targeted therapy. The doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were also less sensitive to the effects of daunorubicin and paclitaxel than the non-selected cells. Activation of Raf-1 but not Akt-1 increased the IC50 for doxorubicin, as determined by sensitive annexin V/apoptosis assays, approximately 10-fold in the drug sensitive and 80-fold in the drug resistant cells (Fig. 6).
The doxorubicin-selected cells were better able to utilize the Raf/MEK/ERK pathway in preventing apoptosis than the non-selected cells. The downstream targets of the Raf/MEK/ERK pathway which are activated and confer resistance to chemotherapeutic drugs are being investigated.
The non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were sensitive to MEK and mTOR inhibitors (Fig. 8). However, the doxorubicin-selected cells were more resistant to the effects of the MEK and mTOR inhibitors than the non-selected cells. As reported in other studies, the mTOR inhibitor rapamycin was effective in inducing approximately 50% apoptosis over a broad concentration range (1 nM to > 5,000 nM).1,3,36 Although growth (Fig. 4), drug sensitivity (Fig. 5), apoptotic (Fig. 6) and caspase activation (Fig. 7) all indicated that activation of Akt-1 by itself was not sufficient to induce growth or prevent apoptosis, clearly Akt-1 activation was very important in the growth and prevention of apoptosis in these cells. Furthermore, constitutive endogenous Akt activation was detected in the doxorubicin-selected cells. In addition, inhibition of downstream mTOR had a profound effect on the induction of apoptosis.
The effects of classical chemotherapy on the inhibition of cell growth and the induction of apoptosis were also compared with signal transduction inhibitors which target the Raf/MEK/ERK or PI3K/PTEN/Akt/mTOR pathways. Doxorubicin is a potent inducer of p53 in cells that have WT p53. Signal transduction inhibitors which target the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways may prove to enhance chemotherapy.6,22,30,33,35,36,38-46 Furthermore targeting of the bone marrow microenvironment and the cancer stem cells with combinations of these signal transduction inhibitors and chemotherapy may enhance cancer treatment.32-36,47 There are also complicated interactions between p53 and PI3K/PTEN/Akt/mTOR, Raf/MEK/ERK and other signaling and apoptotic pathways.4-6,22-24,46,47 Some of these interactions may result in novel therapeutic targets.48-53 For example, Akt phosphorylates MDM-2 which regulates p53 stability.4-6 Furthermore ERK can phosphorylate p53 and regulate its activity. A side effect of doxorubicin treatment is the induction of reactive oxygen species (ROS) which in turn can activate the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways.4-6,22-24,54 p53 can also regulate the activity of the discoidin domain receptor (DDR) that can regulate Ras activity and subsequently the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK cascades.4-6,22-24 A side effect of paclitaxel treatment is the induction of ERK. Hence, the effects of certain drugs such as doxorubicin and paclitaxel may be enhanced by inhibitors which target the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways. The studies presented in this manuscript have demonstrated that targeting the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways allows lower concentrations of chemotherapeutic drugs to induce apoptosis. This is important as one of the important deleterious side effects of doxorubicin is cardiotoxicity, thus lowering the effective concentration of doxorubicin may enhance cancer therapy.
These studies document the benefit of understanding which signal transduction pathway is promoting growth in the transformed cells. Knowledge of the particular pathway affected may allow more effective therapeutic options. The apoptosis-inducing effects of doxorubicin could be enhanced when the cells were treated with MEK or mTOR inhibitors. Our investigation illuminates the potential benefit of combination of targeted therapeutic with classical chemotherapeutic approaches.
Combinations of the MEK and mTOR inhibitors resulted in synergistic induction of apoptosis in both the non-selected and doxorubicin-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells. Hence, inhibition of both the Raf/MEK/ERK and PI3K/Akt/mTOR pathways may be a therapeutic option for cancers which proliferate in response to abnormal activation of these pathways. Many cancers proliferate in response to mutations or elevated receptors or key downstream signaling molecules (Src-family kinases) which transduce their effects through Raf/MEK/ERK and sometimes PI3K/Akt/mTOR.4-6,21-24,42,46
The effects of combining targeted therapy with classical chemotherapy were examined to determine whether we could enhance the anti-proliferative effects of chemotherapeutic drugs. Although therapy based on chemotherapeutic drugs is often viewed with apprehension, these agents are still widely prescribed in the treatment of diverse cancers. Thus continued use of chemotherapy may be in part due to its broad effects that are often potent anti-proliferative in nature. Treatment with the MEK inhibitors decreased the IC50 for doxorubicin greater than 10-fold in the non-selected FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells. Treatment with rapamycin increased the effectiveness of doxorubicin in inducing apoptosis in both non-selected and doxorubicin-selected cell lines (Fig. 9). Furthermore, the combination of MEK, mTOR and doxorubicin was very effective in inducing apoptosis in both drug sensitive and resistant cells. In summary, the effectiveness of conventional chemotherapy can be improved with targeted therapy.
