

Abstract
HIV-infected subjects on highly active antiretroviral therapy (HAART) are susceptible to comorbid microbial infections in the oral cavity. We observed that primary oral epithelial cells (POECs) isolated from HIV+ subjects on HAART grow more slowly and are less innate immune responsive to microbial challenge when compared with POECs from normal subjects. These aberrant cells also demonstrate epigenetic differences that include reduction in histone deacetylase 1 (HDAC-1) levels and reduced total DNA methyltransferase (DNMT) activity specific to enzymes DNMT1 and DNMT3A. The DNMT activity correlates well with global DNA methylation, indicating that aberrant DNMT activity in HIV+ (on HAART) POECs leads to an aberrantly methylated epithelial cell phenotype. Overall, our results lead us to hypothesize that, in patients with chronic HIV infection on HAART, epigenetic changes in key genes result in increased vulnerability to microbial infection in the oral cavity.
Keywords: oral epithelium, HIV, HAART, DNMTs, HDAC-1, hBD-2
Introduction
The oral epithelium, the most abundant structural tissue lining the oral mucosa, is an important line of defense against infectious microorganisms. With the advent of highly active anti-retroviral therapy (HAART), HIV-infected individuals are living longer. While many pathological sequelae have decreased among HAART-treated HIV positive patients, the incidence of certain oral infections, such as oral warts, increase the risk of oral cancer and remain a major concern.1-4 Proteomic analysis of primary oral epithelial cells (POEC) in HIV patients compared with uninfected individuals confirms specific molecular alterations of proteins involved with protein folding, stress regulation, and pro- and anti-inflammatory responses.5 These results, derived from an examination of expanded oral epithelial cells, suggest that potential epigenetic changes as a function of HIV and/or HAART may be the molecular basis for altered susceptibility of HIV patients to oral complications. The well- documented adverse effects of HIV protease inhibitors (PIs) on the orofacial complex also suggests that epithelial cell biology in the oral cavity could be compromised by the effects of these agents.6-9 Danaher et al.10 demonstrated that PIs significantly inhibit the viability of immortalized oral keratinocyte cell-lines as well as primary oral keratinocytes. Moreover, the anti-proliferative effects of PIs on POECs have been reported by several groups.10-14 However, whether HAART therapy and/or HIV chronicity is/are responsible for the changed phenotype in epithelial cells isolated from HIV+ (on HAART) individuals has not yet been determined. Nevertheless, as long-term HAART treatment is the standard of care worldwide, understanding the combined effects of HIV chronicity and HAART treatment is essential to reducing morbidity and mortality of HIV-infected individuals.
The epigenetic landscape is modulated by multiple factors, including modifications of DNA and histones and the role and importance of epigenetic regulation in understanding disease is increasing dramatically.15 Epigenetic mechanisms play important roles during normal development, aging and in a variety of disease states. Numerous studies have implicated aberrant methylation in the etiology of common human diseases.16-22 A multitude of modern molecular biology and next generation sequencing techniques have revolutionized our understanding of the complexity of epigenetic factors and their potential interrelationships. Of increasing biological interest, of course, is the link epigenetics plays between the gene and the organism’s environment. Viral infection is a well-known cause of DNA and histone modifications and HIV in particular has well-established effects in T-cells that regulate the expression of both viral and host genes.23 In addition, drug treatment and diet are also known to modulate gene expression through epigenetic effects.24,25 However, to date, the epigenetic effects (or defects) caused by HIV and/or HAART on oral epithelia and their role in mediating pathogenesis are not well established.
