

Abstract
Squalene synthase catalyzes the conversion of two molecules of (E,E)-farnesyl diphosphate to squalene via the cyclopropylcarbinyl intermediate, presqualene diphosphate (PSPP). Since this novel reaction constitutes the first committed step in sterol biosynthesis, there has been considerable interest and research on the stereochemistry and mechanism of the process and in the design of selective inhibitors of the enzyme. This paper reports the synthesis and characterization of five racemic and two enantiopure aziridine analogues of PSPP and the evaluation of their potencies as inhibitors of recombinant yeast squalene synthase. The key aziridine-2-methanol intermediates (6-OH, 7-OH, and 8-OH) were obtained by N-alkylations or by an N-acylation–reduction sequence of (±)-, (2R,3S)-, and (2S,3R)-2,3-aziridinofarnesol (9-OH) protected as tert-butyldi-methylsilyl ethers. SN2 displacements of the corresponding methanesulfonates with pyrophosphate and methanediphosphonate anions afforded aziridine 2-methyl diphosphates and methanediphosphonates bearing N-undecyl, N-bishomogeranyl, and N-(α-methylene)bishomogeranyl substituents as mimics for the 2,6,10-trimethylundeca-2,5,9-trienyl side chain of PSPP. The 2R,3S diphosphate enantiomer bearing the N-bishomogeranyl substituent corresponding in absolute stereochemistry to PSPP proved to be the most potent inhibitor (IC50 1.17 ± 0.08 μM in the presence of inorganic pyrophosphate), a value 4-fold less than that of its 2S,3R stereoisomer. The other aziridine analogues bearing the N-(α-methylene)bishomogeranyl and N-undecyl substituents, and the related methanediphosphonates, exhibited lower affinities for recombinant squalene synthase.
Introduction
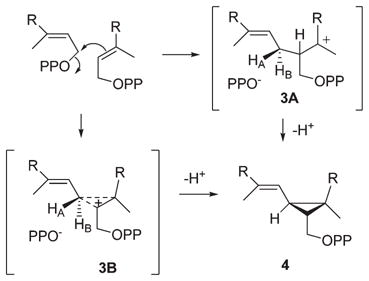
Squalene synthase (SS, EC 2.5.1.21) catalyzes the reductive head-to-head coupling of two molecules of (E,E)-farnesyl diphosphate (FPP, 1-OPP) to squalene (2, eq 1).1 The acyclic polyene product is a precursor to most polycyclic triterpenes, and its formation is the first committed step in the biosynthesis of cholesterol and other essential sterols. The conversion of FPP to squalene occurs in two steps by way of the stable, enzyme-bound intermediate, presqualene diphosphate (PSPP, 4 in eq 2).2 In the first and nonreductive step, a formal stereospecific 1,1-elimination of HOPP from one FPP is effected with two alkylations of the 2,3-double bond of the second FPP molecule to form the cyclopropane ring. Rearrangements and ring cleavage via carbocation intermediates terminated by hydride capture from the NADPH cofactor constitute a chemically reasonable mechanism for the second step that now has considerable experimental support.3
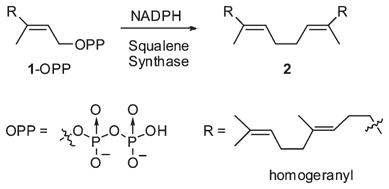 |
(1) |
PSPP and its analogues are intermediates in other related biochemical processes. This vinyl cyclopropylcarbinyl diphosphate is considered to be the progenitor of the botryococcenes, a family of acyclic triterpenes formed by an unusual 1′-3 connection of farnesyl units.4 PSPP serves as an intercellular signal for down-regulation of superoxide formation in neutraphils. 5 The corresponding C40 compound prephytoene diphosphate is an intermediate in phytoene formation in the biosynthesis of the carotenoid plant pigments.6 Chrysanthemyl diphosphate, the C10 relative, is a key intermediate in biosynthesis of irregular monoterpenes such as artemesia ketone.7
 |
(2) |
The location of squalene synthase just after a key branch point on the divergent pathway to essential isoprenoid products led to the expectation that its inhibition would selectively block triterpene and sterol biosynthesis.8 This rationale has motivated a worldwide search for new inhibitors of squalene synthase9 as potential anticholesterolemic,10a–c antiparasitic,10d and antimicrobial agents.11 Some examples of natural products discovered in screening programs are the zaragozic acids,12a,b phomoidrides,12c,d viridiofungin,11b schizostatin,12e and bisabaquals.12f Potent synthetic SS inhibitors include FPP mimics (bisphosphonates, (phosphinyl-methyl) phosphonates, and α-(phosphono)sulfonates)13 as well as some heterocyclic compounds.14 Various ammonium 15,16 and sulfonium17 ion inhibitors designed to mimic the substrate, carbocationic intermediates, or the product of the conversion of FPP to squalene have been reported. Crystal structures of human squalene synthase18a,b and S. aureous dehydrosqualene synthase18c are now available to guide inhibitor design and to comprehend the role of the enzymes in the catalytic mechanism.
We have been interested in the synthesis and evaluation of aza analogues of carbocation intermediates proposed to occur in biosynthetic processes catalyzed by terpene synthases. 15,19,20 These isoprenoid amines are potentially useful as selective inhibitors and as mechanistic probes to elucidate the structure, stereochemistry, charge delocalization, and active-site interactions of the fleeting carbocation intermediates. In the case of PSPP, it is reasonable to propose that the cyclopropane ring is formed by electrophilic attack of C1 of one FPP molecule on the 2,3-double bond of the other, generating a carbocation/OPP− ion pair within the enzyme active site (eq 2). In one mechanistic scenario, an acyclic, tertiary carbocation (3A) with localized positive charge would undergo 1,3 elimination of proton HB (C1 pro-S hydrogen). Alternatively, the intermediate might be better represented as a bridged ion (protonated cyclopropane 3B) with partial bonding to C2 and C3 and more charge delocalization.
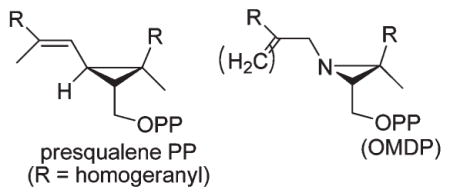
The aza bifarnesyl and aziridinyl diphosphates (±)-5-OPP, (±)-6-OPP, and (±)-7-OPP (Figure 1) were synthesized previously in racemic form as mechanistic probes for the first step in the squalene synthase reaction, and preliminary results on their inhibitory properties were reported.15c,20 The protonated form of the acyclic amino PP was intended to mimic tertiary ion 3A whereas the aziridinium PPs would resemble more closely bridged ion 3B. The nordihydro and β-methylene isoprenoid substituents on nitrogen in 6 and 7 were chosen as more stable surrogates for the N-vinyl group in the exact aziridine aza analogue of PSPP.
FIGURE 1.

Aza analogues of hypothetical acyclic and bridged carbocation/OPP− ion pairs 3A and 3B, respectively, proposed as intermediates in different mechanisms for squalene synthase-catalyzed formation of the three-membered ring of presqualene PP from two molecules of farnesyl PP (eq 2). R=homogeranyl.
In this paper we report syntheses of aziridines 6-OPP and 7- OPP together with the corresponding hydrolytically more stable methanediphosphonates 6-OMDP and 7-OMDP, and assays of their inhibition of a truncated, soluble form of recombinant squalene synthase from yeast. The more potent aziridine inhibitor 6-OPPwas synthesized in both racemic and enantiopure forms to assess the stereoselectivity of its interaction with the enzyme. The related N-undecylaziridines (±)-8-OPP and (±)-8-OMDP were also prepared as simpler models to evaluate the importance of the isoprenoid polyene chains in the binding interaction.
Synthesis of 2,3-Aziridinofarnesol (9-OH)
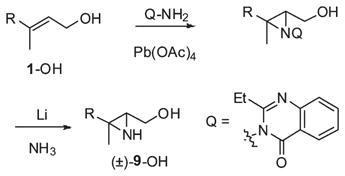
The aziridine ring in (±)-9-OH was introduced by Atkinson’s hydroxyl-directed imination21 of (E,E)-farnesol with 3-amino-4-(3H)- quinazolone (Pb(OAc)4, CH2Cl2, −23 °C, 2 h, 73%) (eq 3). The quinazolone substituent was removed by reductive cleavage of the N–N bond (6 equiv of Li, NH3, THF, −33 °C, 67%), conditions found to be general in a series of N-quinazolonylaziridines. 22
 |
(3) |
The enantiopure forms of 2,3-aziridino farnesol 9-OH were prepared from their 2,3-epoxy precursors according to a method developed for the synthesis of a series of racemic trisubstituted isoprenoid aziridines (eq 4).23 (E,E)-Farnesol was converted to both enantiomeric 2,3-epoxides by the Sharpless asymmetric epoxidation24 in excellent yields and high stereopurities (96:4 ± 0.4 er) according to 1H NMR analyses of the (1S)-camphanate derivatives (see Supporting Information). (+)-2,3-Epoxyfarnesol, (+)-10, was converted to the corresponding azidohydrin by regio- and stereospecific azide opening of the epoxide with diethylaluminum azide25 (Et2AlCl, NaN3, toluene, −78 to 0 °C, 80%). Selective silylation at the primary hydroxyl position (TBDMSCl, imidazole, DMAP, DMF, 0 °C, 82%) followed by conversion to the corresponding mesylate (MsCl, NEt3, CH2Cl2, 0 °C, 81%) and reductive cyclization (LiAlH4, ether, 83%) afforded enantiopure aziridine (−)-9-OH [α]D −19.2 (c 1.42, CHCl3). The enantiomeric NH-aziridino alcohol (+)-9-OH [α]D +19.3 (c 1.21, CHCl3) was prepared from epoxide (−)-10 in the same way.