Clearly signal transduction pathways interact to result in the promotion of cell cycle progression, growth and prevention of apoptosis. Our studies indicate that they can also interact to promote drug resistance. Devising approaches to inhibit these interactions may result in more effective anti-cancer therapies. Elucidating the mechanisms by which signaling pathways cross regulate each other and how they result in the prevention of apoptosis and promote drug resistance may yield more effective therapy. These studies document the effects the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways have on drug resistance and sensitivity to targeted therapy. Clearly, targeting these pathways should be considered as potential therapeutic options in drug resistant cancers.
Methods
Cell lines and growth factors
Cells were maintained in a humidified 5% CO2 incubator with Iscoves Modified Eagles Medium [(IMEM) Invitrogen, Carlsbad, CA, USA] supplemented with 5% fetal bovine serum (FBS) (Atlanta Biologicals, Atlanta, GA, USA). Conditionally-transformed FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were grown in 5% FCS + 500 nM 4 hydroxyl tamoxifen (4HT) (Sigma-Aldrich, St. Louis, MO, USA), an estrogen receptor antagonist which activates the ΔAkt-1:ER*(Myr+) and 100 nM testosterone (Test, Sigma-Aldrich), which activates the androgen receptor (AR) hormone binding domain contained in ΔRaf-1:AR.30 ΔAkt:ER*(Myr+) contains a mutated ER domain (ER*) which responds to 4HT 100-fold more efficiently than β-estradiol. Thus these cells were maintained in 4HT as apposed to β-estradiol.30 In some experiments, the FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells were treated with IL-3 (10% WEHI-3B conditioned medium). The parent FL5.12 IL-3 dependent cell line was maintained in IMEM + 5% FCS + 10% IL3 as described.25-29
In order to determine the importance of certain signaling pathways in prevention of apoptosis, cells were treated with the MEK inhibitor U0126 or the mTOR inhibitor Rapamycin. All were purchased from Calbiochem, San Diego, CA and dissolved in DMSO.
Limiting dilution analysis in medium containing doxorubicin
Limiting dilution analysis of FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells was performed in medium containing 4HT + Test and various concentrations of doxorubicin (1, 10, 25, 50, 100, 1,000 nM) as described.20 Fresh medium containing the drugs was added every three days and clones were isolated from those plates with the least number of colonies after approximately 1 mo in culture. After isolation of clones, they were first expanded in 1 ml cultures in 24 well plates, and then subsequently expanded into 5 ml cultures in 25 cm2 tissue culture flasks. The drug resistant cells were cultured in medium containing 4HT + Test and 10 nM doxorubicin.
Annexin V apoptotic and caspase 3 activation assays
Annexin V/PI binding and Caspase 3 activation assays were performed as previously described with kits purchased from Roche (Indianapolis, IN, USA) and Cell Technologies (Minneapolis, MN, USA) respectively.30,35
Analysis of cell sensitivity to anticancer agents
The sensitivities of FL/ΔAkt-1:ER*(Myr+) + ΔRaf-1:AR cells to doxorubicin, paclitaxel, daunorubicin, cisplatin or 5-flurouracil were investigated by characterizing the effects of these agents on proliferation as described.22 Inhibitory concentration 50% (IC50) is defined in this context as the drug dose that causes the cells to proliferate at a rate that is half as rapid as cells incubated in the absence of drugs as determined by MTT assays as described.22
Western blot analysis
Cells were washed twice with PBS and then cultured in the absence of doxorubicin in the presence of 4HT + Test for 24 h. Cells were then treated with the different concentrations of doxorubicin for 4 h. Western blots were performed with antibodies specific for ERK, Akt, p53, Bcl-XL, JNK, p38MAPK, as we have previously described.22 All antibodies used in this study were purchased from Cell Signaling (Beverly, MA).
Acknowledgments
This work was supported in part by grants from the US Public Health Service, National Institutes of Health, National Cancer Institute (NCI R01CA098195) and a Brody Brothers Endowment Fund (MT7826) to J.A.M. A.M.M. was supported by grants from Progetti Strategici Unibo EF 2006 and MIUR PRIN 2008.