DNA methylation is a common epigenetic tool controlling gene expression and, ultimately, cellular phenotype. Methylation of the 5′ carbon of cytosine is a common epigenetic tag in many eukaryotes that does not affect the primary DNA sequence, but alters secondary interactions of the transcriptional machinery that play a critical role in gene expression. DNA methylation is facilitated by dedicated DNA methyltransferases (DNMTs) with highly conserved catalytic motifs that are essential for the establishment of cytosine methylation patterns, as well as for their maintenance throughout cell replication.26 Of the five DNMT family members, only DNMT1, DNMT3a and DNMT3b possess methyltransferase activity in mammals. Mice that lack a particular DNMT have reduced methylation levels and die early in development.27
Regulating protein function is often accomplished through posttranslational mechanisms. Histones serve as a central target for modification resulting in the alteration of all chromatin-mediated processes. Histones H3, H4, H2A and H2B form the nucleosomal core surrounded by 147bp of DNA stabilized by histones H1 and H5. Modification of histones through covalent changes occurs primarily on histones that protrude from the nucleosome and include common alterations, such as methylation, acetylation, phosphorylation, ubiquitination and ADP-ribosylation.28 Indeed, acetylation of Lys by histone acetyltransferase and deacetylation by histone deacetylases (HDACs) have been shown to alter gene regulation including increased transcription and repression.29 HIV viral latency has also been linked to histone modifications.30
In this communication, we report differences in cellular proliferation rates, changes in DNA methyltransferase (DNMT1 and DNMT3A) activity, changes in histone deacetylase 1 (HDAC-1) activity, targeted proteomics changes and variation in innate immune responsiveness to microbial challenge of POECs derived from the oral mucosa of HIV+ on HAART subjects when compared with healthy control POECs. These observations, coupled to our previous proteomics studies, lead us to suggest that POECs from subjects with chronic HIV on HAART (HIV+O/H) exhibit heritable epigenetic changes modulating their molecular phenotype.
Results and Discussion
Previous in vitro studies have reported retarded epithelial cell growth in the presence of HAART.10-14 All of those studies were performed using epithelial cells isolated from healthy subjects. In the present study, we compared the growth of POECs isolated from 3 healthy subjects with those isolated from 3 HIV+O/H subjects. The POECs isolated from HIV+O/H subjects had reduced (p < 0.05, Mann–Whitney t-test) doubling times compared with POECs isolated from healthy subjects (Fig. 1A). Healthy control POECs expand approximately 6-fold during the 10 d of growth while the cells from the HIV+O/H subjects expand only 2–3 fold. Isolated keratinocytes cells are cultured in normal keratinocyte basal growth media (KBM) with no exogenous nucleoside reverse transcriptase inhibitors (NRTIs), protease inhibitors (PI) or non-nucleoside reverse transcriptase inhibitors. Therefore, changes in proliferation are entirely due to heritable properties regulating cellular duplication.
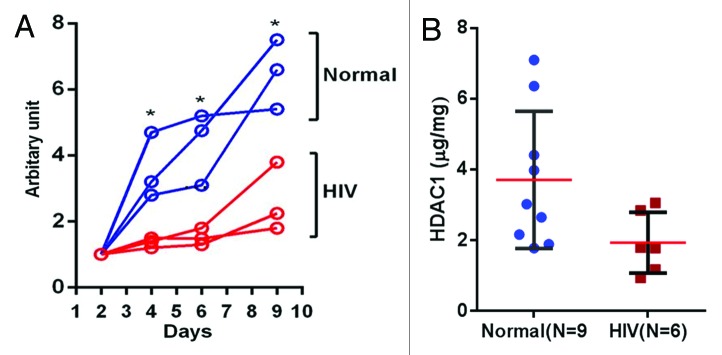
Figure 1. (A) POEC growth comparison (HIV+O/H vs. Normal): Cell growth assays for POECs isolated from 3 HIV+O/H and three normal subjects were performed using PrestoBlue® Cell Viability Reagent. (*p < 0.05). (B) Comparison of HDAC1 protein levels in the nuclear extract of POECs isolated from 9 normal and 6 HIV+O/H subjects. (*p < 0.05, Mann–Whitney t test)
Our previous proteomics study indicated significant dysregulation of multiple protein targets related to cell cycle and proliferation, such as increased levels of phospho-MEK1/2 in epithelial cells of HIV+ (O/H) POECs compared with normal.5 MEK can cause inhibition of cyclin dependent kinase activity31 and can affect cell cycle arrest.32,33 Thus, overall these previous results are consistent with our present data. Since there is evidence implicating the transcription factor NF-κB as a positive mediator of cell growth,34,35 we measured the levels of total NF-κB p65 and phospho-p65 NF-κB in cell lysates of POECs from 9 HIV and 10 healthy subjects. No differences in the endogenous levels of total NF-κB p65 were observed between the two cohorts (Fig. S1). Although endogenous phospho-p65 NF-κB levels were decreased in HIV, the difference was not significant. Thus, our observed proliferation differences are not related to NF-κB signaling.