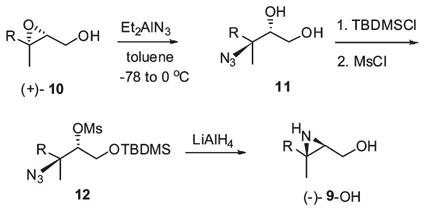 |
(4) |
(E)-6,10-Dimethyl-5,9-undecadienoic acid (14b), the precursor to the nordihydroisoprenoid chain in 6-OH, was synthesized by homologation of homogeraniol (13a) (eq 5).26 Conversion to the iodide (I2, Ph3P, imidazole, THF; 88%)27 followed by alkylation of tert-butyl lithioacetate (HMPA, THF, −78 °C, 77%)28 afforded tert-butyl ester 14a. Saponification under strongly basic conditions, namely Claisen’s alkali29 (KOH, H2O, EtOH, 3 h), provided isoprenoid acid 14b. The corresponding acid chloride 14c (oxalyl chloride, pyridine, benzene, 0 °C, 24 min) was used immediately in the N-acylation of the racemic and enantiopure forms of silyl ether aziridine 9-OTBDMS (eq 6).
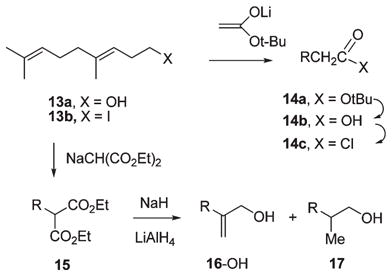 |
(5) |
The (E)-6,10-dimethyl-2-methylene-5,9-undecadienol precursor 16-OH for the N-substituent in aziridino alcohol 7-OH was prepared by means of Marshall’s α-methylene alcohol synthesis.30 Alkylation of diethyl sodiomalonate with iodide 13b provided homogeranyl malonate 15 (NaOEt, EtOH, reflux, 1 h, 62%) accompanied by a substantial amount of the cyclopropylcarbinyl ethyl ether (31%, structure not shown) presumably arising from 13b by competing homoallylic participation and nucleophilic attack of ethanol on the cyclopropylcarbinyl ion. Deprotonation of malonate 15 with 1.2 equiv of NaH (DME, reflux) followed by hydride reduction (2.6 equiv of LiAlH4, reflux, 3 h, 79%) provided a 3:1 mixture allylic alcohol 16-OH and its dihydro derivative 17 based on 1H NMR integrations. The resulting mixture of alcohols was converted to the corresponding mesylate derivatives (MsCl, NEt3, ether, −78 to −15 °C). Immediate N-alkylation of the TBDMS ether (9-OTBDMS)31 of (±)-aziridine 9-OH followed by fluoride-induced desilation afforded (±)-7-OH in 34% yield (eq 6).
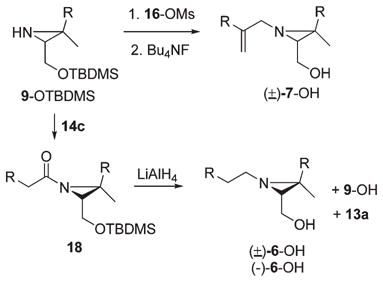 |
(6) |
Installation of the N-alkyl substituent in 6-OH was effected by a three-step sequence: O-silylation–N-acylation–hydride reduction (eq 6). Racemic aziridine 9-OH was again O-silylated to 9-OTBDMS (TBDMSCl, imidazole, DMF, 75%)20,31 and converted to the N-acylaziridine 18 by reaction with acid chloride 14c (Et3N, CH2Cl2). Although 18 was purified once by rapid silica gel flash column chromatography, this material was used in crude form immediately in most reactions owing to its instability on silica gel or to prolonged storage even at −20 °C. Hydride reduction (10 equiv of LiAlH4, ether, reflux, 48 h) was accompanied by concomitant removal of the silyl group. Purification by silica gel chromatography afforded N-alkylaziridine 6-OH (32%), recovered NH-aziridino alcohol 9-OH(17%), and trishomogeraniol 13a (27%) as shown in eq 6. The acylation–reduction approach was used to prepare aziridine 6-OH in both enantiomerically pure forms (−)-6-OH [α]D −3.7 (c 0.85, CHCl3) and (+)-6-OH [α]D+4.9 (c 1.09, CHCl3) from aziridine (−)-9-OH and its enantiomer (+)-9-OH, respectively.
The formation of substantial amounts of acyl cleavage products 9-OH and trishomogeraniol (13a) in the LiAlH4 reduction of N-acyl-O-silyl aziridine 18 prompted some experiments to determine whether this side reaction could be suppressed and to try to understand the competing processes. Reductions of 18 with other hydride reducing agents and/or conditions had no significant effect on the product ratios and yields. The model substrate N-pentanoyl- O-silyl aziridino alcohol 19 was prepared in the same manner by sequential silylation and acylation with pentanoyl chloride (80%). An attempt to remove the TBDMS protecting group with Bu4NF afforded the O-pentanoyl ester 20 (68%) instead of the N-pentanoyl aziridino alcohol isomer (eq 7). Evidently, rapid N-to-O acyl migration followed attack of fluoride on the silyl group. Prolonged reduction of 19 (10 equiv LiAlH4, ether, reflux, 60 h) furnished the N-pentyla-ziridino alcohol 21 (46%) together with cleavage byproduct 9-OH (28%) in accord with the previous results with 18.
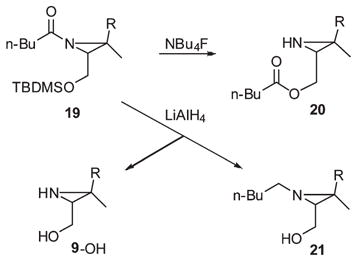 |
(7) |
LiAlH4 reductions of the unstable N,O-dipentanoyl derivative 22, prepared by acylation of aziridino alcohol 9-OH with pentanoyl chloride (2.1 equiv of Et3N, ether, 0 °C, 80%), proved to be time dependent (eq 8). After 1 h, the sole product obtained was the starting NH-aziridino alcohol 9- OH (79%). No effort was made to isolate the C5 alcohol coproduct. However, after 24 h, the N-pentyl aziridino alcohol 21 was isolated in 32% yield, along with 51% of recovered 9-OH. Continuation of the slow reduction for 48 h resulted in further conversion to 21 (67%), and the amount NH, OH aziridino alcohol 9-OH decreased to 13%.
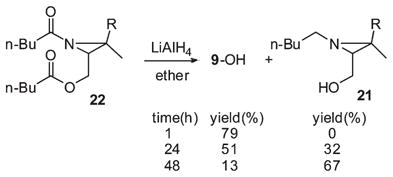 |
(8) |
The relatively slow conversions of the N-acylaziridines to the N-alkylaziridines in the preceding reductions is presumably attributable to angle strain associated with formation of the required aziridinium ion intermediates. This allows time for the initially formed aziridino alkoxides to undergo fragmentation to the corresponding aldehydes and aziridino alcohol 9-OH, and the former would be rapidly reduced to the primary alcohol byproduct. In fact, the partial hydride reduction of N-acyl derivatives of the parent ethyleneimine is known to give aldehydes.32 The facile N-to-O acyl transfer observed in the desilylation of 19 suggests the possibility of similar silyl transfers in the hydride reductions of the N-acyl-O-silyl aziridino alcohols (eq 9). The simultaneous loss of the TBDMS groups in these reductions and the known stability of this protecting group to LiAlH4 33 provide support for this assumption. It therefore seems reasonable to propose the sequence of reactions shown in eq 9 (23a → 23b → 23c), i.e., hydride addition to the aziridinamide carbonyl, silyl transfer, C–O bond heterolysis to the aziridine iminiumion, and finally, a second hydride attack to form the N-alkylaziridino alkoxide.