02/05/10
02/10/10
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/11483
References
- 1.Carracedo A, Baselga J, Pandolfi PP. Deconstructing feedback-signaling networks to improve anticancer therapy with mTORC1 inhibitors. Cell Cycle. 2008;7:3805–9. doi: 10.4161/cc.7.24.7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krymskaya VP, Goncharova EA. PI3K/mTORC1 activation in hamartoma syndromes: therapeutic prospects. Cell Cycle. 2009;8:403–13. doi: 10.4161/cc.8.3.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foster DA, Toschi A. Targeting mTOR with rapamycin: one dose does not fit all. Cell Cycle. 2009;8:1026–9. doi: 10.4161/cc.8.7.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JT, Lehmann BD, Terrian DM, Chappell WH, Stivala F, Libra M, Martelli AM, Steelman LS, McCubrey JA. Targeting prostate cancer based on signal transduction and cell cycle pathways. Cell Cycle. 2008;7:1745–62. doi: 10.4161/cc.7.12.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M, Tafuri A, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- 6.McCubrey JA, Steelman LS, Abrams SL, Bertrand FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M, Tafuri A, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22:708–22. doi: 10.1038/leu.2008.27. [DOI] [PubMed] [Google Scholar]
- 7.Misaghian N, Ligresti G, Steelman LS, Bertrand FE, Bäsecke J, Libra M, Nicoletti F, Stivala F, Milella M, Tafuri A, et al. Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia. 2009;23:25–42. doi: 10.1038/leu.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hambardzumyan D, Becher OJ, Holland EC. Cancer stem cells and survival pathways. Cell Cycle. 2008;7:1371–8. doi: 10.4161/cc.7.10.5954. [DOI] [PubMed] [Google Scholar]
- 9.Blagosklonny MV. Cancer stem cell and cancer stemloids: from biology to therapy. Cancer Biol Ther. 2007;6:1684–90. doi: 10.4161/cbt.6.11.5167. [DOI] [PubMed] [Google Scholar]
- 10.Stuart SA, Minami Y, Wang JYJ. The CML stem cell: evolution of the progenitor. Cell Cycle. 2009;8:1338–43. doi: 10.4161/cc.8.9.8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Borodyansky L, Yang Y. Genomic instability en route to and from cancer stem cells. Cell Cycle. 2009;8:1000–2. doi: 10.4161/cc.8.7.8041. [DOI] [PubMed] [Google Scholar]
- 12.Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res. 2008;11:801–8. doi: 10.1089/rej.2008.0722. [DOI] [PubMed] [Google Scholar]
- 13.Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008;7:3344–54. doi: 10.4161/cc.7.21.6965. [DOI] [PubMed] [Google Scholar]
- 14.Blagosklonny MV, Campisi J. Cancer and aging: more puzzles, more promises? Cell Cycle. 2008;7:2615–8. doi: 10.4161/cc.7.17.6626. [DOI] [PubMed] [Google Scholar]
- 15.Blagosklonny MV. Program-like aging and mitochondria: instead of random damage by free radicals. J Cell Biochem. 2007;102:1389–99. doi: 10.1002/jcb.21602. [DOI] [PubMed] [Google Scholar]
- 16.Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–61. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- 17.Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009;8:1896–900. doi: 10.4161/cc.8.12.8809. [DOI] [PubMed] [Google Scholar]
- 18.Demidenko ZN, Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009;8:1901–4. doi: 10.4161/cc.8.12.8810. [DOI] [PubMed] [Google Scholar]
- 19.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–95. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 20.Blagosklonny MV. Aging-suppressants: cellular senescence (hyperactivation) and its pharmacologic deceleration. Cell Cycle. 2009;8:1883–7. doi: 10.4161/cc.8.12.8815. [DOI] [PubMed] [Google Scholar]
- 21.Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA, Libra M. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle. 2009;8:1352–8. doi: 10.4161/cc.8.9.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCubrey JA, Abrams SL, Ligresti G, Misaghian N, Wong EW, Steelman LS, Bäsecke J, Troppmair J, Libra M, Nicoletti F, et al. Involvement of p53 and Raf/MEK/ERK pathways in hematopoietic drug resistance. Leukemia. 2008;22:2080–90. doi: 10.1038/leu.2008.207. [DOI] [PubMed] [Google Scholar]