HDAC1 has been shown to be associated with cell growth; for example its knockdown in HeLaS3 cells results in reduced proliferation.36 Thus, we compared the levels of HDAC1 in the nuclear extracts of POECs isolated from 9 healthy and 6 HIV+O/H subjects. We found that HDAC1 levels are reduced approximately 2-fold in the nuclear extracts of HIV+O/H subject POECs when compared with healthy volunteers (p < 0.05, Mann–Whitney t-test) (Fig. 1B). Thus, HDAC1 reduction and its effects on histone modifications may be related to the reduced proliferation potential of POECs in HIV+O/H individuals.
In order to probe the changes in global DNA methyltransferase (DNMT) activity, nuclear proteins were extracted from POECs of 9 HIV+O/H and 10 healthy volunteers, and total DNMT activity was measured. Nuclear extracts from HIV+O/H subjects exhibited decreased DNMT activity compared with normal subjects (p < 0.05, Mann–Whitney t-test) (Fig. 2A). Several studies suggest multiple functional roles for DNA methylation, including silencing of transposable elements, mediating developmental gene regulation and reducing transcriptional noise.37-39 DNA methylation in mammals is also essential for differentiation and cell cycle control.40,41 Thus, methylation defects in HIV+O/H subjects may contribute to a multitude of molecular alterations of POECs,

Figure 2. Comparison of DNMT activity (A), DNMT1 (B), DNMT3A (C) and DNMT3B (D) protein levels in the nuclear extract of POECs isolated from 10 normal subjects vs. 9 HIV+O/H subjects. (E) Correlation between DNMT activity and the levels of three individual DNMTs.
Different members of the DNMT family of enzymes act either as de novo DNMTs, i.e., responsible for the initial pattern of methyl groups in place on a DNA sequence, or as maintenance DNMTs, i.e., copying the methylation from an existing DNA strand to its new partner after replication. Lower levels of DNMT activity in HIV+O/H subjects is indicative of lower levels of one or more of DNMT1, DNMT3A and 3B, which play major roles in the establishment and maintenance of methylation patterns.15,42 We determined the levels of these three DNMTs in the same nuclear extracts that were used to determine total DNMT activity. Levels of DNMT1 and DNMT3A, but not DNMT3B, were significantly lower in POECs from HIV+O/H subjects when compared with healthy controls (p < 0.05, Mann–Whitney test) (Fig. 2B–D). A correlation analysis between DNMT protein levels and DNMT activity among all samples revealed a significant correlation between DNMT1 protein expression and DNMT activity (Fig. 2E). This correlation was weaker but still significant for DNMT3A and DNMT3B.
It is important to note that the observed decrease in DNMT activity is a decrease in total DNMT activity and does not distinguish the relative contributions of the maintenance methyltransferase (DNMT1) vs. de novo methyltransferases (DNMT3A and 3B). Relative contributions of DNMTs and how they may mediate a decrease in DNMT activity in POECs from HIV+ subjects requires further investigation. However, to determine if any correlation between DNMT activity and total DNA methylation exists, we measured total global DNA methylation and DNMT activity in genomic DNA and nuclear extracts of additional POEC samples from eight HIV+ (O/H) subjects, respectively. As shown in Figure 3, DNMT activity correlates well (p < 0.02) with global DNA methylation, confirming that aberrant DNMT activity in HIV+ (O/H) POECs will lead to an aberrantly methylated epithelial cell phenotype.