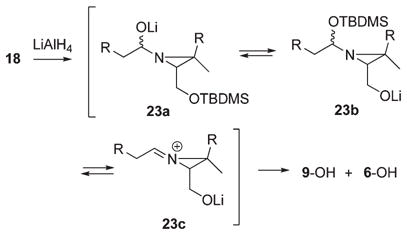 |
(9) |
Formation of Diphosphates and Methanediphosphates
Phosphorylations of N-alkylaziridino alcohols were carried out by activation of the primary alcohols followed by SN2 displacement with pyrophosphate or methanediphosphonate anions (eq 10).34 Reaction of enantiopure and racemic aziridines (±)-6-OH, (−)-6-OH, and (+)-6-OH and racemic 7-OH with MsCl provided the corresponding mesylates 6-OMs and 7-OMs (Et3N, CH2Cl2, −15 °C, 0.5 h, 85–97%). Displacement by inorganic pyrophosphate (2 equiv of (Bu4N)3 OPP, CH3CN, molecular sieves, rt, 48 h) afforded the aziridinyl diphosphates. 31P NMR analysis of the crude products revealed a 1:1 mixture of aziridinyl diphosphates to inorganic diphosphate. Similar reactions with methanediphosphonate anion (2 equiv ((Bu4N)3-OMDP, CH3CN, molecular sieves, rt, 60 h) were considerably slower, and in most cases, 1:3 mixtures of the aziridine methanediphosphates to methanediphosphonate were obtained. The N-undecylaziridine analogues 8-OH, 8-OPP, and 8-OMDP were prepared from (±)-9-OH in the same way.
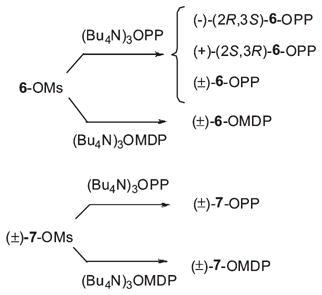 |
(10) |
The crude aziridinyl diphosphates obtained from these reactions were 1:1 mixtures of aziridine diphosphates and inorganic pyrophosphate according to 31P NMR spectra. Purification and characterization of organic diphosphates prepared by the displacement method are typically carried out after ion exchange to the more tractable ammonium salts, at which stage remaining inorganic pyrophosphate can be separated by precipitation from methanol.34 Unfortunately, initial attempts to effect ion exchange caused partial conversion to a new compound tentatively assigned as the 5- or 6-membered cyclic monophosphates 24 or 25 (δP −1.75, s) arising from aziridinium ion opening of 8-OPP, cyclization onto the proximal phosphate, and hydrolysis of the terminal phosphate. Accordingly we opted to forego ion exchange and to conduct the enzyme inhibition assays with the unpurified aziridinyl PPs as their tetrabutylammonium salts, with the consequences that 1H NMR characterization would be limited since the spectra are dominated by the intense resonances for the butyl groups and that approximately 1 equiv of inorganic OPP− would necessarily be present in the incubation medium. Nevertheless, it proved possible to discern the peaks for the CH2OPP protons in the 1H NMR spectra of 6-OPP· (Bu4N)3 salt, as well as resonances ascribable to the ring C-CH3, two CHN protons, and three vinyl protons.
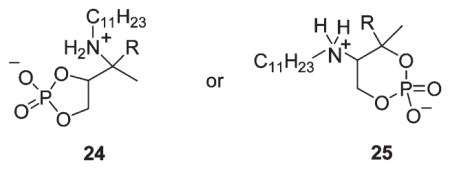
Ion-exchange chromatography (AG 50W-X8, 100–200 mash, NH4+) of 8-OPP· Bu4N using 25 mM NH4CO3 as buffer effected complete conversion to the NH4+ salt. The crude material was washed with CH3OH, and inorganic pyrophosphate was removed by filtration. 31P NMR analysis of the methanol-soluble material showed a 1:1 mixture of the aziridinyl diphosphate (δP −5.85, d, 1P, J=20.7 Hz and δP −7.19, d, 1P, J= 20.7 Hz) and an upfield resonance (δP −1.75, 1P, singlet) attributed to a ring-opened monophosphate product. A 2:1 mixture (31P NMR) favoring the aziridinyl diphosphate was obtained when 1 M (NH4)2CO3 was used as ion exchange buffer, and the receiving tubes were basified immediately with NH4OH. Further purification of the mixture by cellulose chromatography (8:1:1 of 2-propanol/concd. NH4OH/H2O as eluent) provided a white powder, the structure of which was assigned as an organic monophosphate by MS [(+)FAB, m/z 472.4 (M+1)]. Cyclic amino monophosphates 24 or 25 are tentatively proposed as possible structures for this byproduct.
Enzyme Inhibition Assays
The apparent instability of the aziridine PPs during ion exchange in the presence of aqueous NH4HCO3 buffer, and the known tendency of aziridines to undergo solvolytic ring-opening under acidic conditions,35,36 prompted control experiments to evaluate the stability of the compounds in the standard squalene synthase assay conditions (pH 7.2, Mg2+). 31P NMR spectra of solutions (D2O, NH4OD, pH 9) of 8-OMDP were unchanged after 3 days, whereas the spectra of diphosphate 8-OPP showed a new singlet (δP −1.8, s) similar to those seen in the spectra of the aziridinyl PPs after ion exchange. Fortunately, the rate was slow enough (t1/2 ~4 days at pH 9 and 18 h at pH 7) that this process was unlikely to occur to a significant extent during the typical 1-h incubations.
Kinetic assays of recombinant yeast squalene synthase activity37 with [3H]FPP (100 μM, 7.5 μCi/μmol) as substrate at pH 7.2 were carried out at various concentrations of the aziridines as Bu4N+ salts. Product [3H]squalene was isolated by extraction with hexanes, purified by filtration over silica gel, and analyzed by liquid scintillation spectrometry. The IC50 values for each compound (Table 1) were measured in the absence and presence of inorganic pyrophosphate (1.5 mM) on the expectation that the aziridinium/pyrophosphate ion pairs might exhibit stronger interaction with the catalytic site on the enzyme. However ca. 1 equivalent of OPP− anion was already present in the unpurified NBu4+ salts of 6-OPP, 7-OPP, and 8- OPP, and 2–3 equiv of OMDP− anion was present in the corresponding methanediphosphonates. In contrast, the inhibition of squalene synthase by the acyclic aza analogue 5-OPPwas determined on the purified ammonium salt established to be free of OPP− anion.15c,20
TABLE 1.
Inhibition of Recombinant Yeast Squalene Synthase by Azir-idino Diphosphates and Methanediphosphonates (Figure 1) in the Absence and Presence of Added Inorganic Pyrophosphate (PPi) at pH 7.2a
| inhibitor | IC50 b (μM) | Ki d (μM) | |
|---|---|---|---|
| −PPi | +PPic | ||
| (±)-5-OPP | 9.7 ± 2.7 | 11.4 ± 2.2 | |
| (±)-6-OPP | 7.2 ± 0.8 | 1.9 ± 0.2 | 0.21 |
| (2R,3S)-6-OPP | 1.19 ± 0.05 | 1.17 ± 0.08 | |
| (2S,3R)-6-OPP | 18.7 ± 4.9 | 4.8 ± 0.9 | |
| (±)-6-OMDP | 3.2 ± 0.2 | 13.8 ± 3.8 | |
| (±)-7-OPP | 6.9 ± 1.6 | 4.0 ± 0.4 | |
| (±)-7-OMDP | 99.8 ± 18.5 | 17.4 ± 5.3 | |
| (±)-8-OPP | 55% inhibition at 219 μM | 80% inhibition at 219 μM | |
| (±)-8-OMDP | no inhibition | no inhibition | |
Assay conditions: 50 mM MOPS, pH 7.2, 20 mM MgCl2, 2% (v/v) Tween 80, l mM DTT, 1 img/mL bovine serum albumin. 1.0 mM NADPH, 0 or 1.5 mM PPi, 100 uM [3H]FPP (7.5 μCi/μmol). 0–300 μM inhibitor, and 0.1 μg of protein in a total volume of 200 μM. Samples were incubated at 30 °C for 5 min before addition of enzyme and stopped after an additional 10 min by addition of 40% aqueous KOH in methanol. Product [3H] squalene was isolated by extraction with hexanes, purified by filtration over alumina, and analyzed by liquid scintillation spectrometry.
IC50 values were interpolated from plots of specific activity at [I] = 0.001, 0.50, 1.00, 3.00, 10.0, 30.0, and 300 μM using Graf it (Erithicus Software, Staines, UK). Deviations shown are standard errors.
Incubations in the presence of 1.5 mM PPi.
5–10 μM [3H]FPP (7.5 μCi/μmol).
In the absence of added PPi, (2R,3S)-6-OPP obtained from (−)-6-OH resembling the proper enantiomer of PSPP was a much stronger inhibitor of squalene synthase (about 16-fold) than the (2S,3R)-enantiomer obtained from (+)-6-OH. Although the synergistic effect of the PPi additive on the inhibitory properties of the enantiopure aziridino diphosphates was more pronounced for the “wrong” enantiomer (2S,3R)-6-OPP (about 4-fold vs almost no change), the 2R,3S enantiomer remained 4 times more potent under these conditions. Additional kinetic experiments established a Ki of 0.21 μM (competitive against FPP, Km for FPP=20 μM) for racemic 6-OPP in the presence of PPi. The selectivity of the interactions of 6- OPP and 7-OPP with the recombinant enzyme was revealed by the much lower affinity exhibited by 8-OPP bearing the NC11H23 substituent (55% inhibition of squalene synthase at 219 μM in the absence of PPi) instead of the unsaturated isoprenoid groups in 6-OPP and 7-OPP. Among the methane–diphosphonate derivatives, aziridine 6-OMDP and 7-OMDP inhibited squalene synthase in the presence of PPi additive with IC50 values of 13.8 and 17.4 μM, respectively.