- 23.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCubrey JA, Abrams SL, Stadelman K, Chappell WH, Lahair M, Ferland RA, Steelman LS. Targeting signal transduction pathways to eliminate chemotherapeutic drug resistance and cancer stem cells. Adv Enzyme Regul. 2010;50:285–307. doi: 10.1016/j.advenzreg.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKearn JP, McCubrey JA, Fagg B. Enrichment of hematopoietic precursor cells and cloning of multipotential B-lymphocyte precursors. Proc Natl Acad Sci U S A. 1985;82:7414–8. doi: 10.1073/pnas.82.21.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCubrey JA, Holland G, McKearn J, Risser R. Abrogation of factor-dependence in two IL-3-dependent cell lines can occur by two distinct mechanisms. Oncogene Res. 1989;4:97–109. [PubMed] [Google Scholar]
- 27.Mayo MW, Wang X-Y, Algate PA, Arana GF, Hoyle PE, Steelman LS, McCubrey JA. Synergy between AUUUA motif disruption and enhancer insertion results in autocrine transformation of interleukin-3-dependent hematopoietic cells. Blood. 1995;86:3139–50. [PubMed] [Google Scholar]
- 28.Wang X-Y, McCubrey JA. Malignant transformation induced by cytokine genes: a comparison of the abilities of germline and mutated interleukin 3 genes to transform hematopoietic cells by transcriptional and posttranscriptional mechanisms. Cell Growth Differ. 1996;7:487–500. [PubMed] [Google Scholar]
- 29.Wang XY, McCubrey JA. Differential effects of retroviral long terminal repeats on interleukin-3 gene expression and autocrine transformation. Leukemia. 1997;11:1711–25. doi: 10.1038/sj.leu.2400793. [DOI] [PubMed] [Google Scholar]
- 30.Shelton JG, Steelman LS, Lee JT, Knapp SL, Blalock WL, Moye PW, Franklin RA, Pohnert SC, Mirza AM, McMahon M, et al. Effects of the RAF/MEK/ERK and PI3K/AKT signal transduction pathways on the abrogation of cytokine-dependence and prevention of apoptosis in hematopoietic cells. Oncogene. 2003;22:2478–92. doi: 10.1038/sj.onc.1206321. [DOI] [PubMed] [Google Scholar]
- 31.Tazzari PL, Cappellini A, Ricci F, Evangelisti C, Papa V, Grafone T, Martinelli G, Conte R, Cocco L, McCubrey JA, et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia. 2007;21:427–38. doi: 10.1038/sj.leu.2404523. [DOI] [PubMed] [Google Scholar]
- 32.Follo MY, Finelli C, Bosi C, Martinelli G, Mongiorgi S, Baccarani M, Manzoli L, Blalock WL, Martelli AM, Cocco L. PI-PLCbeta-1 and activated Akt levels are linked to azacitidine responsiveness in high-risk myelodysplastic syndromes. Leukemia. 2008;22:198–200. doi: 10.1038/sj.leu.2404855. [DOI] [PubMed] [Google Scholar]
- 33.Chiarini F, Del Sole M, Mongiorgi S, Gaboardi GC, Cappellini A, Mantovani I, Follo MY, McCubrey JA, Martelli AM. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia. 2008;22:1106–16. doi: 10.1038/leu.2008.79. [DOI] [PubMed] [Google Scholar]
- 34.Chang F, McCubrey JA. p21Cip1 induced by Raf is associated with increased Cdk4 activity in hematopoietic cells. Oncogene. 2001;20:4353–64. doi: 10.1038/sj.onc.1204564. [DOI] [PubMed] [Google Scholar]
- 35.Bertrand FE, Steelman LS, Chappell WH, Abrams SL, Shelton JG, White ER, Ludwig DL, McCubrey JA. Synergy between an IGF-1R antibody and Raf/MEK/ERK and PI3K/Akt/mTOR pathway inhibitors in suppressing IGF-1R-mediated growth in hematopoietic cells. Leukemia. 2006;20:1254–60. doi: 10.1038/sj.leu.2404217. [DOI] [PubMed] [Google Scholar]
- 36.Steelman LS, Navolanic PM, Sokolosky ML, Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Wong EW, Stadelman KM, Terrian DM, et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene. 2008;27:4086–95. doi: 10.1038/onc.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lagadinou ED, Ziros PG, Tsopra OA, Dimas K, Kokkinou D, Thanopoulou E, Karakantza M, Pantazis P, Spyridonidis A, Zoumbos NC. c-Jun N-terminal kinase activation failure is a new mechanism of anthracycline resistance in acute myeloid leukemia. Leukemia. 2008;22:1899–908. doi: 10.1038/leu.2008.192. [DOI] [PubMed] [Google Scholar]
- 38.Gomes AR, Brosens JJ, Lam EWF. Resist or die: FOXO transcription factors determine the cellular response to chemotherapy. Cell Cycle. 2008;7:3133–6. doi: 10.4161/cc.7.20.6920. [DOI] [PubMed] [Google Scholar]