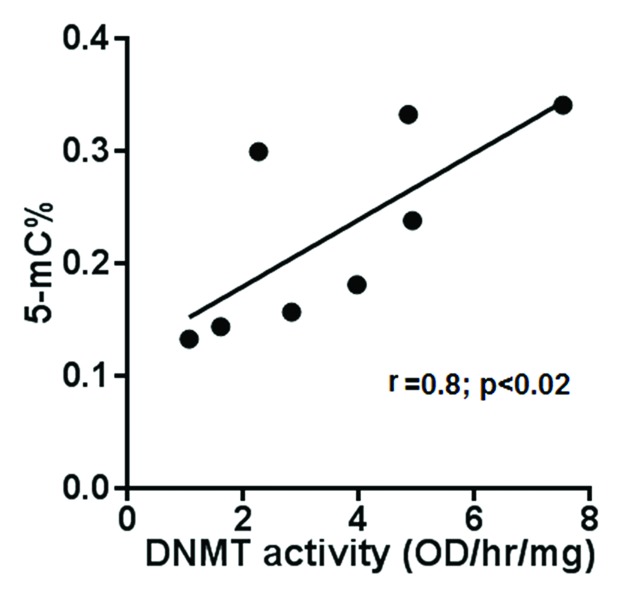
Figure 3. Correlation between DNMT activity and global DNA methylation. Total global DNA methylation and DNMT activity in nuclear extract of eight subjects were measured. DNA methylation (expressed as 5-mC% in total DNA) and DNMT activity (expressed as OD/hr/mg) were plotted against each other for each of the subjects.
Yin and Chung43 have demonstrated that epigenetic modifications play a critical role in the regulation of innate immune responses of POECs where DNMT1 expression is decreased in response to two periodontopathogenic bacteria Porphyromonas gingivalis and Fusobacterium nucleatum. Exposure to different oral bacteria results in differential methylation profiles and bacteria-induced expression of epithelial cell derived antimicrobial peptides, such as human β defensin 2 (hBD-2). We and others have shown that the F. nucleatum cell wall (FnCW) fraction can induce hBD-2 in HOECs.44-46 Here, we compared the induction of hBD-2 by FnCW in POECs isolated from HIV+O/H subjects and healthy controls, where ELISA was used to measure levels of released hBD-2 in culture media. We observed significantly lower (p < 0.05, Mann–Whitney Test) levels of hBD-2 released from FnCW challenged POECs derived from HIV+O/H subjects when compared with FnCW challenged POECs of healthy control subjects (Fig. 4A) indicating a reduced innate immune defense of HIV+O/H individuals. This result supports a previous observation by Sun et al.47 demonstrating lower levels of hBD-2 in the oral epithelium of HIV+ subjects compared with healthy controls. Since p38 regulates induction of hBD-2 by FnCW in POECs44 and, since our previous study,5 suggests aberrant expression and/or activation of MAPK, including p38, in POECs from HIV subjects, we reasoned that the differential induction of hBD-2 in HIV+ on HAART subjects might be due to differences in endogenous p38 MAPK levels in POECs of HIV+O/H and healthy controls. We discovered that phosphorylated p38 (pp38) levels, but not total p38 were significantly reduced (p < 0.05, Mann–Whitney Test), in the cytoplasm of POECs derived from HIV+O/H subjects when compared with POECs from healthy controls (Fig. 4B–D). Moreover, a significant positive correlation was observed between pp38 protein levels and hBD-2 induction by F. nucleatum within both HIV-positive and healthy subjects (Fig. 4E). Thus, lower levels of endogenous pp38 in POECs from HIV subjects may account for reduced F. nucleatum induced hBD-2 levels.

Figure 4. POECs from 9 normal and 7 HIV+O/H subjects were grown to semi-confluence (80%) and treated with FnCW (10 μg/ml) for 18 h, respectively. The levels of hBD-2 in media supernatant of FnCW treated and untreated POECs from HIV+O/H and normal subjects were measured by ELISA. Mean (± SD) fold changes in FnCW induced hBD-2 release for the two cohorts, i.e., HIV+O/H and HIV- subjects, were compared (A). Mean (± SD) values of total (B) and phophorylated p38 (p-p38) (C) levels in the cytoplasmic extracts of POECs from the same two cohorts of subjects were measured and compared. The ratios of p-p38 to total p38 were also compared (D). (E) The correlation between the levels of pp38 and the induction of hBD-2 by FnCW.
The p38 groups of MAP kinases serve as a nexus for signal transduction and play a vital role in numerous biological processes. While p38 MAPK has classically been associated with the induction of apoptosis, p38 MAPK can also mediate cell growth in specific situations.48,49 Therefore, in order to determine if p38 has any role in the regulation of cellular growth of POECs, we pre-treated POECs isolated from healthy subjects with the p38 specific inhibitor (SB203580; Cell Signaling) for 2 h and compared cell growth for 1 week in treated vs. vehicle (DMSO) control. As shown in Figure S2, the pretreatment of POECs with SB203580 did not significantly alter their growth indicating decreased phosphorylation of p38, as observed in HIV+ (O/H) subjects, may not be responsible for reduced cell growth rates observed in POECs from HIV+ (OH) subjects.