Discussion
Aziridine 6-OPP proved to be one of the most potent mechanism-based squalene synthase inhibitors known. The IC50 value of 1.2 μM for the 2R,3S enantiomer and the submicromolar Ki determined previously for the racemate20 suggest quite strong interactions with the enzyme, despite the absence of the proximal double bond and the methyl group in the side chain on the aziridine nitrogen. The heightened inhibition exerted by the 2R,3S enantiomer matching the configuration of PSPP over that of the “wrong” 2S,3R stereoisomer in both the absence and presence of the PPi additive (16-fold and 4-fold, respectively) signify substantial stereoselectivity in their association with the protein. The enhanced affinities of these aza analogue inhibitors for the enzyme compared to those of FPP substrate (S0.5 FPP=19 ± 6 μM) and the PSPP intermediate (Ki PSPP= 75 ± 20 μM)38 encourage the view that the compounds may be valid mimics of transient carbocation intermediates. Furthermore, the greater potency of the aziridine inhibitors 6-OPP and 7-OPP, compared to that of the acyclic aza bifarnesyl compound 5- OPP, is consistent with a protonated cyclopropane intermediate 3B analogous to that proposed in the biosynthesis of related C10 isoprenoids.7b,c Nevertheless, the kinetics and mechanism of the squalene synthase reaction are quite complex,38 and the precise orientation of the compounds in the catalytic site is uncertain. The reduced affinity of the three methanediphosphonates (±)-6-OMDP, (±)-7-OMDP, and (±)-8-OMDP should presumably be attributed to the higher pKa values for methanediphosphonic acids and their weaker interaction with the PP binding pocket.39
Squalene synthase has two nonequivalent PP binding sites that accommodate both FPP substrates. A strong interaction of the PP group of the inhibitors with either one would effectively prevent squalene synthesis. The rate-determining step of the normal enzymatic reaction is proposed to be either PSPP formation or a conformational change of the enzyme during the catalytic cycle.38 The protonation state, hybridization, and charge at the amino nitrogen of the aza analogues no doubt affect the resemblance of the inhibitors to the carbocations they are intended to mimic, i.e., 3A and 3B. Aza-bifarnesyl PP 5-OPP very likely exists predominantly in the tetrahedral ammonium form at pH 7.2 (pKa ca. 10–11). However, the pKa values for highly substituted aziridines are rather low (pKa 4–6),35,40 and the aziridine inhibitors 6-OPP and 7-OPP probably associate with the protein in their unprotonated, but still tetrahedral, forms. Clearly subtle differences such as these will influence the interactions of the inhibitors with the catalytic apparatus of the protein.
Some of the ambiguities are highlighted by the crystal structure of bornyl diphosphate synthase complexed with amine analogues of substrate, a carbocation intermediate, and the bornyl PP product.19g 3-Aza-2,3-dihydrogeranyl PP is bound in the ammonium form with the dimethylallyl tail in an orientation differing from that predicted for the geranyl PP substrate. (R)-7-Aza-7,8-dihydrolimonene, an amine corresponding in structure and stereochemistry to the (R)-α-terpinyl carbocation intermediate, is observed in the active site in an inverted position. Hydrogen bonds with an incorporated water molecule may be one important factor in the distortions observed in the crystal structures of these inhibitor–enzyme complexes.
The stability of the enzyme-bound aziridine PPs is also uncertain. The facility with which aziridinium ions undergo ring-opening to potential electrophiles,35,36 and the observed conversion of 8-OPP to proposed cyclic monophosphates 24 or 25 suggest that similar transformations might occur under the influence of the enzyme. Although the kinetic behavior was inconsistent with appreciable inactivation during the measurements, the structural integrity of the aziridines under the incubation conditions remains unknown.
Despite the forgoing imponderables, aziridine analogues of bridged, nonclassical carbocations proposed as intermediates in various terpene synthase-catalyzed reactions are novel, mechanism-based inhibitors for investigation of these enzymes.41,42 This concept for inhibitor design may lead to useful active site probes for characterization of protein structure by affinity-labeling or X-ray crystallography.43
Experimental Section
The purity of purified products was established by analysis of 1H NMR spectra to be ≥95% pure unless stated otherwise. Complete experimental procedures and spectral data are outlined below for synthesis of enantiopure (−)-(2R,3S)-6-OH and (2R,3S)-6-OPP. Yields or abbreviated procedures, and in some cases optical rotations, are presented for intermediates and the final product in synthesis of ent-6-OH and ent-6-OPP in the Supporting Information. NMR spectra, physical data, and purity evaluations agree well with those reported previously for the racemic series.20,23 Physical and spectral data for the enantiomers were identical.
(+)-(2R,3R)-trans-2,3-Epoxy-3,7,11-trimethyl-(E)-6,10-dodecadien- 1-ol (trans-2,3-Epoxyfarnesol) ((+)-10)
The asymmetric epoxidation was carried out according to a procedure reported for (+)-2,3-epoxygeraniol.24 A suspension of powdered molecular sieves (1.83 g, 3A°, activated) in CH2Cl2 (50 mL) and D-(−)- diethyl tartrate (6.16 g, 28.9 mmol) was stirred and cooled at −20 °C under N2 as titanium (IV) isopropoxide (7.07 g, 24.9 mmol) in CH2Cl2 (7 mL) and 5–6 M tert-butyl hydroperoxide in isooctane (12 mL, 70 mmol) were added sequentially. After 30 min at −20 °C, trans, trans-farnesol (10.06 g, 45.2 mmol, dried over 3 A ° molecular sieves for 40 min) in CH2Cl2 (10 mL) was added dropwise, and the mixture was stirred at −20 °C for 2 h and 1 h at rt to generate a pale yellow oil after workup. Purification by flash column chromatography (50% EtOAc/ hexanes) afforded 10.78 g (97%) of epoxy alcohol (+)-10 as a colorless oil. The 1H and 13C NMR spectral data and assignments correspond to those reported in the literature.44 1H NMR analysis of the (S)-camphanate ester derivative indicated that the enantiomeric ratio was 94:6 (see Supporting Information): [α]D=+6.35, (c 1.73, CHCl3). The magnitude of the optical rotation corresponds to that of enantioenriched (+)-2,3-epoxyfarnesol (10) with minor deviations due to concentration differences [lit.44 [α]D = +6.53 (c 4.21, CHCl3)].
(+)-(2S,3S)-3-Azido-3,7,11-trimethyl-(E)-6,10-dodecadiene- 1,2-diol (11)
The following procedure is based on those reported by Davis23 and Benedetti.25 A suspension of NaN3 (2.41 g, 37.0 mmol) in toluene (42 mL) was stirred and cooled at 0 °C as a solution of Et2AlCl (18.8 mL, 33.6 mmol, 1.8 M in toluene) was added dropwise over 10 min via syringe. After 6 h at rt, the resulting suspension was cooled to −78 °C under N2, and a solution of (+)-2,3-epoxyfarnesol ((+)-10, 4.00 g, 16.8 mmol) was added dropwise over 10 min. The mixture was stirred for 1 h at −78 °C and for 16 h at rt, cooled to 0 °C, and diluted with EtOAc (70 mL). NaF (31.8 g) and H2O (3.9 mL) were added sequentially, and the heterogeneous mixture was allowed to stir for 4 h at rt. Filtration through a pad of anhyd NaSO4 (ca. 100 g) to remove salts and water followed by solvent evaporation gave a pale yellow oil. Purification by flash column chromatography (50% EtOAc/hexanes) afforded 3.80 g (80%) of azido diol (+)-11 as a colorless oil: 1H NMR(500 MHz, CDCl3) δ 1.36 (s, 3H), 1.47 (ddd, J=14.0, 10.7, 6.1 Hz, 1H), 1.59 (s, 3H), 1.62 (s, 3H), 1.65 (m, 1H), 1.68 (s, 3H), 1.97 (3 peak m, 2H), 2.08 (8 peak m, 4H), 2.25 (br s, 1H), 2.75 (br s, 1H) 3.58 (app ddd, J=7.9, 3.2, 1.9 Hz, 1H), 3.6 g (app dd, J=10.7, 2.8 Hz, 1H), 3.76 (app ddd, J = 10.9, 7.1, 3.2 Hz, 1H), 5.09 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 16.2, 17.9, 19.6. 22.4, 25.9, 26.8, 36.7, 39.8, 62.7, 65.8, 76.4, 123.3, 124.4, 131.7, 136.4; IR (neat) νmax 2104 (N3), 3392 (OH) cm−1; [α]D +34.7, (c 1.62, CHCl3).