- 39.Nimbalkar D, Quelle FW. Phosphoinositide 3-kinase signaling overrides a G2 phase arrest checkpoint and promotes aberrant cell cycling and death of hematopoietic cells after DNA damage. Cell Cycle. 2008;7:2877–85. doi: 10.4161/cc.7.18.6675. [DOI] [PubMed] [Google Scholar]
- 40.Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle. 2008;7:965–70. doi: 10.4161/cc.7.8.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papa V, Tazzari PL, Chiarini F, Cappellini A, Ricci F, Billi AM, Evangelisti C, Ottaviani E, Martinelli G, Testoni N, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia. 2008;22:147–60. doi: 10.1038/sj.leu.2404980. [DOI] [PubMed] [Google Scholar]
- 42.Martelli AM, Nyåkern M, Tabellini G, Bortul R, Tazzari PL, Evangelisti C, Cocco L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006;20:911–28. doi: 10.1038/sj.leu.2404245. [DOI] [PubMed] [Google Scholar]
- 43.Tazzari PL, Tabellini G, Bortul R, Papa V, Evangelisti C, Grafone T, Martinelli G, McCubrey JA, Martelli AM. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia. 2007;21:886–96. doi: 10.1038/sj.leu.2404643. [DOI] [PubMed] [Google Scholar]
- 44.Fernandez-Vidal A, Mazars A, Gautier EF, Prévost G, Payrastre B, Manenti S. Upregulation of the CDC25A phosphatase down-stream of the NPM/ALK oncogene participates to anaplastic large cell lymphoma enhanced proliferation. Cell Cycle. 2009;8:1373–9. doi: 10.4161/cc.8.9.8302. [DOI] [PubMed] [Google Scholar]
- 45.Chiarini F, Fala F, Tazzari PL, Ricci F, Astolfi A, Pession A, et al. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009;69:3520–8. doi: 10.1158/0008-5472.CAN-08-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skladanowski A, Bozko P, Sabisz M, Larsen AK. Dual inhibition of PI3K/Akt signaling and the DNA damage checkpoint in p53-deficient cells with strong survival signaling: implications for cancer therapy. Cell Cycle. 2007;6:2268–75. doi: 10.4161/cc.6.18.4705. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Elf SE, Asai T, Miyata Y, Liu Y, Sashida G, Huang G, Di Giandomenico S, Koff A, Nimer SD. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8:3120–4. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle. 2009;8:2810–8. doi: 10.4161/cc.8.17.9503. [DOI] [PubMed] [Google Scholar]
- 49.Vaseva AV, Marchenko ND, Moll UM. The transcription-independent mitochondrial p53 program is a major contributor to nutlin-induced apoptosis in tumor cells. Cell Cycle. 2009;8:1711–9. doi: 10.4161/cc.8.11.8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sohr S, Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle. 2008;7:3448–60. doi: 10.4161/cc.7.21.7014. [DOI] [PubMed] [Google Scholar]
- 51.París R, Henry RE, Stephens SJ, McBryde M, Espinosa JM. Multiple p53-independent gene silencing mechanisms define the cellular response to p53 activation. Cell Cycle. 2008;7:2427–33. doi: 10.4161/cc.6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wade M, Rodewald LW, Espinosa JM, Wahl GM. BH3 activation blocks Hdmx suppression of apoptosis and cooperates with Nutlin to induce cell death. Cell Cycle. 2008;7:1973–82. doi: 10.4161/cc.7.13.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia M, Knezevic D, Tovar C, Huang B, Heimbrook DC, Vassilev LT. Elevated MDM2 boosts the apoptotic activity of p53-MDM2 binding inhibitors by facilitating MDMX degradation. Cell Cycle. 2008;7:1604–12. doi: 10.4161/cc.7.11.5929. [DOI] [PubMed] [Google Scholar]
- 54.Bykov VJ, Lambert JM, Hainaut P, Wiman KG. Mutant p53 rescue and modulation of p53 redox state. Cell Cycle. 2009;8:2509–17. doi: 10.4161/cc.8.16.9382. [DOI] [PubMed] [Google Scholar]
- 55.Varmark H, Sparks CA, Nordberg JJ, Koppetsch BS, Theurkauf WE. DNA damage-induced cell death is enhanced by progression through mitosis. Cell Cycle. 2009;8:2951–63. doi: 10.4161/cc.8.18.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhuang Z, Lu J, Lonser R, Kovach JS. Enhancement of cancer chemotherapy by simultaneously altering cell cycle progression and DNA-damage defenses through global modification of the serine/threonine phospho-proteome. Cell Cycle. 2009;8:3303–6. doi: 10.4161/cc.8.20.9689. [DOI] [PubMed] [Google Scholar]