Additionally, to see if p38 has any role in the epigenetic modification observed in the POECs isolated from HIV+ (O/H) subjects, we pre-treated POECs from healthy subjects with SB203580 and measured the levels of HDAC1, DNMT activities and global DNA methylation. Pretreatment with the p38 inhibitor did not alter HDCA1 levels, DNMT activity or global DNA methylation (Fig. S2), indicating that p38 does not affect the epigenetic changes observed in POECs from HIV+ (O/H) subjects. Indeed, Yin and Chung (2011) showed that F. nucleatum, which is known to cause phosphorylation of p38 in POECs, did not affect the expression of HDAC1 and DNMT proteins in POECs. This observation supports our present finding that p38 inhibition does not directly affect HDAC1 levels or DNMT activity.
As reported in Table S1, there was variation in the HAART regimen of our HIV+ subjects. However, this variation did not alter the variation in the epigenetic markers measured in this study; as similar degrees of variation were noted in the HIV negative subjects. The variation within each cohort might be due to interpersonal variability that is often seen with primary cells from different subjects. Moreover, the viral loads of all the subjects on HAART were similar.
From the novel observations reported herein it is apparent that POECs isolated from HIV+ (O/H) subjects represents a molecular phenotype that is different from those isolated from healthy controls and that the retarded growth phenotype is stable upon cell duplication, consistent with epigenetic alterations. Further research is needed to determine the specific nature of the epigenetic defects in POECs induced by HIV infection per se and those induced by HAART. This would require enrolling subjects who are HIV+ and HAART naïve. However, enrolling subjects with these qualifications has become increasingly difficult due to new medical guidelines for treating all newly diagnosed HIV+ subject with HAART as soon as possible following diagnosis (aidsinfo.nih.gov/contentfile/lvguidelines /adultandadolescentgl.pdf). To best address this important question, a redesigned study using subjects from countries where HIV+ HAART naïve patients are more prevalent would be required, along with in vitro experiments using POECs from HIV negative subjects exposed to various regimens of HAART. We are currently pursuing both approaches.
In summary, our results reveal key phenotypic changes in POECs from HIV+O/H subjects that include diminished cell growth and proliferation and reduced responsiveness to microbial challenge as demonstrated by F. nucleatum induction of hBD-2. Aberrant POEC proliferation in HIV+O/H subjects could result in lesion development and/or altered healing. Reduced DNA methylation activity and reduced levels of DNMT1 in POECs from HIV+ subjects may also be associated with the increased incidence of HPV warts in HIV positive subjects on HAART.50
Materials and Methods
Clinical samples
Human gingival tissue behind the last maxillary or mandibular molars from HIV-infected and healthy control subjects were collected after written informed consent was provided by study participants and/or their legal guardians. University Hospital Case Medical Center Institutional Review Board (IRB) Protocol #: 19981017 approved this study. No diagnosis of gingivitis, i.e., inflammation of the gingival tissue, or periodontitis, i.e., alveolar bone loss, was observed in the biopsy sites from healthy or HIV-infected subjects. For all the HIV+ subjects, CD4+ T-cells counts at the closest date to tissue collection, as well as viral load per ml of blood were determined (Table S1).
Epithelial cells isolation
Primary human oral epithelial cells (HOECs) were isolated and expanded in serum free keratinocyte growth medium with supplements as previously described by Krisanaprakornkit et al.44 Briefly, the epithelial layer was separated from the underlying fibrous connective tissue with dispase. A single cell suspension was prepared from the epithelial sheets by trypsinization and repeated pipetting. Cells were suspended in serum-free EpiLife media (Cascade Biologics Inc.) and plated on 10 cm Petri dishes and grown to near-confluence (~80%). Cells were then detached from the Petri dish, pelleted, frozen and stored in liquid nitrogen until further use.