(2S,3S)-3-Azido-1-(tert-butyldimethylsilyloxy)-3,7,11-trimethyl-(E)-6,10-dodecadien-2-ol (11-OTBDMS)
The following procedure is based on ones reported by Davis23 and Fujii and Ibuka.45 A solution of the azido diol (11, 3.42 g, 12.17 mmol), imidazole (4.98 g, 73.2 mmol), and 4-DMAP (147 mg, 1.20 mmol) in DMF(20 mL) was stirred and cooled at 0 °C under N2 as TBDMSCl (5.52 g, 36.7 mmol) in DMF (18 mL) was added dropwise over 5 min. The solution was warmed to rt, and after 30 min, ethanolamine (2.33 g, 37.7 mmol) was added to scavenge excess TBDMSCl. After 2 h, H2O was added, and the product was extracted with benzene. The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to give a colorless oil. Purification by flash column chromatography (5% EtOAc/hexanes) afforded 3.22 g (66%) of the primary silyl ether and 0.675 g (14%) of the secondary silyl ether isomer. Data for primary silyl ether 11-OTBDMS: 1H NMR (500 MHz, CDCl3) δ 0.09 (s, 3H), 0.10 (s, 3H), 0.91 (s, 9H), 1.32 (s, 3H), 1.46 (ddd, J=14.1, 11.1, 6.0 Hz, 1H), 1.60 (s, 3H), 1.62 (s, 3H), 1.63 (m, 1H), 1.68 (d, J=1.1Hz, 3H), 1.98 (m, 2H), 2.08 (m, 4H), 2.77 (br s, 1H), 3.56 (ABX, JAX=3.2Hz, JBX=8.1Hz, 1H), 3.61 (ABX, JAB= 9.6 Hz, JBX=8.1 Hz, 1H), 3.74 (ABX, JAB=9.6Hz, JAX=3.2 Hz, 1H), 5.10 (m, 2H); 13C NMR (126 MHz, CDCl3) δ −5.2, 16.2, 17.9, 18.4, 19.4, 22.3, 25.9, 26.0, 26.9, 36.8, 39.9, 62.0, 65.1, 75.9, 123.6, 124.5, 131.7, 136.1; IR (neat) νmax 2105 (N3), 3593 (OH) cm−1.
(2S,3S)-3-Azido-1-(tert-butyldimethylsilyloxy)-3,7,11-trimethyl-(E)-6,10-dodecadien -2-yl Methanesulfonate (12)
The following procedure was based on that reported by Crossland.46 A solution of the primary silyl ether 11-OTBDMS (3.13 g, 7.91 mmol) and Et3N (2.54 g, 25.32 mmol) in CH2Cl2 (30 mL) was stirred and cooled at 0 °C under N2 as neat MsCl (2.66 g, 23.7 mmol) was added dropwise over 10 min. After 1 h, ice-cold H2O(10 mL) was added, and the aqueous phase was extracted with Et2O (3 × 30 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure. Purification by flash column chromatography (5% EtOAc/hexanes) afforded 2.95 g (78%) of azido mesylate 12 as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 0.09 (s, 3H), 0.10 (s, 3H), 0.90 (s, 9H), 1.40 (s, 3H), 1.59 (m, 1H), 1.60 (s, 3H), 1.62 (s, 3H), 1.64 (m, 1H), 1.67 (d, J = 1.1 Hz, 3H), 1.97 (m, 2H), 2.10 (m, 4H), 3.15 (s, 3H), 3.85 (ABX, JAB=11.8Hz, JBX=7.7 Hz, 1H), 3.94 (ABX, JAB=11.8 Hz, JAX=2.8 Hz, 1H), 4.57 (ABX, JAX=2.8Hz, JBX=7.7Hz, 1H), 5.08 (m, 2H); 13C NMR(126 MHz,CDCl3) δ −5.2, 16.2, 17.9, 18.6, 20.0, 22.3, 25.9, 26.1, 26.8, 37.2, 39.3, 39.9, 62.8, 64.5, 88.1, 122.9, 124.4, 131.7, 136.6; IR (neat) νmax 2108 (N3) cm−1.
(−)-(2R,3S)-trans-3-((E)-4,8-Dimethyl-3,7-nonadienyl)-3-methylaziridine- 2-methanol (9-OH)
The following procedure is based on those reported by Forestier et al.47 and Davis et al.23 A suspension of LiAlH4 (602 mg, 15.6 mmol) in Et2O (20 mL) was stirred and cooled at 0 °C under N2 as a solution of the azido mesylate 13 (2.95 g, 6.23 mmol) in Et2O (10 mL) was added dropwise over 2 min via syringe. After 4 h, the gray suspension was cooled to 0 °C and stirred as H2O (0.602 mL), 15% NaOH (0.602 mL), and H2O (1.8 mL) were added sequentially with occasional additions of Et2O to maintain the volume at ca. 3 mL. After 2 h, the white solid was filtered and washed with EtOAc (4 × 5 mL). The filtrates were combined, dried (MgSO4), and concentrated under reduced pressure. Purification by flash column chromatography (10% MeOH/CH2Cl2) afforded 1.04 g (70%) of (−)-9-OH as a pale, yellow oil: 1H NMR (500 MHz, CDCl3) δ 1.17 (s, 3H), 1.34 (m, 1H), 1.58 (s, 3H), 1.58 (m, 1H), 1.60 (s, 3H), 1.66 (s, 3H), 2.05 (m, 7H), 3.48 (dd, J = 11.8, 4.3 Hz, 1H), 3.50 (br s, 1H), 3.74 (ddd, J = 11.8, 4.9, 2.4 Hz, 1H), 5.08 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 16.2, 17.3, 17.9, 24.8, 25.9, 26.8, 39.7, 39.9, 41.7, 43.6, 61.6, 123.4, 124.4, 131.7, 136.1; [α]D −19.2 (c 1.42, CHCl3).
(2R,3S)-trans-3-[(tert-Butyldimethylsilyl)oxy]methyl-2-((E)-4,8-dimethyl-3,7-nonadienyl)-2-methylaziridine (6-OTBDMS)
The following procedure is based on that reported by Koohang et al.20 A solution of the (−)-aziridino alcohol ((−)-9-OH, 0.510 g, 2.15 mmol) in DMF (1.1 mL) was stirred and cooled at 0 °C under N2, while imidazole (0.374 g, 5.49 mmol) and TBDMSCl (0.392 g, 2.60 mmol) were added. The ice bath was removed, and the solution was allowed to stir for 30 min after which time EtOAc (10 mL) and H2O (10 mL) were added. The aqueous layer was extracted with EtOAc (3×10 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure to give a yellow oil. Purification by flash column chromatography (10%MeOH/CH2Cl2) afforded 0.327 g (62%) of silyl ether, 9-OTBDMS, and 150 mg of recovered (−)-9-OH. Data for 9-OTBDMS: 1H NMR(400 MHz, CDCl3) δ 0.06 (s, 3H), 0.07 (s, 3H), 0.90 (s, 9H), 1.18 (s, 3H), 1.33 (ddd, J=13.5, 9.4, 6.6 Hz, 1H), 1.53 (m, 1H), 1.59 (s, 3H), 1.60 (s, 3H), 1.67 (d, J=1.1Hz, 3H), 2.03 (m, 7H), 3.57 (dd, J=10.9, 6.4Hz, 1H), 3.73 (ddd, J= 10.9, 5.4, 1.1Hz, 1H), 5.11 (m, 2H); 13C NMR(101 MHz, CDCl3) δ −5.1, −4.9, 16.1, 17.4, 17.9, 18.5, 24.9, 25.9, 26.13, 26.9, 39.9, 41.7, 43.2, 63.3, 123.8, 124.5, 131.6, 135.8.
(2R,3S)-trans-2-((E)-4,8-Dimethyl-3,7-nonadienyl)-1-((E)-6, 10-dimethyl-5,9-undecadienyl)-3-[(tert-butyldimethylsilyl)oxy]-methyl-2-methylaziridine (18)
The following procedure is based on that reported by Koohang et al.20 A solution of bishomogeranic acid (14b, 110 mg, 0.523 mmol) and pyridine (46 μL, 45 mg, 0.646 mmol) in benzene (4.5 mL) was stirred and cooled at 0 °C under N2 as oxalyl chloride (50 μL, 73 mg, 0.573 mmol) was added dropwise. The suspension was allowed to warm to rt, and stirring was continued until no bubbles were observed (ca. 25 min). The crude acid chloride was transferred immediately via syringe to a rapidly stirring solution of 9-OTBDMS (183 mg, 0.521 mmol) obtained from the previous step and Et3N(218 μL, 158 mg, 1.56 mmol) in Et2O (4.5 mL) at 0 °C under N2. After 40 min, H2O (10 mL) was added, and the product was extracted with EtOAc (3 × 20 mL). The organic layers were combined, dried (MgSO4), and concentrated by rotary evaporation with the water bath maintained at 30 °C. The crude N-acylaziridine (288 mg) was immediately taken on to the next step.
(−)-(2R,3S)-trans-3-((E)-4,8-Dimethyl-3,7-nonadienyl)-1-((E)- 6,10-dimethyl-5,9-undecadienyl)-3-methylaziridine-2-methanol (6-OH)
The following procedure is based on that reported by Koohang et al.20 A solution of LiAlH4 (202 mg, 5.32 mmol) in Et2O (18 mL) was stirred and cooled at 0 °C under N2 as the crude chiral N-acylaziridine (288 mg, 0.529 mmol) in Et2O (5 mL) was added dropwise via syringe. The pale gray suspension was heated at reflux for 48h, cooled to 0°C, and stirred as sequential dropwise additions of H2O (0.202 mL), 15% NaOH (0.202 mL), and H2O (0.606 mL) were performed. After 2 h, the white solid was filtered and washed with EtOAc (3× 10 mL). The filtrates were combined, dried (MgSO4), and concentrated under reduced pressure to give a pale yellow oil. Purification by flash column chromatography (50% EtOAc/hexanes) afforded 69 mg (32%) of the N-alkylaziridino alcohol as a colorless oil, 21 mg (17%) of the NH aziridino alcohol (9-OH), and 28 mg (27%) of trishomogeraniol. Data for (−)-6-OH: 1H NMR(500 MHz, CDCl3) δ 1.17 (s, 3H), 1.40 (m, 3H), 1.55 (m, 4H), 1.58 (s, 3H), 1.59 (s, 6H), 1.60 (s, 3H), 1.67 (s, 6H), 2.05 (m, 12H), 2.36 (dt, J = 11.8, 7.7 Hz, 1H), 2.69 (dt, J = 11.8, 7.7 Hz, 1H), 3.53 (dd, J = 11.4, 6.0 Hz, 1H), 3.69 (dd, J = 11.4, 6.0 Hz, 1H), 5.10 (m, 4H); 13CNMR(126 MHz,CDCl3) δ 16.2, 17.9, 19.1, 25.4, 25.9, 26.8, 26.9, 27.8, 28.0, 30.2, 32.80, 39.9, 39.9, 43.5, 50.7, 52.1, 60.1, 123.6, 124.4, 124.5, 124.6, 131.5, 131.7, 135.5, 136.0; [α]D −3.67 (c 0.85, CHCl3). The spectral data and assignments correspond to those reported in the literature for the racemic form.20
(2R,3S)-trans-3-((E)-4,8-Dimethyl-3,7-nonadienyl)-1-((E)-6, 10-dimethyl-5,9-undecadienyl)-3-methylaziridine-2-methanol Methanesulfonate (6-OMs)
The following procedure is based on ones reported by Koohang et al.20 and by Crossland and Servis.46 A solution of 6-OH (46 mg, 0.111 mmol) obtained from the previous step and Et3N (23 μL, 17 mg, 0.166 mmol) in CH2-Cl2 (1.2 mL) was stirred at −20 °C under N2 as methanesulfonyl chloride (10 μL, 15 mg, 0.133 mmol) was added dropwise via syringe. After 30 min, satd NaHCO3 (1 mL) was added, and the solution was stirred vigorously for 5 min. The organic phase was removed, and the aqueous phase was extracted with EtOAc (3 × 4 mL), dried (MgSO4), and concentrated under reduced pressure to give 52 mg of a yellow oil. Purification by flash column chromatography (50% EtOAc/hexanes) gave 46 mg (86%) of the mesylate as a yellow oil: 1H NMR(500 MHz, CDCl3) δ 1.18 (s, 3H), 1.43 (m, 7H), 1.59 (s, 3H), 1.60 (s, 6H), 1.61 (s, 3H), 1.67 (s, 3H), 1.97–2.22 (m, 12H), 2.31 (dt, J=11.8, 7.6 Hz, 1H), 2.65 (dt, J = 11.6, 7.4 Hz, 1H), 3.02 (s, 3H), 4.21 (m, 2H), 5.10 (m, 4H). The spectral data correspond to those reported in the literature for the racemic form.20
(2R,3S)-trans-[2-((E)-4,8-Dimethyl-3,7-nonadienyl)-1-((E)-6, 10-dimethyl-5,9-undecadienyl)-2-methylaziridin-3-yl]methyl Diphosphate, Tris (tetrabutylammonium) Salt (6-OPP)
The following procedure was based on those reported by Koohang et al.20 and Poulter et al.34 A solution of the enantiopure mesylate 6-OMs (44 mg, 0.091 mmol) obtained from previous step in dry acetonitrile (1 mL) containing molecular sieves (3A °, powdered, activated) was stirred at rt under N2 as a solution of (NBu4)3-HP2O7 (165 mg, 0.183 mmol) in dry acetonitrile (1 mL) was added dropwise over 5 min via syringe. After 60 h, the molecular sieves were filtered and washed with dry acetonitrile (5 mL), and the filtrate was extracted with hexane (3×7 mL). The hexane-soluble extracts were combined and concentrated under reduced pressure to give ca. 10 mg of recovered starting material. Concentration of the acetonitrile-soluble extracts under reduced pressure at <30 °C afforded 103 mg of a mixture of the aziridino diphosphate (2R,3S)-6-OPP (73%) and (NBu4)3 HP2O7 (1:1 by 31P NMR analysis) as a brown viscous oil that was analyzed by NMR spectroscopy. Some high-field 1H NMR peaks for 6-OPP were obscured by overlap with the following intense resonances for the NBu4+ counterions: 1.02 (t, J=7.3 Hz, 12H), 1.41 (sextet, J=7.6 Hz, 8H), 1.66 (app quintet, J=ca. 7.8 Hz, 8H), 3.20–3.27 (m, 4× 8H); (2S, 3R)-6-OPP: 1H NMR (400 MHz, CD3-OD) δ 1.22 (s, 3H) 1.51–1.58 (br m, ca. 4H), 1.59 (br s, 12H), 1.63 – 1.70 (m), 2.05 (m, 12H) 2.16 (app br quintet, 1H, J=7.6 Hz), 2.48–2.63 (m, 1H), 3.95–4.17 (app 12 line ABX, assumed 2H), 5.08 (br t, 2H, J=ca. 7 Hz), 5.14 (br t, 2H, J=ca. 7.5 Hz). (2R, 3S)-6-OPP: 31P NMR (162 MHz, CD3OD) δ −6.0 (d, J= 19.5 Hz, 1P), −5.2 (d, J=19.5 Hz, 0.97P), −3.9 (s, 1.78 P), 6.2 (s, 0.14 P). The spectral data correspond to those reported in the literature20 with minor deviations due to solvent differences (CD3OD vs CD3CN).
Squalene Synthase Assays
The assay procedure was modified from that of Zhang and co-workers.37 The assay mixture contained 50 mM MOPS (3-(N-morpholino) propanesulfonic acid), pH 7.2, 20 mM MgCl2, 2% (v/v) Tween 80, 1 mM DTT (dithiothreitol), 1 mg/mL of BSA (bovine serum albumin), 1.0 mM NADPH, 0 or 1.5 mM PPi, 100 μM [3H]FPP (7.5 μCi/μmol), 0–300 μM of inhibitor generally made by dilution of a 3-mM stock solution of the inhibitor in methanol, and enzyme (0.1 μg/reaction) to a final volume of 200 μL. The mixture was equilibrated at 30 °C for 5 min before the addition of the enzyme, and was incubated for an additional 5 min after the reaction was started by the addition of the enzyme. The reaction was stopped by the addition of 300 μL of 1:1 (v/v) 40% aq KOH/CH3OH). Sufficient solid NaCl was added to saturate the reaction mixture, and then 2 mL of ligroine containing 0.2% squalene was added. The mixture was vortexed vigorously for 30 s. A1-mL portion of the organic layer was applied to a 0.5× 6 cm alumina column packed in a Pasteur pipet and pre-equilibrated with 2 mL of ligroine containing 0.2% squalene. The eluent was collected in a scintillation vial, and 5 mL of Cytoscint scintillation fluor was added. The sample was analyzed for tritium on a liquid scintillation spectrometer.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for financial support of this work through Grant Nos. GM 13956 (R.M.C.) and GM 21328 (C.D.P.) and the National Science Foundation for providing support for the VOICE NMR facility at the University of Illinois at Urbana–Champaign.