Cell growth assay
Cell growth assays were performed using PrestoBlue® Cell Viability Reagent (Life Technologies), which is a cell permeable resazurin-based solution that functions as a cell viability indicator by using the reducing power of living cells to quantitatively measure the proliferation of cells. Briefly, 60–70% confluent cells from were seeded onto 96 well plates. Starting from day 2 until day 12, three replicate wells were treated with 10 μL of PrestoBlue and 90 μL of Epilife for 30 min and fluorescence readings (at 530 nm) were taken every 2 d.
Extraction of nuclear proteins
Nuclear proteins from the epithelial cells were extracted using NE-PER reagents (Thermo-Scientific) according to the vendor’s instruction
Determination of HDAC1 levels
Levels of HDAC-1 in nuclear extracts were determined using a kit from Epigentek.
DNMT activity assay
DNMT activity in the nuclear extract was determined using kits from Epigentek, following the vendor’s instructions.
Determination of the levels of DNMTs
Levels of DNMTs (DNMT1, DNMT3A and DNMT3B) in the nuclear extracts were determined using respective kits from Epigentek, following the vendor’s instructions.
Global methylation of DNA in POECs
Genomic DNA was extracted from the POECs with a commercially available kit (Epigentek). Levels of methylated DNA were assessed using the Methyl Flash Methylated DNA Quantification Kit (Epigentek). The relative values of methylation status of the DNA samples were calculated as percentage of 5-mC in total DNA.
Preparation of F. nucleatum cell wall fractions
Cell wall from F. nucleatum (FnCW) was prepared as we described previously.45
Detection of hBD-2 peptides in supernatant
HBD-2 was measured in supernatants from FnCW-challenged and negative control HOECs following our previously published protocol.45,51
Determination of total and phopho-p38 levels
Endogenous levels of total and phospho-p38 in the cytoplasmic extract of HOECs were determined using ELISA kits from R&D Systems.
Statistical analysis
Graphpad Prism version 6 was used for all statistical analyses. Non-parametric Mann–Whitney t tests were done for comparison between groups.
Supplementary Material
Acknowlegdgments
This work was supported by National Institutes of Health grants P01DE019759 (A.W.) and R01DE018276 (A.W.) and the Center for AIDS Research Proteomics Core grant P30AI036219 (M.R.C.). We thank Drs. F. Faddoul, J.R. Blakemore, E.K. Schneider, W.S. Blood and S. Alperin for providing us with human oral tissue and E. Hill, our clinical coordinator, who was instrumental in obtaining oral biopsies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental material may be found here: http://www.landesbioscience.com/journals/epigenetics/article/25028
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/25028
References
- 1.Patton LL, McKaig R, Strauss R, Rogers D, Eron JJ., Jr. Changing prevalence of oral manifestations of human immuno-deficiency virus in the era of protease inhibitor therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:299–304. doi: 10.1016/S1079-2104(00)70092-8. [DOI] [PubMed] [Google Scholar]
- 2.Greenspan D, Canchola AJ, MacPhail LA, Cheikh B, Greenspan JS. Effect of highly active antiretroviral therapy on frequency of oral warts. Lancet. 2001;357:1411–2. doi: 10.1016/S0140-6736(00)04578-5. [DOI] [PubMed] [Google Scholar]
- 3.Tappuni AR, Fleming GJ. The effect of antiretroviral therapy on the prevalence of oral manifestations in HIV-infected patients: a UK study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001;92:623–8. doi: 10.1067/moe.2001.118902. [DOI] [PubMed] [Google Scholar]
- 4.Nittayananta W, Talungchit S, Jaruratanasirikul S, Silpapojakul K, Chayakul P, Nilmanat A, et al. Effects of long-term use of HAART on oral health status of HIV-infected subjects. J Oral Pathol Med. 2010;39:397–406. doi: 10.1111/j.1600-0714.2009.00826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yohannes E, Ghosh SK, Jiang B, McCormick TS, Weinberg A, Hill E, et al. Proteomic signatures of human oral epithelial cells in HIV-infected subjects. PLoS One. 2011;6:e27816. doi: 10.1371/journal.pone.0027816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scully C, Diz Dios P. Orofacial effects of antiretroviral therapies. Oral Dis. 2001;7:205–10. doi: 10.1034/j.1601-0825.2001.70401.x. [DOI] [PubMed] [Google Scholar]