Footnotes
Supporting Information Available: General experimental and instrumentation, preparative procedures, and characterization data for camphanate derivatives, aziridines, aziridino diphosphates, and methanediphosphonates, experimental data for squalene synthase purification, and reproductions of 1H, 13C, and/or 31P NMR spectra of selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Shechter I, Guan G, Boettcher BR. Isoprenoids Including Carotenoids and Steroids. In: Cane DE, Barton D, Nakanishi K, Meth-Cohn O, editors. Comprehensive Natural Products Chemistry. Vol. 2. Elsevier; Amsterdam: 1999. pp. 245–266. [Google Scholar]; (b) Tansey TR, Shechter I. Biochim Biophys Acta. 2000;1529:49–62. doi: 10.1016/s1388-1981(00)00137-2. [DOI] [PubMed] [Google Scholar]; (c) Poulter CD. Acc Chem Res. 1990;23:70–77. [Google Scholar]
- 2.Rilling HC, Epstein WW. J Am Chem Soc. 1969;91:1041–1042. [Google Scholar]
- 3.(a) Jarstfer MB, Zhang DL, Poulter CD. J Am Chem Soc. 2002;124:8834–8845. doi: 10.1021/ja020410i. [DOI] [PubMed] [Google Scholar]; (b) Blagg BSJ, Jarstfer MB, Rogers DH, Poulter CD. J Am Chem Soc. 2002;124:8846–8853. doi: 10.1021/ja020411a. [DOI] [PubMed] [Google Scholar]
- 4.Huang Z, Poulter CD. J Am Chem Soc. 1989;111:2713–2715. [Google Scholar]
- 5.Levy BD, Petasis NA, Serhan CN. Nature (London) 1997;389:985–990. doi: 10.1038/40180. [DOI] [PubMed] [Google Scholar]
- 6.(a) Altman LJ, Kowerski RC, Laungani DR. J Am Chem Soc. 1978;100:6174–6182. [Google Scholar]; (b) Iwata-Reuyl D, Math SK, Desai SB, Poulter CD. Biochemistry. 2003;42:3359–3365. doi: 10.1021/bi0206614. [DOI] [PubMed] [Google Scholar]; (c) Camara B, Dogbo O, Bardat F, Moneger R. C R Acad Sci, Ser III. 1985;301:749–752. [Google Scholar]
- 7.(a) Rivera SB, Swedlund BD, King GJ, Bell RN, Hussey CE, Jr, Shattuck-Eidens DM, Wrobel WM, Peiser GD, Poulter CD. Proc Natl Acad Sci USA. 2001;98:4373–4378. doi: 10.1073/pnas.071543598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thulasiram HV, Erickson HK, Poulter CD. Science. 2007;316:73–76. doi: 10.1126/science.1137786. [DOI] [PubMed] [Google Scholar]; (c) Thulasiram HV, Erickson HK, Poulter CD. J Am Chem Soc. 2008;130:1966–1971. doi: 10.1021/ja0771282. [DOI] [PubMed] [Google Scholar]
- 8.Biller SA, Neuenschwander K, Ponipom MM, Poulter CD. Curr Pharm Design. 1996;2:1–40. [Google Scholar]
- 9.For recent reviews, see: Do R, Kiss RS, Gaudet D, Engert JC. Clin Genetics. 2009;75:19–29. doi: 10.1111/j.1399-0004.2008.01099.x.Tavridou A, Manolopoulos VG. Curr Med Chem. 2008;15:792–802. doi: 10.2174/092986708783955482.Elsayed RK, Evans JD. Expert Opin Emerging Drugs. 2008;13:309–322. doi: 10.1517/14728214.13.2.309.Charlton-Menys V, Durrington PN. Exp Physiol. 2008;93:27–42. doi: 10.1113/expphysiol.2007.035147.Davidson MH. Curr Atherosclerosis Rep. 2007;9:78–80. doi: 10.1007/BF02693932.Charlton-Menys V, Durrington PN. Drugs. 2007;67:11–16. doi: 10.2165/00003495-200767010-00002.
- 10.(a) Abe I, Tomesch JC, Wattanasin S, Prestwich GD. Nat Prod Rep. 1994;11:279–302. doi: 10.1039/np9941100279. [DOI] [PubMed] [Google Scholar]; (b) Bergstrom JD, Dufresne C, Bills GF, Nallin-Omstead M, Byrne K. Annu Rev Microbiol. 1995;49:607–639. doi: 10.1146/annurev.mi.49.100195.003135. [DOI] [PubMed] [Google Scholar]; (c) Watson NS, Procopiou PA. Prog Med Chem. 1996;33:331–378. doi: 10.1016/s0079-6468(08)70308-4. [DOI] [PubMed] [Google Scholar]; (d) Urbina JA, Concepcion JL, Rangel S, Visbal G, Lira R. Mol Biochem Parasitol. 2002;125:35–45. doi: 10.1016/s0166-6851(02)00206-2. [DOI] [PubMed] [Google Scholar]
- 11.(a) Fairlamb IJS, Dickinson JM, O’Connor R, Cohen LH, van Thiel CF. Bioorg Chem. 2003;31:80–97. doi: 10.1016/s0045-2068(03)00025-7. [DOI] [PubMed] [Google Scholar]; (b) Onishi JC, Milligan JA, Basilio A, Bergstrom J, Curotto J, Huang L, Meinz M, Nallin-Omstead M, Pelaez F, Rew D, Salvatore M, Thompson J, Vicente F, Kurtz MB. J Antibiot. 1997;50:334–338. doi: 10.7164/antibiotics.50.334. [DOI] [PubMed] [Google Scholar]
- 12.Zaragozic acids: Bergstrom JD, Kurtz MM, Rew DJ, Amend AM, Karkas JD, Bostedor RG, Bansal VS, Dufresne C, VanMiddlesworth FL, Hensens OD. Proc Natl Acad Sci USA. 1993;90:80–84. doi: 10.1073/pnas.90.1.80.Dufresne C, Wilson KE, Singh SB, Zink DL, Bergstrom JD, Rew D, Polishook JD, Meinz M, Huang L, Silverman KC, Lingham RB, Marina M, Cascales C, Pelaéz F, Gibbs JB. J Nat Prod. 1993;56:1923–1929. doi: 10.1021/np50101a009.. Phomoidrides: Dabrah TT, Harwood HJ, Huang LH, Jankovich ND, Kaneko T, Li JC, Lindsey S, Moshier PM, Subashi TA, Therrien M, Watts PC. J Antibiot. 1997;50:1–7. doi: 10.7164/antibiotics.50.1.Dabrah TT, Kaneko T, Massefski W, Jr, Whipple EB. J Am Chem Soc. 1997;119:1594–1598.Schizostatin: Tanimoto T, Onodera K, Hosoya T, Takamatsu Y, Kinoshita T, Tago K, Kogen H, Fujioka T, Hamano K, Tsujita Y. J Antibiot. 1996;49:617–623. doi: 10.7164/antibiotics.49.617.Bisabosquals: Minagawa K, Kouzuki S, Nomura K, Kawamura Y, Tani H, Terui Y, Nakai H, Kamigauchi T. J Antibiot. 2001;54:896–903. doi: 10.7164/antibiotics.54.896.
- 13.1,1-Bisphosphonates: Magnin DR, Dickson JK, Logan JV, Lawrence RM, Chen Y, Sulsky RB, Ciosek CP, Biller SA, Harrity TW, Jolibois KG, Kunselman LK, Rich LC, Slusarchyk DA. J Med Chem. 1995;38:2596–2605. doi: 10.1021/jm00014a012.Biller SA, Sofia MJ, DeLange B, Forster C, Gordon EM, Harrity T, Rich LC, Ciosek CP., Jr J Am Chem Soc. 1991;113:8522–8524.Lawrence MR, Biller SA, Dickson JK, Jr, Logan JVH, Magnin DR, Sulsky RB, DiMarco JD, Gougoutas JZ, Beyer BD, Taylor SC, Lan SJ, Ciosek CP, Jr, Harrity TW, Jolibois KG, Kunselman LK, Slusarchyk DA. J Am Chem Soc. 1996;118:11668–11669.Song Y, Lin F-Y, Yin F, Hensler M, Rodrigues Poveda CA, Mukkamala D, Cao R, Wang H, Morita CT, Gonzalez Pacanowska D, Nizet V, Oldfield E. J Med Chem. 2009;52:976–988. doi: 10.1021/jm801023u.
- 14.4,1-Benzoxazepine: Miki T, Kori M, Mabuchi H, Tozawa R, Nishimoto T, Sugiyama Y, Teshima K, Yukimasa H. J Med Chem. 2002;45:4571–4580. doi: 10.1021/jm020234o.Quinuclidines: Ishihara T, Kakuta H, Moritani H, Ugawa T, Sakamoto S, Tsukamoto S-I, Yanagisawa I. Bioorg Med Chem. 2003;11:2403–2414. doi: 10.1016/s0968-0896(03)00143-3.Other heterocycles: Miki T, Kori M, Fujishima A, Mabuchi H, Tozawa R-i, Nakamura M, Sugiyama Y, Yukimasa H. Bioorg Med Chem. 2002;10:385–400. doi: 10.1016/s0968-0896(01)00289-9.
- 15.(a) Poulter CD, Capson TL, Thompson MD, Bard RS. J Am Chem Soc. 1989;111:3734–3739. [Google Scholar]; (b) Sandifer RM, Thompson MD, Gaughan RG, Poulter CD. J Am Chem Soc. 1982;104:7376–7378. [Google Scholar]; (c) Steiger A, Pyun HJ, Coates RM. J Org Chem. 1992;57:3444–3449. [Google Scholar]
- 16.(a) Prashad M. J Med Chem. 1993;36:631–632. doi: 10.1021/jm00057a013. [DOI] [PubMed] [Google Scholar]; (b) Prashad M, Kathawala FG, Scallen T. J Med Chem. 1993;36:1501–1504. doi: 10.1021/jm00062a026. [DOI] [PubMed] [Google Scholar]
- 17.Oehlschlager AC, Singh SM, Sharma S. J Org Chem. 1991;56:3856–3861. [Google Scholar]
- 18.(a) Pandit J, Danley DER, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ., Jr J Biol Chem. 2000;275:30610–30617. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]; (b) Thompson JF, Danley DE, Mazzalupo S, Miloa PM, Lira ME, Harwood HJ., Jr Arch Biochem Biophys. 1998;350:283–290. doi: 10.1006/abbi.1997.0502. [DOI] [PubMed] [Google Scholar]; (c) Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AHJ, Oldfield E. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Cane DE, Yang G, Coates RM, Pyun HJ, Hohn TM. J Org Chem. 1992;57:3454–3462. [Google Scholar]; (b) Sagami H, Korenga T, Ogura K, Steiger A, Pyun HJ, Coates RM. Arch Biochem Biophys. 1992;297:314–320. doi: 10.1016/0003-9861(92)90678-p. [DOI] [PubMed] [Google Scholar]; (c) McGeady P, Pyun HJ, Coates RM, Croteau R. Arch Biochem Biophy. 1992;299:63–72. doi: 10.1016/0003-9861(92)90244-q. [DOI] [PubMed] [Google Scholar]; (d) Peters RJ, Ravn MM, Coates RM, Croteau RB. J Am Chem Soc. 2001;123:8974–8978. doi: 10.1021/ja010670k. [DOI] [PubMed] [Google Scholar]; (e) Szabo CM, Matsumura Y, Fukura S, Martin MB, Sanders JM, Sengupta S, Cieslak JA, Loftus TC, Lea CR, Lee HJ, Koohang A, Coates RM, Sagami H, Oldfield E. J Med Chem. 2002;45:2185–2196. doi: 10.1021/jm010412y. [DOI] [PubMed] [Google Scholar]; (f) Ravn MM, Peters RJ, Coates RM, Croteau R. J Am Chem Soc. 2002;124:6998–7006. doi: 10.1021/ja017734b. [DOI] [PubMed] [Google Scholar]; (g) Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Proc Natl Acad Sci USA. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koohang A, Coates RM, Owen D, Poulter CD. J Org Chem. 1999;64:6–7. doi: 10.1021/jo981833z. [DOI] [PubMed] [Google Scholar]
- 21.(a) Atkinson RS, Kelly BJ. J Chem Soc, Chem Commun. 1988:624–625. [Google Scholar]; (b) Atkinson RS, Grimshire MJ, Kelly BJ. Tetrahedron. 1989;45:2875–2886. [Google Scholar]
- 22.Koohang A, Stanchina CL, Coates RM. Tetrahedron. 1999;55:9669–9686. [Google Scholar]
- 23.Davis CE, Bailey JL, Lockner JW, Coates RM. J Org Chem. 2003;68:75–82. doi: 10.1021/jo026506c. [DOI] [PubMed] [Google Scholar]
- 24.(a) Gao Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless KB. J Am Chem Soc. 1987;109:5765–5780. [Google Scholar]; (b) Katsuki T, Martin VS. Org React. 1996;48:1–299. [Google Scholar]
- 25.Benedetti F, Berti F, Norbedo S. Tetrahedron Lett. 1998;39:7971–7974. [Google Scholar]
- 26.Leopold EJ. Org Synth. 1986;64:164–174. [Google Scholar]; Organic Synthesis. VII. Wiley; New York: 1990. pp. 258–263. [Google Scholar]
- 27.Marshall JA, Dehoff BS. Tetrahedron. 1987;43:4849–4860. [Google Scholar]
- 28.(a) Heathcock CH, Piettre S, Ruggeri RB, Ragan JA, Kath JC. J Org Chem. 1992;57:2554–2556. [Google Scholar]; (b) Heathcock CH, Hansen MM, Ruggeri RB, Kath JC. J Org Chem. 1992;57:2544–2453. [Google Scholar]; (c) Heathcock CH, Piettre S, Kath JC. Pure Appl Chem. 1990;62:1911–1920. [Google Scholar]
- 29.Claisen L, Eisleb O, Kremers F. Liebigs Ann Chem. 1919;418:96–120. [Google Scholar]
- 30.Marshall JA, Andersen NH, Hochstetler AR. J Org Chem. 1967;32:113–118. [Google Scholar]
- 31.Corey EJ, Venkateswarlu A. J Am Chem Soc. 1972;92:6190–6191. [Google Scholar]
- 32.Brown HC, Tsukamoto A. J Am Chem Soc. 1961;83:4549–4552. [Google Scholar]
- 33.Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 3. Wiley; New York: 1999. p. 710. [Google Scholar]
- 34.(a) Davisson VJ, Woodside AB, Poulter CD. Methods Enzymol. 1984;110:130–144. doi: 10.1016/s0076-6879(85)10068-6. [DOI] [PubMed] [Google Scholar]; (b) Woodside AB, Huang Z, Poulter CD. Org Synth. 1988;66:211–219. [Google Scholar]; Organic Synthesis. VIII. Wiley; New York: 1993. pp. 616–620. [Google Scholar]; (c) Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, Poulter CD. J Org Chem. 1986;51:4768–4779. [Google Scholar]; (d) Davisson VJ, Davis DR, Dixit VM, Poulter CD. J Org Chem. 1987;52:1794–1801. [Google Scholar]
- 35.(a) Lacourciere GM, Vakharia VN, Tan CP, Morris DI, Edwards GH, Moos M, Armstrong RN. Biochemistry. 1993;32:2610–2616. doi: 10.1021/bi00061a019. [DOI] [PubMed] [Google Scholar]; (b) Tomasz M, Lipman R. J Am Chem Soc. 1979;101:6063–6067. [Google Scholar]; (c) Beijnen JH, Van der Houwen OAGJ, Rosing H, Underberg WJM. Chem Pharm Bull. 1986;34:2900–2913. doi: 10.1248/cpb.34.2900. [DOI] [PubMed] [Google Scholar]
- 36.(a) Earley JE, O’Rourke CE, Clapp LB, Edwards JO, Lawes B. J Am Chem Soc. 1958;80:3458–3462. [Google Scholar]; (b) Schatz VB, Clapp LB. J Am Chem Soc. 1955;77:5113–5116. [Google Scholar]
- 37.Zhang D, Jennings SM, Robinson GW, Poulter CD. Arch Biochem Biophys. 1993;304:133–143. doi: 10.1006/abbi.1993.1331. [DOI] [PubMed] [Google Scholar]
- 38.Radisky ES, Poulter CD. Biochemistry. 2000;39:1748–1760. doi: 10.1021/bi9915014. [DOI] [PubMed] [Google Scholar]
- 39.(a) Blackburn GB, England DA, Kolkmann F. Chem Commun. 1981:930–931. [Google Scholar]; (b) Stremler KE, Poulter CD. J Am Chem Soc. 1987;109:5542–5544. [Google Scholar]
- 40.(a) O’Rourke CE, Clapp LB, Edwards JO. J Am Chem Soc. 1956;78:2159–2160. [Google Scholar]; (b) Searles S, Tamers M, Block F, Quarterman LA. J Am Chem Soc. 1956;78:4917–4920. [Google Scholar]
- 41.(a) Giner JL, Djerassi C. J Am Chem Soc. 1991;113:1386–1393. [Google Scholar]; (b) Fujimoto Y, Morisaki M, Ikekawa N, Horie Y, Nakasone S. Steroids. 1974;24:367–375. doi: 10.1016/0039-128x(74)90034-8. [DOI] [PubMed] [Google Scholar]; (c) Nes WD, Guo D-a, Zhou W. Arch Biochem Biophys. 1997;342:68–81. doi: 10.1006/abbi.1997.9984. [DOI] [PubMed] [Google Scholar]; (d) Janssen GG, Nes WD. J Biol Chem. 1992;267:25856–25863. [PubMed] [Google Scholar]; (e) Pinto WJ, Nes WR. J Biol Chem. 1983;258:44724476. [PubMed] [Google Scholar]
- 42.(a) Roy A, Roberts FG, Wilderman PR, Zhou K, Peters RJ, Coates RM. J Am Chem Soc. 2007;129:12453–12460. doi: 10.1021/ja072447e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu H, Faraldos JA, Coates RM. J Am Chem Soc. 2009;131:11998–12006. doi: 10.1021/ja9044136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christianson DW. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 44.Kigoshi H, Ojika M, Shizuri Y, Niwa H, Yamada K. Tetrahedron. 1986;42:3789–3792. [Google Scholar]
- 45.Fujii N, Nakai K, Habashita Y, Hotta Y, Tamamura H, Otaka A, Ibuka T. Chem Pharm Bull. 1994;42:2241–2250. [Google Scholar]
- 46.Crossland RK, Servis KL. J Org Chem. 1970;35:3195–3196. [Google Scholar]
- 47.Forestier MA, Ayi AI, Condom R, Boyde BP, Conlin JN, Selway J, Challand R, Guedj R. Nucleosides Nucleotides. 1993;12:915–924. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.