- 7.Hodgson TA, Greenspan D, Greenspan JS. Oral lesions of HIV disease and HAART in industrialized countries. Adv Dent Res. 2006;19:57–62. doi: 10.1177/154407370601900112. [DOI] [PubMed] [Google Scholar]
- 8.Goodgame JC, Pottage JC, Jr., Jablonowski H, Hardy WD, Stein A, Fischl M, et al. Amprenavir in combination with lamivudine and zidovudine versus lamivudine and zidovudine alone in HIV-1-infected antiretroviral-naive adults. Amprenavir PROAB3001 International Study Team. Antivir Ther. 2000;5:215–25. [PubMed] [Google Scholar]
- 9.Roca B. Adverse drug reactions to antiretroviral medication. Front Biosci. 2009;14:1785–92. doi: 10.2741/3340. [DOI] [PubMed] [Google Scholar]
- 10.Danaher RJ, Wang C, Roland AT, Kaetzel CS, Greenberg RN, Miller CS. HIV protease inhibitors block oral epithelial cell DNA synthesis. Arch Oral Biol. 2010;55:95–100. doi: 10.1016/j.archoralbio.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chow WA, Jiang C, Guan M. Anti-HIV drugs for cancer therapeutics: back to the future? Lancet Oncol. 2009;10:61–71. doi: 10.1016/S1470-2045(08)70334-6. [DOI] [PubMed] [Google Scholar]
- 12.Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13:5183–94. doi: 10.1158/1078-0432.CCR-07-0161. [DOI] [PubMed] [Google Scholar]
- 13.Jiang W, Mikochik PJ, Ra JH, Lei H, Flaherty KT, Winkler JD, et al. HIV protease inhibitor nelfinavir inhibits growth of human melanoma cells by induction of cell cycle arrest. Cancer Res. 2007;67:1221–7. doi: 10.1158/0008-5472.CAN-06-3377. [DOI] [PubMed] [Google Scholar]
- 14.Pyrko P, Kardosh A, Wang W, Xiong W, Schönthal AH, Chen TC. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007;67:10920–8. doi: 10.1158/0008-5472.CAN-07-0796. [DOI] [PubMed] [Google Scholar]
- 15.Jiang YH, Bressler J, Beaudet AL. Epigenetics and human disease. Annu Rev Genomics Hum Genet. 2004;5:479–510. doi: 10.1146/annurev.genom.5.061903.180014. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura N, Takenaga K. Hypomethylation of the metastasis-associated S100A4 gene correlates with gene activation in human colon adenocarcinoma cell lines. Clin Exp Metastasis. 1998;16:471–9. doi: 10.1023/A:1006589626307. [DOI] [PubMed] [Google Scholar]
- 17.Shahbazian MD, Zoghbi HY. Molecular genetics of Rett syndrome and clinical spectrum of MECP2 mutations. Curr Opin Neurol. 2001;14:171–6. doi: 10.1097/00019052-200104000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Akiyama Y, Maesawa C, Ogasawara S, Terashima M, Masuda T. Cell-type-specific repression of the maspin gene is disrupted frequently by demethylation at the promoter region in gastric intestinal metaplasia and cancer cells. Am J Pathol. 2003;163:1911–9. doi: 10.1016/S0002-9440(10)63549-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta A, Godwin AK, Vanderveer L, Lu A, Liu J. Hypomethylation of the synuclein γ gene CpG island promotes its aberrant expression in breast carcinoma and ovarian carcinoma. Cancer Res. 2003;63:664–73. [PubMed] [Google Scholar]
- 20.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 21.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 22.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–72. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 23.Paschos K, Allday MJ. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 2010;18:439–47. doi: 10.1016/j.tim.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Csoka AB, Szyf M. Epigenetic side-effects of common pharmaceuticals: a potential new field in medicine and pharmacology. Med Hypotheses. 2009;73:770–80. doi: 10.1016/j.mehy.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 25.Choi SW, Friso S. Epigenetics: A New Bridge between Nutrition and Health. Adv Nutr. 2010;1:8–16. doi: 10.3945/an.110.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 28.Morales V, Richard-Foy H. Role of histone N-terminal tails and their acetylation in nucleosome dynamics. Mol Cell Biol. 2000;20:7230–7. doi: 10.1128/MCB.20.19.7230-7237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–26. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 30.Imai K, Ochiai K. Role of histone modification on transcriptional regulation and HIV-1 gene expression: possible mechanisms of periodontal diseases in AIDS progression. J Oral Sci. 2011;53:1–13. doi: 10.2334/josnusd.53.1. [DOI] [PubMed] [Google Scholar]
- 31.Pumiglia KM, Decker SJ. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 1997;94:448–52. doi: 10.1073/pnas.94.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JI, Strock CJ, Ball DW, Nelkin BD. The Ras/Raf/MEK/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/JAK/STAT pathway. Mol Cell Biol. 2003;23:543–54. doi: 10.1128/MCB.23.2.543-554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu PP, Shen X, Huang D, Liu Y, Counter C, Wang XF. The MEK pathway is required for stimulation of p21(WAF1/CIP1) by transforming growth factor-beta. J Biol Chem. 1999;274:35381–7. doi: 10.1074/jbc.274.50.35381. [DOI] [PubMed] [Google Scholar]
- 34.Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-kappa B RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185:953–61. doi: 10.1084/jem.185.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Köntgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–77. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 36.Glaser KB, Li J, Staver MJ, Wei RQ, Albert DH, Davidsen SK. Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem Biophys Res Commun. 2003;310:529–36. doi: 10.1016/j.bbrc.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 37.Walsh CP, Bestor TH. Cytosine methylation and mammalian development. Genes Dev. 1999;13:26–34. doi: 10.1101/gad.13.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 39.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–9. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 40.Panning B, Jaenisch R. DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev. 1996;10:1991–2002. doi: 10.1101/gad.10.16.1991. [DOI] [PubMed] [Google Scholar]
- 41.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–6. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 42.Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, et al. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829–38. doi: 10.1016/S0092-8674(01)00280-X. [DOI] [PubMed] [Google Scholar]
- 43.Yin L, Chung WO. Epigenetic regulation of human β-defensin 2 and CC chemokine ligand 20 expression in gingival epithelial cells in response to oral bacteria. Mucosal Immunol. 2011;4:409–19. doi: 10.1038/mi.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krisanaprakornkit S, Kimball JR, Weinberg A, Darveau RP, Bainbridge BW, Dale BA. Inducible expression of human beta-defensin 2 by Fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect Immun. 2000;68:2907–15. doi: 10.1128/IAI.68.5.2907-2915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta S, Ghosh SK, Scott ME, Bainbridge B, Jiang B, Lamont RJ, et al. Fusobacterium nucleatum-associated beta-defensin inducer (FAD-I): identification, isolation, and functional evaluation. J Biol Chem. 2010;285:36523–31. doi: 10.1074/jbc.M110.133140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh SK, Gupta S, Jiang B, Weinberg A. Fusobacterium nucleatum and human beta-defensins modulate the release of antimicrobial chemokine CCL20/macrophage inflammatory protein 3α. Infect Immun. 2011;79:4578–87. doi: 10.1128/IAI.05586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun L, Finnegan CM, Kish-Catalone T, Blumenthal R, Garzino-Demo P, La Terra Maggiore GM, et al. Human beta-defensins suppress human immunodeficiency virus infection: potential role in mucosal protection. J Virol. 2005;79:14318–29. doi: 10.1128/JVI.79.22.14318-14329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takenaka K, Moriguchi T, Nishida E. Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science. 1998;280:599–602. doi: 10.1126/science.280.5363.599. [DOI] [PubMed] [Google Scholar]
- 49.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–8. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 50.Shetty K, Leigh J. The Role of human papillomavirus (HPV) and the increasing incidence of oral pathology in the era of highly active antiretroviral therapy (HAART) The Internet Journal of Dental Science. 2005;2:2. [Google Scholar]
- 51.Ghosh SK, Gerken TA, Schneider KM, Feng Z, McCormick TS, Weinberg A. Quantification of human beta-defensin-2 and -3 in body fluids: application for studies of innate immunity. Clin Chem. 2007;53:757–65. doi: 10.1373/clinchem.2006.081430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.